
Abstract
Acquired drug resistance to anticancer targeted therapies remains an unsolved clinical problem. Although many drivers of acquired drug resistance have been identified1,2,3,4, the underlying molecular mechanisms shaping tumour evolution during treatment are incompletely understood. Genomic profiling of patient tumours has implicated apolipoprotein B messenger RNA editing catalytic polypeptide-like (APOBEC) cytidine deaminases in tumour evolution; however, their role during therapy and the development of acquired drug resistance is undefined. Here we report that lung cancer targeted therapies commonly used in the clinic can induce cytidine deaminase APOBEC3A (A3A), leading to sustained mutagenesis in drug-tolerant cancer cells persisting during therapy. Therapy-induced A3A promotes the formation of double-strand DNA breaks, increasing genomic instability in drug-tolerant persisters. Deletion of A3A reduces APOBEC mutations and structural variations in persister cells and delays the development of drug resistance. APOBEC mutational signatures are enriched in tumours from patients with lung cancer who progressed after extended responses to targeted therapies. This study shows that induction of A3A in response to targeted therapies drives evolution of drug-tolerant persister cells, suggesting that suppression of A3A expression or activity may represent a potential therapeutic strategy in the prevention or delay of acquired resistance to lung cancer targeted therapy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
Whole-genome and -exome sequencing data on clinical tumour samples are available at dbGaP under accession no. phs003256.v1.p1 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003256.v1.p1). DNA (whole-genome and whole-exome), RNA (whole-transcriptome) and ATAC–seq data on experimental models are available from the NIH Sequence Read Archive under BioProject ID PRJNA941908 (https://www.ncbi.nlm.nih.gov/bioproject/) and at the GEO (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE75602. Sequencing metrics and mutational analyses are provided as Supplementary Tables 4–9. Uncropped immunoblot images and flow cytometry data are provided as Supplementary Data Figs. 1–5. The code used to run the ApoTrack analysis is available at https://github.com/m0s0lawrence/ApoTrack.
References
Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005).
Engelman, J. A. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 (2007).
Sequist, L. V. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl Med. 3, 75ra26 (2011).
Gainor, J. F. et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 6, 1118–1133 (2016).
Salter, J. D., Bennett, R. P. & Smith, H. C. The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci. 41, 578–594 (2016).
de Bruin, E. C. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 (2014).
Roberts, S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976 (2013).
Burns, M. B. et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013).
Cortez, L. M. et al. APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 15, e1008545 (2019).
Law, E. K. et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. https://doi.org/10.1084/jem.20200261 (2020).
Hata, A. N. et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 22, 262–269 (2016).
Shaw, A. T. et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med. 374, 54–61 (2016).
Yoda, S. et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 8, 714–729 (2018).
Nik-Zainal, S. et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012).
Mas-Ponte, D. & Supek, F. DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nat. Genet. 52, 958–968 (2020).
Endesfelder, D. et al. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+ breast cancer. Cancer Res. 74, 4853–4863 (2014).
Petljak, M. et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell 176, 1282–1294 (2019).
Jalili, P. et al. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat. Commun. 11, 2971 (2020).
Chan, K. et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 47, 1067–1072 (2015).
Buisson, R. et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science https://doi.org/10.1126/science.aaw2872 (2019).
Ito, F., Fu, Y., Kao, S. A., Yang, H. & Chen, X. S. Family-wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J. Mol. Biol. 429, 1787–1799 (2017).
Petljak, M. et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature 607, 799–807 (2022).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Sharma, S. et al. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 6, 6881 (2015).
Sharma, S., Patnaik, S. K., Kemer, Z. & Baysal, B. E. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol. 14, 603–610 (2017).
Sharma, S. & Baysal, B. E. Stem-loop structure preference for site-specific RNA editing by APOBEC3A and APOBEC3G. PeerJ 5, e4136 (2017).
Oh, S. & Buisson, R. A digital PCR-based protocol to detect and quantify RNA editing events at hotspots. STAR Protoc. 3, 101148 (2022).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Lee, J. K. et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. 35, 3065–3074 (2017).
