
Abstract
Vertebrate organs require locally adapted blood vessels1,2. The gain of such organotypic vessel specializations is often deemed to be molecularly unrelated to the process of organ vascularization. Here, opposing this model, we reveal a molecular mechanism for brain-specific angiogenesis that operates under the control of Wnt7a/b ligands—well-known blood–brain barrier maturation signals3,4,5. The control mechanism relies on Wnt7a/b-dependent expression of Mmp25, which we find is enriched in brain endothelial cells. CRISPR–Cas9 mutagenesis in zebrafish reveals that this poorly characterized glycosylphosphatidylinositol-anchored matrix metalloproteinase is selectively required in endothelial tip cells to enable their initial migration across the pial basement membrane lining the brain surface. Mechanistically, Mmp25 confers brain invasive competence by cleaving meningeal fibroblast-derived collagen IV α5/6 chains within a short non-collagenous region of the central helical part of the heterotrimer. After genetic interference with the pial basement membrane composition, the Wnt–β-catenin-dependent organotypic control of brain angiogenesis is lost, resulting in properly patterned, yet blood–brain-barrier-defective cerebrovasculatures. We reveal an organ-specific angiogenesis mechanism, shed light on tip cell mechanistic angiodiversity and thereby illustrate how organs, by imposing local constraints on angiogenic tip cells, can select vessels matching their distinctive physiological requirements.
Similar content being viewed by others

Main
In vertebrates growing beyond the limit of oxygen diffusion, a vascular system branches through the body to supply nutrients and oxygen, remove cellular waste products, allow immune cell transport and support coagulation1,6. Contrasting with the apparent uniformity of their ancestral functions, blood vessels exhibit considerable phenotypic heterogeneity. In particular, the inner lining of blood vessels is made of highly malleable endothelial cells (ECs) that engage in a variety of two-way communications with local microenvironments1,2,7. By adopting organ-specific structural and molecular profiles, ECs have essential roles in organ development, metabolism, regeneration and repair. As angiogenesis is almost invariably initiated by an environmental trigger common to all expanding organs, that is, the shortage of oxygen, the cellular and molecular logic of blood vessel branching through sprouting angiogenesis and organotypic endothelial adaptations are generally viewed as distinct events, regulated by independent molecular machineries.
The central nervous system (CNS) offers a notable exception to this rule. From early developmental stages onwards, its functional complexity requires isolation and protection from peripheral chemical variations and toxins. Embryonic cerebral ECs therefore mature into a neuroprotective blood–brain barrier (BBB) phenotype, an archetypical example of EC specialization, that is initiated by endothelial Wnt–β-catenin signalling3,4,5. In response to Wnt ligands, CNS ECs reduce paracellular permeability through tight junctions, repress transcytosis, express numerous solute transporters such as GLUT1 and recruit pericytes. While, as in any other organ, brain angiogenesis is VEGF-dependent, the BBB-inductive Wnt–β-catenin signalling pathway is also required for brain angiogenesis, thereby making a direct, yet enigmatic, link between organ vascularization and organotypic endothelial specialization.
Through this coupling mechanism, no leaky blood vessels penetrate the delicate neuroepithelium, accounting for an efficient endothelial quality-control mechanism. However, how Wnt signalling impinges on the angiogenic cascade selectively in the brain and, thereby, more generally, how organs can ensure that their perfusing vessels match their specific metabolic requirements remain to be determined.
Brain-specific angiogenesis control
To investigate the organotypic control of brain angiogenesis by endothelial Wnt–β-catenin signalling, we recorded this process using time-lapse confocal microscopy in genetically mosaic zebrafish embryos. As in mice8,9,10, zebrafish brain vascularization is controlled by neural-derived Wnt7a/b ligands, recognized by the atypical Gpr124–Reck complex of perineural ECs11,12,13,14 (Extended Data Fig. 1a). Mid-blastula transplantation of wild-type (WT) Tg(kdrl:EGFP) cells into gpr124 morpholino (MO)-injected Tg(kdrl:ras-mCherry) hosts resulted in mosaic perineural primordial hindbrain channels (PHBCs) from which central artery (CtA) sprouts invariably invaded the brain with a WT cell at the tip cell (TC) position11 (Fig. 1a and Supplementary Video 1). By contrast, peripheral intersegmental vessels (ISVs) were led at equal frequencies by WT or gpr124-morphant TCs (Fig. 1a). Similarly, after the CNS-invasive step, gpr124− TCs guiding secondary intraneural vessels could readily be detected (Extended Data Fig. 1b and Supplementary Video 2). Wnt–β-catenin signalling therefore appears to be selectively required in perineural TCs during the initial event of brain invasion. Within this narrow spatiotemporal window, Wnt signalling could control either TC identity or TC behaviour.

a, The TC genotype in mosaic sprouts during brain vascular invasion (36 hpf, n = 23 sprouts) and trunk ISV formation (24 hpf, n = 43 ISVs) of embryos obtained by five transplantation experiments of WT kdrl:EGFP donor cells into gpr124 MO-injected kdrl:ras-mCherry hosts. b, In vivo photoconversion design of pre-angiogenic PHBCs. LDA, lateral dorsal aorta. c, t-Distributed stochastic neighbour embedding (t-SNE) analysis of PHBC EC clusters. d, t-SNE expression profiles of Wnt–β-catenin target genes and TC markers. Max., maximum; min., minimum. e, Time-lapse recordings of calcium oscillations in Tg(fli1a:Gal4FF);(UAS:GCaMP7a) PHBCs (31 to 31.5 hpf). f, Wnt-dependent transcripts in 30 hpf PHBCs or 36 hpf CtAs (β-catenin LOF, IWR-1 treatment). Grey labels below the heat map indicate conditions in which candidate genes are not statistically downregulated. μ, mean expression. g, Fluorescent mmp25b WISH and anti-EGFP staining of Tg(kdrl:EGFP) embryos. DA, dorsal aorta. h, Angiogenic sprouts (arrowheads) in the hindbrain and trunk region of Tg(kdrl:EGFP) embryos. n ≥ 10 embryos from 4 independent experiments. Data are median ± interquartile range. P values were calculated using nonparametric two-tailed Mann–Whitney U-tests. Scale bars, 400 μm (a (left) and b (left)), 100 μm (a (right), e and h), 50 μm (b (right)) and 20 μm (g).
To address this, we transcriptionally profiled fluorescence-activated cell sorting (FACS)-isolated 30 hours post-fertilization (30 hpf) PHBC WT or gpr124 MO ECs after in vivo photoconversion of Tg(fli1a:Gal4FF);(UAS:Kaede) transgenic embryos (Fig. 1b). Single-cell RNA-sequencing (scRNA-seq) analysis of 144 ECs from WT PHBCs revealed three major cell clusters (Fig. 1c,d and Extended Data Fig. 1c–e), namely two venous clusters (G2/M and G1/S cell cycle phases, defined by dab2, nr2f2, flt4 and ephb4a) and one arterial cluster (sox7, dll4, nrp1b, hey2 and efnb2a) containing a TC population (cxcr4a, angpt2a and esm1). The arterial cluster exhibited a BBB signature (Extended Data Fig. 1f–h).
The pre-angiogenic PHBCs contained Wnt-positive (lef1, nkd1 and zic2a) and Wnt-negative cells that both contributed to the venous and arterial cell clusters (Fig. 1d). This heterogeneity in Wnt signalling activity was confirmed by the analysis of zebrafish and mouse transgenic reporters (Extended Data Fig. 1i,j and Supplementary Video 3). TC and Wnt marker gene expression did not correlate across the PHBC EC population (Extended Data Fig. 2a). Wnt-independent specification of perineural TCs was confirmed by implementing a similar scRNA-seq approach in gpr124 morphants. While, as anticipated, Wnt–β-catenin marker gene expression was strongly reduced in gpr124 morphants, TC markers were maintained (Extended Data Fig. 2b).
Wnt–β-catenin signalling has been reported to upregulate Vegfr2 transcripts in the postnatal retinal and brain vasculature15. At brain invasion stages, the expression levels of kdrl, the main functional homologue of mammalian VEGFR2 in zebrafish, correlated partially with Wnt signalling activity (Extended Data Fig. 2c,d). However, kdrl expression did not rely on Wnt activity, as it was maintained in gpr124 morphants (Extended Data Fig. 2b,e). Moreover, the formation of the PHBC-derived basilar artery, a kdrl-dependent process, was unaffected in gpr124 mutants (Extended Data Fig. 2f), implying that Wnt-deficient PHBCs remain responsive to VEGF. Furthermore, transgenic endothelial overexpression of kdrl did not rescue gpr124−/− phenotypes, whereas it partially corrected the control kdrl−/− phenotypes (Extended Data Fig. 2g). Finally, in contrast to kdrl−/− vessels, VEGF-induced cytosolic calcium oscillations16 were unaffected in gpr124−/− Tg(fli1a:Gal4FF);(UAS:GCaMP7a) PHBCs (Fig. 1e, Extended Data Fig. 2h and Supplementary Video 4). Together, these data suggest that the Wnt-dependent brain-specific angiogenic defects are explained neither by defective TC specification nor by impaired VEGF signalling.
TC angiodiversity
To identify the angiogenic effectors of Wnt signalling, we performed bulk RNA-seq analysis of 30 hpf laser-photoconverted FACS-isolated PHBC ECs in WT as well as gpr124-, reck- and wnt7aa-morphant embryos, three Wnt–β-catenin loss of function (LOF) conditions (Extended Data Fig. 3a) that result in avascular brains11,12,17. We also analysed the effect of Wnt inhibition at later stages (36 hpf) by exposing embryos to IWR-1 (β-catenin LOF) from 26 hpf onwards (Extended Data Fig. 3b). This strategy identified 40 genes of which the expression was significantly downregulated in at least three Wnt LOF conditions (Fig. 1f and Extended Data Fig. 3c). Among these candidate genes, known Wnt-target genes were found (lef1, fabp11a, slc16a1a, zic2a and zic2b), validating the approach. Five genes were downregulated in all four conditions, including mmp25b, one of the two zebrafish mmp25 paralogues (Fig. 1f and Extended Data Fig. 3d). Low-expressed mmp25a was also downregulated in two Wnt-LOF conditions (Extended Data Fig. 3d). Both paralogues were enriched in the arterial TC cluster (Extended Data Fig. 3e) and correlated with Wnt activity markers (Extended Data Fig. 3f).
Besides in trigeminal, craniofacial and posterior lateral line ganglia18, mmp25b transcripts were detected by chromogenic whole-mount in situ hybridization (WISH) in scattered cells of the 36 hpf hindbrain anatomically compatible with PHBCs and CtA TCs (Extended Data Fig. 3g). The signal in the presumptive ECs was gpr124 dependent, whereas the signal in sensory ganglia was not (Extended Data Fig. 3g). Combined fluorescence WISH and immunofluorescence staining confirmed the EC- and gpr124-dependent nature of the signal (Extended Data Fig. 3h). Notably, mmp25b transcripts were detected in hindbrain TCs but were absent in ISV TCs (Fig. 1g). In the hindbrain, mmp25b expression was higher in TCs compared with in stalk cells (SCs) and PHBC phalanx cells (Extended Data Fig. 3i). Consistent with a potential role in brain vascular invasion, mmp25b expression peaked in 36 hpf CtAs, decreasing thereafter in PHBC and CtAs (Extended Data Fig. 3j).
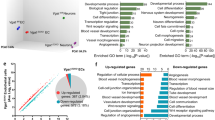
Also known as leukolysin19,20,21,22,23, the glycosylphosphatidylinositol (GPI)-anchored Mmp25 has not been linked to angiogenesis or Wnt signalling to date. To assess Mmp25 function, we generated mmp25a and mmp25b frameshift alleles in zebrafish using CRISPR–Cas9 (Extended Data Fig. 4a,b). Brain angiogenesis was strongly, although not completely, impaired in double homozygous mmp25a−/−mmp25b−/− (mmp25a/b−/−) mutants (Fig. 1h, Extended Data Fig. 4c and Supplementary Video 5). By contrast, peripheral angiogenesis remained unaffected (Fig. 1h, Extended Data Fig. 4c–i and Supplementary Video 6). No other morphological phenotypes were detected in mmp25a/b mutants. Gene-dosage experiments revealed that mmp25 paralogues exhibit partially redundant functions, with mmp25b−/− phenotypes aggravated by the gradual loss of WT mmp25a alleles (Extended Data Fig. 5a–c). By contrast, mmp25a−/− and mmp25a−/−b+/− embryos exhibited no vascular defects, the latter of which were used as controls in subsequent experiments. Genetic inactivation of mmp25a/b did not affect VEGF-induced calcium oscillations or Wnt–β-catenin signalling in PHBCs (Extended Data Fig. 5d,e and Supplementary Video 7). In genetically mosaic embryos, mmp25a/b-mutant cells were outcompeted by WT cells for the TC position selectively in brain-invading sprouts, but not in trunk ISVs or during secondary sprouting events within the brain (Extended Data Fig. 5f,g and Supplementary Video 8). This TC autonomous requirement is reminiscent of the overall function of Wnt signalling in CNS angiogenesis, and compatible with the membrane retention of Mmp25 through a GPI anchor. Mining published datasets revealed an evolutionarily conserved expression pattern in mice, with Mmp25 qualifying as an endothelial-enriched, brain-specific and Wnt-dependent transcript (Extended Data Fig. 6a–j). MMP25 also contributed to brain vascularization in mice, as constitutive knockouts23 exhibited a partial reduction in angiogenic sprouts in the embryonic day 10.5 (E10.5) midbrain and forebrain (Extended Data Fig. 6k). As in zebrafish, the mouse Mmp25 vascular phenotypes were CNS-specific, with no defects detected in peripheral organs (Extended Data Fig. 6l–p).
