
Abstract
The flux balances of carbon and chlorine between subduction into the deep mantle and volcanic emissions into the atmosphere are crucial for the habitability of our planet1,2. However, pervasive loss of fluids from subducting slabs has been thought to cut off the delivery of both carbon and chlorine to the deep mantle owing to their high mobility under hydrous conditions3,4. Our new high-pressure experiments show that most carbonates (>75 wt%) in carbonate-rich crustal rocks—one of the main subducting carbon reservoirs—survive devolatilization and hydrous melting in cold and warm subduction zones, indicating that their subduction has driven the deep carbon cycle since the Mesoproterozoic. We found that KCl and NaCl, respectively, become stable phases crystallizing from hydrous carbonatite melts with low chlorine solubility in warm and hot subduction zones, resulting in the sequestration of chlorine in the solid residue in downwelling slabs. Accordingly, the subduction of carbonate-rich rocks facilitated highly effective recycling of both chlorine and carbon into the deep mantle at intermediate stages of Earth’s history and led to declining atmospheric pCO2 and the formation of carbon-rich and chlorine-rich mantle reservoirs since the Mesoproterozoic. This period of optimal carbon and chlorine subduction may explain the ages of eclogitic diamonds and the formation of the HIMU mantle source.
Similar content being viewed by others


Main
The exchange of volatile elements including carbon and chlorine between the Earth’s deep mantle and the surface dominates the secular evolution of the climate, oceans and life1,2,5. Continual outflux of carbon and chlorine from the mantle through volcanism at mid-ocean ridges, ocean islands and island arcs requires the replenishment of mantle volatile reservoirs by the subduction of surface volatiles6. Otherwise, a persisting imbalance would have led to a volatile-starved mantle and CO2-rich and chlorine-rich atmosphere/ocean, destroying the habitability of our planet7,8. A key issue in deep volatile cycles is the ability of subducting slabs to transport volatiles from the Earth’s surface to the deep mantle. Modern subduction delivers large amounts of crustal carbon and chlorine into subduction channels (68–96 and 13–15 Mt year−1, respectively)4,9. However, fluid loss during metamorphic dehydration in subduction zones has been thought to return most of the subducted carbon and chlorine to the surface via the mantle wedge3,7. This dehydration is a considerable barrier for the deep recycling of both carbon and chlorine3,4, because water dissolves chlorine, drives carbonate dissolution10,11 and depresses the melting temperature of carbonated crustal materials12,13,14,15,16.
The effective return of a large proportion of subducted carbon to the surface has been demonstrated by experiments on the hydrous melting of average global sediment (GLOSS-II (ref. 17)) and altered oceanic crust with CO2 contents of 3.0–5.0 wt% (refs. 15,16) and 2.5–4.1 wt% (ref. 14), respectively (Extended Data Fig. 1), assuming that carbon is spread evenly throughout the slab. However, the distribution of carbonate is highly inhomogeneous in subducting materials. In particular, calcareous sediments (limestones and chalk)17 and highly altered basaltic rocks in the upper 600 m of oceanic crust in fault zones18 are strongly enriched in carbonate (see Methods). Although they are considered to be one of the main carbon reservoirs in modern subduction zones (estimated carbon flux approximately 52 Mt C year−1; see Methods for details), the hydrous melting of such carbonate-rich domains is poorly constrained but is essential for the accurate appraisal of the deep carbon cycle.
Chlorine is hydrophilic and therefore is expected to be highly enriched in any aqueous fluids that leave the subducting slab4. Together with the high solubility in silicate melts19,20, most chlorine is thought to be returned to the mantle wedge during prograde metamorphic devolatilization and melting. This conclusion was based on experiments and studies of chlorine-bearing silicate rocks (for example, sediments and serpentinites). However, the behaviour of chlorine in carbon-rich systems, corresponding to the aforementioned carbonate-rich rocks, has not been explored.
To investigate the fate of both carbon and chlorine together in subducting carbonate-rich domains, we have performed experiments on three mixtures using various proportions of limestone and clastic sediment with different CO2 contents (CS2 = 20.9 wt%, CS1 = 25.9 wt%, CS5 = 36.5 wt%) at temperatures of 700–1,150 °C and pressures of 3–5 GPa for mixture CS2 and 5 GPa for CS1 and CS5 (Extended Data Table 1). In contrast to previous experiments that used global average sediment compositions, our starting materials are similar to natural carbonate-rich sediments and highly altered carbonate-rich oceanic crust and therefore represent substantial components of the inhomogeneously carbonated subducting slab (detailed description in Methods and Extended Data Fig. 1).
Carbonated sediments and basalts are expected to lose most of their water and chlorine during dehydration and metamorphism before reaching arc depths7,21. However, abundant water and chlorine may be transported deeper in the underlying ultramafic layers of the subducting slab as serpentinite22,23,24. Continual dehydration of serpentinized oceanic crust at depths of 100–200 km would form highly saline fluids (>10 wt% Cl (refs. 23,24)) that may infiltrate the overlying carbonated sediments and basalts22,23,24,25. Our starting materials CS2, CS1 and CS5 contain 4.7, 3.7 and 1.7 wt% H2O and 0.66, 0.60 and 0.45 wt% chlorine, respectively. Therefore, our experimental design reconstructs the melting of carbonate-rich crustal rocks in subduction zones facilitated by the infiltration of an aqueous fluid containing about 12–14 wt% Cl.
Carbonate stability during subduction
The subsolidus phases in the experiments are calcite and/or aragonite, garnet, coesite, jadeite, phengite and fluid (cavities) (Extended Data Figs. 2 and 3). Notably, a small amount of carbonate matrix surrounds silicates at 5 GPa and is interpreted to be carbonate precipitated from a fluid, which itself is represented by adjacent cavities (Extended Data Fig. 3b). The fluid is formed by metamorphic devolatilization: calcite ± aragonite + H2O + jadeite = garnet + fluid. Mass-balance calculations show that the fluid carries limited carbon from the starting materials (3.9–7.1%), indicating that little carbon is mobilized during metamorphic devolatilization of carbonate-rich rocks unless the channelled fluid has extremely high water content. This is consistent with both modelling21 and natural observations7.
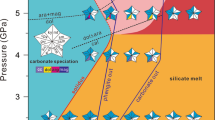
Above the solidus, partial melts are silicate melt at 3 GPa, carbonatite melt at T < 950 °C and 4 GPa, evolving to silicate melt at T ≥ 950 °C and carbonatite melt at 5 GPa (Fig. 1). This is consistent with previous experiments12,26,27, which showed that carbonatite melt is stable only at P > 3 GPa (Extended Data Fig. 4). The melting temperatures of CS2 and CS1 are similar at 5 GPa but lower than that of CS5 (Fig. 2), owing to the relatively low H2O content of CS5. The solidi of CS2 and CS1 are similar to that of H2O-rich carbonated gabbro12 (Extended Data Fig. 4) and match the temperature of the first appearance of carbonatite melts (900 °C at 4.2 GPa) in the simple system CaO–Al2O3–SiO2–CO2 with extremely high H2O contents (5.6–23.5 wt%)13, showing that our experiments represent the melting of hydrous carbonate-rich sediments. Carbonate remains stable during the formation of silicate melts (Extended Data Fig. 5) but contributes substantially to carbonatite melts at higher pressures. Figure 2 shows the detailed melting relationships of the three mixtures at 5 GPa. Near-solidus carbonatite melts are produced mainly by the consumption of aragonite and phengite in CS2 and CS1. With increasing temperature, the carbonate remains relatively stable and the degree of melting is similar for these two mixtures at T < 1,000 °C, consistent with comparable CaO + MgO contents in the carbonatite melts (Extended Data Fig. 6b). Notable consumption of solid carbonate for CS2 and CS1 begins at T ≥ 1,050 °C, resulting in low carbonate modes in their residues (7.0 and 21.3 wt%, respectively). Melting of the CS5 composition starts just below 1000 °C (Fig. 2c) and involves a high proportion of aragonite, resulting in high CaO and low SiO2 contents of the carbonatite melts (Extended Data Fig. 6b). Overall, in contrast to all previous experimental results on average global sediment and altered oceanic crust, even at high pressure (>4 GPa), more than 75 wt% of carbonate is retained in the solid state at T < 1,000 °C (Fig. 2d).

