
Abstract
The stable forms of carbon in Earth’s deep interior control storage and fluxes of carbon through the planet over geologic time, impacting the surface climate as well as carrying records of geologic processes in the form of diamond inclusions. However, current estimates of the distribution of carbon in Earth’s mantle are uncertain, due in part to limited understanding of the fate of carbonates through subduction, the main mechanism that transports carbon from Earth’s surface to its interior. Oxidized carbon carried by subduction has been found to reside in MgCO3 throughout much of the mantle. Experiments in this study demonstrate that at deep mantle conditions MgCO3 reacts with silicates to form CaCO3. In combination with previous work indicating that CaCO3 is more stable than MgCO3 under reducing conditions of Earth’s lowermost mantle, these observations allow us to predict that the signature of surface carbon reaching Earth’s lowermost mantle may include CaCO3.
Similar content being viewed by others


Introduction
Carbon is not the only key to life and Earth’s habitability but also traces and modifies geological processes of subduction, partial melting, degassing, and metasomatism, providing valuable insights into Earth’s evolution1. Over the history of the planet, carbon transport between surface and deep reservoirs has impacted the atmospheric, oceanic, and crustal CO2 budgets in tandem with the composition and redox state of the Earth’s mantle2,3. Carbon is transported from Earth’s surface to its interior mainly as carbonate minerals in subduction zones and is returned in carbon-bearing gas/fluid through volcanic degassing2,3. These processes leave signatures in the mantle, including depletion of incompatible elements4,5, diamond formation (and inclusions)6,7, and isotopic abundances8,9. Carbon flux via subduction to the deep mantle remains uncertain, with estimated magnitudes ranging from 0.0001 to 52 megatons/year3,10. The wide range of these estimates is due in part to limited understanding of the physical and chemical responses of carbonates to mantle pressures, temperatures, and compositional environments.
The dominant carbonates carried into the mantle by subducting slabs, dolomite CaMg(CO3)2, magnesite MgCO3, and calcite CaCO311, undergo changes in crystal structure or state and chemical reactions at depth. Carbonates are likely to be retained as solid minerals in subducting ocean crust until/unless the solidus of carbonated peridotite12,13 or eclogite14,15 intersects with mantle geotherms, initiating melting. These slab-derived carbonatite melts will segregate to the overlying mantle due to low viscosity and density16, or be reduced to diamonds at depths greater than ~250 km via redox freezing7,15,17. However, carbonates are present in the mantle transition zone and possibly lower-mantle depths in some regions, based on direct evidence provided by carbonate minerals found in deep-sourced diamond inclusions18,19. Additional evidence from thermodynamic modeling of devolatilization of carbonate-bearing subducting slab20,21 and melting experiments on carbonates in the MgCO3–CaCO3 system up to 80 GPa22 supports the preservation of solid carbonates along low-temperature geotherms in subducting slabs in the lower mantle. However, the temperature is not the only control on the fate of subducted carbonates: carbonates may also interact chemically with the major phases of the ambient mantle or basalt-rich subducted crust. In these compositions in the lower mantle, the silicates potentially reacting with carbonates are bridgmanite (bdg), post-perovskite (pPv), and Ca-perovskite (Ca-Pv).
The presence of the end-member carbonates, MgCO3 and CaCO3 (note that (Mg,Ca)(CO3)2 dolomite breaks down to these end-members above 5 GPa and 1200 K23), together with lower-mantle silicates depends on the thermodynamics and kinetics of the carbonate–silicate exchange reaction:
Previous experiments24,25 indicate that CaCO3 reacts with silicates to form MgCO3 via the forward reaction up to 80 GPa and 2300 K, i.e., at least to the mid-lower mantle. Theoretical studies further predict that MgCO3 + CaSiO3 are enthalpically favored over CaCO3 + MgSiO3 throughout the lower mantle pressure and temperature regime26,27,28,29,30. However, although many studies have addressed the stability of individual carbonates up to higher pressures31,32,33, no experiments examined the carbonate–silicate cation exchange reaction up to core–mantle boundary conditions.
In this work, to assess the stability of MgCO3 and CaCO3 coexisting with lower-mantle silicates, we conduct a series of experiments on the carbonate–silicate reaction along the lower-mantle geotherm. Thin disks of carbonates and silicates were loaded together in laser-heated diamond-anvil cells (LHDAC, Supplementary Table 1, see “Methods” for details). Laser heating at 1600–2800 K and 33–137 GPa was applied for 10–400 min. Run products were examined by in situ synchrotron X-ray diffraction (XRD) and ex situ energy-dispersive X-ray spectroscopy (EDX) analysis with a scanning transmission electron microscope (STEM, see “Methods” for details).
