
Abstract
Cholangiocarcinoma (CCA) is a rare malignancy that develops at any point along the biliary tree. CCA has a poor prognosis, its clinical management remains challenging, and effective treatments are lacking. Therefore, preclinical research is of pivotal importance and necessary to acquire a deeper understanding of CCA and improve therapeutic outcomes. Preclinical research involves developing and managing complementary experimental models, from in vitro assays using primary cells or cell lines cultured in 2D or 3D to in vivo models with engrafted material, chemically induced CCA or genetically engineered models. All are valuable tools with well-defined advantages and limitations. The choice of a preclinical model is guided by the question(s) to be addressed; ideally, results should be recapitulated in independent approaches. In this Consensus Statement, a task force of 45 experts in CCA molecular and cellular biology and clinicians, including pathologists, from ten countries provides recommendations on the minimal criteria for preclinical models to provide a uniform approach. These recommendations are based on two rounds of questionnaires completed by 35 (first round) and 45 (second round) experts to reach a consensus with 13 statements. An agreement was defined when at least 90% of the participants voting anonymously agreed with a statement. The ultimate goal was to transfer basic laboratory research to the clinics through increased disease understanding and to develop clinical biomarkers and innovative therapies for patients with CCA.
Similar content being viewed by others

Introduction
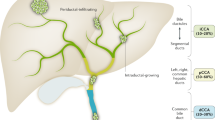
During the past decade, we have witnessed considerable advances in understanding the molecular pathogenesis of cholangiocarcinoma (CCA). However, early diagnosis and effective treatments for this aggressive cancer lag behind those for other fields. To accelerate the development of novel clinical strategies, preclinical models of CCA are essential1. Critical points to consider when using or developing these tools are the tumour anatomical origin (that is, intrahepatic CCA (iCCA), perihilar CCA (pCCA) or distal CCA (dCCA)), the cell or cells of origin (for example, preneoplastic lesions) and the histomorphological tumour features (for example, large versus small bile duct type)2.
Historically, 2D cell cultures have been widely used as in vitro models of CCA. In addition to experimentally immortalized or primary cultures of normal cholangiocytes derived from normal bile ducts, over 50 CCA-derived human cell lines have been established3. A limitation of these models is the lack of resemblance to the original tumours as a result of continuous culturing, making it difficult to infer which therapeutics would have been effective to treat the original neoplasm4. Moreover, 2D monocultures do not accurately mimic the characteristic features of biliary tumours, namely the 3D architecture, cell-to-cell and cell-to-matrix interactions, cellular heterogeneity and the effect of the tumour microenvironment on cancer progression. To overcome these limitations, multicellular 3D models, such as spheroids and organoids, have been developed. Although they are valuable models to study CCA5, spheroids usually do not precisely recapitulate the native tissue architecture and function of the tissue of origin6. By contrast, organoids maintain a higher and more predictable physical order in their cellular self-assembly and display a marked interaction with the extracellular matrix, thereby retaining most of the histological and malignant characteristics of the original neoplasm6,7,8,9. In addition to cell culture-based models, different in vivo CCA models have been developed. Inducing CCA through administering hepatocarcinogens or liver fluke infestation has the advantage of mimicking cancer pathogenesis. However, animal studies are time-consuming, expensive and ethically challenging, and sometimes hepatocellular carcinoma (HCC) rather than CCA preferentially develops10. To give in vivo context to 2D cell lines, CCA cells have been used to generate subcutaneous or orthotopic xenografts in mice10,11. However, these approaches remain limited by poor rates of tumour engraftment. Technological advances have made it possible to grow liver organoids (that is, 3D cultures of bipotent liver precursors) and therefore to develop mouse models based on transplantation of genetically modified liver organoids that undergo in vivo oncogenic transformation along the cholangiocellular lineage12. Alternatively, genetically engineered mouse models (GEMMs) that recapitulate the most frequent genetic alterations detected in CCA have been generated10,12,13.