The distinctive MMP repertoire of brain TCs
Vertebrate genomes encode numerous MMPs, some of which are expressed by ECs. Mmp14 (also known as MT1-MMP) in particular is a well-known TC-enriched angiogenic effector24,25,26,27 that, after TIMP2-dependent activation of Mmp2, amplifies the proteolytic activity of migrating cells24,25,28,29,30. While peripheral TCs exhibited the anticipated MMP repertoire (mmp14b+mmp2+), brain TCs lacked mmp2 transcripts, expressing mmp25b instead (Fig. 2a–d). In comparison to mmp25b, the expression of mmp14b appeared uniform across PHBC ECs (Fig. 2a) and was Wnt–β-catenin independent (Fig. 2b). mmp9 was expressed at low levels in ECs.

a, t-SNE expression profiles of mmp and furin genes. b, Expression levels of mmp genes in WT and gpr124-MO PHBC ECs. Dots represent individual cells. nUMI, normalized unique molecular identifier. P values were calculated using parametric two-tailed Student’s t-tests. c,d, Fluorescent mmp25b, mmp14b, mmp2 or mmp9 WISH and anti-EGFP staining of Tg(kdrl:EGFP) embryos in the hindbrain (c) and trunk region (d). The solid and open arrowheads label mmp-positive and -negative sprouts, respectively. e, CtA sprouts in 38 hpf Tg(kdrl:EGFP) embryos (n ≥ 14 embryos from ≥3 independent experiments), injected at the one-cell stage with 200 pg of mmp25b mRNA or its variants (left). BD, binding domain; H-A, H237A, H241A, H247A. The diagram was created using BioRender. f, Anti-laminin-111 immunofluorescence staining of transverse hindbrain sections, counterstained with DAPI. OV, otic vesicle. g, Hindbrain CtAs in Tg(kdrl:EGFP) embryos (n ≥ 20 embryos from ≥3 independent experiments) injected with control or laminin MOs. In e and g, data are median ± interquartile range. P values were calculated using nonparametric Kruskal–Wallis tests. Scale bars, 100 μm (f (left)), 50 μm (c and d) and 10 μm (f (right)).
Reflecting these distinct expression profiles, brain angiogenesis was largely unaffected by mmp2 or mmp14b genetic inactivation, while robustly reduced in mmp25b or mmp25a/b crispants (Extended Data Fig. 7a,b). However, targeting mmp14b led to a slight but non-significant reduction in CtAs, which was unaffected by the additional mutagenesis of mmp2. The combined deletion of mmp25b and mmp14b modestly aggravated the mmp25b phenotypes, suggesting that mmp14b marginally contributes to brain angiogenesis in an mmp2-independent manner. In peripheral ISVs, mmp2 and mmp14b contributed to angiogenic sprouting, while mmp25a and mmp25b were dispensable (Extended Data Fig. 7c,d). The combined deletion of mmp2 and mmp14b did not exacerbate the individual phenotypes, which is compatible with their function in an Mmp2–Mmp14 complex. In summary, the transcriptional and functional MMP repertoire of TCs differed between CtAs and ISVs.
To determine whether shifting the brain TC repertoire (mmp25+mmp14+) to a peripheral one (mmp2+mmp14+) is compatible with brain angiogenesis, we injected mRNA encoding mmp25b, mmp9, mmp2 or variants thereof into mmp25a/b−/− one-cell stage embryos. While the restoration of mmp25b expression partially rescued mmp25a/b−/− phenotypes, mmp2 or mmp9 did not (Extended Data Fig. 7e). Mmp2, Mmp9, and Mmp25, produced as zymogens, differ in their activation mode. While Mmp2 and Mmp9 activation occurs extracellularly by various proteases, including Mmp1428,29, Mmp25 activation occurs within the secretory pathway, after processing by furin-like proprotein convertases19,20,21. Notably, furina is highly expressed in the PHBC TC cluster, possibly accounting for a robust activation of Mmp25 (Fig. 2a). Moreover, in contrast to the secreted Mmp2 and Mmp9, Mmp25 is retained at the cell surface through a GPI anchor, which may help to concentrate the proteolytic activity31,32. Assessing the brain angiogenic potential of mmp2 mRNA variants encoding a constitutively active form of the enzyme (without the prodomain (Pro−)), a GPI-anchored version (GPI+) or both together revealed that only a variant exhibiting the combination of these properties (Pro−GPI+) was competent for brain angiogenesis (Extended Data Fig. 7e).
The pial basement membrane obstacle
Brain angiogenesis required an EC-autonomous catalytically active form of Mmp25, as revealed by mRNA (Fig. 2e) and transgenic endothelial (fli1a promoter) overexpression rescue experiments (Extended Data Fig. 7f). As Wnt activity, and therefore Mmp25, are selectively required during the initial step of brain vascular invasion, we reasoned that its substrate might reside within the pial (or glia limitans) basement membrane (pBM) enwrapping the developing neuroepithelium. The pBM indeed constitutes a physical barrier that prevents radial overmigration of neurons and glial cells into the meninges33,34,35,36,37. Reciprocally, it could therefore represent an obstacle for vascular ingression into the CNS.
Laminin-111 is a well-established structural component of the pBM34,37,38,39,40. Other laminins are found in distinct BMs, including laminin-411 and laminin-511 around blood vessels39. In 30 hpf zebrafish embryos and E10.5 mice, anti-laminin-111 antibodies decorated the external surface of the developing brain (Fig. 2f and Extended Data Fig. 7g), thereby defining a landmark interface through which perineural endothelial TCs must navigate, a function that is seemingly defective in gpr124- and mmp25a/b-mutant zebrafish (Fig. 2f).
MO- and CRISPR–Cas9-based genetic invalidation of lama1, lamb1a or lamc1, the three constitutive chains of laminin-111, partially rescued brain angiogenesis in mmp25a/b−/− embryos, whereas interfering with lama2, lama4 or lama5 had no effect (Fig. 2g and Extended Data Figs. 7h–l and 8a,b). These observations suggest that weakening the pBM alleviates the need for Mmp25 in brain angiogenesis. The effect of laminin-111 inactivation is probably indirect, through a general destabilization of the pBM, as recombinant catalytic domains of zebrafish and human MMP25 (rzMmp25b and rhMMP25, respectively, Extended Data Fig. 8c) did not cleave laminin-111 in vitro (Extended Data Fig. 8d), as previously reported22. Notably, the activity of the recombinant enzymes was validated on α-1 antitrypsin (Extended Data Fig. 8e), a known substrate of which the genetic inactivation did not affect mmp25 phenotypes (Extended Data Fig. 8a,b,f,g).
Mmp25-substrate identification
To identify the physiologically relevant Mmp25 substrate within the poorly characterized pBM, we transcriptionally characterized the meningeal fibroblasts, the main suppliers of pBM proteins41,42. We first analysed the anatomical relationships between the zebrafish pBM (anti-laminin-111), the meningeal fibroblasts (fluorescence in situ hybridization (FISH) analysis of lama1), a cell population that is to date uncharacterized in this organism, and the kdrl:EGFP+ ECs (Fig. 3a,b). The laminin-111-positive pBM was evident from 18 hpf onwards (Fig. 3a and Extended Data Fig. 9a), with the nearest lama1 signal associated with a ventrolateral population of perineural cells (Fig. 3b and Extended Data Fig. 9b). At 18 hpf, these cells probably represent primary meninx cells, meningeal fibroblast precursors. Between 24 and 30 hpf, the lama1+ cells flattened out on the ventral surface of the hindbrain, with the equatorial plane of their oblong nuclei orienting parallel to the pBM (Fig. 3a,b). Transmission electron microscopy revealed a typical elongated fibroblast cell morphology (Fig. 3c). These cells resemble pial fibroblasts, although we prefer to use the broader meningeal fibroblast terminology, as the molecular diversity of the zebrafish meningeal cell populations remains to be investigated.

a,b, Laminin-111 immunostaining (a) and lama1 FISH analysis and EGFP immunostaining (b) of Tg(kdrl:EGFP) transverse zebrafish hindbrain sections, counterstained with DAPI. The arrowheads indicate meningeal fibroblast nuclei. c, Transmission electron micrograph showing a meningeal fibroblast next to the pBM (arrowhead). N, nucleus. d,e, FISH analysis as in b of col4a5 (d) and col4a6 (e) with EGFP immunostaining. f, Collagen IV and laminin-111 (anti-LAMA1) co-immunostaining on a E10.5 mouse forebrain and midbrain section, counterstained with isolectin B4 and DAPI. g,h, Dorsal views (g) and quantification (h) of Tg(kdrl:EGFP) hindbrain CtAs in embryos (n ≥ 11 embryos from 3 independent experiments), injected with the illustrated sgRNAs and zCas9 mRNA. i, Hindbrain CtAs in embryos (n ≥ 7 embryos from 4 independent experiments) crossed to the dragnet col4a5 allele. j, Anti-HA western blot analysis of zCol4a5–HA-containing HEK293T cell extracts (or control pCS2+ cells) that were treated or not with rzMmp25b or rhMMP25. k, Coomassie blue staining of human placental collagen IV exposed or not to rhMMP25. The solid arrowheads indicate parental fragments (black) and rhMMP25 cleavage products (red) analysed by MS. The open arrowheads indicate additional differences. l, Collagen IV and its most C-terminal non-tryptic peptide identified in rhMMP25-treated samples. NC, non-collagenous. m, Anti-GFP western blot analysis of recombinant GST–GFP fusion proteins (black arrowheads) and cleavage products (white arrowheads). GST and GFP are linked by a PreScission recognition site and the presumptive cleavage site of MMP25 in COL4A1–6 (a1 to a6). C−, GST alone. The diagram was created using BioRender. For h and i, data are median ± interquartile range. P values were calculated using nonparametric Kruskal–Wallis tests. Scale bars, 500 nm (c (bottom)), 200 μm (f (left)), 100 μm (a, b, d and e (top) and g), 50 μm (f (right)), 20 μm (a, b, d and e (bottom)) and 2 μm (c (top)).
Using a candidate-based approach, we identified that zebrafish meningeal fibroblasts expressed col4a5 and col4a6, specific chains of type IV collagen, in a spatiotemporal pattern very similar to lama1 (Fig. 3d,e and Extended Data Fig. 9c–e). Exploration of published mouse transcriptomes confirmed the expression of Col4a5 and Col4a6 by embryonic and adult pial fibroblasts40,42,43 (Extended Data Fig. 9f–j), together with Col4a1 and Col4a2, common components of EC basement membranes40,42,43,44 (Extended Data Fig. 9k). Immunostaining analysis confirmed the presence of type IV collagen in the laminin-111-positive E10.5 mouse pBM (Fig. 3f).
We next inactivated the different zebrafish col4 chains by MOs and somatic CRISPR–Cas9 mutagenesis in mmp25a/b−/− embryos. Selectively targeting col4a5 or col4a6 rescued the cerebrovascular defects partially at 36 hpf and nearly completely at 48 hpf (Fig. 3g,h and Extended Data Figs. 8a,b and 10a,b). This finding was confirmed through Mendelian genetics by crossing the mmp25-mutant alleles to the dragnet allele45, harbouring a premature stop codon in col4a5 (Fig. 3i). The inactivation of col4a5 therefore alleviates the need for Mmp25 in brain angiogenesis. Notably, brain angiogenesis in dragnet mutants proceeded as in WT siblings, despite an initial non-significant reduction in CtA sprouts at 36 hpf (Extended Data Fig. 10c,d).
Recombinant Mmp25 catalytic domains cleaved C-terminally HA-tagged zebrafish Col4a5 (zCol4a5–HA) in HEK293T lysates, yielding at least four distinct fragments including a major ~30 kDa band (Fig. 3j). This suggests that the central triple-helix domain of Col4a5 contains an Mmp25-cleavage site, thereby providing a link between the proteolytic activity of Mmp25 and its function in EC migration across the pBM.