Results
Calcium carbonate reaction to form magnesium carbonate
Experiments assessed thermodynamic stability by using as reactants either (Mg,Ca)CO3 + (Mg,Fe)SiO3 (reactants for the forward reaction, hereafter referred to CaC-to-MgC) and (Mg,Fe)CO3 + CaSiO3 (reactants for the reverse reaction, hereafter referred to MgC-to-CaC). For reaction CaC-to-MgC, the criterion for determining whether the reaction takes place is the presence of newly synthesized CaSiO3-perovskite in the run product. For reaction MgC-to-CaC, newly synthesized MgSiO3 and CaCO3 indicate the reaction is favorable. The silicate reaction products are easier to observe through diffraction than carbonates due to higher diffraction intensity.
Experiments with CaC-to-MgC reactants indicate the forward reaction takes place in runs conducted below 83 GPa (runs #1–4), as determined via both EDX and XRD. For example, ex situ electron microscopic analysis of the sample recovered from 33 GPa and 1650 K (run #1) (Fig. 1a–c) reveals a ~1-μm-thick layer of CaSiO3 between the silicate layer and the carbonate layer, coexisting with SiO2, FeO, MgSiO3, and MgCO3. These observations are consistent with in situ XRD patterns of run products after heating (Supplementary Figs. 3b and 5a), which exhibit several new sharp peaks compared to the pattern before heating (Supplementary Fig. 3a). The diffraction pattern of run products is consistent with the presence of Ca-Pv, magnesite, bdg, wüstite, stishovite, and monoclinic dolomite III (previously observed at pressure above 36 GPa34). Ca-Pv can be observed in the run products of CaC-to-MgC up to 83 GPa (Supplementary Figs. 3c and 4a, b), in agreement with previous experimental observations24,25.

Images of selected recovered sample cross-sections obtained using backscattered scanning electron microscopy (a, d, g), scanning transmitted electron microscopy (b, e, h), and energy-dispersive X-ray mapping (c, f, i) of the cross-section show the silicate layer sandwiched by two carbonate layers, with the reaction region along the contacting interface. a–c Ex situ analysis of sample quenched from 33 GPa and 1650 K heated for 15 min (run #1) demonstrates reaction CaC-to-MgC: CaSiO3 is not present in starting materials but is indicated in EDX map by colocation of Ca and Si, shown in magenta. d–f Ex situ analysis of sample quenched from 88 GPa and 1800 K heated for 150 min (run #9) demonstrates reaction MgC-to-CaC: MgSiO3 is not present in starting materials but is indicated in EDX map by colocation of Mg and Si, shown in blue-green. CaCO3 also appears as a red (Ca, but no Si) ribbon within CaSiO3 starting material. g–i Ex situ analysis of sample quenched from 133 GPa and 2000 K heated for 400 min (run #10) demonstrates reaction MgC-to-CaC: MgSiO3 appears as Ca-depleted, Si-rich region (blue or blue-green) adjacent to CaSiO3 starting material (magenta).
At higher pressures from 91 to 137 GPa, however, we observe no evidence of carbonate–silicate exchange reaction in experiments with CaC-to-MgC reactants. Ca-Pv is not identified in the run products (runs #5-7) through either in situ (Supplementary Figs. 3d, e, 4c, d, and 5b) or ex situ analysis. New, sharp peaks from bdg and pPv can be observed in situ in XRD patterns (Supplementary Fig. 3d, e), indicating the sample was sufficiently heated to transform starting materials to high-pressure silicate structures, but no carbonate–silicate exchange reaction occurs. Two hypotheses can explain these observations: (1) in contrast to theoretical predictions that the reversal of the carbonate-exchange reaction takes place at higher pressures and lower temperatures26,27,28,29,30, CaCO3 + MgSiO3 become more favorable than MgCO3 + CaSiO3 from 91 to 137 GPa and 2100 to 2800 K; (2) the reaction CaC-to-MgC is hindered by reaction kinetics, and metastable starting materials are observed.
Magnesium carbonate reaction to form calcium carbonate
In order to resolve the thermodynamically stable phase assemblage, three separate sets of experiments on the backward reaction (MgC-to-CaC, runs #8–11) were conducted at 35–133 GPa and 1800–2000 K. Elemental mapping of the run products of experiments at 88 GPa (run #10, Fig. 1d–f) and 133 GPa (run #11, Fig. 1g–i) indicates that MgSiO3 layers formed along with the carbonate–silicate interface, and newly formed CaCO3 can be observed as well. At 35 GPa, neither EDX nor XRD shows MgSiO3 formed from MgC-to-CaC reactants (run #8, Supplementary Fig. 9). Observations of the reversal of the reaction confirm that MgCO3 is unstable and reacts with CaSiO3 producing CaCO3 and MgSiO3 at pressures higher than 88 GPa along a lower-mantle geotherm.
