
Abstract
IgG4 is the least abundant subclass of IgG in human serum and has unique functional features. IgG4 is largely unable to activate antibody-dependent immune effector responses and, furthermore, undergoes Fab (fragment antigen binding)-arm exchange, rendering it bispecific for antigen binding and functionally monovalent. These properties of IgG4 have a blocking effect, either on the immune response or on the target protein of IgG4. In this Review, we discuss the unique structural characteristics of IgG4 and how these contribute to its roles in health and disease. We highlight how, depending on the setting, IgG4 responses can be beneficial (for example, in responses to allergens or parasites) or detrimental (for example, in autoimmune diseases, in antitumour responses and in anti-biologic responses). The development of novel models for studying IgG4 (patho)physiology and understanding how IgG4 responses are regulated could offer insights into novel treatment strategies for these IgG4-associated disease settings.
Similar content being viewed by others


Introduction
Humoral (antibody-mediated) immune responses are important for protection against pathogen invasion but can also cause disease. Antibodies recognize and bind specific structures of pathogens through their Fab (fragment antigen binding) arms. In addition to mediating direct neutralization of the pathogen, this opsonization of pathogen structures can result in the activation of various immune effector pathways through the Fc (fragment crystallizable) region of antibodies. The antibody Fc region interacts with Fc receptors on immune cells such as macrophages, neutrophils and natural killer cells, resulting in antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP). The antibody Fc region can also interact with the complement system, resulting in antibody-dependent complement deposition, which further primes pathogens for cellular uptake and destruction. The ability of an antibody to elicit these immune responses depends on the type of Fc tail and modifications thereof (for example, glycosylation)1. In humans, five classes of antibody are recognized based on their Fc tail: IgM, IgD, IgE, IgA and IgG. IgM antibodies are produced in the early stages of a primary, adaptive immune response. IgM and IgD form the B cell receptors (BCRs) on naive B cells. The primary IgM response can be followed by a second, long-lasting wave of IgG or IgA antibodies, the latter being involved particularly in mucosal immune responses. IgE probably evolved as a defence against parasitic worms and is also involved in various allergic diseases. Such mature antibody responses are produced by antigen-stimulated B cells that have undergone a process known as class-switch recombination, in which a B cell rearranges its DNA to produce another class of antibody with the same specificity. B cells can undergo repeated rounds of class-switch recombination until the DNA has been recombined using the most 3′ Fc tail gene segment (Fig. 1).

IgG4 responses are most often observed upon repeated or prolonged exposure to certain classes of antigen, including food antigens, parasites, therapeutic proteins and certain autoantigens. IgG4 responses are T cell dependent, with a role for IL-4 and/or IL-13, IL-10, IL-21 and regulatory T (Treg) cells in their induction. The absence of certain pathogen-associated molecular patterns (PAMPs) associated with viral or bacterial infection may favour IgG4 development for unknown reasons. Class-switching to IgG4 may occur directly from IgM+ B cells, or indirectly via IgG1+ intermediates, but the relative contribution of these routes is not clear. Although IgG4 can be induced by specific immunotherapy to alleviate IgE-mediated allergic symptoms, the shift towards an IgG4-dominated antibody response is not the result of IgE+ B cells class-switching towards IgG4, as this is not possible in light of the class-switch order of immunoglobulin heavy-chain constant region segments in the genome (see inset; once a segment has been removed by prior class-switching, a B cell cannot express it anymore). IgG4+ B cells have phenotypic traits that are distinct from those of IgG1+ B cells, including an altered chemokine receptor profile, with lower levels of expression of CXCR3, CXCR4, CXCR5, CCR6 and CCR7 — chemokine receptors involved in germinal centre reactions and the generation of long-lived plasma cells — which therefore likely results in the increased efflux of IgG4+ B cells from germinal centres and influx into other tissues, and might also have a role in the overall reduced longevity of IgG4-switched B cells. Inset box adapted from Fig. 1 of ref. 157, Springer Nature Limited.
Four subclasses of IgG exist, numbered according to the order of abundance. IgG1 has the largest relative contribution to total IgG, followed by IgG2 then IgG3 and IgG4. Although IgG4 is the least abundant IgG subclass overall, specific responses can be dominated by IgG4, often associated with chronic or repeated antigen exposure. IgG4 has a unique set of properties compared with the other IgG subclasses that has led to IgG4 being widely regarded as an anti-inflammatory, ‘benign’ antibody that may have beneficial functions in allergic disease (Table 1). However, evidence is accumulating that IgG4 also has a pathogenic role in a range of diseases. Research in the past decade has shown that IgG4 can have detrimental roles in IgG4 autoimmune diseases (IgG4-AIDs), in tumour immunology and in IgG4-related diseases (IgG4-RDs). The IgG4-AIDs and IgG4-RDs are chronic conditions and for most patients no cure currently exists. Moreover, the increasing use of biological therapies warrants a better understanding of why certain drugs elicit IgG4 anti-drug responses that limit their efficacy.
In this Review, we highlight how the unique structural and functional characteristics of IgG4 contribute to disease onset and progression in these settings. Furthermore, we provide an overview of our current understanding of how IgG4 responses are regulated. By understanding these processes, future therapeutic strategies could be shaped to prevent pathogenic IgG4 responses or induce beneficial IgG4 responses. Although we appreciate that most antibody responses involve a range of different (sub)classes2, in this Review we focus on IgG4-associated diseases that have a predominant IgG4 (antigen-specific) response.
Development of an IgG4 response
The production of IgG4 requires that B cells undergo class-switch recombination (Fig. 1). A direct switch from IgM to IgG4 can occur, but indirect switching through, for example, IgG1 is also — at least theoretically — possible. Although direct evidence is lacking, in BCR repertoire analyses only limited clonal overlap between IgG1 and IgG4 responses has been observed, which suggests that indirect switching to IgG4 via IgG1 is a minor route in vivo3. In keeping with this, in vitro, naive IgM+ B cells readily switch towards IgG4 production4, whereas IgG1+ memory B cells do not5. Interestingly, substantial clonal overlap between IgG4 and IgA2 was observed, which may reflect that common food antigens often induce both IgG4 and IgA2 responses. This could indicate either that there are similar requirements for the development of IgA2 and IgG4 responses or that substantial sequential class-switching from IgG4 to IgA2 occurs. Class-switching from IgE to IgG4 is not possible owing to the order of class-switch elements in the genome (the heavy-chain constant region segments for IgE being downstream (3′) of those for IgG4). Therefore, the allergen-specific IgG4 that is induced by specific immunotherapy in patients with IgE-mediated allergic disease must be derived from either precursor B cells capable of switching to both IgE and IgG4 (for example, non-switched or IgG1+ memory B cells) or newly recruited (naive) B cells6.
Class-switching towards IgG4 is mostly associated with T helper 2 (TH2) cell responses. The type 2 cytokines IL-4 and/or IL-13 are important for the induction of IgG in general, but class-switching to IgG4 may more strictly depend on these cytokines than does class-switching to IgG1 (ref. 7). IL-10 and regulatory T (Treg) cells may also skew the antibody response towards IgG4 (relative to IgE and, possibly, also IgG1)6 (Fig. 1). However, the role of IL-10 is not fully clarified, and studies in vitro have yielded conflicting results depending on, amongst other factors, which cell types are present in addition to naive B cells4 (Box 1).
IgG4-switched B cells have similar potential for terminal differentiation towards antibody-secreting cells to that of IgG1-switched B cells; hence, limitations in the development of an IgG4 antibody response are not owing to intrinsic limitations of IgG4+ B cells5. However, IgG4-switched B cells differ phenotypically from IgG1-switched B cells in several aspects5,7. In particular, they have an altered chemokine receptor profile with lower levels of expression of CXCR3, CXCR4, CXCR5, CCR6 and CCR7 — chemokine receptors involved in germinal centre reactions and the generation of long-lived plasma cells. In the circulation, numbers of IgG4+ B cells reflect serum IgG4 concentrations5, and their levels follow similar patterns throughout life8. IgG4+ cell numbers in blood are low compared with IgG1+ cells and have a relatively low abundance in secondary lymphoid organs7. Furthermore, IgG4 production by antibody-secreting cells can be markedly shorter lived than for other IgG subclasses, requiring continuous input from newly differentiating B cells. Indeed, rituximab (anti-CD20) therapy for B cell depletion has been shown to be particularly beneficial in autoimmune diseases characterized by pathogenic IgG4 (auto)antibodies9,10,11.