International collaborations to study CCA, spearheaded by the European Network for the Study of Cholangiocarcinoma and the European H2020 COST Action CA18122, have been crucial to fostering advances in this field. To improve the accuracy in obtaining and exchanging information among research groups, it is now essential to establish consensus criteria regarding the minimal standardized characteristics required from preclinical CCA models or when describing a new model. In this Consensus Statement, we detail these criteria for the available and forthcoming in vitro and in vivo models and document the international, interdisciplinary process used for their development.
Methods
Panel of experts
A core group of eight group members, all active researchers with significant contributions to the CCA field, initiated and led a Delphi study to define recommendations on the minimal criteria for experimental CCA models to provide a uniform approach for future studies. Furthermore, core group members identified 27 additional experts to be invited to join the steering committee and to be actively involved in implementing the Delphi process. These core and steering team members filled in the initial Delphi questionnaire and are listed authors, and they proposed ten additional experts to fill in the second and final questionnaire. These ten experts, who were not actively involved in writing the recommendations but provided their important input by filling in the second questionnaire, are listed as one collaborative author: the CCA Model Consortium. Thus, the final panel consisted of 45 experts from ten countries in Europe, Asia and the USA. The names, affiliations, locations and roles of the members of the expert panel are provided in Supplementary Table 1.
Building consensus
We used a modified Delphi method for two rounds of questionnaires. A statement consensus was reached when the agreement was ≥90%. Statements or questions that were agreed upon using this criterion in the first round were omitted from the second round.
Questionnaires
The core team generated the questionnaires using an online form (Google Forms, Alphabet, CA) before sending them out to the experts. The first questionnaire consisted of 47 questions, divided into four parts: defining minimal and advanced criteria for experimental models (part 1); in vivo model for CCA (part 2); in vitro models for CCA (part 3); and preclinical models for CCA (part 4). Based on questionnaire 1 (Supplementary Data 1), a second questionnaire was designed consisting of 13 statements, of which 12 could be solely answered with ‘yes’ or ‘no’ (Table 1). All experts could comment on every question. Both questionnaires and summaries of the outcome are shared in Supplementary Data 1. Through the consensus of experts in the field, we propose overarching criteria to be used when establishing or using preclinical models of CCA and linking this to the clinic (Fig. 1). From the second questionnaire, core recommendations were formulated (Box 1).

a, In vitro models. b, In vivo models. GEMM, genetically engineered mouse model; PDX, patient-derived xenograft.
Clinical features to consider
Experimental models of CCA must reflect the natural history of the known subtypes of CCA, their molecular heterogeneity and the effect of clinical or therapeutic interventions. In the International Classification of Diseases 11th revision (ICD-11), published in 2022, CCA is classified according to its origin as iCCA or extrahepatic CCA (eCCA). iCCA arises from intrahepatic bile ducts, that is, it grows in the liver. Consequently, it is more often surgically resectable than pCCA, the latter of which arises at the liver hilum, where the likelihood of local vascular invasion is greater14. The effect of tumour biology on local invasion is poorly understood and requires further examination.
The biology of CCA subtypes also differs significantly. Approximately 50% of iCCAs have actionable molecular alterations, and targeted therapies against FGFR2 fusions and IDH1 mutation-driven cancers are already approved15,16,17,18. It is not fully understood why iCCAs are more molecularly heterogeneous than pCCAs or dCCAs, and this requires detailed examination. In addition, the influence of biology on the natural history of iCCA and its effect on surgical, local and systemic treatment options require further study19. dCCA more closely resembles pCCA19,20 but, again, the effects of both anatomy and biology on outcome have not been fully elucidated. However, many tools only seek to mimic iCCA, and there is a critical absence of models of pCCA and dCCA.
A second essential requirement of an experimental model is to reflect the interventional outcome. Although chemotherapy remains the standard of care, the increasing use of targeted therapies requires a deeper examination of molecular mechanisms and critical mechanisms of resistance21,22,23,24. As such, any model must reflect molecular changes in the patient that can be measured to provide hypotheses to overcome this commonly occurring resistance. Furthermore, such resistance mechanisms should be unravelled to develop and assess novel interventions to overcome resistance before clinical testing.