Our results agree with previous experimental constraints (Fig. 2) below 80 GPa showing: dolomite is unstable relative to CaCO3 and MgCO3 at lower-mantle conditions23,24,25,35; neither CaO nor MgO are observed in run products, indicating no decomposition of CaCO3 and MgCO3 into oxides plus CO226,27,30; MgCO3 is more favorable in the lower mantle than CaCO3 up to ~80 GPa due to the CaC-to-MgC reaction24,25. Since similar previous studies were limited to pressures below 80 GPa, they did not observe the reversal reaction (MgC-to-CaC). Combining our new results with previous results24,25 and theoretical predictions indicating a positive Clapeyron slope for this reaction28,29,30, we suggest a reaction boundary above 80 GPa with a positive slope (black dashed line in Fig. 2). We note that the experimental data allow for significant uncertainty in this boundary, but are inconsistent with theoretical predictions28,29,30 (yellow region, Fig. 2). This discrepancy may have been produced by theoretical approximations at higher temperatures. If density functional perturbation theory and quasi-harmonic approximation have misestimated the volumes of the carbonate phases expected to be stable at ~80 GPa and higher pressures, this could lead to the systematic overestimation of Gibbs free energy of CaCO3 + MgSiO3 relative to MgCO3 + CaSiO3 at higher temperatures.

The boundary sketched as a black dashed line with gray shadow as uncertainty inferred is based on experimental observations of carbonate–silicate exchange reactions CaC-to-MgC and MgC-to-CaC. Squares represent observations from this work starting with (Ca,Mg)CO3 and (Mg,Fe)SiO3, looking for newly synthesized CaSiO3 to indicate the CaC-to-MgC reaction takes place. Circle symbols represent observations from this work of experiments starting with (Mg,Fe)CO3 + CaSiO3, looking for identification of newly synthesized MgSiO3 to indicate the MgC-to-CaC reaction takes place. Open symbols indicate nonreaction and filled for confirmed reaction, and blue and red colors correspond to the inferred stable phase assemblage based on reaction products. Triangles indicate the P–T conditions for CaC-to-MgC taking place reported by Seto et al.25, and blue-shaded region indicates approximate conditions of four experiments conducted by Biellmann et al.24 using indirect methods for pressure and temperature calibration, which all produced the run products MgCO3 + CaSiO3. The error bars indicate uncertainties of pressure and temperature measurements (see “Methods” for details). The boundaries proposed by previous theoretical predictions are illustrated by yellow-shaded region28,29,30.
Discussion
The pressure/temperature conditions of the reversal reaction as constrained by these experiments are similar to those of polymorphic phase transitions associated with sp2–sp3 bonding changes in both MgCO3 and CaCO3, which suggests these transitions are related to the stabilization of a CaCO3 + MgSiO3 assemblage. The transition from sp2- to sp3 bonds in MgCO3 has been identified at ~80 GPa with the stabilization of the C2/m structure32,33,36, and the resulting densification of MgCO3 supports the forward reaction to MgCO3 + CaSiO3. The transition in CaCO3 from sp2- to sp3 bonds in the P21/c-h structure was experimentally observed at ~105 GPa and 2000 K37. Computational studies predicted this boundary at ~7029 and ~100 GPa38 at mantle-relevant temperatures (red-shaded region in Fig. 3). While an earlier study that did not include the sp3 CaCO3-P21/c-h structure predicted a crossover in silicate–carbonate-exchange reaction at 135 GPa and 0 K26, a later study that predicted the sp3 CaCO3-P21/c-h structure found a silicate–carbonate reaction reversal at 84 GPa and 0 K30. This would correspond to sp2–sp3 crossover and stabilization of CaCO3 + MgSiO3 in the mid-lower mantle.

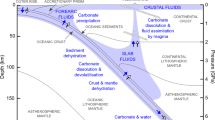
The gray dotted line indicates the reversal boundary of the carbonate–silicate exchange reaction proposed by this study, whereas previous theoretical predictions are illustrated by yellow-shaded region28,29,30. The cyan and orange lines indicate the decarbonation reactions of CaCO3 + SiO240 and MgCO3 + SiO241, respectively. The black dashed line shows the melting curve of MgCO3–CaCO3 system constrained by Thomson et al.22. Four typical mantle geotherms are modified from Maeda et al.36. The red-shaded region indicates the transition boundary of CaCO3 from sp2 to sp3 structure predicted by density functional theory computations29,38.