During infancy, the proportion of IgG4 in circulation rises slowly with age12, and IgG4 titres generally continue to increase throughout life until the fifth decade, after which a small gradual decline is observed8. Serum levels of IgG4 show great variation in the healthy population, although intra-individual levels are generally stable13. One of the hallmarks of most IgG4 responses is that they develop slowly over time for reasons that are not well understood6. Prolonged or repeated exposure to antigen seems to be a necessary — but not sufficient — factor for the development of an IgG4-dominated response. For example, individuals hyperimmunized with tetanus toxoid have an IgG1-dominated response with little IgG4 despite repeated antigen exposure14, whereas individuals repeatedly vaccinated with SARS-CoV-2 mRNA were shown, in some cases, to have increased proportions of IgG4 after a third vaccination, requiring at least 6 months to develop15. IgG4 is not commonly part of the antibody response to bacterial or viral infection. The range of situations in which specific IgG4 is or can be a dominant factor is wide and includes responses to allergens, therapeutically administered proteins, autoantigens and helminth infections. With the exception of helminths, the absence of an infectious agent seems to be a common feature of IgG4 responses and it is tempting to speculate that the absence of certain danger signals such as pathogen-associated molecular patterns (PAMPs) is a prerequisite for B cells to differentiate towards IgG4-secreting cells in vivo. Indeed, the proportion of IgG4 antibodies was smaller in individuals receiving whole-cell pertussis vaccine than in individuals receiving acellular pertussis vaccine (although IgG4 was only a small fraction of the total IgG response even in the latter)16.
Structure and function of IgG4
Despite the high levels of homology between human IgG subclasses, each subclass has a specific set of functional characteristics owing to particular structural features1 (Table 1). IgG4 is unique in that it has a lesser affinity than other IgG subclasses for many effector molecules, such as Fc receptors and complement, and also because of structural features that affect interactions through the Fab region, such as Fab-arm exchange and a greater propensity for acquiring glycosylation in the variable domains4,17 (Fig. 2).

IgG4 has several unique structural features compared with other IgG subclasses, including specific biases in the IgG4 response repertoire (high affinity and increased levels of Fab (fragment antigen binding) glycosylation), functional monovalency (owing to Fab-arm exchange (FAE)) and a reduced ability to induce effector functions mediated by interactions in the Fc (fragment crystallizable) region. Important residues mediating Fab-arm exchange of IgG4 are serine at position 228 (S228) and arginine at position 409 (R409); C1q and Fc receptor binding are particularly reduced by phenylalanine at position 234 (F234), glycine at position 327 (G327) and serine at position 331 (S331) of IgG4, although residues at other positions may also contribute to the altered binding patterns of IgG4. The functional consequences of these structural features include reduced ability to mediate the Fc-dependent effector functions of antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), a poor ability to activate complement through the Fc domain and interference with immune complex formation through the inability to cross-link antigen. SHM, somatic hypermutation.
Fc-dependent effector functions
IgG4 differs in several key amino acid positions from the other IgG subclasses, resulting in a modified binding pattern to Fcγ receptors (Table 1 and Fig. 2). In particular, relative to IgG1, the amino acid changes at L234F, A327G and P331S in IgG4 are implicated in effects on Fcγ receptor binding18,19,20. Binding to most Fcγ receptors is reduced for IgG4 (although not completely abrogated), resulting in IgG4 having poor ADCC activity21,22 and, probably, also ADCP activity (although this has not been studied in detail). Interestingly, binding of IgG4 to the inhibitory receptor FcγRIIb is not affected, which skews Fcγ receptor signalling induced by IgG4 away from cellular activation and towards inhibition. In the case of IgG1, interaction with the activating receptor FcγRIIIa is markedly increased if the conserved Fc glycan does not contain the core fucose moiety. This is also true for IgG4, and an afucosylated variant of IgG4 was found to induce ADCC, albeit still less efficiently than did IgG1 (ref. 23). However, naturally occurring afucosylated antibody responses seem to be restricted to antiviral responses or alloimmunity to blood cells and platelets, which normally do not have high levels of IgG4 (refs. 1,24). The limited signalling of IgG4 through activating Fc receptors might attenuate the impact of certain autoantibody responses; for example, it has recently been shown in a mouse model of thrombotic thrombocytopenic purpura that recombinant IgG1 antibodies to ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13) are more pathogenic than their IgG4 counterparts in an Fcγ-dependent manner25.
In addition to the ‘classical’ Fcγ receptors (FcγRI–FcγRIII), two ‘non-classical’ Fcγ receptors — Fc receptor-like protein 4 (FCRL4) and FCRL5 — have been reported to bind IgG4, albeit weakly26,27. These receptors are mostly expressed on B cells and have been described to either inhibit or enhance BCR signalling, the latter only if CD21 is simultaneously engaged21,28, thereby augmenting or counteracting the role of FcγRIIb. A specific role of IgG4 in this signalling route is as yet unknown.
Furthermore, IgG4 is a poor activator of complement, resulting in a poor capacity for inducing antibody-dependent complement deposition and ADCP. Complement has an important role in clearing pathogens and promoting inflammation, which consequently is limited when IgG4 dominates the antibody response29. This results mainly from reduced binding to C1q, caused mainly by residue S331 of IgG4 (refs. 30,31), which is the counterpart of P331 in IgG1 (the homologous P436S mutation in IgM also markedly affects C1q binding)32. Nevertheless, some studies suggest that IgG4 can activate complement in specific contexts. For example, artificially enforcing the hexamerization of IgG4 — a process that normally would take place ‘spontaneously’ as part of complex formation of antibody with C1 — results in complement activation by the classical route33,34. This shows that IgG4 has a reduced ability to activate complement but is not completely ‘silent’ in this respect. In patients with membranous nephropathy, all of whom have complement deposition in the kidneys35, IgG4 autoantibodies to phospholipase A2 receptor 1 (PLA2R1) are associated with disease. Recent work indicates a possible role for these IgG4 autoantibodies in complement activation via the lectin pathway, whereby decreased galactosylation levels on the autoantibodies allow for mannose-binding lectin (MBL) binding and complement deposition36. This is in contrast to increased levels of galactosylation promoting IgG1 hexamerization and complement activation by the classical route37, and other studies suggest a pathogenic role of IgG4 autoantibodies to PLA2R1 independent of complement38. Complement activation could be shown in vitro for glyco-engineered recombinant IgG4 antibodies, but only at high antigen density and high antibody concentration, and no contribution of the lectin pathway was observed39.
Fab-arm exchange
Uniquely, serum IgG4 typically does not cross-link antigen40. In fact, IgG4 often behaves effectively as a monovalent antibody in the circulation. IgG4 molecules are produced as bivalent, monospecific antibodies but can, subsequently, engage in a process in which half-molecules of IgG4 (heavy chain and light chain) are randomly exchanged with other IgG4 half-molecules, through a process known as Fab-arm exchange17 (Box 2). This makes most IgG4 molecules in the blood bispecific. Early evidence of this process included the ability of serum IgG4 to cross-link two different allergens41. However, in many cases, the second antigen specificity of an IgG4 molecule will be irrelevant because the exchange is random, and the resulting antibody will behave as if monovalent. Thus, effective binding and downstream signalling of functionally monovalent IgG4 will require high affinity for antigen, as IgG4 cannot benefit from the accumulated binding strength (avidity) of multiple Fabs with the same specificity42. Interestingly, in patients with eosinophilic oesophagitis, very high titres of specific IgG4 to cow milk protein have been observed (together with deposits of IgG4 in the oesophageal wall)43, to such a degree that a substantial portion of IgG4 may still be bivalent in this context44,45; this suggests that the effective monovalency of IgG4 is not absolute but depends on the relative levels of specific IgG4 and total IgG4.
The process of Fab-arm exchange is controlled by redox conditions and can be promoted in vitro by choosing an appropriate redox buffer46,47. Comparing IgG4 with IgG1, two mutations in the latter are required to enable Fab-arm exchange: a P228S mutation in the hinge region allowing for the disulfide bonds that normally connect the heavy chains to be easily broken, and a K409R mutation in the carboxy-terminal domains that results in weaker non-covalent interactions between the heavy chains48. Conversely, therapeutic IgG4 monoclonal antibodies often contain a S228P mutation to prevent Fab-arm exchange in vivo49.
A major functional consequence of the effective monovalency of the majority of IgG4 in vivo is that it further reduces the ability for signalling, antigen cross-linking and immune activation (Fig. 2). Furthermore, Fab-arm exchange seems to further decrease the limited potential of IgG4 for complement activation39. Therefore, Fab-arm exchange together with the overall reduced ability of IgG4 to activate Fcγ receptors and complement means that IgG4 is often regarded as a natural type of ‘blocking’ antibody — a high-affinity monovalent binder with limited potential to induce inflammatory responses4. Because of these weak effector functions and blocking ability, IgG4 is the second most widely used antibody format for therapeutic monoclonal antibodies, with examples including natalizumab, nivolumab and reslizumab.

Glycosylation
The glycan structure on IgG molecules can vary. Specific glycan profiles have been associated with (patho)physiological conditions, and the exact glycan structure can affect antibody functions such as Fcγ receptor activation and complement activation. This has been investigated in most detail for the conserved N-linked glycans in the Fc region. In general, Fc galactosylation of IgG4 seems to be decreased in pathological conditions36,50,51. Furthermore, N-linked glycans are also present in the variable regions of immunoglobulins to different degrees52. Variable domain glycosylation is largely dependent on acquiring mutations that introduce glycosylation motifs during somatic hypermutation, such that specific antibody responses may be highly enriched or depleted for variable domain glycans53. In particular, certain autoantibodies, including those against muscle-specific tyrosine kinase, desmoglein 3 (DSG3) and proteinase 3, are found to be highly glycosylated in the variable domain54,55,56. Interestingly, IgG4 antibodies in general have increased levels of Fab glycans compared with other IgG subclasses51,57. This feature seems to be associated with the type 2 response-like characteristics of the IgG4 response, as BCR repertoire analysis of both IgG4 and IgE responses showed increased levels of N-glycosylation motifs57. The functional consequences of these glycans are not well understood but may include attenuation of antibody-mediated signalling by engaging lectins such as CD22 (ref. 52), elimination of autoreactivity58 or enhancement of BCR signalling59. The link between antibody glycosylation and pathogenicity of IgG4 autoantibodies warrants further investigation.