Pathology
Separate classifications (Union for International Cancer Control25, American Joint Committee on Cancer26 and WHO20) exist for iCCA, pCCA and dCCA. Macroscopic features divide iCCA into two subtypes: large duct and small duct20. Large duct iCCAs typically arise near large central ducts and grow along the ductal wall. Small duct iCCAs are usually peripheral mass-forming tumours in the hepatic parenchyma. Four patterns of growth are described for CCA: mass-forming, periductal infiltrating, intraductal and mixed types27.
Histopathology
Small duct iCCAs are typically non-mucin-secreting adenocarcinomas with a ductular or tubular pattern. Large duct iCCAs are generally mucin-secreting tubular adenocarcinomas resembling pCCA and dCCA28. Most pCCAs and dCCAs are adenocarcinomas with pancreaticobiliary morphology, comprising glandular structures and/or small groups of cells within the desmoplastic stroma28.
Immunohistochemistry
No specific immunohistochemical pattern for CCA lesions exists. However, they typically show an upper gastrointestinal or pancreaticobiliary pattern of cytokeratin (CK) expression (CK7+, CK19+, CK20−) when they still exhibit some degree of differentiation. In addition, large duct iCCAs sometimes express intestinal markers (for example, CK20 and CDX2)29. CCA is usually immunonegative for HepPar1, arginase 1 and glypican 3, distinguishing it from HCC and combined HCC–CCA30,31. Transcription factors that mark cell-specific lineages, such as thyroid transcription factor 1 (TTF1) (lung and thyroid cancers)32, PAX8 (renal, thyroid, ovarian and endometrial cancers)33 and GATA3 (breast and urothelial cancers)34, are usually not expressed in CCA.
Biliary precursor lesions
CCA can develop from precursor lesions. Most large duct iCCA as well as pCCA and dCCA presumably originate from biliary intraepithelial neoplasia35. Intraductal papillary neoplasm of the bile duct (IPNB) is an intraductal papillary proliferation; 70% of IPNBs develop in intrahepatic ducts and 30% develop in perihilar ducts36,37. Invasive malignancy is evident in >50% of IPNBs at presentation38,39. Mucinous cystic neoplasm is a cystic epithelial tumour occurring almost exclusively in female patients, and it is debatable whether it represents a true biliary precursor lesion but approximately 5% of these tumours are associated with CCA40,41.
Molecular profiling
Efforts to understand the heterogeneity of CCA have provided insights into the molecular pathogenesis and anatomical complexity of this disease15,42,43,44,45,46,47,48,49. The genetic landscapes are comparable to those of other carcinoma types of the gastrointestinal tract, show substantial similarities to genetic alterations of ductal adenocarcinoma of the pancreas, and exhibit an intermediate degree of alteration counts in the mutational spectrum of cancers50, with shared genetic alterations between iCCA, pCCA and dCCA47. Although we have gained comprehensive insights into the underlying pathobiological processes of resectable invasive tumours, the precise genetic and epigenetic mechanisms involved in the onset of CCA are still unclear.
Integrated genomics approaches have been used to classify patients with CCA based on prognosis39,51,52,53, emphasizing dysregulated oncogenic signalling pathways, including WNT–CTNNB1, MYC, PI3K–AKT–mTOR, ERBB, fibroblast growth factor receptor 2 (FGFR2), RAS–RAF–ERK, tumour necrosis factor (TNF), polo-like kinase 1 (PLK1), transforming growth factor-β (TGFβ), NOTCH, insulin-like growth factor receptor 1 (IGFR1), vascular endothelial growth factor (VEGF) and the Hippo cascade. This predominant molecular classification highlights distinct tumour phenotypes that are either inflammatory or proliferative in nature52. Moreover, iCCA can be classified on the basis of driver gene mutations, which elucidate unique mutational signatures, structural variants and epigenomic alterations46. This approach emphasized specific oncogenetic mechanisms in distinct patient subsets each associated with potential unique drugs such as RNA synthesis inhibitors in IDH-mutant tumours, microtubule modulators in KRAS-mutant tumours, topoisomerase inhibitors in TP53-mutant tumours and mTOR inhibitors in wild-type tumours enriched in FGFR2 fusions15.