To identify the cleavage site, we used a two-step strategy as purified Col4a5–Col4a6 heterotrimer is not commercially available. In step one, we exposed purified human placental collagen IV, which is mostly composed of COL4A1/A2, to rhMMP25. Untreated samples ran as multiple bands, probably a consequence of the harsh pepsin-based placental extraction procedure. rhMMP25 cleaved the main bands (Fig. 3k). The two larger parental bands and their presumptive cleavage products were analysed using mass spectrometry (MS), which revealed a semi-specific (K)GLPGPPGPPGPYD peptide in rhMMP25-treated collagen IV, containing a C-terminal non-tryptic aspartate (Fig. 3l). By contrast, the tryptic (K)GLPGPPGPPGPYDIIK fragment ending with a lysine residue was found in untreated collagen IV samples. MMPs often cleave proteins upstream of two small hydrophobic residues, consistent with the observed (Asp)-Ile-Ile cleavage site in human COL4A1, and the corresponding residues in Col4a1 and Col4a5 across vertebrates (Extended Data Fig. 10e). The peptide mapped to the last short non-collagenous region within the central helical part of the α-chain, upstream of the C-terminal globular domain, essential for collagen IV sheet formation (Fig. 3l). In step two, to confirm that Mmp25 can process Col4a5 or Col4a6 at this site, we expressed tripartite fusion constructs in Escherichia coli, consisting of an N-terminal glutathione S-transferase, a C-terminal GFP and a central linker containing the presumptive cleavage site of Col4 chains in tandem with the recognition site of the human rhinovirus-derived PreScission enzyme (used as a positive control). Recombinant hMMP25 processed all Col4-based fusions, while rhMMP2 did not, revealing qualitative differences in collagen IV cleavage by Mmp2 and Mmp25 (Fig. 3m). Such differences were confirmed on full-length zebrafish Col4a5, which was cleaved by both enzymes, but yielded proteolytic fragments of different sizes (Extended Data Fig. 10f).
Uncoupled angiogenesis and BBB formation
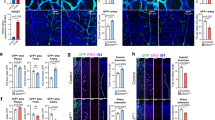
Thus far, we have identified that Wnt–β-catenin signalling, by regulating mmp25 expression, enables the migration of TCs across the Col4a5/6-positive pBM. As Wnt–β-catenin also controls BBB development, this mechanism ensures brain perfusion by vessels led by TCs of adequate properties. The coupling between brain angiogenesis and BBB formation therefore appears to rely on the integrity of the pBM. If true, this model implies that, after impairment of the pBM, Wnt–β-catenin would at least in part become dispensable for brain angiogenesis. Consistent with this prediction, brain angiogenesis was partially restored in gpr124 mutants after col4a5 and/or col4a6 inactivation (Fig. 4a and Extended Data Fig. 10g). Under these conditions, brain vessels remained Wnt–β-catenin negative (Fig. 4b and Extended Data Fig. 10h,i) and, accordingly, did not express BBB markers such as slc2a1a, slc16a1a or fabp11a (Fig. 4c and Extended Data Fig. 10j,k). The vessels lumenized properly (Fig. 4d and Extended Data Fig. 10l), conducted gata1+ red blood cells (Fig. 4e), assembled vasculatures notably similar to WT networks (Fig. 4e), but remained leaky to intracardially injected tracers (Fig. 4f). Tracer accumulation did not result from leakage through the impaired pBM, as it was not observed in the absence of cerebral vessels (Extended Data Fig. 10m). Together, these observations reveal that, after pBM impairment, the organotypic requirement of Wnt signalling for brain vascularization is lost, resulting in properly patterned, yet BBB-defective, cerebrovasculatures. A last prediction from the pBM-mediated quality control on TCs is that not only gpr124−/− cells would gain undue access to the CNS after pBM impairment, but also the naturally occurring Wnt-negative TCs populating the PHBC (Fig. 1d and Extended Data Fig. 2a,b). Consistent with this prediction, Wnt-negative TCs invaded the hindbrain of col4a5 and/or col4a6 morphants (Fig. 4g). The pBM therefore appears to control the genetic competence of the perineural vessels to differentiate into a neuroprotective BBB, by controlling the pioneering TCs. Either vessels are Wnt competent, displaying the right cohort of cell surface receptors, and Mmp25-expressing TCs will guide nascent sprouts into the CNS, or they are not, and they are excluded.

a, Hindbrain CtAs in 48 hpf Tg(kdrl:EGFP) embryos (n ≥ 15 embryos from ≥3 independent experiments). Data are median ± interquartile range. P values were calculated using nonparametric Kruskal–Wallis tests. b, The proportion of 7xTCF-Xla:GFP+ CtAs in Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) 48 hpf embryos. c, The proportion of slc2a1a-positive CtAs was analysed using fluorescent slc2a1a WISH and anti-EGFP immunostaining in Tg(kdrl:EGFP) embryos. d, The percentage of lumenized CtAs in 72 hpf larvae. For b–d, n = total number of CtAs from 3 (b and c) or 5 (d) independent experiments. e, Wire diagrams (top) and dorsal views (bottom) of 72 hpf Tg(gata1:DsRed);(kdrl:EGFP) larvae. f, FITC 150 kDa dextran fluorescence intensity 1 h after intracardial injection in larvae at 4 days post-fertilization (dpf). n ≥ 8 larvae from ≥4 independent experiments. g, The proportion of 7xTCF-Xla:GFP– CtA TCs in Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryos. The solid and open arrowheads label 7xTCF-Xla:GFP+ and 7xTCF-Xla:GFP– sprouts, respectively. n = 6 independent experiments with ≥6 embryos each. For f and g, data are mean ± s.d. P values were calculated using parametric one-way ANOVA. h, Model for brain-specific angiogenesis. The diagram was created using BioRender. Scale bars, 100 μm (a, b, f and g) and 50 μm (c and e).
Discussion
Vascular expansion through angiogenesis is a multicellular migration process that requires the coordinated behaviours of differentially fated TCs and SCs. TCs invariably guide the nascent angiogenic sprouts and therefore display common morphological and molecular adaptations required for tissue exploration. Beyond this apparent uniformity, TCs, in their task to pervade each vertebrate organ, must navigate through extracellular spaces of varying composition likely imposing local constraints on TC function. Here we provide an important example of such TC mechanistic angiodiversity by identifying Mmp25 as a brain-specific TC angiogenic effector.
An alluring aspect of this brain-specific TC machinery is that Wnt–β-catenin signalling, besides conferring organ invasive competence, also instructs endothelial tissue-specific adaptations. This constitutes an elegant quality-control mechanism ensuring that only BBB-differentiating TCs can guide vessels into the brain. However, neuroprotection implies a uniform activity in Wnt–β-catenin among all CNS ECs, not only TCs. How and if a quality-control mechanism operates at the level of naturally occurring Wnt-negative cells other than TCs remains to be investigated. Notably, studies in the postnatal mouse retina detected such a potential process, in which Wnt-signalling-deficient Frizzled4−/− ECs were gradually eliminated from genetically mosaic vessels46.
At the centre of the brain-specific angiogenic program lies the pial (or glia limitans) BM (Fig. 4h). This work extends the function of this critical interface between the brain and the periphery. To date, it has been implicated in proper cortical layering, by acting as a scaffold for radial glia endfeet and avoiding the inside-out overmigration of neurons and glia into the meninges, a condition known in humans as cobblestone lissencephaly34,36,37,47. We show that the pBM also constitutes an important gatekeeper of the brain by hindering the uncontrolled ingression of peripheral cells such as leaky ECs. In doing so, here we reveal a functional connection between two important brain barriers, that is, the BBB and the meningeal barrier to which the pia and its basement membrane belong. This study also illustrates the increasingly recognized importance of fibroblasts in brain barrier function, both within and at the surface of the brain42,43,48. Besides its angiogenic role identified in this study, Mmp25 is better known to be expressed by leukocytes, particularly neutrophils19,23. Whether Mmp25 facilitates immune cell entry across the glia limitans in pathological conditions like infections, trauma and haemorrhage seems worth pursuing in light of the results presented here.
Together, we reveal a mechanism by which endothelial TCs gain brain-specific invasive competence, thereby supporting the existence of specialized TC angiogenic mechanisms in distinct organs, including in the bone, liver, retina and solid tumours, where diverse TC morphologies or gene signatures are increasingly reported49,50. Given the essential role of TCs in guiding new vessels, such organotypic TC functional heterogeneity holds promise for tissue-specific pharmacological control of angiogenesis, at a level of specificity that is unachievable using the current anti-angiogenic strategies.
Methods
Zebrafish strains and husbandry
Zebrafish (Danio rerio) were maintained at 28 °C under a 14 h–10 h light–dark cycle and raised under standard conditions in a certified animal facility (LA1500474) in accordance with European and national ethical and animal welfare guidelines. All of the animal procedures were approved by the corresponding ethical committee (Commission d’Ethique et du Bien Être Animal (CEBEA), Université libre de Bruxelles, protocol approval numbers: CEBEA-IBMM-2016:65 and CEBEA-07 GOS IBMM). Zebrafish staging was performed as described previously51. The following published transgenic and mutant lines have been used in this study: Tg(kdrl:EGFP)s843 (ref. 52), Tg(kdrl:ras-mCherry)s896 (ref. 53), Tg(7xTCF-Xla.Siam:GFP)ia4 (ref. 54), Tg(fli1a:Gal4FF)ubs3 (ref. 55), Tg(UAS:Kaede)rk8 (ref. 56), Tg(UAS:GCaMP7a)zf415 (ref. 57), Tg(gata1:DsRed)sd2 (ref. 58), gpr124s984 (ref. 11), wnt7aaulb2 (ref. 17), reckulb3 (ref. 59), kdrlhu5088 (ref. 60) and col4a5s510 (ref. 45). The mmp25aulb26 and mmp25bulb27 alleles were generated in this study using CRISPR–Cas9 mutagenesis. All of the zebrafish experiments were performed on embryos and larvae younger than 5 days post-fertilization, before they became capable of independent feeding.
Mice
Mice were housed at 20 °C under a 12 h–12 h light–dark cycle under standard conditions and were maintained in a certified animal facility (LA1500474) in accordance with European and national ethical and animal welfare guidelines. The relative ambient humidity level ranged from 45 to 65%. All animal procedures were approved by the corresponding ethical committee (Commission d’Ethique et du Bien Être Animal (CEBEA), Université libre de Bruxelles, protocol approval number: CEBEA-08 GOS IBMM). Mice were maintained on the C57BL/6J background and, for experiments, mice of both sexes were used. BAT-GAL reporter (B6.Cg-Tg(BAT-LacZ)3Picc/J) mice61 and Mmp25-knockout mice23 were provided by S. Piccolo and C. López-Otín, respectively. Vascular networks were quantified as the number of CNS-invading sprouts in the E10.5 midbrain and forebrain in five consecutive 60 μm sections, and as the organ surface-normalized vascular density (length or surface, depending on the vascular morphologies) in 60 μm sections of E10.5 forelimbs and E12.5 intestine, stomach, liver and lung.
CRISPR–Cas9-mediated gene disruption in zebrafish
Germline zebrafish mmp25aulb26 and mmp25bulb27 alleles were generated using CRISPR–Cas9 as described previously62. Target sites were selected using CRISPOR (v.5.01)63. The following primers were annealed and cloned into the pT7-gRNA vector (Addgene, 46759): 5′-TAGGGGCAATGCCCTGCGAGTG-3′ and 5′-AAACCACTCGCAGGGCATTGCC-3′ for mmp25a; 5′-TAGGGGACAGCTACAGAGCAAAGA-3′ and 5′-AAACTCTTTGCTCTGTAGCTGTCC-3′ for mmp25b. sgRNAs were synthesized by in vitro transcription (HiScribe T7 Quick High Yield RNA Synthesis Kit; New England Biolabs) from BamHI-linearized pT7-gRNA vectors. Mmp25a was targeted in exon 4 (catalytic domain) and mmp25b was targeted in exon 2 (pro-domain). Synthetic capped zCas9 mRNA was transcribed from the XbaI-linearized pT3TS-nls-zCas9-nls vector (Addgene, 46757) using the mMESSAGE mMACHINE T3 Kit (Ambion). Co-injection of the sgRNAs (30 pg each) and nls-zCas9-nls mRNA (150 pg) was performed at the one-cell stage.