Physiological roles of IgG4
In general, the ‘blocking’ nature of an IgG4 response may be beneficial when it prevents excessive immune activation. In particular, in both allergic responses and parasitic infections, IgG4 responses are beneficial for the host by inducing tolerance and limiting inflammation (Fig. 3).

The effects of IgG4 can be beneficial to the host. a, In the case of allergy, repeated exposure to an allergen such as bee venom may at first elicit an antibody response dominated by IgE and IgG1, which stimulate inflammation and mast cell degranulation, causing a hypersensitivity response. Continued exposure to the same allergen may result in an increase in type 2 cytokines such as IL-4 and IL-13 and in regulatory cytokines such as IL-10, which stimulate class-switching to IgG4. The increased amounts of IgG4 compete with IgE for binding to allergen, thus preventing mast cell degranulation and inhibiting antigen presentation to T cells by IgE on B cells, which blocks the inflammatory loop and results in tolerance. b, In parasitic infections, the IgE-mediated and IgG1-mediated inflammatory process can be detrimental to host tissue. Worms can secrete immune modulators that skew host cytokine production towards a type 2 profile (also including the regulatory cytokine IL-10), resulting in class-switching to IgG4 and, hence, obstruction of the inflammatory processes directed at the worm as well as reduced inflammatory damage to the host. Such events generally result in asymptomatic symbiosis. APC, antigen-presenting cell.
Allergic responses
Allergy is characterized by hypersensitivity reactions that lead to symptoms including rash, swelling, itching, upper respiratory tract sensitivity and, in severe cases, shock. These reactions result from allergens that trigger IL-13 and IL-4 release by TH2 cells and subsequent class-switching of B cells to IgE production. In response to FcεRI stimulation by IgE bound to allergen, mast cells and basophils release histamine, cytokines and chemokines, which have effects on the vasculature and tissues to cause the hypersensitivity symptoms. Peripheral tolerance to allergens can be achieved by specific immunotherapy or by regular exposure to them, which may induce allergen-specific IgG4 that can contribute to reducing hypersensitivity reactions by competing with IgE for binding to allergen and by other mechanisms60,61,62,63,64,65 (Fig. 3a). Allergen-specific IgG4 responses have been described for a range of allergies, including to grass and birch pollen, cats, bee venom, peanuts and milk4.
In individuals who are allergic, IgG4 can constitute more than 75% of allergen-specific IgG after continuous exposure to antigen66. After specific immunotherapy, IgG1 and, in particular, IgG4 allergen-specific responses can increase in the range 10- to 100-fold, with levels starting to increase after 1 month of therapy66,67. An increase in the titre of allergen-specific IgG4 generally correlates with increased tolerance and reduced hypersensitivity symptoms. The protection against symptoms of allergy mediated by IgG4 is thought to be the result of at least three modes of action: blocking the activity of IgE by competing for allergen binding and preventing mast cell and basophil degranulation68,69,70,71; inhibiting antigen presentation to T cells by IgE on B cells and dendritic cells66; and preventing immune complex formation through the functional monovalency of IgG4. The induction of allergen-specific IgG4 responses is thought to result from prolonged exposure to the allergen and increased production of IL-10 (refs. 62,68). IL-10 not only induces T cell tolerance but also regulates antibody production, resulting in increased IgG production relative to IgE production72,73. IgE-induced CD4+ T cell activation is a very potent route to maintain chronic inflammation. Antigen presentation to T cells via B cells and dendritic cells may be facilitated by IgE. By blocking these effects, IgG4 halts this positive-feedback loop, which limits IgE production and puts a brake on the inflammatory response.
Parasitic infections
IgG4 responses can also occur during parasitic infection (Fig. 3b). The host usually develops a broad B cell-mediated and T cell-mediated immune response against the parasite. In an attempt to evade the host immune response, the parasite stimulates production of cytokines such as IL-10 and induction of Treg cells. As a consequence, in a subset of patients, the anti-parasite B cell response may undergo class-switching towards IgG4. IgG responses can consist of up to 90% IgG4 in these asymptomatic patients74. A dual role for IgG4 in these infections can be envisaged. On the one hand, the ratio of IgG4 to IgE may positively correlate with asymptomatic infection, potentially by preventing ongoing inflammation and damage to host tissues, for example in the case of Brugia malayi infection74. On the other hand, the ratio of specific IgG4 to IgE may correlate with the intensity of infection75 and might represent escape of the parasite from host immunity. Indeed, histamine release by IgE-opsonized basophils from patients with filariasis challenged with filarial antigen was blocked by patient-derived IgG4, and histamine release inversely correlated with IgG4 levels76. Interestingly, it has been suggested that a consequence of the IgG4-mediated attenuation of host immunity by the parasite may be protection of the host from autoimmune disease and allergies (see next section), although the role of parasitic infections in dampening allergy has not been unambiguously determined77. Such a role would be consistent with the ‘hygiene hypothesis’, which suggests that lack of parasite exposure and the improved standards of hygiene in higher-income countries may cause increased prevalence of autoimmune and allergic diseases.
IgG4-dependent pathology
Specific deficiency of IgG4 can occur either as an isolated phenomenon (in ~30% of cases) or in combination with a deficiency of other antibody (sub)classes, such as IgG2, IgA or IgG1 (ref. 78). A selective lack of IgG4 or severely reduced levels of IgG4 are extremely rare. In some individuals (mostly children), IgG4 deficiency is associated with recurrent respiratory tract infections, allergies, candidiasis, chronic diarrhoea and chronic fungal infections78,79. These observations suggest that IgG4 may have an unexplored physiological role in mucosal immunity. In line with this, a recent retrospective study observed IgG4 deficiency in ~20% of patients with inflammatory bowel disease, which was associated with worse disease outcome80. Future studies should elucidate whether there is a causal relationship between these observations.
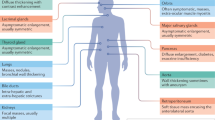
IgG4 hypergammaglobulinaemia occurs in ~5% of the healthy population and seems without consequence. Several diseases are associated with pathogenic IgG4 responses (Fig. 4). Here, we discuss examples of pathology that are directly dependent on IgG4, including autoimmune diseases, antitumour responses and anti-biologic responses.

a, Pemphigus is one of the first-described IgG4 autoimmune diseases (IgG4-AIDs). There are indications that for some forms of pemphigus, exposure to a fly antigen causes an initial, non-pathogenic antibody response that is found also in asymptomatic individuals. In some individuals, epitope spreading occurs and a secondary, pathogenic antibody response develops in which the antibodies cross-react with desmogleins, proteins that are crucial for skin cell adhesion. Further affinity maturation of the autoimmune response results in high-affinity, predominantly IgG4 antibodies that physically obstruct the interaction of desmogleins with other cell adhesion molecules, leading to skin blistering. In some cases, the high-affinity IgG4 antibodies to desmogleins still cross-react with the fly antigen. b, In melanoma, tumour growth can be obstructed by IgG antibodies that cause inflammation and destruction or growth inhibition of tumour cells. Some tumours, in turn, produce IL-4 and IL-10, which stimulate class-switching of local B cells and increased IgG4 production. High levels of high-affinity IgG4 compete with other antibody (sub)classes for binding to the tumour and prevent further inflammation and tumour destruction owing to their anti-inflammatory nature, leading to increased disease progression. c, In patients with chronic inflammatory diseases such as rheumatoid arthritis, the tumour necrosis factor (TNF) inhibitor adalimumab is often used to reduce inflammation. In some individuals, continuous exposure to adalimumab induces an IgG4 response against the variable domain of the therapeutic antibody, which thereby blocks the activity of the biologic and limits its therapeutic efficacy.