Offin, M. et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J. Thorac. Oncol. 14, 1784–1793 (2019).
Niederst, M. J. et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 6, 6377 (2015).
Piotrowska, Z. et al. Heterogeneity and coexistence of T790M and T790 wild-type resistant subclones drive mixed response to third-generation epidermal growth factor receptor inhibitors in lung cancer. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00263 (2018).
Oh, S. et al. Genotoxic stress and viral infection induce transient expression of APOBEC3A and pro-inflammatory genes through two distinct pathways. Nat. Commun. 12, 4917 (2021).
Bivona, T. G. et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 471, 523–526 (2011).
Blakely, C. M. et al. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 11, 98–110 (2015).
Burrell, R. A. et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013).
Choudhury, N. J. et al. Molecular biomarkers of disease outcomes and mechanisms of acquired resistance to first-line osimertinib in advanced EGFR-mutant lung cancers. J. Thorac. Oncol. https://doi.org/10.1016/j.jtho.2022.11.022 (2022).
Grillo, M. J., Jones, K. F. M., Carpenter, M. A., Harris, R. S. & Harki, D. A. The current toolbox for APOBEC drug discovery. Trends Pharmacol. Sci. 43, 362–377 (2022).
Crystal, A. S. et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486 (2014).
Bailey, M. H. et al. Retrospective evaluation of whole exome and genome mutation calls in 746 cancer samples. Nat. Commun. 11, 4748 (2020).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Lin, J. J. et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis. Oncol. 4, 21 (2020).
Dardaei, L. et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 24, 512–517 (2018).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Ellrott, K. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281 (2018).
Drier, Y. et al. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 23, 228–235 (2013).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Chen, Y., Lun, A. T. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 5, 1438 (2016).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Khan, A. et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 (2018).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Buisson, R., Lawrence, M. S., Benes, C. H. & Zou, L. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 77, 4567–4578 (2017).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Gyori, B. M., Venkatachalam, G., Thiagarajan, P. S., Hsu, D. & Clement, M. V. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2, 457–465 (2014).
Raoof, S. et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 38, 6399–6413 (2019).
Acknowledgements
We thank all members of the Hata and Benes Lab for helpful discussions and feedback. This study was funded by support from the NIH: nos. K08 CA197389 (A.N.H.), R01 CA249291 (A.N.H.), R37 CA252081 (R.B.), R01 CA137008 (L.V.S. and A.N.H.), R01 CA164273 (J.J.L. and A.N.H.), U01CA220323 (N.J.D.) and P50 CA265826 (A.N.H. and L.V.S.); by the Doris Duke Charitable Foundation Clinical Scientist Development Award (A.N.H.), Smith Family Foundation Award (A.N.H.), Rullo Family Innovation Award (M.S.L.), SU2C/NSF/V Foundation Convergence Award (A.N.H., L.V.S. and C.S.C.), the Ludwig Center at Harvard (A.N.H.), Tosteson & FMD Award (H.I.), Lung Cancer Research Foundation (H.I.), the Lungstrong foundation, Targeting a Cure for Lung Cancer, Be a Piece of the Solution, the Landry Family and the Suzanne E. Coyne Family.