For somatic gene disruptions, two sgRNAs targeting the same exon were synthesized using the following primer pairs: mmp2 sgRNA1: 5′-TAGGGGGAACTTTATGATGGGTG-3′ and 5′-AAACCACCCATCATAAAGTTCCC-3′; mmp2 sgRNA2: 5′-TAGGGGAACTTTATGATGGGTGA-3′ and 5′-AAACTCACCCATCATAAAGTTCC-3′; mmp14b sgRNA1: 5′-TAGGCCAGTCCATTTGATGGAGA-3′ and 5′-AAACTCTCCATCAAATGGACTGG-3′; mmp14b sgRNA2: 5′-TAGGATTCCCTGGGAAGTAAGCAT-3′ and 5′-AAACATGCTTACTTCCCAGGGAAT-3′; mmp25a sgRNA1: 5′-TAGGGGCAATGCCCTGCGAGTG-3′ and 5′-AAACCACTCGCAGGGCATTGCC-3′; mmp25a sgRNA2: 5′-TAGGGTCTGGTGAGGCTTATTTT-3′ and 5′-AAACAAAATAAGCCTCACCAGAC-3′; mmp25b sgRNA1: 5′-TAGGTAGGACTGGTTGAGCCGGTA-3′ and 5′-AAACTACCGGCTCAACCAGTCCTA-3′; mmp25b sgRNA2: 5′-TAGGAGGAGGCAGATATCCATAC-3′ and 5′-AAACGTATGGATATCTGCCTCCT-3′; lama1 sgRNA1: 5’-TAGGGAACGGCCGTCAGTTCCACT-3′ and 5′-AAACAGTGGAACTGACGGCCGTTC-3′; lama1 sgRNA2: 5′-TAGGCGGACTCTGCCACCACAGGT-3′ and 5′-AAACACCTGTGGTGGCAGAGTCCG-3′; lama1 sgRNA1-scrambled: 5′-TAGGGAACGGCCGTCAGTTACCTC-3′ and 5′-AAACGAGGTAACTGACGGCCGTTC-3′; lama1 sgRNA2-scrambled: 5′-TAGGCGGACTCTGCCACCGATGAC-3′ and 5′-AAACGTCATCGGTGGCAGAGTCCG-3′; lama2 sgRNA1: 5′-TAGGCGCAGACAGGCTCCGGTCAG-3′ and 5′-AAACCTGACCGGAGCCTGTCTGCG-3′; lama2 sgRNA2: 5′-TAGGTCAGCGGGTCACAGCTCAG-3′ and 5′-AAACCTGAGCTGTGACCCGCTGA-3′. lama2 sgRNA1-scrambled: 5′-TAGGCGCAGACAGGCTCCACGGGT-3′ and 5′-AAACACCCGTGGAGCCTGTCTGCG-3′; lama2 sgRNA2-scrambled: 5′-TAGGTCAGCGGGTCACATGCAGC-3′ and 5′-AAACGCTGCATGTGACCCGCTGA-3′; col4a1 sgRNA1: 5′-TAGGATAGGTCCTGGCGGTCCGGG-3′ and 5′-AAACCCCGGACCGCCAGGACCTAT-3′; col4a1 sgRNA2: 5′-TAGGCAGGTCCCAAAGGAACTGAT-3′ and 5′-AAACATCAGTTCCTTTGGGACCTG-3′; col4a2 sgRNA1: 5′-TAGGTGGCAGTCCCGGATCTCCAG-3′ and 5′-AAACCTGGAGATCCGGGACTGCCA-3′; col4a2 sgRNA2: 5′-TAGGAGGTTTGGATGGAGCTTCAG-3′ and 5′-AAACCTGAAGCTCCATCCAAACCT-3′; col4a3 sgRNA1: 5′-TAGGAAGGTTGTGCTGGGGTTCA-3′ and 5′-AAACTGAACCCCAGCACAACCTT-3′; col4a3 sgRNA2: 5′-TAGGAAGGATTCCCAGGATTGTGT-3′ and 5′-AAACACACAATCCTGGGAATCCTT-3′; col4a4 sgRNA1: 5′-TAGGTGGGTCGACAGGGCCCCCAG-3′ and 5′-AAACCTGGGGGCCCTGTCGACCCA-3′; col4a4 sgRNA2: 5′-TAGGAGAACCTTGGGGCCCCTGG-3′ and 5′-AAACCCAGGGGCCCCAAGGTTCT-3′; col4a5 sgRNA1: 5′-TAGGCCTGGGAAACCTGGAACACC-3′ and 5′-AAACGGTGTTCCAGGTTTCCCAGG-3′; col4a5 sgRNA2: 5′-TAGGCCGGGTTTAAAGGGTCAGCC-3′ and 5′-AAACGGCTGACCCTTTAAACCCGG-3′; col4a6 sgRNA1: 5′-TAGGCTTGGACCAGTGGGCAGCGG-3′ and 5′-AAACCCGCTGCCCACTGGTCCAAG-3′; col4a6 sgRNA2: 5′-TAGGATGGGGGCCCGGGACCAGTT-3′ and 5′-AAACAACTGGTCCCGGGCCCCCAT-3′; serpina1 sgRNA1: 5′-TAGGTGCTGCCTTGCTGGTAGCAA-3′ and 5′-AAACTTGCTACCAGCAAGGCAGCA-3′; serpina1 sgRNA2: 5′-TAGGCTGGTAGCAACGGCCTGGG-3′ and 5′-AAACCCCAGGCCGTTGCTACCAG-3′.
The efficiency of somatic gene disruption was scored by high-resolution melt analysis (HRMA) using the Illumina Eco real-Time PCR system, and further characterized using Illumina amplicon deep sequencing (Azenta Life Sciences).
Genotyping
Zebrafish gpr124s984, wnt7aaulb2, kdrlhu5088, reckulb3 and col4a5s510 and mouse Mmp25 alleles were genotyped as described previously11,17,23,45,59,60. The mmp25aulb26 and mmp25bulb27 alleles were genotyped by high-resolution melt analysis (Eco Illumina real-time PCR system) using the following primers: 5′-TTTCCACCTCCCTCAGTGTC-3′ and 5′-GTGGAAACGCAGAGGTGTGT-3′ for mmp25a; 5′-CGCACAGGACAGCTACAGAG-3′ and 5′-CTGCATTTCTCTAATGGCTCTCTCG-3′ for mmp25b.
MO, RNA and DNA microinjection in zebrafish
MOs targeting gpr124 (4 ng; splice blocking; ACTGATATTGATTTAACTCACCACA)11, reck (0.4 ng; splice blocking; CAGGTAGCAGCCGTCACTCACTCTC)64, wnt7aa (4 ng; splice blocking; TTCCATTTGACCCTACTTACCCAAT)17, lama1 (0.5 ng; translation blocking; ATCTCCATCATCGCTCAAACTAAAG), lama2 (1 ng; translation blocking GCCACTAAACTCCGCGTGTCCATGT), lama4 (0.5 ng; translation blocking; GCCATGATTCCCCCTGCAACAACTT), lama5 (0.25 ng; translation blocking; CTCGTCCTGATGGTCCCCTCGCCAT)65, lamb1a (0.125 ng; translation blocking; TATTTCCAGTTTCTTTCTTCAGCGG), lamc1 (0.125 ng; translation blocking; TGTGCCTTTTGCTATTGCGACCTC)66, col4a1 (1 ng; translation blocking; ACACATGGAAGCCGCATCTTCACAC)67, col4a2 (2 ng; translation blocking; TTCTCACCCTCCATGCGAGCCTAAA), col4a5 (2 ng; translation blocking; ATGTTCCTCTGTTAAGCTAACTGCA), col4a6 (2 ng; translation blocking; AGGTAAAGTAGGCTATCCTCCTCGT) were obtained from Gene Tools and were injected at the zygotic stage at the indicated doses. Injection of a standard control MO (CCTCTTACCTCAGTTACAATTTATA, up to 8 ng) did not affect the brain vasculature.
Transgenic mosaic endothelial overexpression was achieved by co-injecting at the one-cell stage 25 pg of Tol2 transposase mRNA and 25 pg of the pTol2-fli1a:kdrl-2A-nls-mtagBFP2, pTol2-fli1a:mmp25b-2A-tagRFP, pTol2-fli1a:mmp25bΔZn2+-BD-2A-tagRFP or pTol2-fli1a:mmp25bZn2+-BDH237A,H241A,H247A-2A-tagRFP constructs68.
Capped mRNAs were transcribed in vitro from NotI-linearized pCS2+ constructs, using the mMessage mMachine SP6 Kit (Thermo Fisher Scientific) and injected at the one-cell stage at a dose of 200 pg. The fragment encoding the Zn2+-binding domain (Zn2+-BD; His237–His247) was deleted in the ΔZn2+-BD mmp25b variant. Three histidines, essential for Zn2+ chelation, were substituted by alanines in the Zn2+-BDH237A, H241A, H247A variant, abbreviated as Zn2+-BDH-A. In the Pro− mmp2 mRNA variant, the sequences encoding the prodomain (Ala30–Val107) were deleted. The sequences corresponding to the GPI-anchoring site of Mmp25b (Ser658–Gln697) were fused 3′ to the mmp2 ORF in the GPI+ mmp2 variant.
Transplantations
Host Tg(kdrl:ras-mCherry)s896 and donor Tg(kdrl:EGFP)s843 embryos were dechorionated with pronase (Millipore, 53702; 1 mg ml−1) during 5 min at 28 °C in 1/3 Ringer solution, supplemented with penicillin (50 U ml−1) and streptomycin (50 µg ml−1). The embryos were subsequently incubated on agarose-coated dishes in the same medium. At the mid-blastula stage, 20 to 50 donor cells were transplanted into the blastoderm margin of stage-matched host embryos. After transplantation, embryos were incubated until the indicated stages. After assessing the contribution of EGFP+ transplanted cells using the Leica M165 stereomicroscope, mosaic vessels were recorded using time-lapse confocal microscopy. The contribution of cells of a defined genotype to the TC position was calculated as the fraction of the total number of mosaic vessels (CtAs or ISVs). The contribution to TC position in intraneural secondary branches was scored as the fraction of the stalk cell genotype in the initial brain-invading CtA.
Immunofluorescence and in situ hybridization
Zebrafish and mouse embryos were fixed in 4% paraformaldehyde (PFA) in PBS. For sections, embryos were washed in PBS and equilibrated in 30% sucrose in PBS (w/v) overnight at 4 °C. The embryos were then mounted in 7.5% gelatin (w/v), 15% sucrose (w/v) in PBS and stored at −80 °C. Zebrafish and mouse embryos were cut into 20 and 60 µm frozen sections, respectively, using the Leica CM1850 Cryostat (Leica) at −30 °C.
For immunofluorescence staining, the sections were washed three times with PBS Triton X-100 (0.4%; PBST) for 5 min, blocked using blocking buffer (PBST, 5% goat serum) for 1 h and then incubated with primary antibodies in blocking buffer solution overnight at 4 °C. After three washing steps in PBST for 5 min, the sections were exposed to secondary antibodies diluted in blocking buffer containing 0.001% DAPI overnight at 4 °C. After three washing steps in PBST for 5 min, the sections were mounted in DAKO fluorescence mounting medium (Agilent, S3023). The following primary antibodies and lectin were used: rabbit anti-laminin-111 (Merck, L9393, 1:250, used for zebrafish immunostaining, polyclonal immunization with an Engelbreth–Holm–Swarm mouse sarcoma extract), rat anti-laminin-111 (R&D systems, MAB4656, 1:250, used for mouse immunostainings, monoclonal reactivity towards LAMA1/B1), chicken anti-GFP (Aves Labs, GFP-1020, 1:200), rabbit anti-collagen type IV (Sigma-Aldrich, AB756P, 1:300), chicken anti-β-galactosidase (Abcam, ab9361, 1:300), anti-Erg1-Alexa Fluor (AF) 647 conjugate (Abcam, ab196149, 1:250) and isolectin B4-AF594 conjugate (Thermo Fischer Scientific, I21413, 1:200). The following secondary antibodies were used: goat anti-chicken AF488 (Thermo Fischer Scientific, A11039, 1:500), goat anti-rabbit AF594 (Thermo Fischer Scientific, A11012, 1:500), and donkey anti-rat AF647 (Thermo Fischer Scientific, A48272, 1:500).
For in situ hybridization, digoxigenin (DIG)-labelled antisense riboprobes were produced by in vitro transcription using the DIG RNA labelling kit and SP6 RNA polymerase (Roche). The templates were amplified from 48 hpf WT embryo cDNA, and cloned into NcoI/SacI-digested pGEMT using the following primers: kdrl: 5′-GCATGCTCCCGGCCGCCATGGTGGCAGGATTCACTTTGAGTGG-3′ and 5′-CATCCAACGCGTTGGGAGCTCTAGTGTAGGGCTCAATCCGCAG-3′; mmp25b: 5′-ATGAGTTTCTCAGGATATCTTGGTCTGG-3′ and 5′-TTATTGCGAGTTGAAGCCAATATGAAGC-3′; mmp14b: 5′-GCATGCTCCCGGCCGCCATGGTGGATGCAGCTCTTCTCTACACG-3′ and 5′-CATCCAACGCGTTGGGAGCTCCATGAGGCTGCTGGAAATGTGC-3′; mmp2: 5′-GCATGCTCCCGGCCGCCATGGTGCTCACACAGACAAAGAAGTGG-3′ and 5′-CATCCAACGCGTTGGGAGCTCTTTCCTGACATCAGCCGTCC-3′; mmp9: 5′-GCATGCTCCCGGCCGCCATGGCAAATCTGTGTTCGTGACGTTTCC-3′ and 5′-CATCCAACGCGTTGGGAGCTCCTCCTTGATTTGGCAGGCATCG-3′; lama1: 5′-GCATGCTCCCGGCCGCCATGGGTCACAACAAAGCCGACGACTG-3′ and 5′-CATCCAACGCGTTGGGAGCTCTGAGCGTTCCCTCAGCGCTGT-3′; col4a1: 5′-GCATGCTCCCGGCCGCCATGGGGTTCTAAGGGTGAAGGAGGTG-3′ and 5′-CATCCAACGCGTTGGGAGCTCCCCTCTTCATGCACACTTGAC-3′; col4a2: 5′-GCATGCTCCCGGCCGCCATGGCCTAAAGGAGATACCGGACCC-3′ and 5′-CATCCAACGCGTTGGGAGCTCCTACAGGTTCTTCATGCACAC-3′; col4a3: 5′-GCATGCTCCCGGCCGCCATGGGGACAAAAAGGACAGTGTGGTC-3′ and 5′-CATCCAACGCGTTGGGAGCTCGCAAGGTCACCTTGAGGCTGTTG-3′, col4a4: 5′-GCATGCTCCCGGCCGCCATGGCTGGGTCCCAGTGGTGCAAAAG-3′ and 5′-CATCCAACGCGTTGGGAGCTCCATTGGTTGGGGTCATTCATC-3′; col4a5: 5′-GCATGCTCCCGGCCGCCATGGGGTTTTCCAGGATCTAAAGGAG-3′ and 5′-CATCCAACGCGTTGGGAGCTCCGTCCTCTTCATACACACCAC-3′; col4a6: 5′-GCATGCTCCCGGCCGCCATGGCGTCCAGGAATAATAGGACC-3′ and 5′-CATCCAACGCGTTGGGAGCTCCTACAAGATCTTCATGCAGAC-3′; slc2a1a: 5′-GCATGCTCCCGGCCGCCATGGCAACTTGGCATTGTCATTG-3′ and 5′-CATCCAACGCGTTGGGAGCTCGGCTGTGATCTCTTCAAACG-3′; slc16a1a: 5′-GCATGCTCCCGGCCGCCATGGATGCCTCCAGCAACAGGAGG-3′ and 5′-CATCCAACGCGTTGGGAGCTCCTATACGACTCCATCTGCCTCCTTTT-3′; fabp11a: 5′-GCATGCTCCCGGCCGCCATGGGATCAAATCTCAATTTACAGCTGTTG-3′ and 5′-CATCCAACGCGTTGGGAGCTCTTCAAAGCACCATAAAGACTGATAAT-3′. Whole-mount chromogenic in situ hybridizations were performed as previously described69 using anti-DIG-AP antibodies (Merck, 11093274910, 1:10,000). Combined immunostainings and FISH were performed as previously described70, using anti-DIG POD antibodies (Merck, 11207733910, 1:1,000) and the TSA Plus Cy3 detection kit (Akoya Biosciences, NEL744001KT).