IgG4 autoimmune diseases
IgG4-AIDs were first defined as a separate subgroup of antibody-mediated autoimmune disorders in 2015 (ref. 10), and a first attempt at their classification based on the level of evidence for a pathogenic role of IgG4 was proposed soon thereafter11,81. IgG4-AIDs are characterized by autoantibody responses predominantly of the IgG4 subclass against a known antigen. These disorders can affect many organ systems, depending on the major site of action of the targeted antigen, including the kidneys, central and peripheral nervous systems, haematopoietic system and skin. So far, a direct pathogenic role of IgG4 autoantibodies has been established for six IgG4-AIDs through passive transfer of IgG4 in experimental animals (Table 2), but this group is likely to expand in the coming years with more evidence becoming available for 23 other candidate IgG4-AIDs. Diagnosis of an IgG4-AID is based on clinical symptoms and the detection of serum IgG4 autoantibodies to the disease-specific antigen. Antigen-specific IgG4 levels correlate closely with disease severity82,83. As IgG4 is predominantly anti-inflammatory in nature and is not thought to induce pathology through Fc-dependent effector mechanisms, a key mode of action in all IgG4-AIDs is thought to be blocking essential protein–protein interactions of the target antigen10. For example, in muscle-specific kinase (MuSK) myasthenia gravis, which is a prototypical IgG4-AID, IgG4 autoantibodies to MuSK block its interaction with low-density lipoprotein receptor-related protein 4 (LRP4), thereby obstructing a key trophic signalling cascade at the neuromuscular junction and resulting in fatigable skeletal muscle weakness84. Much overlap between the IgG4-AIDs can be found in terms of their inflammatory status and treatment response. For example, B cell depletion therapy with rituximab often results in long-term remission in all of these diseases. These observations suggest that although IgG4-AIDs can present with various symptoms depending on the target antigen, they share an underlying immunophenotype.
Serum levels of IgG4 are, if at all, only slightly increased in patients with IgG4-AIDs (refs. 85,86,87) and the numbers of IgG4+ plasma cells and IgG4+ B cells are normal in circulation (M.G.H., unpublished observations). These observations do not support a hypothesis that these patients have an overall tendency to develop dominant IgG4 responses but, rather, support an antigen-driven aetiology. A recent systematic review reports strong associations of IgG4-AIDs with HLA-DQB1*05 and HLA-DRB1*14, suggesting that these haplotypes predispose to the development of IgG4-AIDs (ref. 88). This may occur through directing B cell development and cytokine production, or by facilitating antigen presentation. In the case of three archetypal IgG4-AIDs — MuSK myasthenia gravis, pemphigus vulgaris and thrombotic thrombocytopenic purpura — increased serum levels of the IgG4-promoting cytokine IL-10 have been reported89,90,91,92. Although some autoimmune diseases mediated by IgG1, IgG2 or IgG3 are associated with tumour development, this has thus far not been reported for IgG4-AIDs, and the aetiology of IgG4-AIDs is expected to be different from that of such paraneoplastic syndromes.
Interestingly, evidence for molecular mimicry resulting in an IgG4-dominated response to desmogleins in the skin epidermis is found in patients with endemic pemphigus foliaceus from Brazilian and Tunisian populations (Fig. 4a). In these patients, antibodies develop against a salivary antigen from flies that are cross-reactive with desmogleins93,94. Moreover, monoclonal antibodies derived from patients with pemphigus vulgaris were shown to be cross-reactive with walnut antigen95. Both walnuts and flies carry allergens that are known to induce IgG4 responses. These observations suggest that exposure to certain IgG4-inducing antigens in combination with a permissive HLA haplotype and an IgG4-promoting immune environment (such as increased IL-10 levels) might have a role in the development of certain IgG4-AIDs. However, it is unclear which factors ultimately cause the IgG4 skewing of these responses.
Autoantibodies of other (sub)classes can also be found in patients with IgG4-AIDs, although usually of much lower titres. For several IgG4-AIDs, the pathogenicity of IgG1, IgG3 and IgM autoantibodies has also been confirmed96,97,98,99,100,101. The mechanisms by which these autoantibodies induce pathology may differ between antibody (sub)classes and target antigens, and could include complement-mediated tissue damage and antigenic cross-linking and internalization causing surface depletion. The functional consequences of IgG4 autoantibodies may also differ between diseases25. The relative contribution of IgG4 to pathology compared with the contribution of other antibody (sub)classes has not been carefully delineated, but the effects of different antibody (sub)classes may function in parallel to increase disease severity. Low titres of IgG1 autoantibodies are also found in non-symptomatic relatives of patients with pemphigus vulgaris, which suggests that having (low levels of) such antibodies alone is not sufficient to precipitate disease symptoms102. It furthermore suggests that there is a subclinical stage in these autoimmune diseases that, upon early detection, could allow for IgG4-AID onset to be prevented. The role of autoantibodies of other (sub)classes in the pathophysiology of IgG4-AIDs requires further investigation.
Interestingly, in autoimmune diseases mediated by IgG1, IgG2 or IgG3 autoantibodies, a switch to an IgG4-dominant response may be therapeutic. Passive transfer of an IgG4 monoclonal antibody targeting acetylcholine receptor (AChR) inhibited subsequent complement-mediated damage and cytotoxicity induced by IgG1 binding to AChR, thus preventing the onset of AChR myasthenia gravis in rhesus macaques17.
Antitumour responses
Antitumour antibody responses can contain or even eliminate malignancies by binding to tumour cells and stimulating ADCC, antibody-dependent complement deposition and/or ADCP. However, in 1977 a prospective study in patients with melanoma identified high levels of IgG4 as having negative effects on survival103. It has since become evident that some malignancies evade host immune defences by inducing class-switching of the antitumour antibody response to IgG4. IgG4 competes with other antibody (sub)classes for binding to tumour antigens and owing to its anti-inflammatory properties blocks the induction of antitumour immune responses104 (Fig. 4b). In the absence of an immune response, tumour cells have increased ability to proliferate and metastasize, resulting in disease progression and decreased survival. Immune evasion through class-switching to IgG4 has been observed in patients with melanoma, cholangiocarcinoma, colon cancer, pancreatic cancer and glioblastoma (reviewed in ref. 105).
Both the total IgG4 level and the number of IgG4+ B cells can be increased in the serum of patients with malignancies and are a negative prognostic indicator103,106,107. These factors are also increased in the tumour microenvironment. The development of antitumour IgG4 responses results from IL-4 and IL-10 production by tumour cells, which directs a modified type 2 response that stimulates class-switching to IgG4 (ref. 104) (Fig. 4b). Furthermore, some tumour microenvironments contain tertiary lymphoid structures with functional germinal centres and Treg cells105. Crosstalk between these chronic inflammatory structures and the tumour may induce increased expression of IL-10 by Treg cells104. Importantly, it is not yet understood why certain tumours are capable of inducing IgG4 responses whereas others are not. In addition, although the reactivity of serum IgG4 antibodies from patients with cancer to tumour cells, for example to melanoma cells, has been confirmed, the precise antigen specificity of these antibodies has not yet been delineated108. Broader study is needed to evaluate whether IgG4 has a pathogenic role in other cancer types. Interestingly, in addition to the proposed role of IgG4 in blocking inflammatory responses, a pro-angiogenic IgG4+ B cell subset (CD49b+CD73+IL-10–) was recently identified109. These B cells were increased in the serum of patients with melanoma and might facilitate tumour angiogenesis.
Anti-biologic responses
IgG4-skewed responses can also occur as a result of chronic exposure to biological therapies. Such responses have been described for clotting factors FVIII and FIX used for the treatment of congenital haemophilia A or haemophilia B110,111,112,113, for interferon-β used for the treatment of multiple sclerosis114 and for the tumour necrosis factor (TNF) inhibitors adalimumab and infliximab used for the treatment of inflammatory disorders such as rheumatoid arthritis and Crohn’s disease115,116. For each of these anti-biologic responses, the primary result is that the therapeutic effect of the biologic is impaired (Fig. 4c). Not all biologics trigger an IgG4-skewed anti-drug response. For example, interferon-β induces prominent IgG4 skewing only in some of the patients who develop an antibody response, which argues against chronic exposure to the biologic being the sole determinant of IgG4 skewing114. It is unclear why certain biologics cause these responses and others do not.
Haemophilia A and haemophilia B are severe clotting disorders caused by an inherited deficiency of FVIII or FIX, respectively. First-line treatment in these patients is chronic replacement of these clotting factors using either plasma-derived or recombinant proteins. Approximately 30% of patients treated with FVIII replacement therapy develop inhibitory antibodies predominantly of the IgG4 subclass117. Low-affinity IgG antibodies to FVIII of all subclasses can be found in both healthy individuals and patients with haemophilia, but high-affinity, high-titre IgG4 blocking antibodies to FVIII are unique to patients65,111. Furthermore, high levels of anti-FVIII IgG4 correlate with decreased efficacy of FVIII replacement therapy118. Although these patients may also have antibodies to FVIII of other subclasses, IgG4 antibodies seem to be particularly detrimental. Interestingly, acquired autoimmune haemophilia A is also associated with a dominant IgG4 response to endogenous FVIII, which would thus classify this form of haemophilia as an IgG4-AID (ref. 119). For unknown reasons, FVIII — both endogenous and exogenous — has the propensity to induce an IgG4 response.
Many inflammatory disorders, including rheumatoid arthritis and Crohn’s disease, can be successfully managed with TNF inhibitor therapy. Monoclonal antibodies are immunogenic to varying degrees, depending, amongst other factors, on their extent of humanization. However, even so-called fully human antibodies, of which the TNF inhibitor adalimumab is an early example, contain parts that are unique and foreign to recipients, namely the complementarity-determining regions responsible for target binding. B cell and T cell epitopes will be present in the biologic that can drive the development of high-affinity, class-switched (IgG4) antibodies120. Similarly, although FVIII is a human protein, it will be seen by the immune system as a partially foreign protein in patients with congenital haemophilia owing to genetic defects in endogenous FVIII. This explains the potential for developing high-affinity, class-switched antibodies, although it is unclear why there is a tendency for these responses to favour class-switching to IgG4.
