
Abstract
Tritium labelling is a critical tool for investigating the pharmacokinetic and pharmacodynamic properties of drugs, autoradiography, receptor binding and receptor occupancy studies1. Tritium gas is the preferred source of tritium for the preparation of labelled molecules because it is available in high isotopic purity2. The introduction of tritium labels from tritium gas is commonly achieved by heterogeneous transition-metal-catalysed tritiation of aryl (pseudo)halides. However, heterogeneous catalysts such as palladium supported on carbon operate through a reaction mechanism that also results in the reduction of other functional groups that are prominently featured in pharmaceuticals3. Homogeneous palladium catalysts can react chemoselectively with aryl (pseudo)halides but have not been used for hydrogenolysis reactions because, after required oxidative addition, they cannot split dihydrogen4. Here we report a homogenous hydrogenolysis reaction with a well defined, molecular palladium catalyst. We show how the thianthrene leaving group—which can be introduced selectively into pharmaceuticals by late-stage C–H functionalization5—differs in its coordinating ability to relevant palladium(II) catalysts from conventional leaving groups to enable the previously unrealized catalysis with dihydrogen. This distinct reactivity combined with the chemoselectivity of a well defined molecular palladium catalyst enables the tritiation of small-molecule pharmaceuticals that contain functionality that may otherwise not be tolerated by heterogeneous catalysts. The tritiation reaction does not require an inert atmosphere or dry conditions and is therefore practical and robust to execute, and could have an immediate impact in the discovery and development of pharmaceuticals.
Similar content being viewed by others

Main
Tritium (3H) labelling allows the direct incorporation of a radioactive tag into pharmaceutical candidates without substantial changes in their chemical and physical properties and biological activity6. However, many reliable hydrogenation or hydrogenolysis reactions cannot be suitably used for tritium labelling owing to a lack of reagents, low molar activity, restricted functional-group tolerance or safety concerns. Tritiated water (3H2O) is problematic owing to fast washout of the label from omnipresent water and safety concerns regarding the potential fast uptake of radioactive water by experimentalists. The preferred source of tritium labels is tritium gas (3H2), which is available in high isotopic purity and practical to handle with commercially available manifolds on the small scale typically used for labelling2,7.
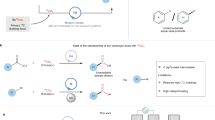
Hydrogenation with hydrogen gas is one of the most extensively studied reactions in chemistry, with numerous important applications ranging from biomass degradation8 to hydrogenolysis of otherwise persistent halogenated pollutants9. Several well defined, homogeneous transition-metal catalysts based on rhodium10, iridium11 and ruthenium12 can split the strong hydrogen–hydrogen bond for countless productive hydrogenation reactions of unsaturated bonds. However, appropriate unsaturated bonds are often not present in pharmaceuticals or would be destroyed by hydrogenation, and the same hydrogenation catalysts are generally not useful for the hydrogenolysis of carbon–halide bonds because most transition-metal hydrides are inactive towards the oxidative addition of carbon–heteroatom bonds. In the presence of both dihydrogen and aryl(pseudo)halides, dihydrogen oxidative addition is commonly faster, which results in metal hydrides in higher oxidation states that are not suitable for aryl (pseudo)halide oxidative addition13,14. Therefore, for the hydrogenolysis of carbon–heteroatom bonds15, chemists select heterogeneous catalysts, such as palladium supported on carbon, which can effectively reduce aryl(pseudo)halides through a mechanistically distinct pathway16,17. The reactivity of the active hydrogen chemisorbed on the catalyst surface results in low chemoselectivity, and the undesired reduction of other functional groups typically found in pharmaceuticals(Fig. 1a)3.

a, Heterogeneous Pd-catalysed hydrogenolysis with H2. b, Homogeneous Pd-catalysed hydrogenolysis of aryl halide. L, neutral 2-electron ligand. c, Homogeneous Pd-catalysed hydrogenolysis of aryl thianthrenium salt. d, Chemo- and site-selective C–H tritiation via arylthianthrenium salt by homogeneous palladium catalysis. X, conventional (pseudo)halide.