Whether a crossover in the carbonate–silicate exchange reaction takes place in the deep Earth depends on whether carbonates are preserved in Earth’s lower mantle to at least 1800-km depth. Previous studies have identified barriers to carbon subduction and stability in the lower mantle, particularly melting15,39 and reduction40,41,42. If carried in cold subducting slabs, MgCO3 and CaCO3 may avoid melting as their melting temperatures22 are higher than some predicted cold slab geotherms36. Any solid carbonate in the mantle will be in contact and may equilibrate with silicates in all mantle environments and with free silica in basalt-rich compositions. MgCO3 and CaCO3 have been observed in experiments40,41,42 to undergo decarbonation reactions with free silica over a pressure range of ~40 to 60 GPa. However, the Clapeyron slope of CaCO3 + SiO2 → CaSiO3 + CO2 is positive and takes place at pressure/temperature conditions warmer than the coolest slab geotherms40. Observations that MgCO3 is less thermally stable than CaCO3 support the survival of CaCO3 rather than MgCO3 along a cold subducted slab geotherm to the lowermost mantle36,41 (Fig. 3). In this study, we report a reversal in the Mg–Ca silicate–carbonate cation exchange reaction at ~90 GPa, making MgCO3 + CaSiO3 favorable in the upper part of the lower mantle, while CaCO3 + MgSiO3 is preferred in the lower part of the lower mantle (Fig. 3). However, the question of whether any carbonate persists to these depths in the coldest subducting slabs remains unresolved. If MgCO3 remains present in cold slabs, and the reaction CaC-to-MgC proceeds throughout most of the mantle eliminating CaCO3, the reversal MgC-to-CaC reaction may transform MgCO3 back to CaCO3 in the lowermost mantle (Fig. 4). CaCO3 could thus be found in the lowermost mantle coexisting with silicates and reduced iron.

Through subduction, the carbonates may undergo melting (red arrow), redox freezing with metallic iron (purple arrow), decarbonation reaction with free silica (blue arrow), and exchange reaction with lower-mantle silicates (green arrow). Based on the observation of reversal of the carbonate–silicate cation exchange reaction at conditions relevant to cold subducted slabs at mid-lower-mantle depths, CaCO3 is the potential stable phase that hosts oxidized carbon in the lowermost mantle.
The reduced nature of the Earth’s mantle, with oxygen fugacity inferred to be near the iron–wüstite buffer in the transition zone and greater depths43, stabilizes diamond or Fe carbide as long-term hosts of carbon, owing to their chemical refractoriness and dynamic immobility44. Similarly, our experimental observations support CaCO3 as a refractory, stable host for oxidized carbon in the middle to the lowermost mantle, in particular, the high-pressure polymorph of CaCO3 (CaCO3-P21/c-h) with tetrahedral bonds37. Experimental observations also suggest that CaCO3 is more resistant to redox breakdown reaction with iron under reduced conditions than MgCO335. In addition, due to the cation exchange between carbonate and silicate, the relative stability of MgCO3 or CaCO3 will change in the lowermost mantle, and depending on conditions one of these phases may buffer the redox state of the mantle through an influx of oxidized carbon in the form of solid carbonate45.
The Mg–Ca silicate–carbonate-exchange reactions along with subduction pressure–temperature (P–T) conditions may impact observable signatures of Mg and Ca isotopes in mantle silicates under certain special conditions, or in carbonate inclusions in diamonds. Subducting carbonates carry low-δ44/40Ca and low-δ26Mg signatures relative to the heavier mantle ratios, but although previous studies have observed heterogeneity in the Ca and Mg isotope signatures in basalts and mantle peridotites, these studies determined that lighter ratios cannot be simply interpreted as evidence of recycled marine carbonates46,47. The Mg–Ca silicate–carbonate-exchange reactions along with subduction P–T conditions may contribute to these variable Mg and Ca isotopic compositions. The reaction CaC-to-MgC in the transition zone and upper part of the lower mantle would transfer light Ca isotopes from subducted CaCO3 to CaSiO3 (Ca-Pv) (Supplementary Note 1 and Supplementary Fig. 12). Isotopically light Ca-Pv can then be trapped in diamond inclusions and return to the surface48, while the Ca isotopic signature of upwelling rocks would remain variable, as it undergoes continuous fractionation within peridotitic mantle lithologies46,49,50,51. The modification of carbonate–silicate phase equilibria observed in this study provides a new process that could alter Mg and Ca isotopic composition in such lithologies (Supplementary Note 1 and Supplementary Fig. 12). While the isotope signature of MgSiO3 produced by reaction MgC-to-CaC would not be observable due to the small masses involved relative to the vast lower-mantle reservoir of MgSiO3, any CaCO3 produced in the deep lower mantle by this reaction would carry a heavier deep mantle δ44/40Ca signature that would distinguish it from surface-derived carbonate. If preserved in diamond inclusions and returned to the surface, heavy CaCO3 could be used to trace the presence of oxidized carbon in the lowermost mantle. The potential of CaCO3 to be a signature of an ultradeep carbon cycle reaching the core–mantle-boundary region may help to reveal other mysteries of the deep mantle, such as heat budget related to radioactive elements stored in Ca-bearing silicates52, and compositions of heterogeneities that may record Earth’s early history48,53.
