
Abstract
We identified a PSEN1 (presenilin 1) mutation carrier from the world’s largest autosomal dominant Alzheimer’s disease kindred, who did not develop mild cognitive impairment until her seventies, three decades after the expected age of clinical onset. The individual had two copies of the APOE3 Christchurch (R136S) mutation, unusually high brain amyloid levels and limited tau and neurodegenerative measurements. Our findings have implications for the role of APOE in the pathogenesis, treatment and prevention of Alzheimer’s disease.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others



Data availability
Anonymized clinical, genetic, and imaging data are available upon request, subject to an internal review by J.F.A.-V., Y.T.Q., E.M.R., and F.L. to ensure that the participants’ anonymity, confidentiality, and PSEN1 E280A carrier or non-carrier status are protected, completion of a data sharing agreement, and in accordance with the University of Antioquia’s and Massachusetts General Hospital’s institutional review board and institutional guidelines. Experimental data are available upon request, subject to Massachusetts General Hospital and Schepens Eye Research Institute of Mass Eye and Ear institutional guidelines. Material requests and data requests will be considered based on a proposal review, completion of a material transfer agreement and/or a data use agreement, and in accordance with the Massachusetts General Hospital and Schepens Eye Research Institute of Mass Eye and Ear institutional guidelines. Please submit requests for participant-related clinical, genetic, and imaging data and samples to Y.T.Q. (yquiroz@mgh.harvard.edu) and requests for experimental data, DNA and single-cell RNA sequencing data, and antibodies to J.F.A.-V. (joseph_arboleda@meei.harvard.edu).
References
Quiroz, Y. T. et al. JAMA Neurol. 75, 548–556 (2018).
Llado, A. et al. Eur. J. Neurol. 17, 994–996 (2010).
Thordardottir, S. et al. Alzheimers Res. Ther. 10, 45 (2018).
Knight, W. D. et al. Alzheimer Dis. Assoc. Disord. 23, 410–414 (2009).
Velez, J. I. et al. Mol. Psychiatry 21, 916–924 (2016).
Acosta-Baena, N. et al. Lancet Neurol. 10, 213–220 (2011).
Wardell, M. R., Brennan, S. O., Janus, E. D., Fraser, R. & Carrell, R. W. J. Clin. Invest. 80, 483–490 (1987).
Smedley, D. et al. Am. J. Hum. Genet. 99, 595–606 (2016).
Lalli, M. A. et al. Mol. Psychiatry 20, 1294–1300 (2015).
Corder, E. H. et al. Nat. Genet. 7, 180–184 (1994).
Mahley, R. W., Huang, Y. & Rall, S. C. Jr. J. Lipid Res. 40, 1933–1949 (1999).
Preische, O. et al. Nat. Med. 25, 277–283 (2019).
Hashimoto, T. et al. J. Neurosci. 32, 15181–15192 (2012).
Lalazar, A. et al. J. Biol. Chem. 263, 3542–3545 (1988).
Rauch, J. N. et al. Sci. Rep. 8, 6382 (2018).
Yamauchi, Y. et al. Biochemistry 47, 6702–6710 (2008).
Aguirre-Acevedo, D. C. et al. Validez y. fiabilidad de. la batería neuropsicológica CERAD-Col. 45, 655–660 (2007).
Fisher, S. et al. Genome Biol. 12, R1 (2011).
Zhong, L. et al. Mol. Neurodegener. 11, 2 (2016).
Fleisher, A. S. et al. JAMA Neurol. 72, 316–324 (2015).
Becker, J. A. et al. Ann. Neurol. 69, 1032–1042 (2011).
Braak, H. & Braak, E. Neurobiol. Aging 18, S85–S88 (1997).
Braak, H., Rüb, U., Schultz, C. & Tredici, K. D. J. Alzheimers Dis. 9, 35–44 (2006).
Johnson, K. A. et al. Ann. Neurol. 79, 110–119 (2016).
Shoup, T. M. et al. J. Labelled Comp. Radiopharm. 56, 736–740 (2013).
Chien, D. T. et al. J. Alzheimers Dis. 34, 457–468 (2014).
Wang, L. et al. JAMA Neurol. 73, 1070–1077 (2016).
Logan, J. Nucl. Med. Biol. 27, 661–27 (2000).