Tritium for hydrogen exchange is a desirable way for tritium incorporation because prior functionalization is not required18. Several impressive hydrogen isotope exchange reactions have been developed with transition-metal catalysts, maybe most notably those based on iridium19,20 and nickel21; however, they require the presence of directing groups or heterocycles for efficient transformations. An aromatic hydrogen isotope exchange reaction that does not require directing groups was developed based on an iron catalyst, which provides successful tritiation complementary to the iridium-catalysed methods22. At present, the low-valent iron catalyst does not tolerate protic functional groups and requires an inert atmosphere, which complicates its routine, practical application in pharmaceutical development23,24. In addition, all hydrogen isotope exchange reactions typically afford several constitutional isomers with more than one label incorporated, which can be advantageous because high molar activity can be reached25, but may also render interpretation of imaging studies difficult when the labelled molecule is metabolized26. In that sense, a robust, practical, site-selective functionalization would be complementary to the hydrogen exchange reactions and could add significant value to the field27.
Metal-catalysed hydrogenolysis proceeds through metal-hydride intermediates28 that can be formed by dihydrogen oxidative addition. Transition-metal complexes that react with aryl (pseudo)halides through oxidative addition do not engage with dihydrogen in a second oxidative reaction to form metal hydrides. Heterolytic cleavage of the hydrogen–hydrogen bond can also result in the formation of metal hydrides29. For example, electrophilic transition-metal complexes of iridium30 and ruthenium31 can substantially acidify dihydrogen through complexation owing to a strong σ donor–acceptor interaction29,32, which can result in the formation of metal hydrides through deprotonation of the metal η2-dihydrogen complexes33. Only a few examples of palladium (Pd) dihydrogen complexes are known4, and no electrophilic Pd(II) catalysts—generated through oxidative addition of an aryl (pseudo)halide to low-valent palladium—have been shown to heterolyse dihydrogen34,35,36. Upon oxidative addition to a ligated Pd(0), no appropriate coordination site is available at square planar d8 Pd(II) tetracoordinate complexes for dihydrogen coordination because (pseudo)halides outcompete dihydrogen from coordination to Pd(II) (Fig. 1b). Aryl thianthrenium salts can be more reactive than aryl halides and aryl triflates in cross-coupling reactions5,37 and photoredox catalysis38,39,40, and they are readily accessed by direct, late-stage C–H functionalization of complex small-molecule arenes in exquisite site selectivity. The fundamental novelty reported here entails that dihydrogen can be split by a palladium catalyst as part of a productive catalytic cycle that includes oxidative addition of aryl thianthrenium salts (Fig. 1c). The ability to engage structurally complex arenes and small-molecule pharmaceuticals at a late stage without directing groups, excellent functional group tolerance, exquisite site selectivity and high isotopic purity, in combination with operational simplicity, distinguish this protocol from heterogeneous hydrodehalogenation and hydrogen isotope exchange reactions for practical and robust tritiation (Fig. 1d).