Phases in experiments on composition CS2 are shown by the squares: SiM, silicate melt (dark blue); CbM, carbonatite melt (red); Cc(arag) (grey, calcite; black, aragonite + calcite); chloride is light blue. Grey-shaded region is subsolidus; carbonatite melt is stable in the pink-shaded area; silicate melt is present at low pressure (no shading). Text indicates main phases hosting C and Cl in corresponding regions. Modern-style subduction zones are dominated by cold subduction (dashed grey line)31 (<800 Ma: occurrence of blueschist, jadeitites and exhumed ultrahigh-pressure eclogites38 and decreasing average juvenile continental thickness32,33), whereas hot subduction slab-top geotherms (dashed turquoise line) were prevalent before the Mesoproterozoic (>1,600 Ma)32,33 and are based on an average mantle cooling rate of about 70 °C Ga−1 (refs. 48,49). Subduction zones at intermediate ages were dominated by warm subduction regimes (dashed purple line), as indicated by constant average juvenile continental thickness32,33.

a, CS2. b, CS1, c, CS5. CbF, carbonate precipitated from fluid; CbM, carbonatite melt; Grt, garnet; Jd, jadeite; KCl, sylvite; Ky, kyanite; NaCl, halite; (Mg, Fe)-Cc, (Mg, Fe)-calcite; Phen, phengite; Sili. D, silicate droplet. d, Fraction of carbon (grey lines) and chlorine (blue lines) retained in the solid residue. Experimental results on hydrous global average sediments (Ave. sediment)50 and basalts (Ave. basalt)14 are shown for comparison. Cold, warm and hot subduction surface geotherm ranges at 5 GPa are shown as shaded backgrounds (see Fig. 1 for references).
Chlorides coexist with carbonate melts
No chlorides were found in subsolidus experiments nor in experiments that contain silicate melts (Figs. 1 and 2), which is fully consistent with previous studies. Under subsolidus conditions, chlorine is preferentially concentrated in the fluid. The lack of chlorides in silicate-melt-bearing experiments at 3–4 GPa is attributed to the high solubility of Cl in silicate melts (Fig. 3a). However, KCl and NaCl crystals occur in all experiments that contain carbonatite melts at 4–5 GPa. The chlorides occur in both quenched carbonatite melts and residues (Extended Data Figs. 2h,i and 3f–i). Notably, KCl crystals form at relatively low temperature (750 < T ≤ 900 °C at 4 GPa and 850 < T < 1,000 °C at 5 GPa), whereas KCl is replaced by NaCl at high temperatures (900 °C at 4 GPa and >1,000 °C at 5 GPa). At relatively low temperature, the decomposition of phengite releases abundant K into the melt. These initial carbonatite melts have much lower K2O/P2O5 than the starting mixtures (P2O5 is not carried by any mineral at 5 GPa and can be used to evaluate the dilution effect caused at increased degree of melting; Fig. 3b), which is consistent with K occurring as KCl. These large, nearly pure KCl crystals coexist with carbonatite melt and a residual mineral assemblage of aragonite + coesite (Extended Data Fig. 3f,g). In experiments at higher temperature, the progressive breakdown of jadeite contributes abundant Na to the melt so that NaCl replaces KCl as the chloride phase. Accordingly, the K2O/P2O5 ratios of carbonatite melts gradually approach those of the starting materials. Both KCl and NaCl are equilibrium phases coexisting with carbonatite melts and can be clearly distinguished from chlorides formed during quenching of melts, in which they normally form small crystals with complex intermediate (Na, K)Cl compositions28.

a, Chlorine contents of carbonatite (CbM) and silicate melts in this study (filled symbols) compared with previous experiments28,29 (open symbols). The error bars are 1 standard error. The dashed lines represent the chlorine contents of starting materials in previous experiments. b, K2O/P2O5 of carbonate precipitated from fluid (CbF) in subsolidus experiments and of carbonatite melts in above-solidus experiments at 5 GPa on CS2 and CS1 compositions. The error bars are 1 standard error. Residual minerals hosting potassium and chlorine in the experiments are shown at the top. Subduction surface geotherm regions as in Fig. 2d.
Generally, the melting behaviour of a chlorine-bearing carbonate–silicate system strongly depends on the Cl content29. At high chlorine contents (>3 wt%), initial melts are carbonatite. They evolve to coexisting immiscible chlorine–carbonate and carbonated silicate melts and finally to a single Cl-rich carbonated silicate melt with increasing temperature28,29,30. However, melts generated in a silicate–carbonate system with low chlorine content do not intersect this miscibility gap29. All previous experimental carbonatite melts at T < 1,400 °C have chlorine contents (<1 wt%) much lower than those of the starting materials (Fig. 3a), indicating relatively low solubility of chlorine in carbonatite melts28. Only at high temperatures (>1,400 °C) are chlorine-rich carbonated silicate melts produced. Consequently, the low solubility of chlorine in carbonatite melts induces the stabilization of separate chlorides (sylvite and halite) up to 100–300 °C above the solidus28,29. Our first melting experiments on hydrous carbonate-rich sediments with low bulk chlorine content (0.45–0.66 wt%) produced chlorine-poor hydrous carbonatite melts (Cl content <0.60 wt%) at low temperature (Fig. 3a). This shows that chlorine solubility in carbonatite melts is low regardless of the water content.
Mass-balance calculations indicate that the KCl and NaCl coexisting with carbonatite melts in our experiments host 82–92 wt% and 63–82 wt% of the bulk Cl, respectively. This is very different from the well-established behaviour of Cl in fluids and silicate melts. Thus, generation of low-Si carbonatite melts provides simple straightforward mechanisms for chlorine sequestration (as well as carbon and, potentially, further volatile components: F, N, S) in the solid residues and their transport further into the deep mantle by subduction.
Deep subduction of C and Cl through time
Combining our experimental results with estimates of the evolution of subduction geotherms31,32,33 and the proportion of carbonate rocks in marine sediments through time34 (Fig. 4a,b), we can constrain the deep subduction of surface carbon and chlorine throughout Earth’s history. Although there are many proposed ages for the beginning of plate tectonics ranging from the Hadean to 700 million years ago (Ma), a global-scale initiation of plate tectonics is generally accepted to have occurred at about 2.9–3.0 billion years ago (Ga) (ref. 35) (Fig. 4a). Before the Mesoproterozoic (around 1.6 Ga), the subduction process was dominated by hot subduction36,37, as gauged by increasing average juvenile continental thickness32,33. This predated the main production of biogenic carbonates, so that the proportion of carbonate rocks in sediments was low at the time (Fig. 4b). Furthermore, carbonate rocks underwent a high degree of melting in hot subducting slabs with only NaCl and a low proportion of carbonates retained (Figs. 1 and 4b). Consequently, as solid chlorides only become stable when carbonatite melts are present, the extensive recycling of carbon and chlorine would not have occurred before the Mesoproterozoic. At intermediate ages of the Earth (about 800–1,600 Ma), when subduction geotherms lay between cold, modern-style and hot, Archaean subduction geotherms (Fig. 4a), the proportion of carbonate rocks in marine sediments steadily increased (Fig. 4b) and both carbonate and KCl were stable during subduction owing to the intermediate geothermal gradients (Figs. 1 and 4b). Under these conditions, extensive recycling of carbonate and particularly chloride into the deep mantle were enabled (Fig. 4b). Since the late Neoproterozoic (<800 Ma), cold subduction has predominated31,38 (>95% of geotherms <900 °C at 5 GPa (ref. 31)) and the proportion of carbonate rocks (and particularly carbonate-rich rocks) in marine sediments was the highest in geological history. Therefore, most carbonates in carbonate-rich crustal rocks resisted devolatilization along cold geotherms and did not undergo melting when infiltrated by chlorine-rich fluid (Fig. 1). This optimized the effective transport of carbon, but not chlorine, to the deep mantle (Fig. 4b). The chlorine remains in the fluid under these conditions and leaves the slab.