Photoconversion and FACS isolation of zebrafish brain ECs
Photoconversion of Tg(fli1a:Gal4FF)ubs3;(UAS:Kaede)rk8 PHBC or CtA ECs was performed using the Zeiss LSM710 confocal microscope (Carl Zeiss, objective lenses: Plan-Apochromat ×20/0.8 M27), as described previously71. In brief, anaesthetized embryos were mounted laterally in 1% low-melting-point agarose and the fluorescent Kaede protein was photoswitched by scanning the selected region of interest (ROI) using a 405 nm laser (five iterations of 50 s). After isolation from the agarose, the embryos were washed in Ca2+/Mg2+-free Hank’s Balanced Salt Solution (HBSS, Gibco) and dissociated at 28.5 °C for 30 min in TrypLE select (Thermo Fischer Scientific, 12563011). Dissociation was stopped by the addition of FBS and centrifugation. The cell pellet was resuspended in HBSS containing Ca2+/Mg2+ and 5% FBS, filtered and submitted for FACS analysis (BD Biosciences FACSAria III).
For scRNA-seq analyses, single photoswitched (red fluorescent) WT ECs were distributed in individual wells of 384-well plates containing 2.3 µl of Smart-seq2 lysis buffer (0.2% Triton X-100, 2 U µl−1 RNase inhibitor, 2 mM dNTP mix and 1 µM Smart-seq2 primer (5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′). The plates were stored at −80 °C before mRNA-seq using the Smart-Seq2 protocol72 and analysis using the Seurat v4 toolkit in Rstudio (v.1.1.463)73. In brief, single-cell fastq files were demultiplexed by applying standard parameters of the Illumina pipeline (bcl2fastq v.2.19.0.316) using Nextera XT index kit v2 adapters. Mapping was performed to the zebrafish reference genome build GRCz11, with TopHat v.2.1.1 and Bowtie1 or Bowtie2 option. Adapter sequences were removed using Trim Galore v.0.4.4 before read mapping and doublets were removed using Samtools v.1.16.1 software. The generated BAM files containing the alignment results were sorted according to the mapping position, and raw read counts for each gene were calculated using the FeatureCounts function from the Subread package v.1.4.6-p5. For technical control, 92 ERCC RNAs were included in the lysis buffer and in the mapping.
For bulk RNA-seq analyses, Tg(fli1a:Gal4FF)ubs3;(UAS:Kaede)rk8 embryos were injected, or not, at the one-cell stage with gpr124, reck or wnt7aa MOs and PHBC ECs were isolated at 30 hpf as described above. Alternatively, embryos were treated with IWR-1 from 26 hpf onwards and CtA ECs were photoconverted and sorted at 36 hpf, as described above. Photoconverted PHBC ECs of 80 embryos were pooled and submitted for RNA extraction and RNA-seq, as previously described71. Transcriptomes were analysed and compared using DESeq2 (v.1.12)74.
Light microscopy image acquisition and processing
All images were acquired using the Leica M165 stereomicroscope, the Zeiss LSM710 or the Zeiss LSM900 confocal microscope equipped with the Leica Application Suite (LAS) v.4.2 or ZEN Blue v.3.1 microscopy software. Image analysis was performed using ImageJ v.1.53c. Zebrafish embryos were imaged live or after fixation in 4% PFA in PBS overnight at 4 °C. Mouse embryos were fixed (4% PFA in PBS), and stained after sectioning. Live imaging of dechorionated zebrafish embryos was performed after embryo immobilization with a low dose of tricaine in low-melting-point agarose (1% in E3 zebrafish medium supplemented with N-phenylthiourea and tricaine) in a glass-bottom Petri dish (MatTek Corporation). Confocal time-lapse images were recorded at a stable temperature of 28.5 °C, using an incubation chamber. Ca2+-oscillations were recorded by time-lapse imaging of Tg(fli1a:Gal4FF);(UAS:GCaMP7a) embryos, taking a z stack every 5 s during the 30 min before CtA sprouting (31–31.5 hpf). Circular ROIs (<5 µm diameter) were centred on oscillating PHBC ECs. F/F0 was calculated to quantify changes in fluorescence, where F0 is the baseline fluorescence. Ca2+ spikes were identified as events of F/F0 ≥ 1.5.
For angiography, imaging was performed 1 h after injection of 1 nl of tetramethylrhodamine dextran 2,000,000 Da molecular mass (Thermo Fisher Scientific, D7139, 25 μg µl−1 in PBS) in the heart of 72 hpf larvae using a micromanipulator. Tracer leakage assays were performed by injecting 1 nl of 150,000 Da FITC-labelled dextran (FD150S, 25 μg µl−1 in PBS) intracardially and imaging 1 h after injection. Three-dimensional reconstructions were performed using the Imaris Filament Tracer software (Bitplane) before manual false-colouring to highlight extra- and intracerebral vessels exhibiting or not BBB properties.
Transmission electron microscopy
WT zebrafish embryos (32 hpf) were fixed overnight in 2.5% glutaraldehyde (Electron Microscopy Sciences), 4% PFA at 4 °C and post-fixed with 1% osmium tetroxide (Electron Microscopy Sciences) and 1.5% ferrocyanide (Electron Microscopy Sciences) in 0.15 M cacodylate buffer. The embryos were further stained with 1% uranyl acetate (Electron Microscopy Sciences), serially dehydrated and embedded in epoxy resin (Agar 100 resin; Agar Scientific). Resin blocks containing the processed embryos were trimmed to reach the ROI, which was evaluated by toluidine staining of thin sections (15 μm). Ultrathin 70 nm sections were then produced with a Leica EM UC6 ultramicrotome and mounted onto copper-Formvar-carbon grids (Electron Microscopy Sciences). Observations were made using the Tecnai 10 transmission electron microscope (FEI), and images were captured with a Veleta camera and processed using SIS iTEM v.5.1 software (Olympus).
Western blot analysis
Samples were denatured in Bolt LDS sample buffer and reducing agent (Thermo Fischer Scientific, B0007 and B0009) at 70 °C for 10 min. Gel electrophoresis was performed using 4–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad, 4561085). Proteins were transferred to nitrocellulose membranes. After blocking in 5% milk in Tris-buffered saline (TBS), the membranes were incubated with primary antibodies (1% milk in 0.05% Tween-20 TBS, TBST) overnight at 4 °C. After washing in TBST, membranes were incubated with secondary antibodies in 1% BSA in TBST, for 1 h at room temperature. Blots were revealed using Western Lightning Plus ECL (PerkinElmer, NEL103001EA).
The following primary antibodies were used: rabbit anti-HA (Merck, H6908, 1:1,000), chicken anti-GFP (Aves Biolabs, GFP-1020, 1:10,000), rat anti-laminin-111 (R&D systems, MAB4656, 1:250, monoclonal reactivity towards LAMA1/B1). The following secondary antibodies were used: goat anti-rabbit IgG HRP conjugate (Promega, W401B, 1:5,000), goat anti-chicken IgY HRP conjugate (Thermo Fischer Scientific, A16054, 1:40,000) and rabbit anti-rat IgG HRP conjugate (Merck, A9542, 1:5,000). Uncropped blots are provided in Supplementary Fig. 1.
Recombinant protein expression and purification
The human MMP25 and MMP2 catalytic domains were amplified from HUVEC cDNA and the zebrafish Mmp25b catalytic domain was synthesized after codon optimization. The fragments were cloned into the NcoI and XhoI restriction sites of pET21d. The catalytic domains span residues Tyr113 to Gly284 of zebrafish Mmp25b (UniProtKB: E7F1N5), Tyr108 to Gly280 of human MMP25 (UniProtKB: Q9NPA2) and Tyr110 to Asp452 of human MMP2 (UniProtKB: P08253). BL21 (DE3) E. coli cells were transformed with pET21d-zMmp25b-6xhis, pET21d-hMMP25-6xhis or pET21d-hMMP2-6xhis and grown in 100–300 ml LB medium (supplemented with 100 µg ml−1 ampicillin). Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) when the culture reached an optical density at 600 nm (OD600) of 0.9. After overnight incubation at 37 °C under agitation, cells were collected by centrifugation (5,000g, 20 min, 4 °C) and frozen at −80 °C. After resuspension in 50 mM Tris (pH 8), cells were mechanically lysed on ice (Microfluidics, 110SCE, 3 cycles). Inclusion bodies were recovered from the lysate by centrifugation (16,000g, 20 min, 4 °C) and solubilized in 8 M urea, 50 mM Tris (pH 7.6), 150 mM NaCl, 5 mM CaCl2 and 50 µm ZnCl2. The insoluble fraction was removed by centrifugation (16,000g, 20 min, 4 °C) and the supernatant was incubated overnight with 100 µl of Ni+/nitrilotriacetic acid agarose beads (Qiagen) at 4 °C. The beads were washed with 20 mM imidazole in TBS 8 M urea and elution was performed with 500 mM imidazole in TBS 8 M urea. Recombinant protein purity was assessed by SDS–PAGE and Coomassie blue staining, and protein concentrations were measured by the BCA protein assay (Thermo Fischer Scientific, 23223). Catalytic domains were refolded by dilution (1/20, v/v) in 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, 50 µM ZnCl2, 0.005% Brij-35 (Thermo Fischer Scientific, 20150) for 1 h at 12 °C. The insoluble fraction was removed by centrifugation (21,400g, 10 min, 4 °C). Uncropped gels are available in Supplementary Fig. 1.
Mmp25 cleavage assays
For α-1 antitrypsin, 2 µM of α-1 antitrypsin (Athens Research and Technology, 16-16-0011609) was incubated with 2 µM rzMmp25b or 75 nM rhMMP25 overnight at 28 °C and 37 °C, respectively, in 50 µl Mmp25 cleavage buffer (50 mM Tris (pH 7.6), 150 mM NaCl, 5 mM CaCl2, 0.005% Brij-35 (Thermo Fischer Scientific, 20150)).
For laminin-111, 15 µg of Matrigel (Corning, 354230) was incubated overnight at 37 °C with 1 µM of rhMMP25 in 50 µl Mmp25 cleavage buffer. The samples were concentrated by acetone precipitation before SDS–PAGE and western blot analysis for LAMA/B1 (R&D systems, MAB4656).
For collagen IV, 20 µg of collagen IV purified from human placenta (Merck, C7521) was incubated with 1 µM rhMMP25 overnight at 37 °C in 50 µl Mmp25 cleavage buffer. The samples were concentrated by acetone precipitation before SDS–PAGE and Coomassie blue staining.
For recombinant HA-tagged Col4a5 expressed in HEK293T cells, Zebrafish col4a5 was amplified from 48 hpf zebrafish cDNA, cloned in fusion to a C-terminal HA tag into pCS2+ (digested with BamHI and XhoI) and transiently expressed using PEI (polyethylenimine) in HEK293T cells (ATCC CRL-3216, authenticated by ATCC STR profiling, tested negative for mycoplasma contamination). The empty pCS2+ was used as negative control. Then, 48 h after transduction, the cells were washed twice in PBS, before collection and cell disruption using a disposable grinding pestle in Mmp25 cleavage buffer. After centrifugation (21,400g, 10 min, 4 °C), 4 µg of the supernatant was incubated overnight with 2 µM of rzMmp25b at 28 °C or 75 nM of rhMMP25 at 37 °C in 50 µl Mmp25 cleavage buffer.