Hydrogenolysis of aryl thianthrenium salts was performed with 2H2 as a more convenient and safer 3H2 surrogate. Palladium-catalysed hydrogenolysis of biphenyl-derived thianthrenium salt 1-TT with 2H2 afforded the desired 2H-labelled product [2H]1 with more than 99% deuterium (D) incorporation (Fig. 2a). No synthetically useful reaction was observed with aryl bromides, aryl iodides and aryl triflates, nor with other arylsulfonium salts such as those derived from dibenzothiothene (DBT) and diphenyl sulfide (DPS), respectively. The lack of reactivity of the aryl halides could not be addressed through the use of other commonly used, highly active monodentate phosphine ligands (Fig. 2a, Supplementary Table 11). Likewise, the addition of (pseudo)halide anions to the productive reaction of arylthianthrenium salts poisons the reactivity, probably due to palladophilic halide outcompeting dihydrogen for binding; the starting material 1-TT is recovered in these cases (Fig. 2b). Although sulfur-containing molecules are often responsible for catalyst poisoning owing to strong coordination41, we could not identify any coordination of thianthrene to cationic Pd(II), which is consistent with an open coordination site on the metal centre for interaction and coordination of H2 during catalysis. As measured by infrared spectroscopy, even triflate outcompetes thianthrene from coordination to Pd(II) (Supplementary Scheme 2, Supplementary Fig. 23). Cationic Pd(II) complexes can commonly be obtained from Pd(II) aryl halides by addition of silver(I) (Ag(I)) salts with non-coordinating anions during catalysis, as exemplified by Overman in Heck reactions that proceed through a mechanistically relevant cationic reaction pathway42. However, hydrogenolysis of aryl bromide in the presence of Ag(I) additives did not result in a productive reaction as observed with the arylthianthreniums, which may be due to the incompatibility of oxidizing Ag(I) and the presumed in situ-generated palladium hydride (Fig 2a).

a, Hydrogenolysis of aryl (pseudo) halides. dppf, 1,1'-bis(diphenylphosphino)ferrocene. aFor an additional evaluation of highly active monodentate phosphine ligands, see Supplementary Table 11. b, Effect of halide anions in hydrogenolysis of aryl thianthrenium salt.
(Tetrafluoro)thianthrenium (TFT and TT, respectively) salts of electron-deficient (2, 7), electron-neutral (4, 14, 24) and electron-rich arenes (3, 8, 11), as well as arenes bearing ortho substituents (15, 17, 22) successfully participate in Pd-catalysed reductive deuteration (Fig. 3). Functional-group compatibility is high, as evidenced by substrates containing unprotected hydroxyl groups (4), carbamates (5), amides (10, 20, 25), heterocycles (6, 13, 18) and acid-sensitive glycosidic linkages (21). In contrast to heterogeneous catalysis, the homogeneous catalyst system tolerates reducible functional groups such as aryl (pseudo)halides (2, 3, 6, 7, 8), nitriles (3), alkenes (20, 26, 27), esters (14, 15), aldehydes (23), sulfones (26), nitro groups (19) and benzyl ethers (23). Primary amines must be protected. No 1H–2H scrambling was observed when the reaction was performed in the presence of water, and high levels of deuterium incorporation were detected throughout. When compared with heterogeneous deuterodehalogenation, where significant isotopic scrambling can occur43, the new catalytic platform with aryl thianthrenium salts provides a viable approach for the synthesis of deuterated and tritiated drug molecules (vide infra) with high isotopic purity in a practical setting that does not require the rigorous absence of water or oxygen.

Deuterium-labelling of aryl thianthrenium salts. The general reaction conditions are as follows: (tetrafluoro)thianthrenium salt (0.20 mmol, 1.0 equiv.), K3PO4 (1.0 equiv.), Pd(OAc)2 (1 mol%), dppf (1 mol%) or Pd[(PtBu3)]2 1 mol%), THF (0.1 M or 0.2 M), 2H2 (1 atm, about 5 equiv.), 23 °C, 12 h. aYield of the isolated product. bDeuterium incorporation determined by 1H NMR. cH incorporation determined by mass analysis. d5 mol% of catalyst. e2.5 mol% of catalyst. f48 h. g10 mol% of catalyst. h2 h. iIsolated as triflimide adduct. TFT, 2,3,7,8-tetrafluorothianthrene.
Given our design of electrophilic, cationic Pd(II) species for H2 activation and the observed poisoning of the active catalyst by anions as weakly coordinating as triflate, successful reductive deuteration of substrates featuring coordinating groups such as tertiary amines (6, 20), pyridines (11, 25), pyrazoles (18), quinolines (22), anilines (10, 16) and thiophenes (17) is non-trivial. We observed that the appropriate choice of counterion for the arylthianthrenium salt starting material can reduce its solubility in tetrahydrofuran (THF), with the reservoir of starting material in the solid phase, leaching material for conversion as the reaction progresses. The ability to tune solubility is another yet-unappreciated advantage of the arylthianthrenium salts when compared with arenes with conventional leaving groups that lack such a handle.