IgG4-related diseases
IgG4-RDs are a heterogeneous group of inflammatory disorders characterized by massive influx of IgG4+ B cells in affected organs and increased serum levels of IgG4 (refs. 9,121,122). Similar to IgG4-AIDs, a wide variety of organs can be affected in IgG4-RDs, including the thyroid, pituitary gland, pancreas, lungs, kidneys, gastrointestinal system and vasculature, with symptoms varying according to the organ affected. Diagnosis of an IgG4-RD is based on the histopathological finding of an IgG4+ B cell infiltrate, resulting in swelling of the organ, storiform fibrosis and obliterative phlebitis in a tissue biopsy. In addition, increased serum levels of IgG4 and of IgG4+ plasmablasts are often a good biomarker for both diagnosis and monitoring disease progression123; 70–80% of patients have increased serum levels of IgG4. IgG4-AIDs and IgG4-RDs are currently considered to be separate disease entities as there is no evidence for large-scale influx of IgG4+ B cells in the affected organs in IgG4-AIDs or for significantly increased serum levels of IgG4 (refs. 85,87). The typical histology observed in IgG4-RDs involving fibrosis and tissue damage does not seem to have a major role in IgG4-AIDs, although biopsy data in the latter are limited. Furthermore, the pathogenic role of specific IgG4 and IgG4+ B cells in IgG4-RDs remains enigmatic. BCR repertoire sequencing confirmed the clonal expansion of IgG4+ B cells in patients with IgG4-RDs, suggesting that some clones may contribute specifically to disease onset and progression124, although their antigen specificity was not determined. Further research is needed to determine the similarities and differences between IgG4-RDs and IgG4-AIDs in terms of their pathology, aetiology, histology and clinical features.
There are three hypotheses to explain the role of IgG4 in the pathophysiology of IgG4-RDs. First, IgG4-RDs, similar to IgG4-AIDs, are caused by IgG4 antibodies targeting autoantigens in the specific organ that is affected. Second, patients with IgG4-RDs have a type 2-skewed inflammatory environment, for as yet unknown reasons, which triggers pleiotropic IgG4 responses and impaired homing of IgG4+ B cells. Third, IgG4 is present in IgG4-RDs simply to dampen an ongoing immune response and, as such, does not contribute to the pathology.
Clinical clues to support an autoimmune hypothesis in these patients are the responsiveness to immunosuppressants, chronic disease course, presence of autoantibodies and HLA type II associations. Passive transfer of IgG1 and IgG4 from patients with IgG4-RDs can induce similar pathology in experimental animals125. Although IgG antibodies to autoantigens (for example, nuclear antigens, lactoferrin, carbonic anhydrases II and IV, pancreatic secretory inhibitor, trypsinogens and annexin A11) have been found, none of these consistently correlates with an IgG4-RD (refs. 126,127,128). Furthermore, these autoantibodies are mainly of the IgG1 subclass and target intracellular proteins that are unlikely to be the initial autoimmune trigger owing to lack of accessibility. By contrast, and in keeping with the second hypothesis, patients with IgG4-RDs generally have increased IgG4 reactivity against environmental antigens, suggesting that increased levels of IgG4 may be the result of a pleiotropic activation of IgG4+ B cells independent of their antigen specificity129. Type 2 cytokines are increased in the serum of patients with IgG4-RDs, which could fit with both the first and second hypotheses130.
Given the diverse nature of immune cells infiltrating affected organs in patients with IgG4-RDs, some argue that the IgG4 is induced in response to chronic immune stimulation and does not contribute to the pathology. Indeed, passive transfer of IgG1 purified from a patient with pancreatitis reproduced disease in mice, whereas symptoms were markedly reduced upon co-transfer of IgG1 and IgG4 isolated from the same patient125. Furthermore, in addition to IgG4, levels of IgE are often increased in IgG4-RDs (refs. 131,132). Upon treatment with rituximab, both IgG4 and IgE levels have a tendency to decrease131,133. Interestingly, this was also observed upon treatment with abatacept, which interferes with T cell activation, albeit in a limited number of patients134. Furthermore, dupilumab, which blocks the receptors for IL-4 and IL-13, has recently been considered for the treatment of IgG4-RDs (ref. 134). These studies point to a role of T cells (possibly TH2 cells) and IL-4 and/or IL-13 in the pathogenesis of IgG4-RDs, as well as indicating an apparent lack of persistence of IgG4 (and IgE) responses in this disease setting, which, at least for IgG4, seems to be a more general phenomenon7. Future research should focus on determining the sequence of events that lead to the production of IgG4 and confirming or acquitting a pathogenic role for IgG4 in IgG4-RDs.
Some reports suggest an increased incidence of malignancies in patients with IgG4-RDs, through the effects of IgG4 on suppressing antitumour immune responses, although this is still a matter of debate135,136. Having a tumour may also predispose to developing an IgG4-RD through cytokines secreted by the tumour that induce class-switching to IgG4 (ref. 137). Whether having one type of IgG4-associated disease (IgG4-AID or IgG4-RD) can lead to a second type of IgG4-associated disease should be further investigated. Case reports of co-occurrence of IgG4-AIDs with IgG4-RDs do exist but are generally rare138,139.
Future directions
The role of IgG4 antibody responses in physiological and pathological settings is gaining increasing attention. Although, historically, the anti-inflammatory nature of IgG4 was associated with dampening ongoing immune responses, it is increasingly recognized that these antibodies can also cause pathology. The first steps towards understanding the pathological mechanisms underlying these IgG4-associated diseases have been taken, but little is still known regarding what triggers and maintains these IgG4 responses. There is a clear need to better understand how IgG4 responses are regulated. This knowledge could then form the basis for novel therapeutic strategies targeting these responses. Specifically, the aim would be to stimulate beneficial IgG4 responses, for example in allergy, or to inhibit IgG4 responses in autoimmune diseases and antitumour and anti-biologic responses. Supporting these (pre)clinical ambitions will require better models to study the development of ‘natural’ human IgG4 responses (Box 1).
References
Vidarsson, G., Dekkers, G. & Rispens, T. IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 5, 520 (2014).
Volkov, M. et al. Comprehensive overview of autoantibody isotype and subclass distribution. J. Allergy Clin. Immunol. 150, 999–1010 (2022).
Kitaura, K. et al. Different somatic hypermutation levels among antibody subclasses disclosed by a new next-generation sequencing-based antibody repertoire analysis. Front. Immunol. 8, 389 (2017).
Lighaam, L. C. & Rispens, T. The immunobiology of immunoglobulin G4. Semin. Liver Dis. 36, 200–215 (2016).
Lighaam, L. C. et al. Phenotypic differences between IgG4+ and IgG1+ B cells point to distinct regulation of the IgG4 response. J. Allergy Clin. Immunol. 133, 267–270.e1–e6 (2014). This paper is the first to report the isolation and phenotyping of IgG4+ B cells.
Aalberse, R. C., Stapel, S. O., Schuurman, J. & Rispens, T. Immunoglobulin G4: an odd antibody. Clin. Exp. Allergy 39, 469–477 (2009).
Unger, P. A. et al. Divergent chemokine receptor expression and the consequence for human IgG4 B cell responses. Eur. J. Immunol. 50, 1113–1125 (2020).
Blanco, E. et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 141, 2208–2219.e16 (2018). This study provides comprehensive quantification of absolute serum levels of antibody and numbers of B cells and plasma cells in an isotype-specific and subclass-specific manner in humans.
Katz, G. & Stone, J. H. Clinical perspectives on IgG4-related disease and its classification. Annu. Rev. Med. 73, 545–562 (2022).
Huijbers, M. G. et al. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur. J. Neurol. 22, 1151–1161 (2015).
Koneczny, I. A new classification system for IgG4 autoantibodies. Front. Immunol. 9, 97 (2018).
Lepage, N., Huang, S. H., Nieuwenhuys, E. & Filler, G. Pediatric reference intervals for immunoglobulin G and its subclasses with Siemens immunonephelometric assays. Clin. Biochem. 43, 694–696 (2010).
Aucouturier, P. et al. Measurement of serum IgG4 levels by a competitive immunoenzymatic assay with monoclonal antibodies. J. Immunol. Methods 74, 151–162 (1984).
Unger, P. P. et al. Repeated vaccination with tetanus toxoid of plasma donors with pre-existing specific IgE transiently elevates tetanus-specific IgE but does not induce allergic symptoms. Clin. Exp. Allergy 48, 479–482 (2018).