Methods
Starting materials
To investigate phase equilibria in the carbonate–silicate system in Earth’s lower mantle and control for effects of reaction kinetics, both CaC-to-MgC and MgC-to-CaC experiments were carried out in symmetric diamond-anvil cells (DAC) with flat-top double-sided laser heating54. For CaC-to-MgC, natural dolomite with homogeneous composition of (Mg0.38Ca0.59Fe0.03)CO3 was used as a carbonate reactant, the composition, and structure of which has been characterized by X-ray fluorescence spectroscopy and X-ray diffraction, respectively35. Fe-bearing enstatite synthesized at École Polytechnique Fédérale de Lausanne with a composition of (Mg0.5Fe0.5)SiO3 was used as a silicate reactant55. For MgC-to-CaC, natural ferromagnesite (sample from Princeton University) was used as a carbonate reactant, with composition determined to be (Mg0.87Fe0.13)CO3 by wavelength dispersive X-ray spectroscopy in a Cameca SX100 Electron Probe Microanalyzer at the University of Michigan. Pure calcium silicate (CaSiO3, Alfa Aesar) was used as a silicate reactant. The chief advantages to the abovementioned starting compositions are that recognition of a carbonate–silicate exchange reaction only requires identification of the presence of newly synthesized silicates in quenched run products, i.e., Ca-perovskite (Ca-Pv) in CaC-to-MgC and bridgmanite (bdg) in MgC-to-CaC; and Fe-bearing enstatite and ferromagnesite can serve as laser absorber during the forward CaC-to-MgC and reversal MgC-to-CaC experiments, respectively.
LHDAC experiments
The dolomite, enstatite, and calcium silicate samples were separately ground under acetone in an agate mortar for ~2 h each to achieve homogenous, finely powdered samples with grain size typically less than ~2 µm. A single ferromagnesite crystal was double-side polished to ~10-micron thickness. All starting materials were dried in an oven at 120 °C overnight before loading, and the powder samples were subsequently pressed in a DAC to form thin foils approximately ~8–10-µm thick. The enstatite foils and ferromagnesite crystals were sandwiched between iron-free dolomite and calcium silicate, respectively, serving as thermal insulators in symmetric DACs for CaC-to-MgC and MgC-to-CaC (Supplementary Figs. 1 and2). No other pressure standard or medium was loaded to prevent reactions with other components and contamination of the chemical system. The sample sandwiches were loaded in sample chambers with diameters approximately halves of the anvil culet sizes drilled into Re gaskets pre-indented to a thickness of ∼30 μm, by using the laser drilling system at HPCAT (Sector 16) of the Advanced Photon Source (APS), Argonne National Laboratory (ANL)56. Diamond anvils with flat culets of 300 μm were used for experiments under 60 GPa, beveled culets of 150/300 μm for experiments under 100 GPa, and beveled culets of 75/300 μm for experiments up to 140 GPa.
Before laser heating, each sample was compressed to the target pressure at 300 K, and after heating each sample was quenched to ambient pressure at 300 K to limit and preserve reactions at target conditions. Pressures were determined from the Raman shift of the singlet peak of the diamond anvil at the culet surface57, and post-heating pressures were typically within 3% of the pre-heating pressure. Thermal pressure during heating may be estimated to be ~10% GPa higher than the pre-heating pressure at the modest temperatures58,59. High-temperature conditions were achieved by using a double-sided ytterbium fiber laser heating system at beamline 13-ID-D (GeoSoilEnviroCars) of APS, ANL54, with two 1.064 μm laser beams focused down to a flat-top spot with a diameter of 10–12 μm on both sides of the sample. Temperatures of the heated samples were determined by fitting the measured thermal radiation spectra using the Planck radiation function under the graybody approximation54. The temperature reported in Supplementary Table 1 is the temporal average of multiple temperature measurements over the heating duration. Temperature fluctuations over this timescale were less than the specified uncertainty, which is derived from a standard deviation of temperature measurements from both sides of the laser-heated sample (typically ± 100 K below 2000 K and ±150 K above 2000 K) (Supplementary Figs. 10 and 11). Experiments were held at temperatures between 1600 and 2800 K for ~30 min in CaC-to-MgC experiments and up to 400 min in MgC-to-CaC experiments.
In situ XRD
Phases synthesized at high P/T and achievement of chemical steady-state were determined by in situ angle-dispersive X-ray diffraction (XRD) measurements performed before, during, and after heating at beamline 13-ID-D (GeoSoilEnviroCars) of APS, ANL. The incident X-ray beam was focused down to less than 3 × 4 μm2 with a monochromatic wavelength λ = 0.3344 Å. Diffracted X-rays were recorded using a MAR 165 detector or Pilatus 1 M CdTe pixel array detector. NIST standard LaB6 was used to calibrate the detector distance, tilt angle, and rotation angle of the image plane relative to the incident X-ray beam. Exposure times were typically 30 s. The XRD patterns were integrated to produce 2θ plots using the software DIOPTAS60.
Ex situ EDX
After complete pressure release, each sample was recovered from the LHDAC, and then sectioned along the compression axis through the laser-heated spot and over the entire thickness of the DAC sample (~5–20 μm), using a focused ion beam (FIB) coupled with a field-emission scanning electron microscope (FE-SEM) at IPGP (Paris, France) or the Michigan Center for Materials Characterization at the University of Michigan (Ann Arbor, USA). A ~30-nm-thick Au layer was coated on each sample to reduce charging in the scanning electron microscope, and a 2-μm-thick Pt layer was deposited across the center of each heated spot to protect the sample from damage by the Ga+ ion beam. Thin sections of each heated spot were extracted and polished to electron transparency (∼100-nm thickness).