a, Summary diagram showing global geodynamics35 and slab-top geotherms (shaded backgrounds). Onset of global plate tectonics is suggested by increasing juvenile continental crust thickness32,33, seawater 87Sr/86Sr (ref. 51) and mineral inclusions in diamonds from subcontinental mantle52 at 2.9–3.0 Ga. Evolution of subduction slab-top geotherms is constrained by juvenile continental crust thickness and blueschist record (for details, see Fig. 1). b, Proportion of carbonate rocks in marine sediments through time34 for qualitative evaluation of the carbon input flux. Efficiency of carbon and chlorine subduction into the deep mantle is estimated from average proportions of carbon and chlorine in the residues of our melting experiments (Fig. 2d). c, Overview of atmospheric pCO2 based on data from natural palaeosols39,41, calcified cyanobacteria40 and altered basalt53, and modelling by one-dimensional climate model (grey dotted line)54 and a geological carbon cycle model (green dashed line)55. d, Age distribution of eclogitic diamonds constrained by Sm–Nd and Re–Os isochrons for inclusions in diamonds (Supplementary Data 1).
Atmospheric pCO2 and mantle volatiles
Atmospheric pCO2 estimated from the chemical compositions of natural samples39,40,41 (for example, palaeosols) shows a decrease since the Mesoproterozoic (Fig. 4c) but is poorly constrained for the Archaean. Atmospheric pCO2 is controlled by the sources of CO2 (for example, volcanic emissions) and by the efficiency of the CO2 sinks42. Since the Mesoproterozoic, the carbon sink in the form of increased proportion of carbonate rocks in marine sediments has grown34 (Fig. 4b). However, this carbon sink could not lead to a decrease in atmospheric pCO2 over a long timescale if extensive metamorphic degassing and/or melting occurred during subduction, because these processes return CO2 to the atmosphere3,7. The fate of carbonate-rich rocks during subduction plays a key role in the evolution of atmospheric pCO2 owing to the considerable carbon input flux compared with volcanic emissions (modern flux: about 52 Mt C year−1 versus about 79 Mt C year−1)9. Our results indicate that subduction of carbonate-rich crustal rocks into the deep mantle has been a highly efficient long-term carbon sink since the Mesoproterozoic, and particularly during the Phanerozoic, and may explain the long-term decrease in atmospheric pCO2 since the Mesoproterozoic.
We suggest that subduction of carbonate-rich rocks transports carbon and chlorine to the deep mantle on the modern Earth, resisting fluid infiltration from deeper levels in subducting slabs and thus replenishing mantle volatile reservoirs. One reservoir with marked enrichment in carbonate and chloride is the lower cratonic lithosphere mantle1,5, as indicated by highly saline inclusions in fibrous diamonds43, alkali chlorides in kimberlites44,45 and appreciable chlorine contents of peridotite xenoliths sampled by kimberlites1. Most lithospheric diamonds grow here because of the concentration of diamond-forming chemical components (chloride and carbonate melt/fluid)46. Our work indicates that the effective deep subduction of both carbonate and KCl at intermediate stages of Earth’s history (Fig. 4b) plays a key role in the formation of this lower cratonic reservoir, promoting diamond formation (Extended Data Fig. 7). This is in perfect agreement with the compiled age distribution of the global eclogitic diamonds (Supplementary Data 1), showing that this intermediate period of Earth’s history was the heyday for the formation of eclogitic diamonds (Fig. 4d). Another reservoir enriched in recycled carbon and chlorine is the mantle source of HIMU-type ocean island basalts, in which recycled chlorine accounts for at least 13–26% of the total mantle Cl (ref. 8). Although interpreted to originate from the recycling of carbonated oceanic crust8,47, no chlorine-bearing phases have so far been identified in subducted oceanic crust beyond arc depth8. Our data unambiguously indicate that melting of carbonate-rich and highly altered oceanic rocks during infiltration of chlorine-bearing fluids from deeper levels of the oceanic crust should produce stable chlorides in residual carbonated eclogites, potentially forming the source of HIMU-type ocean island basalts (Extended Data Fig. 7).
Methods
Choice of starting materials and comparison with previous studies
Both sediments and altered basaltic crust in subducting slabs show a large variation in carbonate content (Extended Data Fig. 1). Calcareous sediments (or limestones and chalk) are widespread in subducting sedimentary layers, as indicated by carbonate-rich sediments in the Lesser Antilles (Site 144) and high CO2 contents (9.1–26.5 wt%) of average sediments in some trenches17. Given an average carbonate content of 7 wt% for global subducted sediments (GLOSS-II) and a high fraction of silicate sediments in subducting sediments17, carbonate-rich sediments are one of the main carbon reservoirs.
The oceanic crust experiences hydrothermal alteration along the ridge axis during its formation and low-temperature hydrothermal circulation/seawater alteration away from the ridge axis in the upper approximately 600 m of oceanic crust56,57. Hydrothermally altered crust along the ridge axis has a low CO2 content (mostly <0.6 wt%) that originated mainly from magmatic degassing58. The precipitation of carbonate during alteration by penetrative low-temperature hydrothermal fluid/seawater (<60 °C) forms carbonate veins in the upper 600 m of oceanic crust. The average CO2 content of the upper oceanic crust older than 100 Ma is 2.5–4.1 wt% and is higher than that of the Cenozoic upper crust (about 0.5 wt%) because all substantial low-temperature exchange is completed in old oceanic crust56,57. Therefore, only old altered oceanic crust represents the subducting altered oceanic crust. The distribution of carbonate in the altered oceanic crust is highly heterogeneous with enrichment in fault zones in which low-temperature hydrothermal circulation frequently causes several stages of alteration, forming carbonate-rich veins up to 50 m wide, as in the Southwest Indian Ocean59. Individual altered samples from the Western Central Atlantic (118 Ma, DSDP Sites 417 and 418) show highly variable CO2 contents from 0.37 to 29.16 wt%, indicating a key role for carbonate-rich rock in hosting carbon in the oceanic crust18. In summary, carbonate-rich rocks are a key carbon reservoir in both subducting sediments and altered basaltic crust.
Previous high-pressure experimental studies have considered the global weighted average composition of subducted sediments and altered oceanic crust (Extended Data Fig. 1a), corresponding to clastic sediments with 3.5 wt% CO2 (refs. 26,27,60,61) and basalts/gabbros with 2.5–4.1 wt% CO2 (refs. 12,14,62,63,64,65). However, this ignores the role of carbonate-rich sediments and altered basalts rich in carbonate veins: this is an important distinction because the melting behaviour of carbonated sediments and basalts depends strongly on the CO2 content in the system50. Skora et al.50 investigated hydrous melting of calcareous sediment at 3.0 GPa, but the CO2 content in their starting material (12.5 wt%) is much lower than in carbonate-rich sediments (Extended Data Fig. 1).