As the three anatomical CCA subtypes differ in their molecular alterations47 and potentially in their cell of origin54,55,56,57, the CCA subtypes should be studied in separate experimental models2. However, the step-wise progression of human CCA and, thus, the accumulation of a wide variety of molecular alterations might not be reflected in the mouse models in which tumours develop most rapidly. Furthermore, the available experimental models represent specific subsets of patients with CCA, and it is essential to consider the molecular heterogeneity of patients with CCA when using these models. With this in mind, integrative transcriptomics might represent a relevant strategy to define the best-fit models, as previously demonstrated for HCC58,59.
In vivo CCA models
Engrafted models
Xenografts
Xenografts are grafts of tissue or cells transplanted from a different species into an immunodeficient host60. Xenograft CCA models are generated by either implanting human neoplastic CCA cells subcutaneously into the flanks of immunodeficient or athymic mice (ectopic grafts) or directly into the liver (orthotopic grafts). These experimental animal models help to evaluate the therapeutic efficacy and safety of novel candidate drugs or physical-based therapies for treating CCA in vivo. They are highly reproducible, cost-efficient, technically easy and feasible, with limited adverse effects related to the procedure, and they only require short periods for evaluation60,61,62,63. Furthermore, when engrafted subcutaneously, the generated tumours are easily accessible throughout the duration of the in vivo model, which enables the real-time measurement of tumour volume growth with a caliper. Several studies have investigated the therapeutic efficacy and safety of different compounds such as sorafenib or an epigenetic inhibitor61,64,65,66,67. Additionally, the role of various proteins68,69,70,71,72,73 and microRNAs74,75,76,77,78 were evaluated in ectopic xenograft models by implanting genetically manipulated CCA cells. Nevertheless, ectopic xenografts also have intrinsic limitations. Xenografts usually reflect advanced tumour stages, growing rapidly and making the study of early CCA challenging. At the same time, distinct CCA cell lines display different tumorigenic activity, with some being unable to generate tumours after injection. Furthermore, these tumours are implanted in a non-physiological site, seldom metastasize, and might lose the molecular heterogeneity characteristic of human CCA. Most importantly, they do not enable study of the crosstalk between tumour cells, the multicellular microenvironment milieu and the immune system10,60,62,63.
The use of orthotopic xenograft models might overcome some of these limitations by developing tumours directly in the organ of origin. Orthotopic grafts are more likely to trigger tumour dissemination, with the development of distant metastases79. Intrahepatic implantation of CCA cells can be achieved either by injecting cells directly into the liver parenchyma using ultrasound-guided injection80 or through the portal or splenic vein60. Small fragments of CCA tumours previously generated in subcutaneous xenografts or cancer stem cell-derived spheroids can also be orthotopically implanted81,82. Although intrasplenic injection is technically easier than intraportal administration and carries fewer postoperative complications, the implantation of CCA cells by intrasplenic injection resulted in successful engraftment in the liver and the spleen83. Notably, intrasplenic injection of EGI-1 CCA cells also induced the development of lung metastases84. Still, generating orthotopic models is more time-consuming, and some postoperative complications can arise. Furthermore, the assessment of tumour development and growth and metastases requires imaging techniques or is performed only when the animal is killed60,63. In this sense, using luciferase-expressing CCA cells is an excellent choice to monitor tumour growth over time83. However, this tool might not be accessible to all.