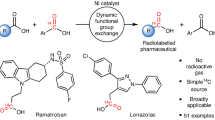
Translation of the hydrogenolysis reaction to tritium labelling was accomplished on micromole scales at a subatmospheric pressure of 3H2 gas to reduce the risk of tritium gas leakage and with a higher loading of catalyst to achieve faster reaction rates (Fig. 4). No special care is required to exclude air or moisture during the radiosynthesis, and, in contrast to purification after hydrogen exchange reactions, the radiolabelled product can be readily separated from the starting material owing to the pronounced polarity difference due to the cationic nature of the thianthrenium salts. No isotope scrambling and straightforward purification result in high molar activity, which is often required for receptor binding and occupancy studies44. Higher molar activities can be achieved when more than one label is introduced, as shown for 3H2-etofenprox ([3H2]29). In all cases, high, predictable positional selectivity enables single, well defined, labelled molecules that are generally not accessible with hydrogen isotope exchange reactions unless directing groups are used.

Tritium labelling of aryl (tetrafluoro)thianthrenium salts. The general reaction conditions are as follows: (tetrafluoro)thianthrenium salt (3.0 µmol, 1.0 equiv.), K3PO4 (1.0 equiv.), Pd(OAc)2 (10 mol%), dppf (10 mol%), THF (6 mM), 3H2 (0.3 atm, about 20 equiv.), 23 °C, 19 h. a68 h. bK3PO4 (4 equiv.), 3H2 (0.7 atm, about 20 equiv.), 4 h.
A plausible reaction pathway that is consistent with all experimental data is outlined in Fig. 5. The generation of a catalytically active, monoligated Pd(0) catalyst from the only observed resting state Pd[(PtBu3)]2 during catalysis is consistent with the observed reaction order of 0.5 in Pd[(PtBu3)]2. We determined a primary kinetic isotope effect (KIE) of kH/kD = 3.1, where k is the rate constant, by measuring the initial rates of independent reactions with H2 and 2H2, respectively, at the same pressure, and an equilibrium isotope effect of 1.1 from an intermolecular competition experiment between H2 and 2H2 present at the same partial pressure, which is inconsistent with Ar–TT+ oxidative addition preceding dihydrogen binding but consistent with reversible dihydrogen association before oxidative addition to Ar–TT+. The observed first order in Ar–TT+ and positive order in H2 are in agreement with this proposal. Although we cannot exclude dihydrogen oxidative addition from A based on our kinetic data, it would need to be reversible, as indicated by the competition KIE, and the Pd(0) intermediate A could be expected to react with Ar–TT+ faster in concerted oxidative-addition or single-electron-transfer pathways than a putative off-cycle Pd(II) dihydride. Irrespective of the exact mechanism of irreversible—measured zero-order in TFT—oxidative addition, which may also differ depending on the ancillary ligand(s), intermediate B distinguishes the proposed pathway from those that would be accessible with other aryl(pseudo)halides. No H/D scrambling into the product was observed when hydrogenolysis was performed in the presence of H2O (Supplementary Figs. 4, 5), consistent with irreversible proton transfer from B. As the base is not fully dissolved in the liquid phase, its concentration does not appear in the rate law, but the primary KIE excludes turnover-limiting oxidative addition. Similarly, an inverse KIE or a primary KIE much smaller than 3.1 would be observed for turnover-limiting reductive elimination45 from C; moreover, C–H reductive elimination is expected to be fast46,47. The data are consistent with turnover-limiting dihydrogen splitting to C, followed by fast reductive elimination to product. The addition of halides and pseudohalides or other soluble Lewis bases may prevent formation of B in sufficient quantities. Although aryldiazonium salts are typically not accessible through late-stage functionalization, a productive albeit less efficient (Supplementary Scheme 1), hydrogenolysis of biphenyldiazonium tetrafluoroborate under our reaction conditions is consistent with our mechanism hypothesis, as no coordinating anion is generated upon oxidative addition. Although some homogeneous transition-metal complexes can function as precursors for heterogeneous nanoparticles, we have excluded active heterogeneous catalysts as the major contributor to catalysis by means of observing the reaction in the presence and absence of mercury (Supplementary Fig. 22)48.