Textural and chemical characterization of recovered samples was performed with scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDX) in a JEOL 2200FS field-emission TEM (Center for Advanced Microscopy, MSU), operated at 200 kV to image the sample in brightfield. EDX maps were scanned over 512 × 384 pixel areas with a pixel dwell time of 50 μs. Typical count rates were ~2000 counts per second. Chemical mapping rather than point measurement approach prevents migration of elements due to damage by the electron beam. Uncertainties in compositions were determined from standard deviations of EDX measurements obtained from selected regions within multiple grains.
Data availability
Additional diffraction and spectroscopy data and metadata are available in the Supplementary Information and from the corresponding authors upon request.
References
Hazen, R. M. & Schiffries, C. M. Why deep carbon? Rev. Mineral. Geochem. 75, 1–6 (2013).
Plank, T. & Manning, C. E. Subducting carbon. Nature 574, 343–352 (2019).
Kelemen, P. B. & Manning, C. E. Reevaluating carbon fluxes in subduction zones, what goes down, mostly comes up. Proc. Natl Acad. Sci. USA 112, E3997–E4006 (2015).
Stachel, T. et al. The trace element composition of silicate inclusions in diamonds: a review. Lithos 77, 1–19 (2004).
Thomson, A. R. et al. Trace element composition of silicate inclusions in sub-lithospheric diamonds from the Juina-5 kimberlite: evidence for diamond growth from slab melts. Lithos 265, 108–124 (2016).
Palyanov, Y. N. et al. Mantle-slab interaction and redox mechanism of diamond formation. Proc. Natl Acad. Sci. USA 110, 20408–20413 (2013).
Rohrbach, A. & Schmidt, M. W. Redox freezing and melting in the Earth’s deep mantle resulting from carbon-iron redox coupling. Nature 472, 209–212 (2011).
Cartigny, P., Palot, M., Thomassot, E. & Harris, J. W. Diamond formation: a stable isotope perspective. Annu. Rev. Earth Planet. Sci. 42, 699–732 (2014).
Teng, F.-Z. Magnesium isotope geochemistry. Rev. Mineral. Geochem. 82, 219–287 (2017).
Dasgupta, R. & Hirschmann, M. M. The deep carbon cycle and melting in Earth’s interior. Earth Planet Sci. Lett. 298, 1–13 (2010).
Poli, S. & Schmidt, M. W. Petrology of subducted slabs. Annu. Rev. Earth Planet. Sci. 30, 207–235 (2002).
Ghosh, S., Litasov, K. & Ohtani, E. Phase relations and melting of carbonated peridotite between 10 and 20 GPa: a proxy for alkali- and CO2-rich silicate melts in the deep mantle. Contrib. Miner. Pet. 167, 23 (2014).
Dasgupta, R. & Hirschmann, M. M. Melting in the Earth’s deep upper mantle caused by carbon dioxide. Nature 440, 659–662 (2006).
Kiseeva, E. S. et al. An experimental study of carbonated eclogite at 3.5-5.5 GPa-implications for silicate and carbonate metasomatism in the cratonic mantle. J. Petrol. 53, 727–759 (2012).
Thomson, A. R., Walter, M. J., Kohn, S. C. & Brooker, R. A. Slab melting as a barrier to deep carbon subduction. Nature 529, 76–79 (2016).
Sun, C. & Dasgupta, R. Slab–mantle interaction, carbon transport, and kimberlite generation in the deep upper mantle. Earth Planet Sci. Lett. 506, 38–52 (2019).
Stagno, V., Ojwang, D. O., McCammon, C. A. & Frost, D. J. The oxidation state of the mantle and the extraction of carbon from Earth’s interior. Nature 493, 84–88 (2013).
Brenker, F. E. et al. Carbonates from the lower part of transition zone or even the lower mantle. Earth Planet Sci. Lett. 260, 1–9 (2007).
Wirth, R., Kaminsky, F., Matsyuk, S. & Schreiber, A. Unusual micro- and nano-inclusions in diamonds from the Juina Area, Brazil. Earth Planet Sci. Lett. 286, 292–303 (2009).
Kerrick, D. M. & Connolly, J. A. Metamorphic devolatilization of subducted marine sediments and the transport of volatiles into the Earth’s mantle. Nature 411, 293–296 (2001).
Poli, S., Franzolin, E., Fumagalli, P. & Crottini, A. The transport of carbon and hydrogen in subducted oceanic crust: an experimental study to 5 GPa. Earth Planet Sci. Lett. 278, 350–360 (2009).
Thomson, A. R., Walter, M. J., Lord, O. T. & Kohn, S. C. Experimental determination of melting in the systems enstatite-magnesite and magnesite-calcite from 15 to 80 GPa. Am. Miner. 99, 1544–1554 (2014).