Engrafting cells or tissues obtained directly from patients can result in the development of patient-derived xenografts (PDXs). The original genetic and epigenetic features and surrounding stroma as observed in the initial mass are usually maintained in subcutaneous or orthotopic tumours, which are thus the ideal model to predict therapeutic responses and are excellent tools in personalized medicine. Indeed, several studies have already used PDXs to examine CCA that harbour specific mutational patterns and to test the use of specific targeted therapies85,86,87,88,89,90,91. Nevertheless, the success of PDX engraftment is relatively low, depending on the primary tumour itself and the experimental design for tumour engraftment. Thus, they constitute a time-intensive and resource-intensive model and might require several months for successful implantation60. Based on the available data and unanimous agreement, the expert panel strongly suggests that the type of CCA should be defined by a pathologist for PDX models, with the histology of the tumour shown in publications (Box 1).
Allografts (syngeneic)
Syngeneic models enable the implantation of murine CCA cells into an immunocompetent host, and thereby display a fully functional immune system. The first syngeneic model was developed when two rat CCA cell lines (BDEneu and BDEsp) were directly implanted into the biliary tract of Fischer 344 rats. While BDEsp engraftment induces the development of non-metastatic iCCA, BDEneu-derived tumours are more aggressive, with the rapid and consistent formation of CCA lesions and metastases92,93. This model was used to elucidate the mechanisms that underlie tumour progression and to evaluate the efficacy of novel drug candidates93,94,95,96,97. A development of this approach using a novel syngeneic murine model was reported in which the malignant mouse cell lines SB1-7, obtained from a bile duct ligation (BDL) and transposon-based CCA model, were engrafted into the mice98,99. The obtained cell lines were successfully implanted, leading to CCA lesions resembling human CCAs99. In addition, fetal liver cells obtained from genetically modified mouse embryos can be implanted into mouse liver, inducing CCA formation100. Furthermore, the CCA cells can be genetically manipulated before engraftment, revealing insights into the mechanisms that govern cholangiocarcinogenesis and enabling implantation of the cells into already established knockout mouse strains, thereby permitting the study of alterations in specific genes in the tumour stroma101. In this line, unpublished observations from the SB1 orthotopic model indicate that extending the frequently used end point (4 weeks) by 2 additional weeks enables the formation of extrahepatic metastases in the lung (J.V. and E.G.-S., unpublished work). Therefore, further characterization of this timeline in a genetically treatable immunocompetent host, coupled with the isolation of tumour cells from the original site of injection and the metastatic sites, could provide an excellent model to understand, and perhaps even prevent, a rather under-studied process such as CCA metastatic spreading. Overall, these models can overcome the limitations of xenografts, such as the absence of the immune system, are ideal for studying tumour–stroma interactions, and are an excellent alternative for testing immunotherapy-based strategies. Still, they require microsurgical procedures, increasing the probability of procedure-related complications.
Chemically induced models
High levels of inflammation, fibroblast activation and rich extracellular matrix deposition in the tumour typify CCA in patients102. In some cases, these tumours develop in patients with chronic diseases such as chronic liver fluke infection or in patients with primary sclerosing cholangitis2, and the cells associated with these pre-cancerous conditions contribute to cancer formation. Several chemical models that generate chronic and iterative injury, leading to tumour formation, have been developed to recapitulate this complex microenvironment in CCA.
Early work demonstrated that administering thiourea or thioacetamide (TAA) to rats triggers liver cancer formation over 2 years103. TAA is a potent hepatotoxin that induces hepatic fibrosis and cirrhosis in rodents owing to progressive damage of hepatocytes and biliary epithelium. TAA-induced biliary damage reproduces the typical dysplasia–carcinoma sequence, ultimately evolving to invasive iCCA104. Consequently, the use of TAA to induce tumour-initiating injury in rodents has become a cornerstone of CCA research. However, as detailed in this early work, CCA formation in TAA-treated rats is very variable, with only ∼50% of animals developing frank carcinomas. Results are even more variable in wild-type mice. TAA is not mutagenic per se; instead, the initiation of chronic sclerosing inflammation and continuous regeneration drives the spontaneous accumulation of mutations in biliary cells, which then become cancerous, as is observed in patients with chronic cholangiopathies105,106. Therefore, combined with BDL, a classic model of obstructive cholestasis and subsequent bile duct proliferation, TAA accelerates the formation of biliary tumours in rats106,107. In addition to TAA, several other mutagenic models have also been developed to induce CCA in rodents. For instance, diethylnitrosamine and dimethylnitrosamine generate DNA adducts in the liver and are sufficient for liver carcinogenesis108 and, in combination with inflammatory injury (BDL or Opisthorchis viverrini infection), drive CCA development in mice and hamsters109,110,111. Furan is a potent mutagen capable of initiating CCA in rats112. Long-term furan treatment is currently the only chemically induced model of CCA with a tumour incidence of nearly 100%, which results in multi-organ metastases and closely recapitulates the primary and secondary pathologies of human CCA. Available models are summarized in Table 2 and Fig. 2.