Irrgang, P. et al. Class switch toward noninflammatory, spike-specific IgG4 antibodies after repeated SARS-CoV-2 mRNA vaccination. Sci. Immunol. 8, eade2798 (2023).
van der Lee, S., Sanders, E. A. M., Berbers, G. A. M. & Buisman, A. M. Whole-cell or acellular pertussis vaccination in infancy determines IgG subclass profiles to DTaP booster vaccination. Vaccine 36, 220–226 (2018).
van der Neut Kolfschoten, M. et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 317, 1554–1557 (2007). This study shows the therapeutic potential of IgG4 to counteract the effects of IgG1 autoantibodies to AChR in rhesus macaques.
Bruhns, P. et al. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725 (2009).
Sondermann, P., Huber, R., Oosthuizen, V. & Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-FcγRIII complex. Nature 406, 267–273 (2000).
Shields, R. L. et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 (2001).
Bruggemann, M. et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 166, 1351–1361 (1987).
Steplewski, Z. et al. Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proc. Natl Acad. Sci. USA 85, 4852–4856 (1988).
Gong, Q. et al. Increased in vivo effector function of human IgG4 isotype antibodies through afucosylation. MAbs 8, 1098–1106 (2016).
Larsen, M. D. et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 371, eabc8378 (2021).
Bi, Y. et al. Distinct impact of IgG subclass on autoantibody pathogenicity in different IgG4-mediated diseases. eLife 11, e76223 (2022).
Franco, A. et al. Human Fc receptor-like 5 binds intact IgG via mechanisms distinct from those of Fc receptors. J. Immunol. 190, 5739–5746 (2013).
Wilson, T. J., Fuchs, A. & Colonna, M. Cutting edge: human FcRL4 and FcRL5 are receptors for IgA and IgG. J. Immunol. 188, 4741–4745 (2012).
Franco, A. et al. CD21 and FCRL5 form a receptor complex with robust B-cell activating capacity. Int. Immunol. 30, 569–578 (2018).
van der Zee, J. S., van Swieten, P. & Aalberse, R. C. Inhibition of complement activation by IgG4 antibodies. Clin. Exp. Immunol. 64, 415–422 (1986).
Tao, M. H., Smith, R. I. & Morrison, S. L. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J. Exp. Med. 178, 661–667 (1993).
Davies, A. M. et al. Structural determinants of unique properties of human IgG4-Fc. J. Mol. Biol. 426, 630–644 (2014). This study reports the high-resolution crystal structure of human IgG4.
Taylor, B. et al. C1q binding properties of monomer and polymer forms of mouse IgM μ-chain variants. Pro544Gly and Pro434Ala. J. Immunol. 153, 5303–5313 (1994).
Smith, R. I., Coloma, M. J. & Morrison, S. L. Addition of a μ-tailpiece to IgG results in polymeric antibodies with enhanced effector functions including complement-mediated cytolysis by IgG4. J. Immunol. 154, 2226–2236 (1995).
Diebolder, C. A. et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 343, 1260–1263 (2014).
Reinhard, L., Stahl, R. A. K. & Hoxha, E. Is primary membranous nephropathy a complement mediated disease? Mol. Immunol. 128, 195–204 (2020).
Haddad, G. et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1-associated membranous nephropathy. J. Clin. Invest. 131, e140453 (2021).
Wei, B. et al. Fc galactosylation follows consecutive reaction kinetics and enhances immunoglobulin G hexamerization for complement activation. MAbs 13, 1893427 (2021).
Li, Y., Yu, J., Wang, M., Cui, Z. & Zhao, M. H. Anti-phospholipase A2 receptor antibodies directly induced podocyte damage in vitro. Ren. Fail. 44, 304–313 (2022).
Oskam, N. et al. Factors affecting IgG4-mediated complement activation. Front. Immunol. 14, 1087532 (2023).
van der Zee, J. S., van Swieten, P. & Aalberse, R. C. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J. Immunol. 137, 3566–3571 (1986).
Schuurman, J. et al. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology 97, 693–698 (1999).
Oostindie, S. C., Lazar, G. A., Schuurman, J. & Parren, P. Avidity in antibody effector functions and biotherapeutic drug design. Nat. Rev. Drug Discov. 21, 715–735 (2022).
Clayton, F. et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology 147, 602–609 (2014).
Schuyler, A. J. et al. Specific IgG4 antibodies to cow’s milk proteins in pediatric patients with eosinophilic esophagitis. J. Allergy Clin. Immunol. 142, 139–148.e12 (2018).
Platts-Mills, T. A. E. et al. An overview of the relevance of IgG4 antibodies in allergic disease with a focus on food allergens. Children 8, 418 (2021).
Rispens, T., Ooijevaar-de Heer, P., Bende, O. & Aalberse, R. C. Mechanism of immunoglobulin G4 Fab-arm exchange. J. Am. Chem. Soc. 133, 10302–10311 (2011).
Labrijn, A. F. et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl Acad. Sci. USA 110, 5145–5150 (2013). This study describes a novel method that extrapolates the unique ability of IgG4 to exchange Fab-arms to create stable, bispecific IgG1 antibodies.
Labrijn, A. F. et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3–CH3 interaction strength. J. Immunol. 187, 3238–3246 (2011).
Chen, Z. et al. Design of next-generation therapeutic IgG4 with improved manufacturability and bioanalytical characteristics. MAbs 12, 1829338 (2020).
Konno, N. et al. Changes in N-glycans of IgG4 and its relationship with the existence of hypocomplementemia and individual organ involvement in patients with IgG4-related disease. PLoS ONE 13, e0196163 (2018).
Culver, E. L. et al. Unique patterns of glycosylation in immunoglobulin subclass G4-related disease and primary sclerosing cholangitis. J. Gastroenterol. Hepatol. 34, 1878–1886 (2019).
van de Bovenkamp, F. S., Hafkenscheid, L., Rispens, T. & Rombouts, Y. The emerging importance of IgG fab glycosylation in immunity. J. Immunol. 196, 1435–1441 (2016).
van de Bovenkamp, F. S. et al. Adaptive antibody diversification through N-linked glycosylation of the immunoglobulin variable region. Proc. Natl Acad. Sci. USA 115, 1901–1906 (2018).
Rombouts, Y. et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 75, 578–585 (2016).
Lardinois, O. M. et al. Immunoglobulins G from patients with ANCA-associated vasculitis are atypically glycosylated in both the Fc and Fab regions and the relation to disease activity. PLoS ONE 14, e0213215 (2019).
Koers, J. et al. Differences in IgG autoantibody Fab glycosylation across autoimmune diseases. J. Allergy Clin. Immunol. https://doi.org/10.1016/j.jaci.2022.10.035 (2023).
Koers, J. et al. Biased N-glycosylation site distribution and acquisition across the antibody V region during B cell maturation. J. Immunol. 202, 2220–2228 (2019).
Sabouri, Z. et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc. Natl Acad. Sci. USA 111, E2567–E2575 (2014).
Kissel, T. et al. Surface Ig variable domain glycosylation affects autoantigen binding and acts as threshold for human autoreactive B cell activation. Sci. Adv. 8, eabm1759 (2022).
Akdis, C. A. Therapies for allergic inflammation: refining strategies to induce tolerance. Nat. Med. 18, 736–749 (2012).
Holgate, S. T. & Polosa, R. Treatment strategies for allergy and asthma. Nat. Rev. Immunol. 8, 218–230 (2008).
Nouri-Aria, K. T. et al. Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J. Immunol. 172, 3252–3259 (2004).
Eckl-Dorna, J. et al. Two years of treatment with the recombinant grass pollen allergy vaccine BM32 induces a continuously increasing allergen-specific IgG4 response. EBioMedicine 50, 421–432 (2019).
Jones, S. M. et al. Clinical efficacy and immune regulation with peanut oral immunotherapy. J. Allergy Clin. Immunol. 124, 292–300.e1–e.97 (2009).
Montalvao, S. A. et al. A longitudinal evaluation of anti-FVIII antibodies demonstrated IgG4 subclass is mainly correlated with high-titre inhibitor in haemophilia A patients. Haemophilia 21, 686–692 (2015).
Akdis, M. & Akdis, C. A. Mechanisms of allergen-specific immunotherapy: multiple suppressor factors at work in immune tolerance to allergens. J. Allergy Clin. Immunol. 133, 621–631 (2014).
Jutel, M. et al. Allergen-specific immunotherapy with recombinant grass pollen allergens. J. Allergy Clin. Immunol. 116, 608–613 (2005).
Garcia, B. E., Sanz, M. L., Dieguez, I., de las Marinas, M. D. & Oehling, A. Modifications in IgG subclasses in the course of immunotherapy with grass pollen. J. Investig. Allergol. Clin. Immunol. 3, 19–25 (1993).
James, L. K. et al. Long-term tolerance after allergen immunotherapy is accompanied by selective persistence of blocking antibodies. J. Allergy Clin. Immunol. 127, 509–516.e1–e5 (2011).
Santos, A. F. et al. IgG4 inhibits peanut-induced basophil and mast cell activation in peanut-tolerant children sensitized to peanut major allergens. J. Allergy Clin. Immunol. 135, 1249–1256 (2015).
Subbarayal, B. et al. Kinetics, cross-reactivity, and specificity of Bet v 1-specific IgG4 antibodies induced by immunotherapy with birch pollen. Allergy 68, 1377–1386 (2013).