For human COL4A1–6 putative cleavage sites expressed as GST–GFP linkers in E.coli, DNA sequences encoding a N-terminal fusion between a 12 amino acid fragment centred on the putative cleavage site of MMP25 in COL4A1–6 and GFP were cloned into pGEX-6P-1 downstream of the GST and the recognition sequence for site-specific cleavage by the PreScission Protease-encoding sequences. BL21 (DE3) E. coli were transformed with these constructs and protein expression was induced with 1 mM IPTG when OD600 reached 0.7. After overnight incubation at 30 °C under agitation, cells were collected by centrifugation (5,000g, 20 min at 4 °C) and lysed in 50 mM Tris (pH 8) using the FastPrep-24 cell disrupter and Lysing Matrix B Bulk (M.P. Biomedicals). After three cell disruption cycles of 20 s, the cell lysates were clarified by centrifugation (21,400g, 10 min, 4 °C). Protein concentration of the supernatant was determined using BCA (Thermo Fischer Scientific, 23223). A total of 500 ng of the soluble fraction was incubated overnight in Mmp25-cleavage buffer with 75 nM of rhMMP25 or rhMMP2 at 37 °C, or with 1 × 10−3 IU of the control PreScission Protease (GenScript, N02799-100) at 25 °C in 50 µl Mmp25 cleavage buffer. Uncropped gels and blots are available in Supplementary Fig. 1.
MS analysis
For protein digestion, bands of interest were excised from SDS–PAGE gels, washed twice with distilled water and shrunk in 100% acetonitrile. In-gel proteolytic digestion was performed by the addition of 4 µl of trypsin (Promega; in 50 mM NH4HCO3) and overnight incubation at 37 °C.
For MS, protein digests (supernatants) were analysed using nano-liquid chromatography–electrospray ionization–MS/MS on the timsTOF Pro (Bruker v.5.3) system. Peptides were separated by nanoUHPLC (nanoElute, Bruker) on a 75 μm inner diameter, 25 cm C18 column with integrated CaptiveSpray insert (Aurora, IonOpticks) at a flow rate of 200 nl min−1, at 50 °C. LC mobile phase A was 0.1% formic acid (v/v) in H2O, and mobile phase B was 0.1% formic acid (v/v) in acetonitrile. Digests (1 µl) were loaded at a constant pressure of 600 bar, directly on the column. After injection of the digest (1 µl), the mobile phases were linearly increased from 2% B to 13% over 18 min, from 13% B to 19% over 7 min, from 19% B to 22% over 4 min, and from 22% B to 85% in 3 min.
Data acquisition on the timsTOF Pro was performed using Hystar v.5.1 and timsControl v.2.0. The TIMS accumulation time was 100 ms and mobility (1/K0) ranged from 0.6 to 1.6 V s cm−2. Analyses were performed using parallel accumulation serial fragmentation (PASEF) acquisition method75. Per total cycle of 1.1 s, one MS spectrum was followed by ten PASEF MS/MS spectra.
For data processing, tandem mass spectra were extracted, charge-state deconvoluted and deisotoped by Data analysis (Bruker) v.5.3. All MS/MS samples were analysed using Mascot (Matrix Science; v.2.8.1), searching the Human Proteome database (https://www.uniprot.org/uniprotkb?query=(proteome:UP000005640), 101,673 entries) assuming semi-specific trypsin digestion. Three missed cleavages were tolerated. Mascot was searched with a fragment ion mass tolerance of 0.050 Da and a parent ion tolerance of 15 ppm. Carbamidomethyl of cysteine was specified as a fixed modification in Mascot. Oxidation of methionine, hydroxylation of lysine and proline, deamination of asparagine and glutamine, and acetylation of the N-terminus were specified in Mascot as variable modifications.
Peptide and protein identifications were performed using Scaffold (v.Scaffold_5.10.0, Proteome Software). Peptide identifications were accepted by the Scaffold Local FDR algorithm if establishing a probability higher than 96.0% to achieve an FDR lower than 1.0%. Protein identifications were accepted if the probability was higher than 5.0% to achieve an FDR lower than 1.0% and containing at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm76. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters.
Statistics and reproducibility
Seurat v.4 was used to analyse the scRNA-seq datasets. Bulk RNA-seq data were analysed using DESeq2 v.1.12. Statistical analyses were performed using RStudio v.1.1.463 and GraphPad Prism v.9. Pearson correlation analyses and visualizations were performed using ggcorrplot v.0.1.3. Normally distributed data are represented as mean ± s.d. and were analysed using one-tailed one-way ANOVA (with post hoc Dunnett’s test) and two-tailed Student’s t-tests for multiple and single comparisons, respectively. Non-normally distributed data are represented as median ± interquartile range and were analysed using one-tailed Kruskal–Wallis tests (with post hoc Dunn’s test) for multiple comparisons and two-tailed Mann–Whitney U-tests for single comparisons. No statistical methods were used to determine the sample size. The sample size was determined by the technical constraints of the experiments, as well as our and other’s previous work on zebrafish neurovascular development11,12,13,14,17,71. One-cell stage embryos are undistinguishable irrespective of their genotype, and were therefore randomized during injections. The allocation of organisms into experimental groups was randomized. Experimental groups of an experiment were always raised in parallel, under identical conditions. For zebrafish and mouse Mendelian genetics experiments, genotyping was always performed after phenotypic assessment. The researcher is therefore inherently blinded to the experimental conditions. In MO and somatic gene disruption experiments, investigators were not blinded. The sex of animals was not determined (embryonic or larval zebrafish) or was not analysed (embryonic mice) at the developmental stage of interest. The number and nature of observations (n), mean or median, type of error bar and statistical tests used for analysis are indicated in the figure legends. Images of immunofluorescence, in situ hybridization, transmission electron microscopy, and protein gels or blots are representative of experiments that were repeated independently at least three times. All attempts at replication were successful.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq and MS data were deposited at the NCBI Gene Expression Omnibus through GEO Series accession numbers GSE121041, GSE233488 and GSE233662 and in PRIDE with the dataset identifier PXD042613 (Proteomics Identification Database), respectively. Databases used in this study include UniProt (E7F1N5, Q9NPA2 and P08253). The Human Proteome database (https://www.uniprot.org/uniprotkb?query=(proteome:UP000005640)) was used for MS/MS analysis. Publicly accessible datasets used in this study are: ref. 40, (Sequence Read Archive: PRJNA637987 and http://mousebrain.org/); ref. 42 (GEO: GSE150219); ref. 44 (GEO: GSE122564); ref. 77 (GEO: GSE79306); ref. 78 (GEO: GSE111839); ref. 79 (GEO: GSE95401 and GSE95201); ref. 80 (GEO: GSE52564); ref. 81 (GEO: GSE66848); ref. 82 (GEO: GSE74052). All other data are available in the Article and its Supplementary Information. Source data are provided with this paper.
References
Augustin, H. G. & Koh, G. Y. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 357, eaal2379 (2017).
Gomez-Salinero, J. M., Itkin, T. & Rafii, S. Developmental angiocrine diversification of endothelial cells for organotypic regeneration. Dev. Cell 56, 3042–3051 (2021).
Stenman, J. M. et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250 (2008).
Liebner, S. et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 183, 409–417 (2008).
Daneman, R. et al. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl Acad. Sci. USA 106, 641–646 (2009).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779 (2020).
Kuhnert, F. et al. Essential regulation of CNS angiogenesis by the orphan G protein–coupled receptor GPR124. Science 330, 985–989 (2010).
Cho, C., Smallwood, P. M. & Nathans, J. Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron 95, 1056–1073 (2017).
Vallon, M. et al. A RECK-WNT7 receptor-ligand interaction enables isoform-specific regulation of Wnt bioavailability. Cell Rep. 25, 339–349 (2018).
Vanhollebeke, B. et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. eLife 4, e06489 (2015).
Ulrich, F. et al. Reck enables cerebrovascular development by promoting canonical Wnt signaling. Dev. Camb. Engl. 143, 147–159 (2016).
Eubelen, M. et al. A molecular mechanism for Wnt ligand-specific signaling. Science 361, eaat1178 (2018).
America, M. et al. An integrated model for Gpr124 function in Wnt7a/b signaling among vertebrates. Cell Rep. 39, 110902 (2022).
Martowicz, A. et al. Endothelial β-catenin signaling supports postnatal brain and retinal angiogenesis by promoting sprouting, tip cell formation, and VEGFR (vascular endothelial growth factor receptor) 2 expression. Arterioscler. Thromb. Vasc. Biol. 39, 2273–2288 (2019).
Yokota, Y. et al. Endothelial Ca2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. eLife 4, e08817 (2015).
Martin, M. et al. Engineered Wnt ligands enable blood-brain barrier repair in neurological disorders. Science 375, eabm4459 (2022).
Crawford, B. D. et al. Mmp25β facilitates elongation of sensory neurons during zebrafish development. Genesis 52, 833–848 (2014).
Pei, D. Leukolysin/MMP25/MT6-MMP: a novel matrix metalloproteinase specifically expressed in the leukocyte lineage. Cell Res. 9, 291–303 (1999).
Kojima, S., Itoh, Y., Matsumoto, S., Masuho, Y. & Seiki, M. Membrane-type 6 matrix metalloproteinase (MT6-MMP, MMP-25) is the second glycosyl-phosphatidyl inositol (GPI)-anchored MMP. FEBS Lett. 480, 142–146 (2000).
Velasco, G. et al. Human MT6-matrix metalloproteinase: identification, progelatinase A activation, and expression in brain tumors. Cancer Res. 60, 877–882 (2000).
English, W. R., Velasco, G., Stracke, J. O., Knäuper, V. & Murphy, G. Catalytic activities of membrane-type 6 matrix metalloproteinase (MMP25). FEBS Lett. 491, 137–142 (2001).
Soria-Valles, C. et al. MMP-25 metalloprotease regulates innate immune response through NF-κB signaling. J. Immunol. 197, 296–302 (2016).
Haas, T. L., Davis, S. J. & Madri, J. A. Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMP-2 in microvascular endothelial cells. J. Biol. Chem. 273, 3604–3610 (1998).
Haas, T. L. & Madri, J. A. Extracellular matrix-driven matrix metalloproteinase production in endothelial cells: implications for angiogenesis. Trends Cardiovasc. Med. 9, 70–77 (1999).
Zhou, Z. et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl Acad. Sci. USA 97, 4052–4057 (2000).
Yana, I. et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J. Cell Sci. 120, 1607–1614 (2007).
Sato, H. et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370, 61–65 (1994).
Strongin, A. Y. et al. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 270, 5331–5338 (1995).
Itoh, T. et al. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 58, 1048–1051 (1998).
Hotary, K., Allen, E., Punturieri, A., Yana, I. & Weiss, S. J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 149, 1309–1323 (2000).
Chun, T.-H. et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol. 167, 757–767 (2004).
Costell, M. et al. Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol. 147, 1109–1122 (1999).
Halfter, W., Dong, S., Yip, Y.-P., Willem, M. & Mayer, U. A critical function of the pial basement membrane in cortical histogenesis. J. Neurosci. 22, 6029–6040 (2002).
Borrell, V. & Marín, O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat. Neurosci. 9, 1284–1293 (2006).
Zarbalis, K. et al. Cortical dysplasia and skull defects in mice with a Foxc1 allele reveal the role of meningeal differentiation in regulating cortical development. Proc. Natl Acad. Sci. USA 104, 14002–14007 (2007).
Hecht, J. H., Siegenthaler, J. A., Patterson, K. P. & Pleasure, S. J. Primary cellular meningeal defects cause neocortical dysplasia and dyslamination. Ann. Neurol. 68, 454–464 (2010).
Sasaki, T., Giltay, R., Talts, U., Timpl, R. & Talts, J. F. Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp. Cell. Res. 275, 185–199 (2002).
Hannocks, M.-J. et al. Molecular characterization of perivascular drainage pathways in the murine brain. J. Cereb. Blood Flow Metab. 38, 669–686 (2018).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Sievers, J., Pehlemann, F. W., Gude, S. & Berry, M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J. Neurocytol. 23, 135–149 (1994).
DeSisto, J. et al. Single-cell transcriptomic analyses of the developing meninges reveal meningeal fibroblast diversity and function. Dev. Cell 54, 43–59 (2020).
Pietilä, R. et al. Molecular anatomy of adult mouse leptomeninges. Neuron 111, 3745–3764 (2023).
Corada, M. et al. Fine-tuning of Sox17 and canonical Wnt coordinates the permeability properties of the blood-brain barrier. Circ. Res. 124, 511–525 (2019).
Xiao, T. & Baier, H. Lamina-specific axonal projections in the zebrafish tectum require the type IV collagen Dragnet. Nat. Neurosci. 10, 1529–1537 (2007).
Wang, Y. et al. Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151, 1332–1344 (2012).
Siegenthaler, J. A. et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell 139, 597–609 (2009).
Dorrier, C. E., Jones, H. E., Pintarić, L., Siegenthaler, J. A. & Daneman, R. Emerging roles for CNS fibroblasts in health, injury and disease. Nat. Rev. Neurosci. 23, 23–34 (2022).