Schematic summary of available chemical models to initiate cholangiocarcinoma in rodents and induce metastatic dissemination. DEN, diethylnitrosamine; DMN, dimethylnitrosamine.
Although many rat and mouse CCA models that are driven by chemical insults reflect both the pre-cancerous disease history and the molecular and histopathological features of human CCA, their use is becoming less popular, primarily due to their long latency, cost and variability (both in terms of tumour penetrance and high molecular heterogeneity). Advances in CCA modelling have focused on combining the disease-inducing aspects of these models, such as inflammation and fibrosis, with GEMMs, which are discussed in more detail in the next section. A critical point is the choice of the control tissue to compare with malignant biliary cells. Indeed, as the whole liver is inappropriate because hepatocytes are the prevalent cell population, isolated bile ducts should be considered the best control.
Genetically engineered mouse models
GEMMs are advanced animal models of human cancer (Table 3). They are rationally designed to mimic the genetic and epigenetic alterations, the aberrant activation of signalling pathways and the sequence of preneoplastic and early and late tumour stages, including metastasis, in human CCAs. In addition, GEMMs can be coupled to in vivo transfection (hydrodynamic tail vein injection and/or electroporation) or injection (adeno-associated viruses (AAV)) approaches to activate or express transgenes in adult hepatocytes to further expand the mouse model toolbox113.
General concerns precluding the use of GEMMs are their high cost, tumour latency and embryonic Cre expression in non-inducible models that might compromise translation to human disease. However, adopting CRISPR–Cas9 strategies to generate new GEMM strains, and the development of tamoxifen-inducible, organ-specific Cre recombinase strains, circumvented some of these limitations.
Most CCA GEMMs incorporate common oncogenic alterations found in humans, including inactivation of tumour suppressor genes (PTEN, SMAD4 and P53) or induction of oncogenes (KRAS, IDH1/2, AKT1 and NOTCH1) to investigate the consequences of cell-autonomous effects on cholangiocarcinogenesis. In the first reported CCA GEMM, ablation of Pten and Smad4 in fetal bipotential hepatic progenitors (liver progenitor cells (LPCs)) was achieved during embryogenesis using an albumin–Cre (Alb–Cre) strain114. Alb–Cre,Smad4flox/flox,Ptenflox/flox mice displayed the histopathological stages detected in human disease, from bile duct hyperplasia and dysplasia to carcinoma in situ and invasive CCA.
Another model closely recapitulating human cholangiocarcinogenesis consists of concomitant Trp53 abrogation and KrasG12D expression in the Alb–Cre mouse background115. This model features premalignant biliary lesions (IPNB and von Meyenburg complexes), leading to invasive carcinoma and distal metastases. To directly probe the cell of origin in this model, KrasLSL-G12D/+,Tp53flox/flox mice were bred to the tamoxifen-inducible Sox9–CreERT2+ strain (targeting cholangiocytes) or intravenously administered the AAV8 vector expressing Cre under the thyroxine-binding protein (targeting adult hepatocytes)116. KrasG12D activation and Trp53 loss in adult hepatocytes required co-administration of the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) pro-cholestasis diet to form tumours (iCCA and HCC with a similar incidence, in addition to mixed HCC–CCA), highlighting the role of inflammation in liver cancer formation. By contrast, KrasG12D activation and Trp53 loss in the adult ductal compartment in the Sox9–CreERT2+ mouse strain accelerated the development of hepatic tumours, mainly iCCA, from preneoplastic lesions (not found in AAV8-injected mice) without the need for inflammatory cues116.