Mormino, E. C. et al. JAMA Neurol. 71, 1379–1385 (2014).
Hedden, T. et al. J. Neurosci. 29, 12686–12694 (2009).
Gisslén, M. et al. EBioMedicine 3, 135–140 (2015).
Hashimoto, T. et al. J. Neurosci. 32, 15181–15192 (2012).
Walsh, D. M. et al. Nature 416, 535–539 (2002).
Hudry, E. et al. Sci. Transl. Med. 5, 12ra161 (2013).
Futamura, M. et al. J. Biol. Chem. 280, 5414–5422 (2005).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Nat. Biotechnol. 36, 411–420 (2018).
Acknowledgements
The authors thank the Colombian families with ADAD for making this work possible. This study was supported by US National Institutes of Health (NIH) Office of the Director grant DP5 OD019833 and US National Institute on Aging grant R01 AG054671, Claflin Distinguished Scholar Award from the Massachusetts General Hospital Executive Committee on Research, Physician/Scientist Development Award from the Massachusetts General Hospital, and Alzheimer’s Association Research Grant to Y.T.Q.; US National Institute of Neurological Disorders and Stroke and National Institute on Aging co-funded grants UH3 NS100121 and RF1 NS110048 to J.F.A.-V.; Grimshaw-Gudewicz Charitable Foundation grant to J.F.A.-V., J.B.M., and L.A.K.; Banner Alzheimer’s Foundation and Nomis Foundation grants to E.M.R. and P.N.T.; Anonymous Foundation grant to E.M.R.; US National Institute on Aging grants R01 AG031581 and P30 AG19610 to E.M.R.; US National Institute on Aging grant RF01 AG057519 to G.R.J.; and a State of Arizona grant to E.M.R. We also thank A. Koutoulas for technical support with DNA sequencing, B. Hyman from Massachusetts General Hospital for providing expression plasmids for the split-luciferase complementation assay, and Y. Alekseyev, A. LeClerc, M.J. Mistretta, J. Horvath, and J. Campbell from the Boston University Department of Medicine Single Cell Sequencing Core and Boston University Microarray and Sequencing Resource Core Facilities for reagents and technical support for single-cell RNA sequencing.
Author information
Authors and Affiliations
Contributions
J.F.A.-V., E.M.R., F.L., and Y.T.Q. initiated this work, supervised the study, and drafted the manuscript. A.B., S.R.-R., D.A., M.G., E.G.-V., D.N., E.P.-D., A.A., L.A.K., and J.B.M. collected and analyzed phenotypic data. M.J.H., M.N., R.A.R., G.R.J., K.S.K., J.A.-U., M.L., X.G., M.B., J.J., K.L.S.-T., L.S., and S.D.-T. collected and analyzed genetic data. N.C. and D.L.-C. conducted and analyzed molecular and genetic studies. K.C., Y.C., P.N.T., J.L., Y.S., P.T., R.A.S., A.P.S., K.A.J., and J.S.S. analyzed and interpreted imaging data. M.O. and C.M. contributed to the biochemistry experiments and data analysis, and finalized the manuscript. H.Z. and K.B. conducted and analyzed blood biomarker data.
Corresponding authors
Ethics declarations
Competing interests
P.N.T. has received personal compensation for consulting, serving on scientific advisory boards, and other activities from AbbVie, AC Immune, Acadia, Auspex, Boehringer Ingelheim, Chase Pharmaceuticals, Corium, Eisai, GliaCure, INSYS Therapeutics, Pfizer, T3D, AstraZeneca, Avanir, Biogen, Eli Lilly, H. Lundbeck A/S, Merck and Company and Roche; holds a provisional patent on ‘Biomarkers of Alzheimer’s Disease’ at the University of Rochester; holds stock options in Adamas; received research support from AstraZeneca, Avanir, Biogen, Eli Lilly, H. Lundbeck A/S, Merck and Company, Roche, Amgen, Avid, Functional Neuromodulation, GE Healthcare, Genentech, Novartis, Takeda, Targacept, the National Institute on Aging and the Arizona Department of Health Services. H.Z. has served on scientific advisory boards for Roche Diagnostics, Wave, Samumed, and CogRx, has given lectures in symposia sponsored by Biogen and Alzecure and is a co-founder of Brain Biomarker Solutions, a GU Ventures-based platform company at the University of Gothenburg (outside submitted work). R.A.S. has received personal compensation from AC Immune, Eisai, Roche, and Takeda, and research grant support from Eli Lilly and Janssen. All other authors have no competing interests.