Watson, E. C. & Adams, R. H. Biology of bone: the vasculature of the skeletal system. Cold Spring Harb. Perspect. Med. 8, a031559 (2018).
Zarkada, G. et al. Specialized endothelial tip cells guide neuroretina vascularization and blood-retina-barrier formation. Dev. Cell 56, 2237–2251 (2021).
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. & Schilling, T. F. Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 (1995).
Jin, S.-W., Beis, D., Mitchell, T., Chen, J.-N. & Stainier, D. Y. R. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132, 5199–5209 (2005).
Chi, N. C. et al. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 22, 734–739 (2008).
Moro, E. et al. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev. Biol. 366, 327–340 (2012).
Herwig, L. et al. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr. Biol. 21, 1942–1948 (2011).
Hatta, K., Tsujii, H. & Omura, T. Cell tracking using a photoconvertible fluorescent protein. Nat. Protoc. 1, 960–967 (2006).
Muto, A., Ohkura, M., Abe, G., Nakai, J. & Kawakami, K. Real-time visualization of neuronal activity during perception. Curr. Biol. 23, 307–311 (2013).
Traver, D. et al. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 4, 1238–1246 (2003).
Parab, S. et al. Local angiogenic interplay of Vegfc/d and Vegfa controls brain region-specific emergence of fenestrated capillaries. eLife 12, e86066 (2023).
Bussmann, J. et al. Arteries provide essential guidance cues for lymphatic endothelial cells in the zebrafish trunk. Development 137, 2653–2657 (2010).
Maretto, S. et al. Mapping Wnt/β-catenin signaling during mouse development and in colorectal tumors. Proc. Natl Acad. Sci. USA 100, 3299–3304 (2003).
Jao, L.-E., Wente, S. R. & Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl Acad. Sci. USA 110, 13904–13909 (2013).
Concordet, J.-P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Prendergast, A. et al. The metalloproteinase inhibitor Reck is essential for zebrafish DRG development. Development 139, 1141–1152 (2012).
Pollard, S. M. et al. Essential and overlapping roles for laminin alpha chains in notochord and blood vessel formation. Dev. Biol. 289, 64–76 (2006).
Parsons, M. J. et al. Zebrafish mutants identify an essential role for laminins in notochord formation. Development 129, 3137–3146 (2002).
Stratman, A. N. et al. Interactions between mural cells and endothelial cells stabilize the developing zebrafish dorsal aorta. Development 144, 115–127 (2017).
Kwan, K. M. et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099 (2007).
Thisse, C. & Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 (2008).
Kochhan, E. & Siekmann, A. F. Zebrafish as a model to study chemokine function. Methods Mol. Biol. 1013, 145–159 (2013).
Hübner, K. et al. Wnt/β-catenin signaling regulates VE-cadherin-mediated anastomosis of brain capillaries by counteracting S1pr1 signaling. Nat. Commun. 9, 4860 (2018).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Meier, F. et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteom. 17, 2534–2545 (2018).
Nesvizhskii, A. I., Keller, A., Kolker, E. & Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 (2003).
Hupe, M. et al. Gene expression profiles of brain endothelial cells during embryonic development at bulk and single-cell levels. Sci. Signal. 10, eaag2476 (2017).
Sabbagh, M. F. et al. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife 7, e36187 (2018).
Munji, R. N. et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 22, 1892–1902 (2019).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Jensen, L. D. et al. Disruption of the extracellular matrix progressively impairs central nervous system vascular maturation downstream of β-catenin signaling. Arterioscler. Thromb. Vasc. Biol. 39, 1432–1447 (2019).
Chang, J. et al. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 23, 450–460 (2017).
Acknowledgements
We thank C. Lopez-Otin, A. Inbal, S. Piccolo and H. Baier for sharing mutant mice and zebrafish; and P. Tebabi, T. Voet, D. Brown, M. Adam, N. Dumont and E. Dupont for assistance. The illustrations in Figs. 2e, 3m and 4h and Extended Data Fig. 7e were created using BioRender. G.S. and L.D.G. are FRIA fellows, and P.C. is a postdoctoral researcher of the FRS.-FNRS. Work in the B.V. laboratory is supported by FNRS (MIS F.4543.15), the Concerted Research Action, the Fondation ULB, the H2020 ITN “BtRAIN”, the Queen Elisabeth Medical Foundation, the FRFS-WELBIO (CR-2017S-05R) and the ERC (Ctrl-BBB 865176). The CMMI is supported by the European Regional Development Fund and the Walloon Region.
Author information
Authors and Affiliations
Contributions
All of the authors performed research and/or analysed data, discussed results and edited the manuscript. G.S. performed most of the experiments, designed experiments, analysed the data and prepared the figures. P.C., M.A., L.D.G., A.V., B.C., R.F.V.G. and M.M. performed or assisted in some experiments. S.G., L.H., M. Vanlandewijck and C.B. performed the bulk and single-cell RNA-seq and provided assistance with the bioinformatic analysis. M.D. and P.R. performed the MS analyses. M. Vermeersch and D.P.-M. performed the electron microscopy. B.V. designed the study, supervised the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Holger Gerhardt, Ghislain Opdenakker and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Characterization of the pre-angiogenic perineural endothelium.
a, Lateral views and quantifications of angiogenic sprouts in the 36 hpf hindbrain of WT, wnt7aa−/−, gpr124−/−, and reck−/− Tg(kdrl:EGFP) embryos (n ≥ 20 embryos from 3 independent experiments, arrowheads: CtA sprouts). Data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test. b, TC genotype in mosaic angiogenic sprouts during post-invasive secondary branching (42 hpf) of embryos obtained by transplanting wild-type (WT) kdrl:EGFP+ donor cells into gpr124 morpholino-injected kdrl:ras-mCherry hosts. The arrowhead points at a TC within the hindbrain. n = number of analysed mosaic vessels from 10 embryos recorded in 5 independent transplantation experiments. c, Heat map of scRNA-Seq transcriptome analysis of 30 hpf PHBC ECs showing the expression levels of the 25 highest ranked (lowest p-value) transcripts for each cell cluster. Ven: venous. d-h, t-SNE expression profiles of arterial markers (d), venous markers (e), BBB-associated transporters (f), tight junction proteins (g), and plvapb, a transcytosis marker (h). i, Lateral view of a WT Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryo at 30 hpf, counterstained with DAPI. The solid and open arrowheads point at some 7xTCF:GFP+ and 7xTCF:GFP− PHBC ECs, respectively. j, Immunostaining for β-Galactosidase (LacZ) and Erg1 in a transverse spinal cord section of a WT BAT-GAL E9.5 embryo, counterstained with DAPI. The solid and open arrowheads indicate some LacZ+ and LacZ− perineural vascular plexus (PNVP) ECs, respectively.
Extended Data Fig. 2 Contrasting Wnt–β-catenin and VEGF signalling functions during CNS angiogenesis.
a, Expression correlation matrix of Wnt–β-catenin target genes and TC markers based on the scRNA-Seq transcriptome of the 30 hpf PHBC ECs. Pearson correlation coefficients (r) and p-values were calculated in R. b, Density plots of Wnt–β-catenin target genes, TC markers and Vegf receptors in WT and gpr124 MO PHBC ECs. nUMI: normalized unique molecular identifier. p-values were determined using the parametric two-tailed Student’s t-test. c, t-SNE expression profiles of VEGF receptors within PHBC ECs. d, Expression correlation matrix between Wnt–β-catenin activity markers and Vegf receptor genes based on the scRNA-Seq transcriptome of the 30 hpf PHBC ECs. Pearson correlation coefficients (r) and p-values were calculated in R. e, Chromogenic WISH for kdrl in WT, gpr124+/−, and gpr124−/− embryos at 30 hpf. f, Dorsal views of WT, gpr124−/− and kdrl−/− Tg(kdrl:EGFP) embryos at 48 hpf. Black arrowheads point at the basilar artery (BA). g, Dorsal views of 48 hpf WT, gpr124−/−, or kdrl−/− Tg(kdrl:EGFP) embryos, co-injected or not at the one-cell stage with 10 pg of a pTol2-fli1a:kdrl-2A-nls-mTagBFP2 construct and 25 pg Tol2 transposase mRNA. The graphs show CtA numbers and proportion of embryos with a BA. n ≥ 9 embryos from 8 independent experiments. Data represent median ± interquartile range. p-values were determined using the two-tailed non-parametric Mann-Whitney test. h, Characterization of Ca2+ oscillations in WT, gpr124+/−, gpr124−/−, kdrl+/−, or kdrl−/− Tg(fli1a:Gal4FF);(UAS:GCaMP7a) PHBC ECs at pre-angiogenic stages (31 to 31.5 hpf). Frequency, area under peak, area under curve, and peak size data are represented as mean ± standard deviation. p-values were determined using the parametric one-way ANOVA test. The number of Ca2+-oscillating cells is represented as median ± interquartile range and analysed with the non-parametric Kruskal-Wallis test. n ≥ 11 embryos from 3 independent experiments.
Extended Data Fig. 3 RNA-Seq analysis reveals Wnt–β-catenin-dependent expression of mmp25a/b in CNS ECs.
a, Dorsal views of 30 hpf Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryos injected with control, gpr124, reck, or wnt7aa morpholinos and quantification of 7xTCF:GFP+ cells in the PHBCs. b, Left, dorsal views of untreated, DMSO, and IWR-1 (20 µM)-treated Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryos at 36 hpf. Centre, number of 7xTCF:GFP+ cells in the PHBCs at 36 hpf. Right, number of CtA sprouts at 36 hpf. In a,b, n ≥ 10 embryos from 3 independent experiments. Data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test. c, Volcano plots illustrating the differential RNA-Seq expression profiles between WT and Wnt-deficient PHBC ECs. d, Bulk RNA-Seq normalized expression values of mmp25a and mmp25b in WT or Wnt-deficient PHBC ECs. n = brain endothelia analysed by RNA-Seq. Data represent mean ± standard deviation. In c,d, p-values were determined using DESeq2 analysis. e, scRNA-Seq t-SNE expression profile of mmp25a and mmp25b and their combined expression score in PHBC-derived EC clusters. f, Expression correlation matrix between Wnt–β-catenin signalling target genes and mmp25a, mmp25b, or their combined score based on the scRNA-Seq transcriptome of the 30 hpf PHBC ECs. Pearson correlation coefficients (r) and p-values were calculated in R. g, Dorsal and dorso-lateral views of WISH for mmp25b in WT and gpr124−/− embryos at 36 hpf. The white arrowhead points at the PHBC, black arrowheads point at CtA TCs. TG: trigeminal nerve nuclei, CF: craniofacial nerve nuclei, PLL: posterior lateral line nuclei. h, Combined fluorescent in situ hybridization for mmp25b and anti-EGFP immunostaining of 30 hpf WT and gpr124−/− Tg(kdrl:EGFP) embryos. The magnified view shows expression in the PHBC and the underlying CF. i, Combined fluorescent WISH for mmp25b and anti-EGFP immunostaining of Tg(kdrl:EGFP) embryos in 38 hpf hindbrain when CtAs harbour two cells. The normalized EC signal intensity is shown on the right. n = 10 angiogenic sprouts from 10 embryos examined over ≥ 3 independent experiments. j, Normalized mmp25b fluorescence in PHBC and CtAs during embryogenesis. n ≥ 10 embryos examined from ≥ 3 independent experiments. In i,j, Data represent mean ± SD. p-values were determined using the parametric two-tailed Student’s t-test.
Extended Data Fig. 4 Mmp25 mutagenesis and organotypic requirement in zebrafish blood vessels.
a, Sequences of WT mmp25a and the mmp25aulb26 mutant allele. b, Sequences of WT mmp25b and the mmp25bulb27 mutant allele. c, Time-course analysis of the number of CtA and ISV angiogenic sprouts on one side of WT and mmp25a/b−/− embryos (n ≥ 4 embryos from 3 independent experiments). d-i, Comparison of the developing vasculature in WT and mmp25a/b mutant Tg(kdrl:EGFP) zebrafish (n ≥ 10 embryos or larvae from 3 independent experiments) during venous intersegmental vessel sprouting (vISV) at 36 hpf (d), caudal vein plexus angiogenesis (CVP) at 28 hpf (e), sub-intestinal vein (SIV) sprouting at 34 hpf (f) and 3.5 dpf (g), liver vascularization at 3 dpf (h), and formation of the myelencephalic choroid plexus (mCP) irrigating vessels at 54 hpf (i). DLV: dorsal longitudinal vein. PCeV: posterior cerebral vein. Arrowheads point at angiogenic sprouts. In all panels, data represent median ± .two-tailed Mann-Whitney test.