Author information
Authors and Affiliations
Contributions
H.I., A.N.H. and M.S.L. designed the study, analysed data and wrote the paper. H.I., N.N., W.S., S.M., M.S., H.F.C., F.M.S., D.T., H.L.A., V.N. and A.N.H. performed cell line experiments investigating drug sensitivity and evolution of resistance, APOBEC expression analysis and cell signalling pathway studies. S.O., P.J. and R.B. performed RNA editing studies. H.I. and M.S. performed DNA damage experiments. H.I., N.N., H.F.C., N.P., S.B. and M.G.-C. performed tumour xenograft studies. K.D. and A.R. generated patient-derived cell lines from patients with NSCLC. R.S., A.A., A.L., M.L., C.J.O., C.S.C., J.J.-K.L., Y.E.M. and M.S.L. performed computational analysis of WGS and WXS, RNA-seq and ATAC–seq of cell culture experimental models and clinical tumour samples. L.Z., N.J.D., C.B., G.G., R.B. and J.A.E. were involved with experimental design and data interpretation. M.K.B., R.G.C., A.T.S., J.F.G., J.J.L., L.V.S. and Z.P. provided samples from patients with NSCLC and carried out clinical data interpretation. B.Y.Y. performed statistical analysis for patient samples. A.N.H. and M.S.L. contributed equally to the study. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.N.H. has received grants/research support from Amgen, Blueprint Medicines, BridgeBio, Bristol-Myers Squibb, C4 Therapeutics, Eli Lilly, Novartis, Nuvalent, Pfizer, Roche/Genentech and Scorpion Therapeutics; and has served as a compensated consultant for Engine Biosciences, Nuvalent, Oncovalent, TigaTx and Tolremo Therapeutics. C.J.O. has received research support from Gilead, Scorpion Therapeutics and eFFECTOR Therapeutics. G.G. has received research funds from IBM and Pharmacyclics; is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, SignatureAnalyzer-GPU and MinimuMM-seq; and is a founder, consultant and has privately held equity in Scorpion Therapeutics. J.F.G. has served as a compensated consultant and/or had advisory roles for Amgen, Blueprint Medicines Corporation, BMS, Genentech/Roche, Gilead Sciences, Jounce Therapeutics, Lilly, Loxo Oncology, Merck, Mirati, Silverback Therapeutics, Sanofi, GlydeBio, Moderna Therapeutics, Oncorus, Regeneron, Takeda, Merus, Novocure, Curie Therapeutics, AI Proteins, AstraZeneca, Jazz Pharmaceuticals, InterVenn and Pfizer; has stock and ownership in Ironwood Pharmaceuticals (immediate family member) and AI Proteins; has received Honoraria from Merck, Novartis, Pfizer and Takeda; has received research funding from Adaptimmune, ALX Oncology, Array BioPharma, AstraZeneca, Blueprint Medicines Corporation, BMS, Genentech, Jounce Therapeutics, Merck, Novartis and Tesaro; and has an immediate family member who is an employee of Ironwood Pharmaceuticals. J.J.L. has served as a compensated consultant or received honorarium from Chugai Pharma, Boehringer-Ingelheim, Pfizer, C4 Therapeutics, Nuvalent, Turning Point Therapeutics, Blueprint Medicines, Mirati Therapeutics, Novartis, Elevation Oncology, Bayer, Genentech and Regeneron; received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Roche/Genentech, Pfizer, Linnaeus Therapeutics, Elevation Oncology, Nuvalent and Novartis; and received travel support from Pfizer. L.V.S. has served as a compensated consultant for Genentech, AstraZeneca and Janssen, and has received institutional research support from BI, AZ, Novartis, Genentech, LOXO and Blueprint Medicines. M.L. has served as a compensated consulted for C4 Therapeutics. R.B. has served as a compensated consultant for Pfizer and Health Advances. Y.E.M. has served as a compensated consultant for Foresee Genomics. Z.P. has served as a compensated consultant and/or received honoraria from Sanofi, Janssen, Taiho, Takeda, AstraZeneca, Eli Lilly, Daiichi Sankyo, Cullinan Oncology, C4 Therapeutics, Jazz Pharmaceuticals, Blueprint Medicines, InCyte and Genentech, travel support from AstraZeneca and research funding (to institution) from Novartis, Takeda, Spectrum, AstraZeneca, Tesaro/GSK, Cullinan, Daiichi Sankyo, AbbVie, Janssen and Blueprint. M.S. is currently an employee of ImmunoGen, C.B. and A.T.S. are currently employees of Novartis, Inc. and J.A.E. is currently an employee of Treeline Biosciences (their contributions to the manuscript occurred while they were employees of Massachusetts General Hospital). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Mariam Jamal-Hanjani, Charles Rudin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 APOBEC mutational signatures in NSCLC patients with compound resistance mutations.