B, K3PO4. Counterion can also be PF6 or NTf2.
In contrast to (pseudo)halides, the thianthrenyl group can be introduced selectively into complex small molecules. We show here how the lack of strong coordination to palladium and the intrinsic solubility properties enables thianthrenium chemistry to solve the challenge of homogeneous palladium-catalysed hydrogenolysis for the chemoselective synthesis of tritium-labelled small molecules.
Methods
See Supplementary Information for further methods.
Data availability
The data reported in this paper are available in the main text or the Supplementary Information.
References
Isin, E. M., Elmore, C. S., Nilsson, G. N., Thompson, R. A. & Weidolf, L. Use of radiolabeled compounds in drug metabolism and pharmacokinetic studies. Chem. Res. Toxicol. 25, 532–542 (2012).
Voges, R., Heys, J. R. & Moenius, T. in Preparation of Compounds Labeled with Tritium and Carbon-14 109–209 (John Wiley, 2009).
Nishimura S. in Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis 572–663 (Wiley-Interscience, 2001).
Connelly, S. J., Chanez, A. G., Kaminsky, W. & Heinekey, D. M. Characterization of a palladium dihydrogen complex. Angew. Chem. Int. Ed. 54, 5915–5918 (2015).
Berger, F. et al. Site-selective and versatile aromatic C–H functionalization by thianthrenation. Nature 567, 223–228 (2019).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Saljoughian, M. Synthetic tritium labeling: reagents and methodologies. Synthesis 13, 1781–1801 (2002).
Yang, Y. & McCarty, P. L. Biomass, oleate, and other possible substrates for chloroethene reductive dehalogenation. Bioremediat. J. 4, 125–133 (2000).
Nakano, M. M. & Zuber, P. in Strict and Facultative Anaerobes Medical and Environmental Aspects 303–317 (CRC Press, 2004).
Shapley, J. R., Schrock, R. R. & Osborn, J. A. Preparation and catalytic properties of some cationic iridium(III) and rhodium(III) dihydrido complexes. J. Am. Chem. Soc. 91, 2816−2817 (1969).
Vaska, L. & DiLuzio, J. W. Activation of hydrogen by a transition metal complex at normal conditions leading to a stable molecular dihydride. J. Am. Chem. Soc. 84, 679−680 (1962).
Chinn, M. S. & Heinekey, D. M. Synthesis and properties of a series of ruthenium dihydrogen complexes. J. Am. Chem. Soc. 109, 5865−5867 (1987).
Fan, L., Parkin, S. & Ozerov, O. V. Halobenzenes and Ir(I): kinetic C–H oxidative addition and thermodynamic C–Hal oxidative addition. J. Am. Chem. Soc. 127, 16772–16773 (2005).
Douglas, T. M., Chaplin, A. B. & Weller, A. S. Dihydrogen loss from a 14-electron rhodium(III) bis-phosphine dihydride to give a rhodium(I) complex that undergoes oxidative addition with aryl chlorides. Organometallics 27, 2918–2921 (2008).
Alonso, F. Beletskaya, I. P. & Yus, M. Metal-mediated reductive hydrodehalogenation of organic halides. Chem. Rev. 102, 4009–4091 (2002).
van Santen, R. A. in Modern Heterogeneous Catalysis: An Introduction 293–344 (John Wiley, 2017).
Sisak, A. & Simon O. B. in The Handbook of Homogeneous Hydrogenation (eds de Vries J. G. & Elsevier, C. J.) 513–546 (Wiley-VCH, 2007).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C–H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022–3047 (2018).
Heys, R. Investigation of [IrH2(Me2CO)2(PPh3)2]BF4 as a catalyst of hydrogen isotope exchange of substrates in solution. J. Chem. Soc. Chem. Commun. 680−681 (1992).