Extended Data Fig. 5 Phenotypic characterization of zebrafish mmp25a/mmp25b mutants.
a, Number of CtAs in 36, 40, 48, and 72 hpf embryos and larvae with different combinations of mutant mmp25a and mmp25b alleles. b, Number of connections to the basilar artery (BA) at 48 and 72 hpf. Data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test. c, Percentage of lumenized CtAs at 48 and 72 hpf. Data represent mean ± standard deviation. p-values were determined using the parametric one-sided ANOVA. For a-c, n = embryos analysed from ≥ 3 independent experiments. d, Lateral confocal images and quantification of normalized fluorescence (F/F0) of WT, mmp25a−/−/b+/−, and mmp25a/b−/− Tg(fli1a:Gal4FF);(UAS:GCaMP7a) PHBC ECs at pre-angiogenic stages (31 to 31.5 hpf). For each genotype, images and GCaMP intensity profiles are representative of ≥ 6 embryos recorded from 4 independent experiments. e, Dorsal views of 30 hpf WT, gpr124−/−, and mmp25a/b−/− Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryos (n ≥ 8 embryos from 3 independent experiments). The graph shows the number of 7xTCF:GFP+ PHBC ECs at 30 hpf. Data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test. f,g, Lateral views of angiogenic sprouts in the hindbrain at the initial step of brain vascular invasion (f, 36 hpf) or at later intraneural branching steps (g, 42 hpf) and in the trunk region (f, 24 hpf) of genetically mosaic embryos obtained by blastula-stage transplantation of wild-type (WT) kdrl:EGFP+ donor cells into mmp25a/b−/− kdrl:ras-mCherry hosts. White arrowheads point at TCs within the hindbrain. For f and g, n = distinct mosaic vessels from 8 embryos recorded in 3 independent transplantation experiments.
Extended Data Fig. 6 Mmp25 expression and organotypic angiogenic requirement in mice.
a-j, Normalized mouse Mmp25 expression levels in Cdh5+ ECs and whole organ extracts at E14.5 (a)77, ECs from different organs (Tie2+) at p7 (b)78, Cdh5+ adult ECs from various organs (c)79, single brain Tie2+ ECs at p7 (d)78, different cell types in the p7 cortex (e)80, brain ECs (CD31+; CD45−) at E15.5, p5, p9, and in adults (f)44, p7 and adult Tie2+ brain ECs (g)78, brain ECs (CD31+) of control (Cdh5CreERT2;mCherryTRAP) or Axin1iEC-OE (Axin1+/+; Cdh5CreERT2;mCherryTRAP) mice at E14.5 (h)81, adult brain ECs (CD31+) of control (Gpr124flox/+;ROSA-CreER), Gpr124 CKO (Gpr124flox/−;ROSA-CreER), control (Ctnnb1+/+; Cdh5-CreER), and β-catenin activated (Ctnnb1lox(ex3)/+; Cdh5-CreER) mice (i)82, and adult liver and lung ECs (Cdh5+) of control (Rosa-Bcat-GOF) and with β-catenin activated (Rosa-Bcat-GOF; Cdh5-CreERT2) mice (j)79. Data represent mean ± standard deviation. In e,f,h-j, n = number of animals analysed by RNA-seq. k, Isolectin B4 staining of blood vessels on sections (dashed lines) through the E10.5 WT and Mmp25−/− mouse fore- and midbrain. DAPI: 4′,6-diamidino-2-phenylindole. Magenta asterisks indicate CNS-penetrating angiogenic sprouts. Quantifications are provided on the right. Data represent median ± interquartile range, and p-values were determined using the non-parametric two-tailed Mann-Whitney test. l-p, Comparison of the vascular networks of WT and Mmp25 KO mice in the E10.5 forelimb buds (l), and the E12.5 intestinal wall (m), gastric wall (n), lung buds (o) and liver (p). Data represent mean ± SD. p-values were determined using the parametric two-tailed Student’s t-test. In k-p, n ≥ 7 embryos from 3 independent experiments.
Extended Data Fig. 7 Genetic interaction between mmp25 and pial basement membrane components during brain angiogenesis.
a-d, Lateral views and quantification of 36 hpf hindbrain CtAs (a,b) or 24 hpf trunk ISVs (c,d) in WT Tg(kdrl:EGFP) embryos injected at the one-cell stage with 150 pg of zCas9 mRNA and 60 pg of the illustrated sgRNAs. e, Left, schematics of Mmp25, Mmp9 and Mmp2, and its variants. Right, quantification of CtA sprouts in 36 hpf mmp25a/b mutant embryos, injected at the one-cell stage with 200 pg of the indicated mRNA. The diagram was created using BioRender. f, Transgenic fli1a-driven endothelial expression of an active or inactive form of Mmp25 in gpr124−/− Tg(kdrl:EGFP) embryos. Mmp25 and its variant are expressed as P2A-tagRFP fusions. Quantifications of 48 hpf CtAs (arrowheads) are shown on the right. g, Immunostaining of laminin-111 on a section through the E10.5 mouse forebrain and midbrain, counterstained with Isolectin B4 and DAPI. h, Dorsal views of 48 hpf Tg(kdrl:EGFP) mmp25 mutant embryos injected at the one-cell stage with a control or a lama1-targeting morpholino. i, Quantification of hindbrain CtAs in 48 hpf embryos, injected at the one-cell stage with 150 pg of zCas9 mRNA and 60 pg of the illustrated sgRNAs. j, Lateral views of 36 hpf Tg(kdrl:EGFP) embryos injected at the one-cell stage with a control or a lama1-targeting morpholino. k,l, Quantification of hindbrain CtA sprouts in 36 hpf mmp25a/b mutant embryos, injected at the one-cell stage with a control or laminin morpholinos (k), or with 150 pg of zCas9 mRNA and 60 pg of the illustrated sgRNAs (l). In all panels, data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test for multiple comparisons, and the Mann-Whitney test for the single comparison of mmp25b vs. mmp25b/mmp14 in b. In b,d-f,i,k,l n ≥ 10 embryos from 3 (b,d,i, k, l), 5 (e), or 6 (f) independent experiments.
Extended Data Fig. 8 Somatic gene disruption efficiencies in zebrafish and recombinant Mmp25 catalytic domains production in E.coli.
a,b, Targeting efficiencies (a) and SNPs and INDELs distributions (b) of embryos co-injected with 150 pg of zCas9 mRNA and 60 pg of the indicated targeting sgRNAs, as detected by Illumina amplicon deep sequencing. c, Coomassie blue staining of 400 ng of recombinant zebrafish and human his-tagged Mmp25 catalytic domains produced in E.coli, and purified on Ni-NTA resins. rzMmp25b: recombinant catalytic domain of zebrafish Mmp25, rhMMP25: recombinant catalytic domain of human MMP25. d, Western blot showing laminin-111 chains (black arrowheads) in 15 µg of Matrigel, after exposure or not to rhMMP25. e, Coomassie blue staining of 750 ng α1-antitrypsin (α1-AT, 2 µM), after exposure to 300 ng of rzMmp25b (2 µM) or 12 ng of rhMMP25 (75 nM). The black arrowheads point to α1-AT degradation products. The open arrowhead points to rzMmp25b. rhMMP25 is below detection limit. f,g, Quantification of hindbrain CtAs of control (mmp25a−/−;b+/−) and mmp25a/b−/− Tg(kdrl:EGFP) embryos at 36 hpf (f) and 48 hpf (g), injected or not (NI) at the one-cell stage with 150 pg of zCas9 mRNA and 60 pg of a serpina1 (sa1)-targeting sgRNA. Data represent median ± interquartile range. p-values were determined using the non-parametric two-tailed Mann-Whitney test. For f and g, n ≥ 10 embryos from 2 independent experiments.
Extended Data Fig. 9 Collagen IV isoform expression in zebrafish and mouse meningeal fibroblasts.
a, Immunostaining for laminin-111 on transverse hindbrain sections of 18, 24, and 30 hpf WT Tg(kdrl:EGFP) embryos, counterstained with DAPI. b, Combined fluorescent in situ hybridization for lama1 and immunostaining for EGFP of 18, 24, and 30 hpf WT Tg(kdrl:EGFP) transverse hindbrain sections, counterstained with DAPI. In a and b, the 30 hpf images are the same as in Figs. 3a and 3b, to facilitate the direct comparison with the earlier stages. c, Combined fluorescent in situ hybridization for col4a1-6 and immunostaining for EGFP of 30 hpf WT Tg(kdrl:EGFP) transverse hindbrain sections, counterstained with DAPI. The col4a5 and col4a6 images are the same as in Figs. 3d and 3e, respectively, to facilitate the comparisons with the other col4a isoforms. d,e, Combined fluorescent in situ hybridization for col4a5 (d) or col4a6 (e) and immunostaining for EGFP of 18 and 24 hpf WT Tg(kdrl:EGFP) transverse hindbrain sections, counterstained with DAPI. f, Cluster annotation of a whole brain scRNA-Seq analysis during embryonic stages, adapted from La Manno et al.40. g, Cell populations co-expressing the pan-fibroblast marker Col1a2 and the meningeal specific fibroblast markers S100a6 and Lama1. h, Pial fibroblast clusters and their marker genes as determined by unsupervised clustering. i, Expression profiles of Col4a1-6 isoforms. j, Normalized expression values for Col4a1-6 in mouse meningeal fibroblasts (Col1a1+) at E14.542. k, Normalized counts for Col4a1-6 transcripts in ECs (CD31+; CD45−) at E15.544. In j and k, Data represent mean ± standard deviation. n = number of individual meningeal fibroblasts (j) or brain endothelia (k) analysed by transcriptomics.
Extended Data Fig. 10 Genetic interaction between col4a5/6 and mmp25 or gpr124 during brain angiogenesis.
a, Quantification of hindbrain CtA sprouts in 36 hpf mmp25a/b mutant embryos injected at the one-cell stage with 150 pg of zCas9 mRNA and 60 pg of the indicated col4-targeting sgRNAs. b, Lateral views and quantification of hindbrain CtA sprouts in 36 hpf Tg(kdrl:EGFP) mmp25 mutant embryos injected at the one-cell stage with a control or col4-targeting morpholinos. c,d, Quantification of hindbrain CtA sprouts in WT, col4a5+/− or col4a5−/− embryos at 36 (c) and 48 hpf (d). In a-d, n ≥ 6 embryos from 3 independent experiments. e, Amino acid sequence alignment (Jalview v2.11.3.2) of the Mmp25 cleavage site (red arrowhead) within human (h), mouse (m), and zebrafish (z) Col4a1 and Col4a5. f, Anti-HA western blot of zCol4a5-HA-containing HEK293T extracts (or control pCS2+) treated or not with rhMMP25 or rhMMP2. g, Dorsal views of WT, gpr124 and col4a5 single or double mutant Tg(kdrl:EGFP) embryos at 48 hpf. h,i, Dorsal views (h) and quantification (i) of 7xTCF:GFP+ PHBC ECs in 30 hpf Tg(7xTCF-Xla.Siam:GFP);(kdrl:ras-mCherry) embryos injected with control, col4a5 and/or col4a6 MOs. n ≥ 10 embryos from 5 independent experiments. In a-d,i, data represent median ± interquartile range. p-values were determined using the non-parametric Kruskal-Wallis test. j,k, Combined fluorescent WISH for slc16a1a (j) or fabp11a (k) and immunostaining for EGFP in Tg(kdrl:EGFP) embryos injected at the one-cell stage with col4a5 and/or col4a6 MOs. On the right, proportion of slc16a1a or fabp11a-positive 48 hpf CtAs. In j and k, n = number of CtAs from 3 independent experiments. l, 2000 kDa dextran-rhodamine angiography in 72 hpf WT and gpr124−/− Tg(kdrl:EGFP) larvae injected with control or col4a5/6 morpholinos. m, 1 h before imaging, FITC-labelled 150 kDa dextran was injected intracardially into kdrl−/− 4 dpf (kdrl:ras-mCherry) larvae injected or not with a col4a5-targeting MO. Tracer accumulation was normalized to the intra-luminal fluorescence of the lateral dorsal aorta. n = 11 embryos from 3 independent experiments. Data represent mean ± SD. p-values were determined using the parametric two-tailed Student’s t-test.
Supplementary information
Supplementary Fig. 1
Raw, unprocessed images of Coomassie blue gels and western blots relating to Fig. 3j,k,m and Extended Data Figs. 8c–e and 10f.
Supplementary Video 1
Brain vascular invasion is initiated by Wnt–β-catenin signalling-competent TCs in genetically mosaic zebrafish.
Supplementary Video 2
Intraneural vessels can be guided by Wnt–β-catenin signalling-incompetent TCs in genetically mosaic zebrafish.
Supplementary Video 3
PHBCs contain Wnt–β-catenin signalling-positive and -negative ECs, while CNS-invading TCs are always positive.
Supplementary Video 4
Wnt–β-catenin signalling is dispensable for VEGF-dependent endothelial Ca2+ oscillations in the perineural endothelium.
Supplementary Video 5
Intravital imaging of hindbrain angiogenesis in WT and mmp25a/b−/− zebrafish embryos.
Supplementary Video 6
Intravital imaging of intersegmental vessel formation formation in WT and mmp25a/b−/− zebrafish embryos.
Supplementary Video 7
Mmp25 is dispensable for VEGF-dependent endothelial Ca2+ oscillations in the perineural endothelium.
Supplementary Video 8
mmp25a/b−/− ECs are outcompeted by WT cells during brain angiogenesis.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schevenels, G., Cabochette, P., America, M. et al. A brain-specific angiogenic mechanism enabled by tip cell specialization. Nature 628, 863–871 (2024). https://doi.org/10.1038/s41586-024-07283-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07283-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