Targeted KrasG12D activation and Pten deletion triggered the fastest GEMM of CCA in Alb–Cre mice117. In KrasLSL-G12D/+,Ptenflox/flox,Alb–Cre mice, early hyperplastic biliary foci were detected by 4 weeks of age, and mice died by 7 weeks. Tumours were multifocal, stroma-rich localized iCCA. Interestingly, mice with heterozygous Pten deletion and KrasG12D activation developed tumours after a longer latency, showing hepatocyte and cholangiocyte differentiation features. The use of Alb–CreERT2+ or K19–CreERT/+ mouse strains to activate the oncogenic alterations in adult hepatocytes or cholangiocytes, respectively, led to the development of HCC and HCC precursor lesions, but not iCCA precursor lesions, in 8-week-old Alb–CreERT2+,KrasLSL-G12D,Ptenflox/flox mice, whereas tamoxifen injection on day 10 elicited iCCA. The formation of iCCA in Alb–CreERT2+,KrasLSL-G12D,Ptenflox/flox mice might be because Alb–Cre is still active in biliary cells at 10 days of age, and indicates that cholangiocytes are the cell of origin of CCA in these models, which was later independently confirmed using similar approaches118.
IDH1 and IDH2 oncogene modelling in mice was employed119,120. Breeding of Idh2LSL-R172K and KrasLSL-G12D mice in the Alb–Cre background yielded multifocal iCCA-like liver masses with invasive growth and metastatic capacity. Furthermore, adjacent to the tumours, oval cell expansion and biliary intraepithelial neoplasia-like lesions, suggestive of preneoplastic stages, occurred. Subsequently, the same group generated Idh1LSL-R132C mice that developed iCCA upon crossing with KrasLSL-G12D mice in the Alb–Cre background121. Another oncogene investigated in Alb–Cre mice was Notch1, via a mouse strain expressing the NOTCH1 intracellular domain from the Rosa26 locus122. By 8 months after birth, malignant foci were detected, leading to CCA formation in transplanted immunodeficient mice.
Two GEMMs highlighted the importance of a pro-inflammatory environment in cholangiocarcinogenesis. In the first model, severe liver damage by inflammatory cues originating from mitochondrial dysfunction characterized Hspd1flox/flox mice bred to the Alb–Cre strain123. Mice developed hepatocyte and cholangiocyte regenerative foci, the latter resembling human biliary intraepithelial neoplasia. The lesions arose in the context of an injured microenvironment and not through cell-autonomous mechanisms, as most regenerative liver foci exhibited HSPD1 expression. In the second model, KrasG12D expression and deletion of both Tgfbr2 and Cdh1 (encoding E-cadherin) were achieved in adult CK19+ biliary cells, leading to early-onset metastatic tumours in the extrahepatic and hilar bile duct124. In response to E-cadherin ablation, dying cholangiocytes released IL-33 to foster a proliferative phenotype in biliary epithelial cells that contributed to neoplastic transformation. However, after 4 weeks of tamoxifen administration, mice succumbed to liver and/or respiratory failure. In these models, transplantation of liver tissues in immunodeficient mice123 or derivation of tumour organoids from mice124 enabled follow-up experiments otherwise limited by the short lifespan of the mice.
Additional carcinogen-exposed GEMMs that model the consequences of an inflammatory environment, which is a frequent risk factor in human CCA, have also been reported. However, both the low penetrance and the high latency limited their use125,126. Nonetheless, co-exposure with carcinogens might be a strategy in GEMMs to accelerate cholangiocarcinogenesis by providing a pro-inflammatory and pro-fibrogenic environment that recapitulates the human context127.
Orthotopic or subcutaneous allograft models of premalignant liver cells (LPCs or adult liver organoids) or GEMM-derived CCA cell lines provide an alternative experimental strategy to time-consuming GEMMs12,73,100,121. These cellular models are amenable to gene editing, and their orthotopic transplantation into syngeneic mice enables tumour growth in an immune-competent microenvironment. Additionally, the plasticity of LPCs and liver organoids to originate CCA-like or HCC-like tumours, depending on the genetic context, is preserved.