a, Lego plots of the 7 mutational signatures resolved by NMF with assigned biological process. Smoking and ROS (reactive oxygen species) mutations were mixed. For simplicity, MSI (microsatellite instability), POLE (mutant polymerase epsilon), FFPE (formalin-fixed, paraffin-embedded) and Misc (miscellaneous) signatures are combined and plotted as “Other” throughout the manuscript. Base substitutions were classified into six subtypes and each category was represented by a different color. Base substitutions were further divided into 96 possible mutation types according to the flanking nucleotides surrounding the mutated base. b, Treatment history and acquired compound mutations in MGH086. Treatment lines are colored blue (first-line TKI), green (second-line TKI), yellow (third-line TKI). Five ALK amino acid changes result from base substitutions at TpC sites. c, Mutational signatures (right panel) and clonal relationship of pre-treatment biopsies (left panel) and autopsy sites from patient MGH10032 after sequential EGFR TKI therapy (middle panel). Amino acid substitutions are indicated for each resistance mutation. Branches colors depict the fraction of mutations corresponding to an APOBEC signature. Branches length represents APOBEC mutation number. Treatment timelines were colored blue (first-line TKI) and green (second-line TKI). N, normal. d–f, Mutational signatures and clonal relationship of multiple sequential biopsies from patient MGH95313, MGH808 and MGH98713 over the course of sequential TKI therapy. Amino acid substitutions are indicated for each resistance mutation. Branch colors depict the fraction of mutations corresponding to an APOBEC signature. Branch length represents mutation count. Treatment timelines were colored blue (first-line TKI), green (second-line TKI), yellow (third-line TKI). N, normal. g, Clonal relationship of MGH119-1 gefitinib resistant clones.
Extended Data Fig. 2 Detailed clinical histories and mutational heterogeneity of compound mutation cohort.
a, Sample collection timing and resistant driver mutations. Treatment time (month) lines are colored gray (chemotherapy), blue (first-line TKI), green (second-line TKI), yellow (third-line TKI). Samples were derived from surgical resection, biopsy, autopsy or pleural fluid (L lung, left lung; LN, lymph node; PDC, patient-derived cell line; LLL, left lower lobe). b, Heatmaps depict presence of mutations in each sample from MGH086, MGH10032, MGH953, and MGH808. Truncal mutations present in all samples are colored dark gray, shared mutations common to 2 or more samples are colored blue, and private mutations are colored green. APOBEC mutations are indicated in red. The absence of a mutation is denoted by light gray. The primary and resistant driver mutations are shown on the left. The sample numbers correspond to Fig. 1a, Extended Data Fig. 1c–e.
Extended Data Fig. 3 TKI-resistant clones that evolve from drug-tolerant persister cells accumulate APOBEC mutations.
a, Number and percentage of TCT>TGT and TCA>TGA mutations that are highly specific for APOBEC43 in early (E) and late (L) resistant clones (bar: median, two-sided Mann Whitney test). b, Number of omikli (n = 2, 3, or 4 mutations with an intermutational distance < 1 kb) and kataegis (n >= 5 mutations with an intermutational distance of < 1 kb) mutation clusters in early and late resistant clones. c, Shared and private mutations in early and late resistant clones. Shared mutations in early resistant clones refer to mutations shared across early clones from one individual. Private mutations in resistant clones refer to mutations observed in only one sample of an individual and are not shared across other samples. d, Phylogenetic tree depicting evolutionary relationships of early and late resistant clones based on pattern of shared and private mutations. e, Mutational signatures of private and shared mutations in PC9 early and late resistant clones. Late resistant clones exhibited significantly higher private APOBEC mutation percentage compared to early resistant clones (two-sided Mann Whitney test). f, Relationship between timing of EGFRT790M mutation, TKI treatment and APOBEC mutational signatures for shared and private mutations.