Luth, R. W. Experimental determination of the reaction aragonite + magnesite = dolomite at 5 to 9 GPa. Contrib. Miner. Pet. 141, 222–232 (2004).
Biellmann, C., Gillet, P., Peyronneau, J. & Reynard, B. Experimental evidence for carbonate stability in the Earth’s lower mantle. Earth Planet Sci. Lett. 118, 31–41 (1993).
Seto, Y., Hamane, D., Nagai, T. & Fujino, K. Fate of carbonates within oceanic plates subducted to the lower mantle, and a possible mechanism of diamond formation. Phys. Chem. Min. 35, 223–229 (2008).
Oganov, A. R., Ono, S., Ma, Y. M., Glass, C. W. & Garcia, A. Novel high-pressure structures of MgCO3, CaCO3 and CO2 and their role in Earth’s lower mantle. Earth Planet Sci. Lett. 273, 38–47 (2008).
Pickard, C. J. & Needs, R. J. Structures and stability of calcium and magnesium carbonates at mantle pressures. Phys. Rev. B 91, 104101 (2015).
Yao, X., Xie, C. W., Dong, X., Oganov, A. R. & Zeng, Q. F. Novel high-pressure calcium carbonates. Phys. Rev. B 98, 014108 (2018).
Zhang, Z. G., Mao, Z., Liu, X., Zhang, Y. G. & Brodholt, J. Stability and reactions of CaCO3 polymorphs in the Earth’s deep mantle. J. Geophys Res.-Sol. Ea 123, 6491–6500 (2018).
Santos, S. S. M., Marcondes, M. L., Justo, J. F. & Assali, L. V. C. Stability of calcium and magnesium carbonates at Earth’s lower mantle thermodynamic conditions. Earth Planet Sci. Lett. 506, 1–7 (2019).
Isshiki, M. et al. Stability of magnesite and its high-pressure form in the lowermost mantle. Nature 427, 60–63 (2004).
Boulard, E. et al. New host for carbon in the deep Earth. Proc. Natl Acad. Sci. USA 108, 5184–5187 (2011).
Binck, J. et al. Phase stabilities of MgCO3 and MgCO3-II studied by Raman spectroscopy, X-ray diffraction, and density functional theory calculations. Phys. Rev. Mater. 4, 055001 (2020).
Mao, Z. et al. Dolomite III: a new candidate lower mantle carbonate. Geophy. Res. Lett. 38, L22303 https://doi.org/10.1029/2011gl049519 (2011).
Dorfman, S. M. et al. Carbonate stability in the reduced lower mantle. Earth Planet Sci. Lett. 489, 84–91 (2018).
Maeda, F. et al. Diamond formation in the deep lower mantle: a high-pressure reaction of MgCO3 and SiO2. Sci. Rep. 7, 40602 (2017).
Lobanov, S. S. et al. Raman spectroscopy and X-ray diffraction of sp3 CaCO3 at lower mantle pressures. Phys. Rev. B 96, 104101 (2017).
Santos, S. S. M., Marcondes, M. L., Justo, J. F. & Assali, L. V. C. Calcium carbonate at high pressures and high temperatures: a first-principles investigation. Phys. Earth Planet. Inter. 299, 106327 (2020).
Kiseeva, E. S., Litasov, K. D., Yaxley, G. M., Ohtani, E. & Kamenetsky, V. S. Melting and phase relations of carbonated eclogite at 9-21 GPa and the petrogenesis of alkali-rich melts in the deep mantle. J. Petrol. 54, 1555–1583 (2013).
Li, X. Y. et al. New high-pressure phase of CaCO3 at the topmost lower mantle: implication for the deep-mantle carbon transportation. Geophys. Res. Lett. 45, 1355–1360 (2018).
Drewitt, J. W. E. et al. The fate of carbonate in oceanic crust subducted into earth’s lower mantle. Earth Planet Sci. Lett. 511, 213–222 (2019).
Kakizawa, S., Inoue, T., Suenami, H. & Kikegawa, T. Decarbonation and melting in MgCO3–SiO2 system at high temperature and high pressure. J. Mineral. Petrol. Sci. 110, 179–188 (2015).
Frost, D. J. & McCammon, C. A. The redox state of Earth’s mantle. Annu. Rev. Earth Planet. Sci. 36, 389–420 (2008).
Shirey, S. B. et al. Diamonds and the geology of mantle carbon. Carbon Earth 75, 355–421 (2013).
Stagno, V. et al. Carbon-Bearing Phases throughout Earth’s Interior. In Deep Carbon: Past to Present, (eds. Orcutt, B. N., Daniel, I. & Dasgupta, R.) 66–88 https://doi.org/10.1017/9781108677950.004 (Cambridge University Press, UK 2019).
Ionov, D. A. et al. Calcium isotopic signatures of carbonatite and silicate metasomatism, melt percolation and crustal recycling in the lithospheric mantle. Geochim Cosmochim. Acta 248, 1–13 (2019).