Ellames, G. J., Gibson, J. S., Herbert, J. M.; Kerr, W. J. & McNeill, A. H. Deuterium exchange mediated by an iridium–phosphine complex formed in situ. Tetrahedron Lett. 42, 6413−6416 (2001).
Zarate, C., Yang, H., Bezdek, M. J., Hesk, D. & Chirik, P. J. Ni(I)–X complexes bearing a bulky α‑diimine ligand: synthesis, structure, and superior catalytic performance in the hydrogen isotope exchange in pharmaceuticals. J. Am. Chem. Soc. 141, 5034–5044 (2019).
Yang, H. et al. Site-selective nickel-catalyzed hydrogen isotope exchange in N-heterocycles and its application to the tritiation of pharmaceuticals. ACS Catal. 8, 10210−10218 (2018).
Yang, H. & Hesk, D. Base metal-catalyzed hydrogen isotope exchange. J. Labelled Compd Radiopharm. 63, 296−307 (2020).
Yu, R. P., Hesk, D., Rivera, N., Pelczer, I. & Chirik, P. J. Iron-catalysed tritiation of pharmaceuticals. Nature 529, 195–199 (2016).
Yang, H., Dormer, P. G., Rivera, N. R. & Hoover, A. J. Palladium (II)-mediated C–H tritiation of complex pharmaceuticals. Angew. Chem. Int. Ed. 57, 1883–1887 (2018).
Lockley, W. J. S., McEwen, A. & Cooke, R. Tritium: a coming of age for drug discovery and development ADME studies. J. Labelled Compd Radiopharm. 55, 235–257 (2012).
Koniarczyk, J. L., Hesk, D., Overgard, A., Davies, I. W. & McNally, A. A general strategy for site-selective incorporation of deuterium and tritium into pyridines, diazines, and pharmaceuticals. J. Am. Chem. Soc. 140, 1990–1993 (2018).
Sean, M. G. & Gemma, G. The multifarious world of transition metal hydrides. Chem. Soc. Rev. 32, 383–392 (2003).
Kubas, G. J. Fundamentals of H2 binding and reactivity on transition metals underlying hydrogenase function and H2 production and storage. Chem. Rev. 107, 4152–4205 (2007).
Crabtree, R. H., Lavin, M. & Bonneviot, L. J. Some molecular hydrogen complexes of iridium J. Am. Chem. Soc. 108, 4032–4037 (1986).
Gunanathan, C. & Milstein, D. Bond activation and catalysis by ruthenium pincer complexes. Chem. Rev. 114, 12024–12087 (2014).
Crabtree, R. H. Dihydrogen complexation. Chem. Rev. 116, 8750–8769 (2016).
Heinekey, D. M. & Oldham, W. J. Coordination chemistry of dihydrogen Chem. Rev. 93, 913–926 (1993).
Fulmer, G. R., Muller, R. P., Kemp, R. A. & Goldberg, K. I. Hydrogenolysis of palladium(II) hydroxide and methoxide pincer complexes. J. Am. Chem. Soc. 131, 1346–1347 (2009).
Fulmer, G. R., Herndon, A. N., Kaminsky, W., Kemp, R. A. & Goldberg, K. I. Hydrogenolysis of palladium(II) hydroxide, phenoxide, and alkoxide complexes. J. Am. Chem. Soc. 133, 17713–17726 (2011).
Leñero, K. Q. et al. A heterolytic activation of dihydrogen by platinum and palladium complexes. Dalton Trans. 42, 6495–6512 (2013).
Engl, P. S. et al. C–N cross-couplings for site-selective late-stage diversification via aryl sulfonium salts. J. Am. Chem. Soc. 141, 13346–13351 (2019).
Li, J. et al. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat. Chem. 12, 56–62 (2020).
Ye, F. et al. Aryl sulfonium salts for site-selective late-stage trifluoromethylation. Angew. Chem. Int. Ed. 58, 14615–14619 (2019).