Extended Data Fig. 4 Chromosomal aberrations in TKI-resistant clones.
a, Copy number profiles of early and late resistant PC9 clones. b, Copy number changes in each sample are shown on scatter plots where the y-axis is the copy number in the sample, and the x-axis is the copy number in the parental sample it was derived from. Genomic copy number segments are shown for both the minor allele (blue) and major allele (red). Size of points corresponds to the size of the copy-number segments. Points along the diagonal represent genomic segments that did not change in copy number between the parental and derived sample. Points above and below the diagonal represent copy-number gains and losses, respectively, relative to the parental sample (Right panels, two-sided Mann Whitney test). c, Late-evolving PC9 clones have increased SVs compared with early resistant clones. SVs were determined relative to parental cells using dRanger and BreakPointer. d, Circos plots depicting APOBEC mutations, copy number changes, kataegis/omikli mutation clusters and intra/inter-chromosomal interactions in PC9 resistant clones (all relative to parental cells).
Extended Data Fig. 5 APOBEC mutations do not occur spontaneously while culturing cells in the absence of drug.
a, Determination of the number of persister doublings prior to EGFRT790M acquisition (n = 300, Tukey box and whisker plots). b, Experimental schema. Two rounds of single cell cloning were performed using PC9, E1 (GR2, early resistant, no prior APOBEC) and L2 (late resistant, prior APOBEC) cells. Single cells were expanded ~20-25 doublings to match the number of persister doublings prior to acquisition of EGFRT790M. Whole-genome sequencing was performed on expanded clones. c, APOBEC mutation percentage of shared and private mutations. Shared mutations refer to mutations shared between sequential clones from one individual cell line and represent mutations that were acquired in the distant past. Private mutations refer to mutations observed in only one sample of an individual cell line and are not shared with paired sample. The enrichment of APOBEC signature in shared L2 mutations reflects known accumulation of APOBEC mutations that were acquired during TKI treatment prior to this study. Private mutations that were newly acquired in PC9, E1 (GR2) and L2 cells correspond to those that occurred spontaneously during culture in the absence of drug.
Extended Data Fig. 6 Lung cancer targeted therapies induce APOBEC3A expression.
a, mRNA expression of APOBEC family genes in response to osimertinib. PC9 cells were treated with 1 μM osimertinib for up to 14 days and gene expression was determined by quantitative RT-PCR. Data are expressed as log2 fold change (FC) relative to untreated control. b, mRNA expression levels (CPM: counts per million) of APOBEC family genes determined by RNA-seq in EGFR-mutant NSCLC cell lines treated with 300 nM gefitinib for 0, 1 and 14 days. Data correspond to BioProject ID PRJNA941908 (ref. 64) c, Schema for allele specific droplet digital PCR assay for quantifying A3A editing at the DDOST hairpin hotspot (adapted from Jalili et al.18). d, e, Expression of A3A or A3B in PC9 with Tet-On flag-tagged wild-type A3AWT/A3BWT or catalytic inactive mutant A3AE72A/A3BE255Q constructs. Cells were treated with 200 ng/mL doxycycline (DOX) treatment for 72 h. Protein expression was confirmed by western blot (d). mRNA expression levels were determined by quantitative RT-PCR (e). Data are expressed as relative to untreated control (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). f, DDOST mRNA editing in PC9 cells overexpressing wild-type A3AWT/A3BWT and catalytic inactive A3AE72A/A3BE255Q mutants. Cells were treated with 200 ng/mL DOX treatment for 72 h. DDOST editing was determined by ddPCR assay (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). g, Change of tumor volume in EGFR-mutant NSCLC xenograft models treated with osimertinib at the time of harvesting minimal residual disease (MRD) for DDOST hairpin hotspot analysis in Fig. 2c. h, Top edited hairpin hotspot sites (supported by at least 2 edited reads) in PC9 cells in ApoTrack mRNA-seq analysis in Fig. 2e. The majority are predicted to be synonymous mutations. None of the missense mutations have been reported to be recurrently mutated in cancer.