Our study is focused on the melting behaviour of carbonate-rich rocks rather than average subducted sediments or basalts. The three starting materials CS2, CS1 and CS5 are mixtures of a natural limestone and carbonated silicate sediment with the ratios of 1:2, 1:1 and 6:1, respectively. The limestone is from Ocean Discovery Program Expedition 115 Site 714A located in the northern equatorial Indian Ocean (U0714A-13H-2 W) at 5° 3.6′ N and 73° 47.2′ E and the carbonated sediment is from the International Ocean Discovery Project (ODP161-976B-18X3-105-106.5): compositions are reported in previous publications66,67. The chemical compositions of the three starting materials are listed in Extended Data Table 2. Their bulk compositions represent carbonate-rich sediments and are similar to highly altered carbonated basalts in the upper 600 m of oceanic crust owing to the formation of carbonate and clay during low-temperature alteration (Extended Data Fig. 1). Therefore, our experiments were designed to model the subduction processes of both carbonate-rich sediments and highly altered oceanic crust.
Carbon input flux of carbonate-rich crustal rocks in modern subduction
Carbonate-rich rocks are widespread in subducting sedimentary layers and highly altered oceanic crust. Clift68 estimated carbon flux (about 60 Mt C year−1) including carbonate and organic carbon in top sedimentary layers of modern subducting plates from scientific ocean-drilling results. Chen et al.66 compiled the carbon flux of calcareous sediments (or limestones and chalk) from the database of ref. 68. They concluded that carbonate-rich sediments carry about 25% of the subducted sedimentary carbon (about 21.4 Mt C year−1). For the altered oceanic crust, Kelemen and Manning3 estimated the input carbon flux to be roughly 25 Mt C year−1 by averaging young and old altered oceanic crusts. Carbon concentrations and isotope measurements of young and old altered oceanic crusts have yielded a slightly lower flux (18 Mt C year−1)69. However, only the old altered oceanic crust represents the subducting altered oceanic crust. On the basis of data for volcanic rocks from DSDP Sites 417 and 418 in 118-Ma Atlantic crust, Staudigel et al.70 calculated that the average CO2 content of highly altered oceanic crust (upper 600 m) is 2.98 wt%. For the modern subduction zone length (44,454 km) and subduction rate (50 mm year−1)71, the carbon flux of the upper 600 m of highly altered oceanic crust into the subduction zones is about 31.3 Mt C year−1. Finally, we estimated the total carbon input flux of carbonate-rich crustal rocks in modern subduction zones to be about 52 Mt C year−1 (roughly 31.3 Mt C year−1 plus about 21.4 Mt C year−1).
We cannot quantitatively estimate the carbon flux during ancient subduction owing to the lack of data on CO2 contents of ancient sediments and altered oceanic crust and the poor constraints on the length and rate of ancient subduction. However, the temporal variations of the carbon input flux are discussed in the main text on the basis of the proportion of carbonate rocks in marine sediments over time34 (Fig. 4b).
Experimental techniques
Starting powders were dried at 120 °C for 48 h before loading into Ag70Pd30 capsules and then welded shut. Experiments were conducted in a 1,000-t Walker-type multi-anvil apparatus with Hawedia tungsten carbide (7% Co) cubes as secondary anvils at the School of Natural Sciences, Macquarie University. The runs were carried out with 18/11 (octahedral edge length/truncation edge length in mm) assembly. The high-pressure assembly consisted of octahedral spinel-doped MgO pressure medium, ZrO2 sleeve, stepped graphite heater and MgO sleeve containing the sample capsule. Components of the high-pressure assemblies were fired at 1,000 °C for 1 h before experiments to remove moisture. The assembled octahedra were dried in a vacuum oven at 125 °C overnight before each experiment. In all experiments, the temperature was monitored with an axial W97Re3/W75Re25 thermocouple. The samples were first compressed to the target pressure within 3 h and then heated at a rate of 100 °C min−1 to the target temperature. The samples were quenched by switching off the electrical power and the quench rate was approximately 500 °C s−1. The samples were then decompressed over 16 h. The pressure was calibrated by bracketing transformations of SiO2 (quartz–coesite and coesite–stishovite) and CaGeO3 (garnet–perovskite) at 1,000 °C. The duration of all experiments was longer than 48 h (Extended Data Table 1).
The recovered capsules were embedded in epoxy and cut into halves using a slow saw under oil-based lubricant for imaging and chemical analysis. To avoid chloride contamination, only chlorine-free materials were used in sample preparation. One half was mounted in epoxy, polished under oil-based lubricant and repeatedly re-impregnated with epoxy to minimize the loss of minerals and quenched melts during polishing. No water was used during sample preparation to avoid dissolution of alkali carbonates and chlorides. The other half of the capsule was used for direct scanning electron microscopy observation of chlorides without polishing to confirm the primary nature of chloride crystals.
Analytical methods
Major element analyses of phengite, garnet, jadeite, aragonite, (Mg, Fe)-calcite and carbonatite melt were performed using a JEOL JXA-8530F Plus field emission electron microprobe at the Centre for Advanced Microscopy, Australian National University. Operating conditions were 15 kV accelerating voltage and a beam current of 8 nA with peak counting times between 20 and 30 s for analyses of phengite, garnet, jadeite and carbonatite melt. Except for carbonatite melt, the beam diameter was set to 2–5 μm. Owing to the chemical heterogeneity caused by quenching of carbonatite melt, we used a larger beam diameter of 10 μm for analysing major elements in the carbonatite melt. A variety of minerals and synthetic materials were used as reference materials.
The Cl content of the starting materials was measured using an ELEMENT XR inductively coupled plasma sector field mass spectrometry system (Thermo Fisher Scientific) at the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences. The detailed digestion method and instrumentation parameters were described in ref. 72.
Mass-balance calculation
The phase modes (M) were obtained by solving the following equations to minimize the ∑R2:
in which i = SiO2, TiO2, Al2O3, MnO, MgO, CaO, Na2O, K2O and Cl; n = minerals and melts in the experiments; \({C}_{{\rm{bulk}}\,{\rm{C}}}^{i}\) is the calculated bulk concentration of component i in the system; \({C}_{n}^{i}\) is the measured component i concentration of phase n; and \({C}_{{\rm{bulk}}\,{\rm{S}}}^{i}\) is the i concentration of the starting material in the capsule.
Textures, phase assemblages, solidus and melting reactions
We detect the onset of partial melting in our experiments using textural criteria. Below the solidus, fluid is observed at the upper part of the capsules in the form of cavities or vesicles filled by epoxy resin during sample preparation (Extended Data Figs. 2a,d and 3a,b). The fluid pool in subsolidus experiments at 5 GPa contains carbonate matrix surrounding silicates (Extended Data Fig. 3b). The carbonate matrix precipitates from fluid rather than melt as recognized by: (1) absence of lobes, dendrites, cusps or feathery grain boundaries at contacts with silicates interpreted by Poli12 as the quench microstructures of hydrous carbonatite liquid; (2) the presence of cavities or vesicles indicating the existence of fluid or vapour; (3) extremely low SiO2 content (<0.8 wt%) (Extended Data Table 2). Above the solidus at 3 GPa, silicate melt is observed throughout the whole capsule volume. Above the solidus at 4–5 GPa, a carbonatite melt pool segregated at the top of the capsule and consists of a dense mat of carbonate dendrites without any cavities or vesicles (Extended Data Fig. 3d–f). The separation of fluid and carbonatite melt from the solid mineral assemblages at the top of the capsules is attributed to their low density and/or thermal gradient in the multi-anvil assembly. The melt at 4 GPa converts to silicate melt as melting progresses at T ≥ 950 °C (Fig. 1 and Extended Data Fig. 2f,g). The phase modes are given in Extended Data Table 1.