Additional information
Peer review information Brett Benedetti was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
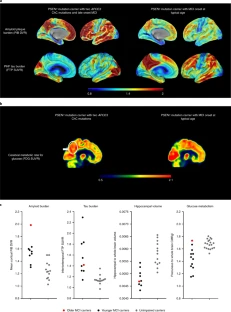
Extended Data Fig. 1 Subject’s genealogy.
Circles represent females, squares represent males, diamonds represent individuals whose gender has been masked for privacy, arrowhead depicts proband individual with MCI and shading indicates individual with history of dementia. Deceased individuals are marked with a crossed bar. APOE genotypes are indicated as appropriate to preserve anonymity. Family links were verified by three family informants. Genotypes of relatives of the R136S homozygote individual were determined by Sanger sequencing (Extended Data Fig. 2).
Extended Data Fig. 2 Sanger DNA sequencing of homozygous APOEch carrier individual.
Representative direct sequencing results of amplicon in APOE gene from control, proband, and descendants’ samples. R136 homozygous sequence is shown in upper panel from a control individual. In the middle panel, proband case is shown. R136S homozygous mutation can be observed, resulting from a change from cytosine (C) to adenine (A). In bottom panel a R136S heterozygous mutation from a descendant is identified. Data representative of n = 2 independent experiments and further validated independently with whole exome and whole genome sequencing.
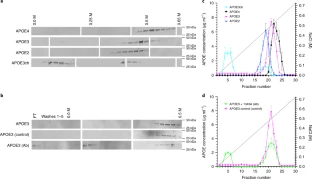
Extended Data Fig. 3 APOE3ch modulates Aβ aggregation.
(a) Rate of Aβ42 fibril formation in the presence of C-terminus fragments of APOE3 wild-type (WT), APOE3ch, or in the absence of APOE as detected by Thioflavin T fluorescence. Changes in relative fluorescence units (RFU) were plotted for time in minutes (min). Aβ42 fibrillation rate was lower in the presence of APOE3ch compared to APOE3 WT (****p=0.00000797 for 55 μM Aβ42 + APOE3WT vs. 55 μM Aβ42 + APOE3ch; ****p=0.0000000042 for 55 μM Aβ42 vs. 55 μM Aβ42 + APOE3WT; ***p=0.00022 for 55 μM Aβ42 vs. 55 μM Aβ42 + APOE3ch, 2-way ANOVA followed by Tukey’s multiple comparisons test; n = 3 represended as mean ± SEM.). (b) Schematic representation of the split-luciferase complementation triggered by amyloid oligomerization in vitro. (c) Percentage of luminescence expressed as relative luminescence units (RLU) was obtained by split-luciferase complementation assay after 24 hours in culture media from 293 T cells transfected with full-length APOE3ch or APOE3 WT cDNA. Luciferase luminescence produced by oligomer formation is significantly reduced in APOE3ch compared to APOE3 WT, indicating lower aggregation. (*p=0.0202, 2-tailed unpaired T-test, n = 6). Representative data presented as individual values ± SEM of n = 3 independent experiments.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and CARE Checklist
Rights and permissions
About this article

Cite this article
Arboleda-Velasquez, J.F., Lopera, F., O’Hare, M. et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med 25, 1680–1683 (2019). https://doi.org/10.1038/s41591-019-0611-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-019-0611-3
This article is cited by
-
Resilience conferred by APOE-R136S: a defense bestowed by nature to combat Alzheimer’s disease
Signal Transduction and Targeted Therapy (2024)
-
Efficient prime editing in mouse brain, liver and heart with dual AAVs
Nature Biotechnology (2024)
-
Molecular and cellular mechanisms of selective vulnerability in neurodegenerative diseases
Nature Reviews Neuroscience (2024)
-
How CRISPR gene editing could help treat Alzheimer’s
Nature (2024)
-
Cell type-specific roles of APOE4 in Alzheimer disease
Nature Reviews Neuroscience (2024)