Extended Data Fig. 7 Induction of A3A is a common response of oncogene-driven lung cancer cells treated with targeted therapies.
a, Expression of APOBEC family genes in ALK fusion-positive NSCLC cells after treatment with 100 nM lorlatinib as determined by quantitative RT-PCR (3 biological replicates each). b, DDOST mRNA editing in ALK fusion-positive NSCLC cell lines treated with 100 nM lorlatinib for up to 14 days. DDOST editing was determined by ddPCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). c, Expression of A3A and A3B in H358 KRASG12C NSCLC cells treated with 1 μM AMG 510, 1 μM ARS-1620 or 100 nM trametinib for up to 72 h (3 biological replicates each). d–e, mRNA-seq was performed on H358 KRASG12C NSCLC cells treated with ARS-1620 or trametinib to quantify DDOST (d) and global transcriptome editing (ApoTrack) (e). f–g, Correlation between DDOST mRNA editing activity and mRNA expression of A3A (f) or A3B (g) in EGFR-mutant and ALK fusion-positive NSCLC cell lines. Data is adapted from Fig. 2a, 2b, Extended Data Fig. 7a, 7b (r = Pearson product-moment correlation coefficient). h, Correlation between log2 fold change and baseline mRNA expression of A3A or A3B. Data was adapted from Fig. 2a, 2b, Extended Data Fig. 7a, 7b (r = Pearson product-moment correlation coefficient).
Extended Data Fig. 8 TKI-induced APOBEC activity and evolutionary history of evolving resistant clones.
a, A3A and A3B expression levels from RNA-seq (GSE75602) performed on parental PC9 cells, PC9 DTP cells after 14 days of gefitinib treatment (GP), early EGFRT790M resistant clone PC9-GR2 and late EGFRT790M resistant clone PC9-GR3 (previously described in Hata and Niederst et al.11). PC9 cells were treated with gefitinib, PC9-GR2/GR3 cells were treated with the third-generation EGFR inhibitor WZ4002 (all for 24 h). b, Percentage of DDOST hotspot reads with A3A editing in PC9 and PC9-GR2/GR3 cells treated with gefitinib or WZ4002, respectively. c, A3A induction and suppression of EGFR downstream signaling. NSCLC cells were treated with or without 1 μM gefitinib, 1 μM osimertinib, 100 nM trametinib or 1 μM GDC0941 for 24 h. Expression of A3A was determined by quantitative RT-PCR (upper panel). Data are expressed as log2 fold change relative to non-treated control (NT) (mean ± s.d. of 3 biological replicates). Western blot of AKT and ERK phosphorylation (lower panel). d, Summary of the relationship between EGFR signaling and A3A mRNA expression during targeted therapy in PC9 early and late resistant clones.
Extended Data Fig. 9 A3A vs A3B character of mutations in resistant tumors derived from NSCLC Patients.
a-d, A3A (YTCA) and A3B (RTCA) character (upper panels) and A3A hairpin motif mutations (lower panels) of pre- and post-treatment MGH119-1 (a), PC9 (b, c) and patient tumors (d). A reference set of ~2600 WGS-analyzed tumors from the International Cancer Genome Consortium (ICGC) Pan-Cancer Analysis of Whole Genomes (PCAWG) project is plotted for comparison. Samples were designated APOBEC+ if ≥10% of total mutations were assigned to the APOBEC mutation signature by NMF analysis, then classified according to A3A vs. A3B character: A3A-dominated samples (enrichment of mutations at YTCA tetranucleotides) are colored red, A3B-dominated samples (enrichment of mutations at RTCA tetranucleotides) are colored blue. Samples with 2.5-10% APOBEC mutations are colored grey. APOBEC- samples (< 2.5% APOBEC mutations) are colored green. e, A3A (YTCA) and A3B (RTCA) character (upper panels) or A3A hairpin motif mutations (lower panels) of transformed small cell lung cancer (SCLC) (red symbols) and corresponding lung adenocarcinoma (LUAD) (gray symbols). Tumors from the same patient are connected by a dashed line. Right panels depict pre- and post-transformation tumors described in Lee et al.29 (PCD, patient-derived cell line; lu, lung metastasis; li, liver metastasis; d, diaphragm metastasis) f, Metastatic sites and mutational signatures from MGH7 autopsy31. g, Clinical history, mutational signatures of serial biopsies from patient MGH77232 (RUL, right upper lobe, PDC, patient-derived cell line).