The subsolidus phases for CS2 are carbonate, garnet, phengite, coesite, kyanite and fluid (3.0–5.7 wt%). The carbonate phase changes with increasing pressure: at 3 GPa, carbonate is a single calcite phase (48.0 wt%), whereas aragonite appears at 4 GPa (about 10.9 wt%) and coexists with calcite (37.4 wt%). At 5 GPa, abundant aragonite (32.9–34.5 wt%) coexists with (Mg, Fe)-calcite (6.5–9.1 wt%) owing to the high XCa in the starting materials73,74. Carbonate precipitation (about 1.8 and 2.0–4.0 wt%) (Extended Data Fig. 3b and Extended Data Table 1) and spherical inclusions of silicate droplets occur at 850 °C. These may have formed by quenching from the solute of a vapour/fluid13 (Extended Data Tables 1 and 2). Relative to CS2, the subsolidus phases of CS1 and CS5 at 5 GPa include more aragonite (46.4–48.1 wt% and 77.9–82.4 wt%), less (Mg, Fe)-calcite (about 5.0 wt% and 5.0–2.3 wt%), garnet (10.6–11.6 wt% and 4.2–5.7 wt%), coesite (8.0–8.6 wt% and 5.3–5.6 wt%), jadeite (6.3–6.5 wt% and 3.5–2.1 wt%) and phengite (12.2–14.1 wt%, 0).
The solidus for CS2 at 3 GPa is between 700 and 800 °C, as gauged by the occurrence of silicate melt. The near-solidus silicate melt is formed by:
in which Phen = phengite, Coe = coesite, Jd = jadeite, Cc = calcite, SiM = silicate melt, Grt = garnet and Ep = epidote. With increasing temperature, the proportion of carbonate contributing to the melt is still low (Extended Data Fig. 5), thus carbonate is stable during melting (for CS2 at T = 900 °C):
We identified the solidus temperature for CS2 to be between 750 and 850 °C at 4 GPa and between 850 and 900 °C at 5 GPa by the appearance of quenched carbonatite melts. The melt at 4 GPa converts to silicate melt at T ≥ 950 °C and all melts are carbonatite at 5 GPa (Fig. 1). At 4 GPa, the near-solidus carbonatite melt at T = 850 °C is produced by:
in which Arag = aragonite, Ky = kyanite, CbM = carbonatite melt and KCl = sylvite. At 5 GPa, the complete breakdown of phengite in near-solidus experiments on CS2 (Fig. 2a) occurred at temperatures much lower than in carbonate-free sediments75. This is consistent with low temperatures for the disappearance of phengite or phlogopite in carbonate-bearing sediments and peridotites, attributed to the effect of high CO2 in previous experiments15,76. The proportion of aragonite decreases slightly in experiments at T < 1,050 °C (Fig. 2a). The near-solidus carbonatite melts are produced for CS2 at 900 °C by:
in which (Mg, Fe)-Cc = (Mg, Fe)-calcite and CbF = carbonate precipitation. High-degree melting of CS2 is observed at T ≥ 1,050 °C owing to the complete breakdown of (Mg, Fe)-calcite and jadeite and consumption of a high proportion of carbonate:
in which NaCl = halite. The solidus and phase relationships for CS1 at 5 GPa are similar to those of CS2:
The solidus of CS5 is 100 °C higher than for CS2 and CS1, which—in turn—are higher than that of hydrous silicate sediments without carbonate75 (Extended Data Fig. 4). This is attributed to the enrichment in fusible components such as Na2O, K2O and particularly H2O in hydrous silicate sediments. This is in agreement with the presence of silicate melt droplets at 850 °C and the substantial contribution of phengite to melts in CS2 and CS1, indicating that the fusible components have depressed the solidus and triggered melting. The proportion of carbonate contributing to the melt is high in the near-solidus experiment on CS5 (1,000 °C) because no hydrous minerals are present in subsolidus conditions:
Phase compositions
Compositions of all run products are compiled in Extended Data Table 2 and Supplementary Data 2. Generally, the compositions of carbonatite melts depend on the starting materials, pressure and temperature (Extended Data Figs. 6 and 8). The compositions of carbonates, garnets and jadeites show systematic variations with both pressure and temperature.
Silicate melts at 3 and 4 GPa in this study have high SiO2 (51.9–63.6 wt%) and Al2O3 (11.7–13.9 wt%) but low CaO (2.8–6.2 wt%) and MgO (0.34–1.01 wt%) contents (Extended Data Fig. 6b). The silicate melts have high Cl content (1.04–1.40 wt%) (Fig. 3a). Carbonatite melts at 4 and 5 GPa have higher SiO2 and Al2O3 contents than melts from previous experiments on carbonated sediments26,27 (Extended Data Fig. 6b), owing to the effect of water. Carbonatite melts in CS5 show higher CaO + MgO and lower SiO2 + Al2O3 contents than those of carbonatite melts for CS2 and CS1 (Extended Data Fig. 6b). All these carbonatite melts have low Cl contents (0.13–0.76 wt%) (Fig. 3a).
Calcite in the subsolidus experiments at 3 GPa contains 45.8 wt% CaO, 2.35 wt% MgO and 3.3 wt% FeO. (Mg, Fe)-calcite at 5 GPa has 7.9–10.3 wt% MgO, 4.6–5.3 wt% FeO and 32.5–37.6 wt% CaO. MgO and FeO contents are higher at 4 and 5 GPa owing to the formation of aragonite (Extended Data Fig. 6a). Aragonite is observed in the low-temperature experiments at 4 and 5 GPa (T ≤ 900 °C and ≤1,000 °C, respectively) and has nearly pure compositions with 51.6–55.02 wt% CaO (Extended Data Fig. 8). It is replaced by calcite at T ≥ 950 °C and T ≥ 1050 °C (4 and 5 GPa, respectively) and the calcite has 0.58–1.44 wt% MgO and 50.0–52.8 wt% CaO.
The carbonate matrix precipitated from the fluid at 5 GPa is characterized by low SiO2 but high CaO and plots on a mixing line between (Mg, Fe)-calcite and aragonite in the ternary CaCO3–MgCO3–FeCO3 diagram (Extended Data Fig. 6a). Furthermore, it differs substantially from aragonite and (Mg, Fe)-calcite in P2O5 (0.88–1.5 wt%), Na2O (0.76–1.4 wt%) and K2O (0.39–1.04 wt%).
Phengite is found at T ≤ 900 °C in the experiments on CS2 and CS1. The temperature of complete breakdown of phengite is much lower than in carbonate-free sediments75. This effect is attributed to high CO2 content15,76. Compositions of phengite are homogeneous and do not change markedly with increasing temperature with Si = 3.13–3.17 a.f.u., K = 0.76–0.82 a.f.u. and Cl contents of 0.04–0.25 wt%.
Garnet appears in all experiments except at T = 1,100 °C and P = 5 GPa for CS5. Its composition changes systematically with temperature but not substantially with pressure (Extended Data Fig. 8). The grossular component shows an increase from 30.3 to 55.8%, 35.5 to 61.8% and 39.2 to 67.4% for CS2, CS1 and CS5, respectively, with increasing temperature (Extended Data Fig. 6). The pyrope component increases from 7.8 to 22.7%, 9.7 to 20.5% and 13.2 to 21.8% for CS2, CS1 and CS5, respectively, with temperature.