Wang, S. J., Teng, F. Z. & Li, S. G. Tracing carbonate-silicate interaction during subduction using magnesium and oxygen isotopes. Nat. Commun. 5, 5328 (2014).
Nestola, F. et al. CaSiO3 perovskite in diamond indicates the recycling of oceanic crust into the lower mantle. Nature 555, 237–241 (2018).
Chen, C. et al. Calcium isotope evidence for subduction-enriched lithospheric mantle under the northern North China Craton. Geochim. Cosmochim. Acta 238, 55–67 (2018).
Kang, J.-T. et al. Calcium isotopic fractionation in mantle peridotites by melting and metasomatism and Ca isotope composition of the Bulk Silicate Earth. Earth Planet Sci. Lett. 474, 128–137 (2017).
Amsellem, E. et al. Calcium isotopic evidence for the mantle sources of carbonatites. Sci. Adv. 6, eaba3269 (2020).
Corgne, A., Liebske, C., Wood, B. J., Rubie, D. C. & Frost, D. J. Silicate perovskite-melt partitioning of trace elements and geochemical signature of a deep perovskitic reservoir. Geochim. Cosmochim. Acta 69, 485–496 (2005).
Howell, D. et al. Deep carbon through time: Earth’s diamond record and its implications for carbon cycling and fluid speciation in the mantle. Geochim. Cosmochim. Acta 275, 99–122 (2020).
Prakapenka, V. B. et al. Advanced flat top laser heating system for high pressure research at GSECARS: application to the melting behavior of germanium. High. Press. Res. 28, 225–235 (2008).
Dorfman, S. M. et al. Effects of composition and pressure on electronic states of iron in bridgmanite. Am. Miner. 105, 1030–1039 (2020).
Hrubiak, R., Sinogeikin, S., Rod, E. & Shen, G. The laser micro-machining system for diamond anvil cell experiments and general precision machining applications at the high pressure collaborative access team. Rev. Sci. Instrum. 86, 072202 (2015).
Akahama, Y. & Kawamura, H. Pressure calibration of diamond anvil Raman gauge to 310GPa. J. Appl. Phys. 100, 043516 (2006).
Nomura, R. et al. Low core-mantle boundary temperature inferred from the solidus of pyrolite. Science 343, 522–525 (2014).
Fiquet, G. et al. Melting of peridotite to 140 gigapascals. Science 329, 1516–1518 (2010).
Prescher, C. & Prakapenka, V. B. DIOPTAS: a program for reduction of two-dimensional X-ray diffraction data and data exploration. High. Press. Res. 35, 223–230 (2015).
Acknowledgements
We thank Feng Huang and Shichun Huang for helpful discussions on isotopes. We thank A. Hunter (University of Michigan’s Michigan Center for Materials Characterization), X. Fan (Center for Advanced Microscopy, Michigan State University), and O.K. Neill (Robert B. Mitchell Electron Microbeam Analysis Lab) for assistance with SEM-FIB, STEM, and EPMA, respectively. This work was supported by new faculty startup funding from Michigan State University, the Sloan Foundation’s Deep Carbon Observatory Grant G-2017-9954, and National Science Foundation (NSF) grant EAR-1751664 to S.M.D. Parts of this work were supported by IPGP multidisciplinary program PARI, and by Region Île-de-France SESAME Grant no. 12015908. GeoSoilEnviroCARS is supported by U.S. Department of Energy (DOE) award DE-FG02-94ER14466 and NSF grant EAR-1634415. The Advanced Photon Source, a DOE Office of Science User Facility, is operated by Argonne National Laboratory under contract DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
M.L.: conceptualization, investigation, formal analysis, writing—original draft, and visualization; S.M.D.: conceptualization, methodology, investigation, writing—review and editing, and supervision; J.B.: conceptualization, supervision, and writing—review and editing; S.B.: investigation; E.G.: resources and writing— review & editing; V.B.P.: resources, methodology, and writing—review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lv, M., Dorfman, S.M., Badro, J. et al. Reversal of carbonate-silicate cation exchange in cold slabs in Earth’s lower mantle. Nat Commun 12, 1712 (2021). https://doi.org/10.1038/s41467-021-21761-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-21761-9
This article is cited by
-
Crystal structure of calcite-type Ca1–xMnxCO3 solid solution by X-ray diffraction and Raman spectroscopy
Physics and Chemistry of Minerals (2024)
-
Deep recycling of crustal materials by the Hainan mantle plume: evidence from Zn–Sr–Nd–Pb isotopes of Hainan Island basalts
Contributions to Mineralogy and Petrology (2024)
-
Recycled carbonates elevate the electrical conductivity of deeply subducting eclogite in the Earth’s interior
Communications Earth & Environment (2023)
-
GeoSoilEnviroCARS (Sector 13) at the Advanced Photon Source: a comprehensive synchrotron radiation facility for Earth science research at ambient and extreme conditions
Physics and Chemistry of Minerals (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