Extended Data Fig. 10 NFκB1 mediates TKI-induced A3A expression.
a, Chromatin accessibility profile of the APOBEC3 locus (chromosome 22) of PC9 cells treated with 300 nM gefitinib for 0 or 14 days (2 biological replicates), with expanded view of the A3A promoter region. Relative fold-change of peak accessibility in treated vs. non-treated control cells is shown. Transcription factors mapping to these peak regions were identified from the ENCODE database. b, Differential z-score (gefitinib vs. control) of global motif accessibility scores for each identified transcription factor family. c, Knockdown efficacy of each SMARTPool siRNA in PC9 cells. (SCR = scrambled control siRNA) d, Expression of A3A in PC9 cells transfected scrambled or individual NFκB1 siRNAs. Cells were treated with osimertinib for 24 h and mRNA expression was determined by quantitative RT-PCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). Knockdown efficacy was confirmed by western blot. e, Knockdown efficacy of each SMARTPool siRNA in PC9 cells. f, Western blot of PC9 and MGH064-1 cells after treatment with TKI (osimertinib or lorlatinib) and IKKβ inhibitors (representative of 3 biological replicates).
Extended Data Fig. 11 TKI-induced APOBEC3A increases DNA damage and emergence of drug-tolerant cells.
a, TKI-induced DDOST RNA editing in PC9 CRISPR scrambled (SCR) or A3A knockout (KO) clones (cl, clone; sg, sgRNA). Cells were treated with or without 1 μM osimertinib for up to 3 days and DDOST editing was determined by digital PCR (SCR cl3, n = 4; others, n = 3 biological replicates, mean ± s.d., two-sided Student’s t-test). b, Acquired SNVs stratified by mutation signature, and shown as count (upper) or percentage (lower) in resistant clones derived from MRD mice tumors (M = MRD mouse; -1, -2, -3 = single cell clone 1, 2, 3). Each MRD clone was compared to its matched pre-treatment clone to identify acquired mutations. c, Expression of A3A in H3122 CRISPR SCR or A3A KO cells treated with 1 μM lorlatinib for up to 3 days. mRNA expression was determined by quantitative RT-PCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). d, Acquired SNVs stratified by mutation signature, and shown as count (upper) or percentage (lower) in clones derived from H3122 DTP cells. H3122 CRISPR SCR or A3A KO cells were treated with 0, 0.3, 1 μM lorlatinib for 8 weeks and single cell clones were expanded for WGS (-1, -2, -3 = single cell clone 1, 2, 3). Each DTP clone was compared to its matched pre-treatment clone to identify acquired mutations. Of note, only a single H3122 A3A KO clone could be recovered after lorlatinib treatment due to the increase in drug sensitivity associated with knockout of A3A. e, Acquired APOBEC mutation number (left) or percentage (right) in DTP clones. Each resistant clone was compared to its matched pre-treatment clone to identify acquired mutations (A3A KO sg2cl1, n = 1; others n = 3, mean ± s.d.). f, Representative images of cells in G1, S and G2 phase; scale bars = 10 μm (upper). Scatter plots of EdU cell cycle assay in Fig. 3i (lower). PC9 cells were treated with 1 μM osimertinib for 14 days and stained with EdU/DAPI to resolve cell cycle phase, and γH2AX to quantify DNA damage. g, Western blot for APOBEC3B (A3B) expression in PC9 CRISPR A3B KO or siRNA A3B. h, Survival of PC9 Tet-On overexpressing A3A. Cells were treated with 1 μM osimertinib for 4 weeks and DTP colonies were visualized by crystal violet. Colony area was quantified by imageJ. The individual and aggregate area of the largest 30 colonies for each experimental condition is shown. i, MGH119-1 A3AWT or A3AE72A cells were treated with 200 ng/mL DOX for 72 h to induce A3A expression. mRNA expression was determined by quantitative RT-PCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). j, Knockdown efficacy of PC9 (left) or H3122 (right) transduced with A3A shRNA. Cells were treated with 1 μM gefitinib or 1 μM lorlatinib. mRNA expression was determined by quantitative RT-PCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). k, Re-expression of A3A in PC9 CRISPR A3A KO cells (“A3A back”). Cells were treated with 200 ng/mL DOX for 72 h to induce A3A expression, which was verified by quantitative RT-PCR (mean ± s.d. of 3 biological replicates, two-sided Student’s t-test). l, Schema of competition assay using fluorescently labeled A3A KO/SCR cells. H3122 CRISPR SCR (GFP labeled) and A3A KO (RFP labeled) cells were mixed in a 1:1 ratio and treated with 1 μM lorlatinib for 4 weeks.