Jadeite is found at T < 800 °C, ≤900 °C and ≤950 °C for 3, 4 and 5 GPa, respectively (Extended Data Fig. 5), for CS2 and CS1 starting mixtures and in all experiments with CS5. At 5 GPa, Na2O and Al2O3 contents decrease from 12.1 to 4.7 wt% and 22.2 to 14.3 wt%, respectively, with increasing temperature and CaO content increases from 3.44 to 14.9 wt% (Extended Data Fig. 8). Jadeites at 3–4 GPa have lower Na2O but higher CaO than those at the same temperature at 5 GPa (Extended Data Fig. 8).
At 4 GPa, KCl crystals are found at T = 850 and 900 °C and NaCl forms at T = 900 °C. At 5 GPa, KCl crystals are observed in above-solidus experiments at T < 1,000 °C (0.7–1.5 wt%) and NaCl crystals at T ≥ 1,050 °C (0.36–1.2 wt%) (Figs. 1 and 2a and Extended Data Fig. 3). These chlorides have nearly pure compositions.
Approach to equilibrium
All minerals are compositionally homogeneous and unzoned. Furthermore, regular compositional trends of minerals and melts throughout the experimental grid (Extended Data Figs. 6 and 8) indicate that equilibrium was reached.
No laboratory contamination of salt during sample preparation
The chlorides found in the above-solidus experiments crystallized from the carbonatite melts and are not because of contamination during sample preparation. We avoided contact with any salt-bearing materials during sample preparation and the chlorides were observed in both polished and unprepared halves of capsules (Extended Data Fig. 9), excluding any possible contamination during sample preparation. Furthermore, the type of chloride in the experiments depends consistently on the experimental temperature, with KCl crystals in the low-temperature experiments and NaCl crystals in the high-temperature experiments (Fig. 1 and Extended Data Fig. 9). The chemical compositions of the carbonatite melts do not correspond to the pure compositions of the crystals, also suggesting that the chlorides are equilibrated with carbonatite melts.
Data availability
The data that support the findings of this study are available in the paper or in the supplementary files and are also available at https://doi.org/10.6084/m9.figshare.22698292.v1. Source data are provided with this paper.
References
Broadley, M. W., Barry, P. H., Ballentine, C. J., Taylor, L. A. & Burgess, R. End-Permian extinction amplified by plume-induced release of recycled lithospheric volatiles. Nat. Geosci. 11, 682–687 (2018).
Dasgupta, R. & Hirschmann, M. M. The deep carbon cycle and melting in Earth’s interior. Earth Planet. Sci. Lett. 298, 1–13 (2010).
Kelemen, P. B. & Manning, C. E. Reevaluating carbon fluxes in subduction zones, what goes down, mostly comes up. Proc. Natl Acad. Sci. 112, E3997–E4006 (2015).
Barnes, J. D., Manning, C. E., Scambelluri, M. & Selverstone, J. in The Role of Halogens in Terrestrial and Extraterrestrial Geochemical Processes: Surface, Crust, and Mantle (eds Harlov, D. E. & Aranovich, L.) 545–590 (Springer, 2018).
Foley, S. F. & Fische, T. P. An essential role for continental rifts and lithosphere in the deep carbon cycle. Nat. Geosci. 10, 897–902 (2017).
Hirschmann, M. M. Comparative deep Earth volatile cycles: the case for C recycling from exosphere/mantle fractionation of major (H2O, C, N) volatiles and from H2O/Ce, CO2/Ba, and CO2/Nb exosphere ratios. Earth Planet. Sci. Lett. 502, 262–273 (2018).
Stewart, E. M. & Ague, J. J. Pervasive subduction zone devolatilization recycles CO2 into the forearc. Nat. Commun. 11, 6220 (2020).
Hanyu, T. et al. Tiny droplets of ocean island basalts unveil Earth’s deep chlorine cycle. Nat. Commun. 10, 60 (2019).
Plank, T. & Manning, C. E. Subducting carbon. Nature 574, 343–352 (2019).
Ague, J. J. & Nicolescu, S. Carbon dioxide released from subduction zones by fluid-mediated reactions. Nat. Geosci. 7, 355–360 (2014).
Farsang, S. et al. Deep carbon cycle constrained by carbonate solubility. Nat. Commun. 12, 4311 (2021).
Poli, S. Carbon mobilized at shallow depths in subduction zones by carbonatitic liquids. Nat. Geosci. 8, 633–636 (2015).
Schettino, E. & Poli, S. in Carbon in Earth’s Interior (eds Manning, C. E., Lin, J.-F. & Mao, W. L.) 209–221 (American Geophysical Union, 2020).
Martin, L. A. J. & Hermann, J. Experimental phase relations in altered oceanic crust: implications for carbon recycling at subduction zones. J. Petrol. 59, 299–320 (2018).
Grassi, D. & Schmidt, M. W. The melting of carbonated pelites from 70 to 700 km depth. J. Petrol. 52, 765–789 (2011).
Chen, X. et al. Melting of carbonated pelite at 5.5–15.5 GPa: implications for the origin of alkali-rich carbonatites and the deep water and carbon cycles. Contrib. Mineral. Petrol. 177, 2 (2021).
Plank, T. in Treatise on Geochemistry 2nd edn (eds Holland, H. D. & Turekian, K. K.) 607–629 (Elsevier, 2014).
Staudigel, H., Plank. T., White, B. & Schmincke, H.-U. in Subduction: Top to Bottom (eds Bebout, G. E., Scholl, D. W., Kirby, S. H. & Platt, J. P.) 19–38 (American Geophysical Union, 2013).
Van den Bleeken, G. & Koga, K. T. Experimentally determined distribution of fluorine and chlorine upon hydrous slab melting, and implications for F–Cl cycling through subduction zones. Geochim. Cosmochim. Acta 171, 353–373 (2015).
Li, H. & Hermann, J. Apatite as an indicator of fluid salinity: an experimental study of chlorine and fluorine partitioning in subducted sediments. Geochim. Cosmochim. Acta 166, 267–297 (2015).
Kerrick, D. M. & Connolly, J. A. D. Metamorphic devolatilization of subducted marine sediments and the transport of volatiles into the Earth’s mantle. Nature 411, 293–296 (2001).
Kendrick, M. A., Scambelluri, M., Honda, M. & Phillips, D. High abundances of noble gas and chlorine delivered to the mantle by serpentinite subduction. Nat. Geosci. 4, 807–812 (2011).
Philippot, P., Agrinier, P. & Scambelluri, M. Chlorine cycling during subduction of altered oceanic crust. Earth Planet. Sci. Lett. 161, 33–44 (1998).
Scambelluri, M. & Philippot, P. Deep fluids in subduction zones. Lithos 55, 213–227 (2001).
Rüpke, L. H., Morgan, J. P., Hort, M. & Connolly, J. A. D. Serpentine and the subduction zone water cycle. Earth Planet. Sci. Lett. 223, 17–34 (2004).
Tsuno, K., Dasgupta, R., Danielson, L. & Righter, K. Flux of carbonate melt from deeply subducted pelitic sediments: geophysical and geochemical implications for the source of Central American volcanic arc. Geophys. Res. Lett. 39, L16307 (2012).
Thomsen, T. B. & Schmidt, M. W. Melting of carbonated pelites at 2.5–5.0 GPa, silicate–carbonatite liquid immiscibility, and potassium–carbon metasomatism of the mantle. Earth Planet. Sci. Lett. 267, 17–31 (2008).