Meiler, F., Klunker, S., Zimmermann, M., Akdis, C. A. & Akdis, M. Distinct regulation of IgE, IgG4 and IgA by T regulatory cells and Toll-like receptors. Allergy 63, 1455–1463 (2008).
Meiler, F. et al. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J. Exp. Med. 205, 2887–2898 (2008).
Kurniawan, A. et al. Differential expression of IgE and IgG4 specific antibody responses in asymptomatic and chronic human filariasis. J. Immunol. 150, 3941–3950 (1993).
Turner, J. D. et al. Allergen-specific IgE and IgG4 are markers of resistance and susceptibility in a human intestinal nematode infection. Microbes Infect. 7, 990–996 (2005).
Hussain, R., Poindexter, R. W. & Ottesen, E. A. Control of allergic reactivity in human filariasis. Predominant localization of blocking antibody to the IgG4 subclass. J. Immunol. 148, 2731–2737 (1992).
Nkurunungi, G. et al. Do helminth infections underpin urban-rural differences in risk factors for allergy-related outcomes? Clin. Exp. Allergy 49, 663–676 (2019).
Heiner, D. C. IgG4 immunodeficiency. N. Engl. Reg. Allergy Proc. 9, 43–50 (1988).
Moss, R. B., Carmack, M. A. & Esrig, S. Deficiency of IgG4 in children: association of isolated IgG4 deficiency with recurrent respiratory tract infection. J. Pediatr. 120, 16–21 (1992).
Koutroumpakis, F. et al. Serum IgG4 subclass deficiency defines a distinct, commonly encountered, severe inflammatory bowel disease subtype. Inflamm. Bowel Dis. 27, 855–863 (2021).
Koneczny, I. Update on IgG4-mediated autoimmune diseases: new insights and new family members. Autoimmun. Rev. 19, 102646 (2020). This work presents a comprehensive review of IgG4-AIDs.
Futei, Y. et al. Predominant IgG4 subclass in autoantibodies of pemphigus vulgaris and foliaceus. J. Dermatol. Sci. 26, 55–61 (2001).
Huijbers, M. G. et al. Longitudinal epitope mapping in MuSK myasthenia gravis: implications for disease severity. J. Neuroimmunol. 291, 82–88 (2016).
Huijbers, M. G. et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc. Natl Acad. Sci. USA 110, 20783–20788 (2013).
Endmayr, V. et al. Anti-neuronal IgG4 autoimmune diseases and IgG4-related diseases may not be part of the same spectrum: a comparative study. Front. Immunol. 12, 785247 (2021).
Funakoshi, T. et al. Enrichment of total serum IgG4 in patients with pemphigus. Br. J. Dermatol. 167, 1245–1253 (2012).
Vergoossen, D. L. E. et al. Enrichment of serum IgG4 in MuSK myasthenia gravis patients. J. Neuroimmunol. 373, 577978 (2022).
Panhuber, A. et al. A systematic review and meta-analysis of HLA class II associations in patients with IgG4 autoimmunity. Sci. Rep. 12, 9229 (2022).
Bhol, K. C., Rojas, A. I., Khan, I. U. & Ahmed, A. R. Presence of interleukin 10 in the serum and blister fluid of patients with pemphigus vulgaris and pemphigoid. Cytokine 12, 1076–1083 (2000).
Satyam, A., Khandpur, S., Sharma, V. K. & Sharma, A. Involvement of TH1/TH2 cytokines in the pathogenesis of autoimmune skin disease – pemphigus vulgaris. Immunol. Invest. 38, 498–509 (2009).
Yilmaz, V. et al. Differential cytokine changes in patients with myasthenia gravis with antibodies against AChR and MuSK. PLoS ONE 10, e0123546 (2015).
Westwood, J. P., Langley, K., Heelas, E., Machin, S. J. & Scully, M. Complement and cytokine response in acute thrombotic thrombocytopenic purpura. Br. J. Haematol. 164, 858–866 (2014).
Li, N. et al. From insect bites to a skin autoimmune disease: a conceivable pathway to endemic pemphigus foliaceus. Front. Immunol. 13, 907424 (2022).
Qian, Y. et al. Cutting edge: Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J. Immunol. 189, 1535–1539 (2012). This study provides the first evidence for molecular mimicry as a cause of the IgG4-AID pemphigus foliaceus.
Lin, L. et al. Walnut antigens can trigger autoantibody development in patients with pemphigus vulgaris through a “hit-and-run” mechanism. J. Allergy Clin. Immunol. 144, 720–728.e4 (2019).
Doppler, K. et al. Anti-CNTN1 IgG3 induces acute conduction block and motor deficits in a passive transfer rat model. J. Neuroinflammation 16, 73 (2019).
Huang, C. C. et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod. Pathol. 26, 799–805 (2013).
Koneczny, I., Cossins, J., Waters, P., Beeson, D. & Vincent, A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1–3 can disperse preformed agrin-independent AChR clusters. PLoS ONE 8, e80695 (2013).
Joubert, B. et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. 73, 1115–1124 (2016).
Kornau, H. C. et al. Human cerebrospinal fluid monoclonal LGI1 autoantibodies increase neuronal excitability. Ann. Neurol. 87, 405–418 (2020).
Sinkovits, G. et al. Concentration and subclass distribution of anti-ADAMTS13 IgG autoantibodies in different stages of acquired idiopathic thrombotic thrombocytopenic purpura. Front. Immunol. 9, 1646 (2018).
Bhol, K., Mohimen, A. & Ahmed, A. R. Correlation of subclasses of IgG with disease activity in pemphigus vulgaris. Dermatology 189, 85–89 (1994).
Daveau, M. et al. IgG4 subclass in malignant melanoma. J. Natl Cancer Inst. 58, 189–192 (1977).
Karagiannis, P. et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J. Clin. Invest. 123, 1457–1474 (2013). This study provides evidence for IgG4+ B cells in the tumour microenvironment and the induction of IL-10 secretion by tumour cells, and shows how IgG4 can block endogenous antitumour immune responses.
Crescioli, S. et al. IgG4 Characteristics and functions in cancer immunity. Curr. Allergy Asthma Rep. 16, 7 (2016).
Jordakieva, G. et al. IgG4 induces tolerogenic M2-like macrophages and correlates with disease progression in colon cancer. Oncoimmunology 10, 1880687 (2021).
Karagiannis, P. et al. Elevated IgG4 in patient circulation is associated with the risk of disease progression in melanoma. Oncoimmunology 4, e1032492 (2015).
Gilbert, A. E. et al. Monitoring the systemic human memory B cell compartment of melanoma patients for anti-tumor IgG antibodies. PLoS ONE 6, e19330 (2011).
van de Veen, W. et al. A novel proangiogenic B cell subset is increased in cancer and chronic inflammation. Sci. Adv. 6, eaaz3559 (2020). This study identifies a novel IgG4+ B cell subset that has pro-angiogenic features.
Andersen, B. R. & Terry, W. D. γG4-globulin antibody causing inhibition of clotting factor 8. Nature 217, 174–175 (1968). This paper is the first description of acquired haemophilia A being caused by IgG4 autoantibodies to clotting factor FVIII.
Hofbauer, C. J. et al. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood 125, 1180–1188 (2015).
Iizuka, A. & Nagao, T. Analysis of IgG heavy chain subclasses of alloantibodies to factor IX by crossed immunoelectrophoresis of factor IX using the intermediate gel technique. Br. J. Haematol. 53, 687–688 (1983).
Whelan, S. F. et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood 121, 1039–1048 (2013).
Deisenhammer, F., Reindl, M. & Berger, T. Immunoglobulin subclasses in patients with neutralizing and nonneutralizing antibodies against IFN-β1b. J. Interferon Cytokine Res. 21, 167–171 (2001).
van Schie, K. A. et al. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 74, 311–314 (2015).
van Schouwenburg, P. A. et al. IgG4 production against adalimumab during long term treatment of RA patients. J. Clin. Immunol. 32, 1000–1006 (2012).
Witmer, C. & Young, G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther. Adv. Hematol. 4, 59–72 (2013).
van Helden, P. M. et al. IgG subclasses of anti-FVIII antibodies during immune tolerance induction in patients with hemophilia A. Br. J. Haematol. 142, 644–652 (2008).
Knobl, P. Prevention and management of bleeding episodes in patients with acquired hemophilia A. Drugs 78, 1861–1872 (2018).
van Schouwenburg, P. A. et al. Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies. J. Biol. Chem. 289, 34482–34488 (2014).
Kamisawa, T. et al. A new clinicopathological entity of IgG4-related autoimmune disease. J. Gastroenterol. 38, 982–984 (2003).
Kamisawa, T., Zen, Y., Pillai, S. & Stone, J. H. IgG4-related disease. Lancet 385, 1460–1471 (2015).
Wallace, Z. S. et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann. Rheum. Dis. 74, 190–195 (2015).
Maillette de Buy Wenniger, L. J. et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology 57, 2390–2398 (2013).
Shiokawa, M. et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut 65, 1322–1332 (2016).