Extended Data Fig. 12 APOBEC mutations are enriched in NSCLCs with acquired TKI resistance.
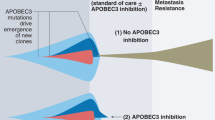
a, Distribution of tumor samples by TKI treatment timepoint. b, Treatment history and sample collection time of 24 NSCLC patients. Time on treatment was counted from the starting date of 1st-line TKI treatment. The following patients received chemotherapy treatment before 1st-line TKI treatment: MGH10032, MGH086, MGH778, MGH130, MGH903, MGH742 and MGH804. MGH patient codes and order correspond to Fig. 5a. The treatment timeline is colored according to specific therapy received. c, Number of post-TKI timepoints per patient. d, Distribution of APOBEC mutation percentages in all samples. APOBEC mutation >= 20% was defined as APOBEC “high” and less than 20% was “low”. e, Frequency of APOBEC-high tumors in all patients (n = 24). f, Schema of early versus late acquired resistance. Tumor samples were divided into two groups based on duration of TKI treatment: less (early) or greater (late) than 18 months. g, Frequency of patients APOBEC-high tumors using different thresholds for defining high versus low (APOBEC mutation >= 15% or 25%). Two-sided McNemar’s test was used to compare the two groups (e,g). h, Conceptual diagram for evolutionary trajectory and APOBEC mutational burden of evolving persistent cancer cells during targeted therapy. i, Examples of acquired (validated) driver mutations in the APOBEC context.
Supplementary information
Supplementary Information
This file contains Supplementary Figs. 1–5.
Supplementary Tables 1–3
Cell line and PDX model characteristics.
Antibodies used in this study.
Oligonucleotides used in this study.
Supplementary Table 4
Mutation signatures and A3A/A3B mutation characteristics of experimental models.
Supplementary Table 5
Mutational signatures and A3A/A3B mutation characteristics of patient tumor samples.
Supplementary Table 6
Individual mutation calls in experimental models.
Supplementary Table 7
Individual mutation calls in patient tumor samples.
Supplementary Table 8
RNA-seq of EGFR mutant NSCLC cell lines treated with gefitinib for 0, 1 or 14 days.
Supplementary Table 9
ATAC-seq of PC9 cells treated with or without gefitinib.
Supplementary Table 10
Potential cancer driver mutations in PC9 Early and Late resistant clones.
Supplementary Table 11
Potential cancer driver mutations in patient tumor samples.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Isozaki, H., Sakhtemani, R., Abbasi, A. et al. Therapy-induced APOBEC3A drives evolution of persistent cancer cells. Nature 620, 393–401 (2023). https://doi.org/10.1038/s41586-023-06303-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06303-1
This article is cited by
-
Mesoscale DNA features impact APOBEC3A and APOBEC3B deaminase activity and shape tumor mutational landscapes
Nature Communications (2024)
-
Distinguishing preferences of human APOBEC3A and APOBEC3B for cytosines in hairpin loops, and reflection of these preferences in APOBEC-signature cancer genome mutations
Nature Communications (2024)
-
Cytidine deaminases APOBEC3C and APOBEC3D promote DNA replication stress resistance in pancreatic cancer cells
Nature Cancer (2024)
-
The role of APOBEC3B in lung tumor evolution and targeted cancer therapy resistance
Nature Genetics (2024)
-
Acute expression of human APOBEC3B in mice results in RNA editing and lethality
Genome Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