Litasov, K. D. & Ohtani, E. Phase relations in the peridotite–carbonate–chloride system at 7.0–16.5 GPa and the role of chlorides in the origin of kimberlite and diamond. Chem. Geol. 262, 29–41 (2009).
Safonov, O. G., Kamenetsky, V. S. & Perchuk, L. L. Links between carbonatite and kimberlite melts in chloride–carbonate–silicate systems: experiments and application to natural assemblages. J. Petrol. 52, 1307–1331 (2010).
Safonov, O. G., Perchuk, L. L. & Litvin, Y. A. Melting relations in the chloride–carbonate–silicate systems at high-pressure and the model for formation of alkalic diamond–forming liquids in the upper mantle. Earth Planet. Sci. Lett. 253, 112–128 (2007).
Syracuse, E. M., van Keken, P. E. & Abers, G. A. The global range of subduction zone thermal models. Phys. Earth Planet. Inter. 183, 73–90 (2010).
Hawkesworth, C. J., Cawood, P. A., Dhuime, B. & Kemp, T. I. S. Earth’s continental lithosphere through time. Annu. Rev. Earth Planet. Sci. 45, 169–198 (2017).
Dhuime, B., Wuestefeld, A. & Hawkesworth, C. J. Emergence of modern continental crust about 3 billion years ago. Nat. Geosci. 8, 552–555 (2015).
Ronov, A. B. Common tendencies in the chemical evolution of the earth’s crust, ocean and atmosphere. Geokhiniiya 8, 715–743 (1964).
Palin, R. M. et al. Secular change and the onset of plate tectonics on Earth. Earth Sci. Rev. 207, 103172 (2020).
Martin, H. Effect of steeper Archean geothermal gradient on geochemistry of subduction-zone magmas. Geology 14, 753–756 (1986).
Komiya, T., Hayashi, M., Maruyama, S. & Yurimoto, H. Intermediate-P/T type Archean metamorphism of the Isua supracrustal belt: implications for secular change of geothermal gradients at subduction zones and for Archean plate tectonics. Am. J. Sci. 302, 806–826 (2002).
Tsujimori, T. & Ernst, W. G. Lawsonite blueschists and lawsonite eclogites as proxies for palaeo-subduction zone processes: a review. J. Metamorph. Geol. 32, 437–454 (2014).
Sheldon, N. D. Precambrian paleosols and atmospheric CO2 levels. Precambrian Res. 147, 148–155 (2006).
Kah, L. C. & Riding, R. Mesoproterozoic carbon dioxide levels inferred from calcified cyanobacteria. Geology 35, 799–802 (2007).
Kanzaki, Y. & Murakami, T. Estimates of atmospheric CO2 in the Neoarchean–Paleoproterozoic from paleosols. Geochim. Cosmochim. Acta 159, 190–219 (2015).
Berner, R. A. The Phanerozoic Carbon Cycle: CO2 and O2 (Oxford Univ. Press, 2004).
Klein-BenDavid, O., Wirth, R. & Navon, O. TEM imaging and analysis of microinclusions in diamonds: a close look at diamond-growing fluids. Am. Mineral. 91, 353–365 (2006).
Kamenetsky, M. B. et al. Kimberlite melts rich in alkali chlorides and carbonates: a potent metasomatic agent in the mantle. Geology 32, 845–848 (2004).
Maas, R., Kamenetsky, M. B., Sobolev, A. V., Kamenetsky, V. S. & Sobolev, N. V. Sr, Nd, and Pb isotope evidence for a mantle origin of alkali chlorides and carbonates in the Udachnaya kimberlite, Siberia. Geology 33, 549–552 (2005).
Palyanov, Y. N., Shatsky, V. S., Sobolev, N. V. & Sokol, A. G. The role of mantle ultrapotassic fluids in diamond formation. Proc. Natl Acad. Sci. 104, 9122–9127 (2007).
Cabral, R. A. et al. Volatile cycling of H2O, CO2, F, and Cl in the HIMU mantle: a new window provided by melt inclusions from oceanic hot spot lavas at Mangaia, Cook Islands. Geochem. Geophys. Geosyst. 15, 4445–4467 (2014).
Herzberg, C., Condie, K. & Korenaga, J. Thermal history of the Earth and its petrological expression. Earth Planet. Sci. Lett. 292, 79–88 (2010).
Dasgupta, R. Ingassing, storage, and outgassing of terrestrial carbon through geologic time. Rev. Mineral. Geochem. 75, 183–229 (2013).
Skora, S. et al. Hydrous phase relations and trace element partitioning behaviour in calcareous sediments at subduction-zone conditions. J. Petrol. 56, 953–980 (2015).
Veizer, J. & Mackenzie, F. T. in Treatise on Geochemistry (eds Holland, H. D. & Turekian, K. K.) 369–407 (Elsevier, 2003).
Shirey, S. B. & Richardson, S. H. Start of the Wilson cycle at 3 Ga shown by diamonds from subcontinental mantle. Science 333, 434–436 (2011).
Driese, S. G. et al. Neoarchean paleoweathering of tonalite and metabasalt: implications for reconstructions of 2.69 Ga early terrestrial ecosystems and paleoatmospheric chemistry. Precambrian Res. 189, 1–17 (2011).
Kasting, J. F. Theoretical constraints on oxygen and carbon dioxide concentrations in the Precambrian atmosphere. Precambrian Res. 34, 205–229 (1987).
Krissansen-Totton, J., Arney, G. N. & Catling, D. C. Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl Acad. Sci. 115, 4105–4110 (2018).
Alt, J. C. & Teagle, D. A. H. The uptake of carbon during alteration of ocean crust. Geochim. Cosmochim. Acta 63, 1527–1535 (1999).
Coogan, L. A. & Gillis, K. M. Evidence that low-temperature oceanic hydrothermal systems play an important role in the silicate-carbonate weathering cycle and long-term climate regulation. Geochem. Geophys. Geosyst. 14, 1771–1786 (2013).
Shilobreeva, S., Martinez, I., Busigny, V., Agrinier, P. & Laverne, C. Insights into C and H storage in the altered oceanic crust: results from ODP/IODP Hole 1256D. Geochim. Cosmochim. Acta 75, 2237–2255 (2011).
Dick, H. J. B., MacLeod, C. J., Blum, P. & the Expedition 360 Scientists. Expedition 360 Preliminary Report: Southwest Indian Ridge Lower Crust and Moho. International Ocean Discovery Program. https://doi.org/10.14379/iodp.pr.360.2016 (2016).
Tsuno, K. & Dasgupta, R. The effect of carbonates on near-solidus melting of pelite at 3 GPa: relative efficiency of H2O and CO2 subduction. Earth Planet. Sci. Lett. 319, 185–196 (2012).
Mann, U. & Schmidt, M. W. Melting of pelitic sediments at subarc depths: 1. Flux vs. fluid-absent melting and a parameterization of melt productivity. Chem. Geol. 404, 150–167 (2015).
Dasgupta, R., Hirschmann, M. M. & Dellas, N. The effect of bulk composition on the solidus of carbonated eclogite from partial melting experiments at 3 GPa. Contrib. Mineral. Petrol. 149, 288–305 (2005).
Hammouda, T. High-pressure melting of carbonated eclogite and experimental constraints on carbon recycling and storage in the mantle. Earth Planet. Sci. Lett. 214, 357–368 (2003).
Kiseeva, E. S. et al. An experimental study of carbonated eclogite at 3·5–5·5 GPa—implications for silicate and carbonate metasomatism in the cratonic mantle. J. Petrol. 53, 727–759 (2012).