Sang, R. et al. Site-selective C–H oxygenation via aryl sulfonium salts. Angew. Chem. Int. Ed. 58, 16161–16166 (2019).
Wise, H. Mechanisms of catalyst poisoning by sulfur species. Stud. Surf. Sci. Catal. 68, 497–504 (1991).
Abelman, M. M., Oh, T. & Overman, L. E. Intramolecular alkene arylations for rapid assembly of polycyclic systems containing quaternary centers. A new synthesis of spirooxindoles and other fused and bridged ring systems. J. Org. Chem. 52, 4130–4133 (1987).
Lockley, W. J. S., Venanzi, N. A. E. & Crane, G. J. Studies of hydrogen isotope scrambling during the dehalogenation of aromatic chloro-compounds with deuterium gas over palladium catalysts. J. Labelled Compd Radiopharm. 63, 531–552 (2020).
Hein, P. et al. in Practical Methods in Cardiovascular Research (eds Dhein, S. et al.) 723–783 (Springer, 2005).
Churchill, D. G., Janak, K. E., Wittenberg, J. S. & Parkin, G. Normal and inverse primary kinetic deuterium isotope effects for C–H bond reductive elimination and oxidative addition reactions of molybdenocene and tungstenocene complexes: evidence for benzene sigma-complex intermediates. J. Am. Chem. Soc. 125, 1403–1420 (2003).
Low, J. J. & Gaddard, W. A. Reductive coupling of hydrogen–hydrogen, hydrogen–carbon, and carbon–carbon bonds from palladium complexes. J. Am. Chem. Soc. 106, 8321–8322 (1984).
Low, J. J. & Gaddard, W. A. Theoretical studies of oxidative addition and reductive elimination. 3. Carbon–hydrogen and carbon-carbon reductive coupling from palladium and platinum bis(phosphine) complexes. J. Am. Chem. Soc. 108, 6115–6128 (1986).
Widegren, J. A. & Finke, R. G. A review of the problem of distinguishing true homogeneous catalysis from soluble or other metal-particle heterogeneous catalysis under reducing conditions. J. Mol. Catal. A 198, 317–341 (2003).
Acknowledgements
We thank S. Marcus, D. Margold, F. Köhler, N. Haupt and D. Kampen for mass spectrometry analysis; M. Kochius, M. Leutzsch, C. Fares, S. Tobegen and C. Wirtz for NMR spectroscopy analysis; F. Kaegi (F. Hoffmann-La Roche Ltd) and RC Tritec for tritium-labelling experiments; and P. Münstermann for high-performance liquid chromatography analysis. We thank Q. Cheng, B. Lansbergen, F. Juliá and L. Zhang for helpful discussions.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
D.Z. developed the reaction chemistry and investigated the mechanism. D.Z., R.P. and J.Y. optimized and explored the substrate scope for reductive deuteration. D.M. optimized and performed the reductive tritiation reaction. D.Z., D.M. and T.R. wrote the manuscript. T.R. directed the project.
Corresponding author
Ethics declarations
Competing interests
T.R. may benefit from royalty payments related to sales from thianthrene-based compounds.
Additional information
Peer review information Nature thanks Viktoria Däschlein-Gessner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains detailed experimental procedures and spectroscopic data, and includes 11 supplementary tables and 33 supplementary figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, D., Petzold, R., Yan, J. et al. Tritiation of aryl thianthrenium salts with a molecular palladium catalyst. Nature 600, 444–449 (2021). https://doi.org/10.1038/s41586-021-04007-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04007-y
This article is cited by
-
Chemoselective umpolung of thiols to episulfoniums for cysteine bioconjugation
Nature Chemistry (2024)
-
A metal-catalysed functional group metathesis approach to the carbon isotope labelling of carboxylic acids
Nature Chemistry (2024)
-
Tritiation gets selective
Nature Synthesis (2022)
-
One-pot H/D exchange and low-coordinated iron electrocatalyzed deuteration of nitriles in D2O to α,β-deuterio aryl ethylamines
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