Lohr, J. M. et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am. J. Gastroenterol. 105, 2060–2071 (2010).
Asada, M. et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas 33, 20–26 (2006).
Hubers, L. M. et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut 67, 728–735 (2018).
Culver, E. L. et al. Increased IgG4 responses to multiple food and animal antigens indicate a polyclonal expansion and differentiation of pre-existing B cells in IgG4-related disease. Ann. Rheum. Dis. 74, 944–947 (2015).
Liu, J. et al. Immune dysregulation in IgG4-related disease. Front. Immunol. 12, 738540 (2021).
Culver, E. L. et al. Increases in IgE, eosinophils, and mast cells can be used in diagnosis and to predict relapse of IgG4-related disease. Clin. Gastroenterol. Hepatol. 15, 1444–1452.e6 (2017).
Della Torre, E. et al. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy 69, 269–272 (2014).
Khosroshahi, A., Bloch, D. B., Deshpande, V. & Stone, J. H. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 62, 1755–1762 (2010).
Matza, M. A. et al. Abatacept in IgG4-related disease: a prospective, open-label, single-arm, single-centre, proof-of-concept study. Lancet Rheumatol. 4, e105–e112 (2022).
Hirano, K. et al. Incidence of malignancies in patients with IgG4-related disease. Intern. Med. 53, 171–176 (2014).
Tang, H. et al. Malignancy and IgG4-related disease: the incidence, related factors and prognosis from a prospective cohort study in China. Sci. Rep. 10, 4910 (2020).
Wallace, Z. S. et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 67, 2466–2475 (2015).
Kindle, S. A. et al. Dermatologic disorders in 118 patients with autoimmune (immunoglobulin G4-related) pancreatitis: a retrospective cohort analysis. Am. J. Clin. Dermatol. 16, 125–130 (2015).
Raibagkar, P., Ferry, J. A. & Stone, J. H. Is MuSK myasthenia gravis linked to IgG4-related disease? J. Neuroimmunol. 305, 82–83 (2017).
Labasque, M. et al. Specific contactin N-glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J. Biol. Chem. 289, 7907–7918 (2014).
Querol, L. et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 82, 879–886 (2014).
Delmont, E. et al. Antibodies against the node of Ranvier: a real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J. Neurol. 267, 3664–3672 (2020).
Manso, C., Querol, L., Mekaouche, M., Illa, I. & Devaux, J. J. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 139, 1700–1712 (2016).
Devaux, J. J. et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 86, 800–807 (2016).
Ogata, H. et al. Unique HLA haplotype associations in IgG4 anti-neurofascin 155 antibody-positive chronic inflammatory demyelinating polyneuropathy. J. Neuroimmunol. 339, 577139 (2020).
Martin-Aguilar, L. et al. Clinical and laboratory features in anti-NF155 autoimmune nodopathy. Neurol. Neuroimmunol. Neuroinflamm. 9, e1098 (2022).
Manso, C. et al. Anti-neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J. Clin. Invest. 129, 2222–2236 (2019).
Evoli, A. et al. Myasthenia gravis with antibodies to MuSK: an update. Ann. N. Y. Acad. Sci. 1412, 82–89 (2018).
Klooster, R. et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 135, 1081–1101 (2012).
Amagai, M. et al. Conformational epitopes of pemphigus antigens (Dsg1 and Dsg3) are calcium dependent and glycosylation independent. J. Invest. Dermatol. 105, 243–247 (1995).
Yokouchi, M. et al. Pathogenic epitopes of autoantibodies in pemphigus reside in the amino-terminal adhesive region of desmogleins which are unmasked by proteolytic processing of prosequence. J. Invest. Dermatol. 129, 2156–2166 (2009).
Sekiguchi, M. et al. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J. Immunol. 167, 5439–5448 (2001).
Kasperkiewicz, M. et al. Pemphigus. Nat. Rev. Dis. Prim. 3, 17026 (2017).
Schaller, M., Vogel, M., Kentouche, K., Lammle, B. & Kremer Hovinga, J. A. The splenic autoimmune response to ADAMTS13 in thrombotic thrombocytopenic purpura contains recurrent antigen-binding CDR3 motifs. Blood 124, 3469–3479 (2014).
Doyle, A. J. et al. Long-term risk of relapse in immune-mediated thrombotic thrombocytopenic purpura and the role of anti-CD20 therapy. Blood 141, 285–294 (2022).
Kremer Hovinga, J. A. et al. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Prim. 3, 17020 (2017).
Xu, Z. et al. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat. Rev. Immunol. 12, 517–531 (2012).
Wagar, L. E. et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 27, 125–135 (2021). This study reports an in vitro model based on tonsil cell extracts that allows for the study of de novo human antibody responses, including IgG4.
Cevhertas, L. et al. IL-10 induces IgG4 production in NOD-scid Il2rγnull mice humanized by engraftment of peripheral blood mononuclear cells. Allergy 76, 3525–3529 (2021).
Gon Y. et al. Invention and phenotypic evaluation of human IgG4-knock-in mice [abstract]. Arthritis Rheumatol. Abstr. 1066, https://acrabstracts.org/abstract/invention-and-phenotypic-evaluation-of-human-igg4-knock-in-mice/ (2017).
Acknowledgements
M.G.H. thanks the neuroimmunology research group, J. Verschuuren, J. Plomp and S. van der Maarel for fruitful discussions on IgG4. T.R. thanks M. van Ham, A. ten Brinke, J. Koers, P. P. Unger and R. Aalberse for inspiring discussions on IgG4. M.G.H and T.R. gratefully acknowledge the Target-to-B (T2B) consortium for supporting their work on IgG4.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
M.G.H. is co-inventor on muscle-specific kinase (MuSK)-related patents. Leiden University Medical Center (LUMC) and M.G.H. receive royalties from these patents. LUMC receives royalties on a MuSK ELISA. T.R. declares no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks P. Bruhns, Y. Tanaka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ORPHAcode: https://www.orpha.net/
Glossary
- Antibody-dependent cell-mediated cytotoxicity
-
(ADCC). An immune response induced by antibodies binding through their Fc (fragment crystallizable) domains to pro-inflammatory, activating Fc receptors on immune cells, causing the immune cells to lyse the antibody-bound antigen or cell.
- Antibody-dependent cellular phagocytosis
-
(ADCP). An immune response induced by antibodies binding through their Fc (fragment crystallizable) domains to pro-inflammatory, activating Fc receptors on immune cells, causing the immune cells to phagocytose the antibody-bound pathogen or cell.
- Antibody-dependent complement deposition
-
An immune response induced by high levels of antigen-bound pro-inflammatory antibodies, causing precipitation of complement factor C1q and subsequent induction of the complement cascade, resulting in lysis of the antibody-bound pathogen or cell.
- Class-switch recombination
-
Process by which B cells edit their immunoglobulin heavy-chain constant region DNA to produce antibodies of a different (sub)class. This results in antibodies that have the same specificity but different effector functions.
- Complementarity-determining regions
-
Loops within the variable domains of an antibody (or B cell receptor) that are highly variable (between antibodies), unique and interact with the antigen.
- Fab-arm exchange
-
A stochastic process in which IgG4 half-molecules recombine with other IgG4 half-molecules, forming an antibody that is bispecific and thus functionally monovalent.
- Fc receptors
-
Immune receptors specific for antibody (sub)classes that can activate or inhibit immune cells upon binding to the Fc (fragment crystallizable) portion of antigen-bound antibodies.
- Fragment antigen binding
-
(Fab). A highly variable antibody segment that confers its specificity for antigen binding.
- Fragment crystallizable
-
(Fc). A constant domain of an antibody molecule that mediates the immunological effector functions of the antibody, including antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis and antibody-dependent complement deposition. Antibody classes are grouped based on structural and functional (amino acid) determinants in this part of the antibody.
- Molecular mimicry
-
Pathogens and endogenous proteins may have highly similar structures or molecular features. The molecular mimicry hypothesis suggests that (auto)immune responses may derive from an initial (antibody) response to a pathogen that cross-reacts with an endogenous protein having a highly similar structure.
- Paraneoplastic syndromes
-
Autoimmune diseases that develop as a result of an antitumour antibody response. Tumour cells often overexpress proteins normally present elsewhere in the body. Antibody responses directed at such tumour proteins may not only contain the tumour but also cause disease at the natural site of activity of the protein.
- Specific immunotherapy
-
A treatment regimen to induce tolerance, based on regular exposure to ultra-low doses of antigen that induce a class-switch to anti-inflammatory IgG4.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rispens, T., Huijbers, M.G. The unique properties of IgG4 and its roles in health and disease. Nat Rev Immunol 23, 763–778 (2023). https://doi.org/10.1038/s41577-023-00871-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-023-00871-z
This article is cited by
-
B-cell and antibody responses to SARS-CoV-2: infection, vaccination, and hybrid immunity
Cellular & Molecular Immunology (2023)
-
Anti-allergen monoclonal antibodies for the treatment of allergies
Allergo Journal International (2023)
-
Monoklonale Anti-Allergen-Antikörper für die Behandlung von Allergien
Allergo Journal (2023)