Thomson, A. R., Walter, M. J., Kohn, S. C. & Brooker, R. A. Slab melting as a barrier to deep carbon subduction. Nature 529, 76–79 (2016).
Chen, C. F., Förster, M. W., Foley, S. F. & Liu, Y. S. Massive carbon storage in convergent margins initiated by subduction of limestone. Nat. Commun. 12, 4463 (2021).
Förster, M. W., Foley, S. F., Marschall, H. R., Alard, O. & Buhre, S. Melting of sediments in the deep mantle produces saline fluid inclusions in diamonds. Sci. Adv. 5, eaau2620 (2019).
Clift, P. D. A revised budget for Cenozoic sedimentary carbon subduction. Rev. Geophys. 55, 97–125 (2017).
Li, K., Li, L., Pearson, D. G. & Stachel, T. Diamond isotope compositions indicate altered igneous oceanic crust dominates deep carbon recycling. Earth Planet. Sci. Lett. 516, 190–201 (2019).
Staudigel, H., Hart, S. R., Schmincke, H.-U. & Smith, B. M. Cretaceous ocean crust at DSDP Sites 417 and 418: carbon uptake from weathering versus loss by magmatic outgassing. Geochim. Cosmochim. Acta 53, 3091–3094 (1989).
Jarrard, R. D. Subduction fluxes of water, carbon dioxide, chlorine, and potassium. Geochem. Geophys. Geosyst. 4, 8905 (2003).
He, T. et al. Determination of Cl, Br, and I in geological materials by sector field inductively coupled plasma mass spectrometry. Anal. Chem. 91, 8109–8114 (2019).
Yaxley, G. M. & Brey, G. P. Phase relations of carbonate-bearing eclogite assemblages from 2.5 to 5.5 GPa: implications for petrogenesis of carbonatites. Contrib. Mineral. Petrol. 146, 606–619 (2004).
Franzolin, E., Schmidt, M. W. & Poli, S. Ternary Ca–Fe–Mg carbonates: subsolidus phase relations at 3.5 GPa and a thermodynamic solid solution model including order/disorder. Contrib. Mineral. Petrol. 161, 213–227 (2011).
Hermann, J. & Spandler, C. J. Sediment melts at sub-arc depths: an experimental study. J. Petrol. 49, 717–740 (2008).
Saha, S. & Dasgupta, R. Phase relations of a depleted peridotite fluxed by a CO2-H2O fluid—implications for the stability of partial melts versus volatile-bearing mineral phases in the cratonic mantle. J. Geophys. Res. Solid Earth 124, 10089–10106 (2019).
Carpentier, M., Chauvel, C., Maury, R. C. & Mattielli, N. The “zircon effect” as recorded by the chemical and Hf isotopic compositions of Lesser Antilles forearc sediments. Earth Planet. Sci. Lett. 287, 86–99 (2009).
Poli, S. Melting carbonated epidote eclogites: carbonatites from subducting slabs. Prog. Earth Planet. Sci. 3, 27 (2016).
Acknowledgements
S.F.F. and C.C. are funded by ARC grant FL180100134 and S.S.S. by Macquarie University support funds for the FL project. M.W.F. is funded by Macquarie University grant MQRF0001074-2020. The Ocean Discovery Project provided the marine limestone and sediment samples. We acknowledge the facilities of the Centre for Advanced Microscopy at the Australian National University, Canberra. We thank Z. Hu and T. He for analysis of Cl contents of the starting materials. We acknowledge I. Ezad for proofreading this manuscript.
Author information
Authors and Affiliations
Contributions
C.C., M.W.F. and S.F.F. designed the study. C.C. and S.S.S. carried out the experiments. C.C. and M.W.F. performed analytical measurements. C.C. wrote the manuscript and all authors contributed to interpreting data and revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Ananya Mallik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 The compositions of starting materials in this study compared with previous high-pressure experiments and natural sediments and altered basalts.
a, CO2 and H2O contents of starting materials from this study (CS2, CS1 and CS5) and from previous high-pressure experiments on carbonated sediments26,27,50,60,61 and oceanic crust12,14,62,63,64. The model CaO–Al2O3–SiO2–CO2–H2O system is shown for comparison13. b–f, Chemical compositions of sedimentary columns subducting at global trenches17, the Lesser Antilles sediments from DODP Site 144 (ref. 77), altered oceanic crust18 and starting materials in this study. The global weighted average composition of subducted sediments (GLOSS-II)17 is shown for comparison.
Extended Data Fig. 2 Representative backscattered electron images of run products at 3 and 4 GPa.
a–c, 3 GPa. d–i, 4 GPa. a,d–e, Subsolidus experiments. b,c,f–i, Above-solidus experiments. Silicate melt at 3 GPa (b,c) and 4 GPa (g). Carbonatite melt at T = 900 °C and 4 GPa (f). KCl at T = 850 °C (h) and both KCl and NaCl at T = 900 °C (i).
Extended Data Fig. 3 Representative backscattered electron images of experimental charges at 5 GPa.
a–c, Subsolidus experiments. d–i, Above-solidus experiments. KCl in carbonatite melt (f) and in the solid residues (g). NaCl in carbonatite melts (h) and in the solid residues (i).
Extended Data Fig. 6 The chemical compositions of carbonate-rich phases in the experiments.
The chemical compositions of carbonate precipitated from fluid and of calcite (a) and compositions of silicate and carbonatite melts (b) in this study. Carbonate precipitates and melts from previous experiments on hydrated carbonated gabbro12 and carbonated sediments26,27,50 are shown for comparison. The stability regions of (Mg, Fe)-calcite and siderite–magnesite solid solution are from ref. 78.
Extended Data Fig. 7 Subduction of carbonate-rich crustal rocks through time.
a, Schematic illustration showing subduction of carbonate-rich crustal rocks and replenishment of carbon and/or chlorine to cratonic mantle roots and deep sources of HIMU-type ocean island basalts (OIBs). Stability of carbonate and chloride during subduction of carbonate-rich crustal rocks influenced by the infiltration of Cl-rich fluid in the cold (b), warm (c) and hot (d) subduction regimes.
Extended Data Fig. 8 The chemical compositions of minerals in the experiments.
Major element chemistry of garnet, jadeite and carbonates showing systematic changes with temperature. Their compositions are independent of the bulk compositions of the starting materials.
Extended Data Fig. 9 Representative backscattered electron images of the experimental charges in the unpolished halves of capsules.
KCl coexists with other residual minerals as inclusions (a) and coexists with carbonatite melts (b) in the experiment at 950 °C. c, NaCl coexists with carbonatite melts in the experiment at 1,100 °C.
Supplementary information
Supplementary Data 1
Compilation of age of eclogitic diamonds constrained by Sm–Nd and Re–Os isochrons for inclusions (n > 2) in diamonds. n = the number of inclusions for the isochron. Note that we compile the isochron ages only when n > 2.
Supplementary Data 2
Major element compositions of phengite, garnet, jadeite, aragonite, (Mg, Fe) calcite, epidote and calcite in the experiments.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, C., Förster, M.W., Foley, S.F. et al. Carbonate-rich crust subduction drives the deep carbon and chlorine cycles. Nature 620, 576–581 (2023). https://doi.org/10.1038/s41586-023-06211-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06211-4
This article is cited by
-
Crystal structure of calcite-type Ca1–xMnxCO3 solid solution by X-ray diffraction and Raman spectroscopy
Physics and Chemistry of Minerals (2024)
-
Fertile upper mantle peridotite xenoliths indicate no wholesale destruction of cratonic root in East Asia
Communications Earth & Environment (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