GEMMs have shown that LPCs, cholangiocytes (intrahepatic and extrahepatic) and mature hepatocytes can be the cell of origin of CCA in mice57,128. However, the relevance of these findings for human CCA remains under evaluation. Indeed, various elements, including the targeted cell population (differentiated versus stem cells and additional cell types only present in humans), the tissue location (intrahepatic versus extrahepatic), the increased complexity of oncogenic alterations, the type, degree and duration of the pro-oncogenic and pro-inflammatory stimuli, the liver status, and other factors, might ultimately affect CCA development.
For all preclinical in vivo models, based on statements on histological assessment and unanimous agreement (Box 1 and Table 1), the expert panel strongly suggests that: the invasion of the basement membrane and tumorigenic capacity of isolated cells engrafted subcutaneously in immunodeficient mice are the most critical malignant features of CCA; morphological examination by H&E and immunohistochemistry should be conducted to characterize an early-stage tumour in the preclinical CCA model; immunohistochemistry of at least one biliary cytokeratin (CK7 or CK19) should always be performed to characterize a lesion as CCA in the absence of hepatobiliary primary lesions in a preclinical model; three histopathological features of human CCA must be assessed in a preclinical model: 1) intra-tumoural heterogeneity (high stroma, inflammatory response, epithelial phenotype); 2) pattern of growth (mass-forming, periductal infiltration, intraductal growth); and 3) immunopositivity for CK7 or CK19; the expert panel recommends classifying preclinical CCA models as iCCA, pCCA and dCCA, and suggests that focal desmoplastic stroma is a morphological feature required to classify a lesion as CCA in a preclinical model; and finally, a drug should be tested in more than one model.
Finally, to adopt a shared tool for homogeneously defining experimental models of CCA, an ‘experimental model sheet’ was generated, based on an initial expert discussion by all members of the CCA Model Consortium in a physical ad hoc meeting (Malta meeting 20189, WG1 meeting, European H2020 COST Action CA18122) (Box 2 and Supplementary Table 3) to provide complete information on animal experimentations to the scientific community through publications.
In vitro CCA models
2D culture with cell lines or primary cells
The urgent need to understand the biological processes of CCA progression and drug resistance has led to the widespread use of in vitro models represented by human and animal primary cultures and established cell lines. In 1985, the first CCA cell line — HChol-Y1 — was established from a patient with iCCA and then characterized129. Later, an assortment of CCA cell lines of intrahepatic and extrahepatic origin were generated from primary tumours, ascites, metastases and PDXs (Supplementary Table 2). In addition to human CCA cells, several lines derived from mouse, rat and hamster models have been described (Supplementary Table 2). Primary cultures of normal cholangiocytes should be used as control cells.
Molecular studies performed in human CCA tissues have uncovered recurring genomic alterations in specific genes such as mutations in TP53, IDH1/2, FGFR2, KRAS, BRAF and SMAD4, FGFR2 receptor fusions and ERBB family gene amplifications130, which qualify as targets for molecular approaches. Although most described CCA cell lines have been studied in terms of phenotypic and functional characterization of some parameters, only in the past few years, with the development of high-throughput sequencing techniques, have three studies used exome sequencing or RNA sequencing analyses to perform deep molecular phenotyping of some of the most widely used CCA cell lines131,132,133 (Supplementary Table 2). This has enabled the selection of cell lines with specific genetic alterations representing valuable drug screening tools, particularly for targeted therapy.
Most cell lines were established before the release of the latest WHO guidelines on the classification of tumours of the digestive system134, and potential misclassification of the origin of some cell lines might affect the clinical translation of some molecular and functional studies. For instance, Mz-ChA-1 cells have traditionally been used as a CCA cell line135,136, but they are classified as a gallbladder carcinoma cell line. Thus, results acquired using this cell line should be helpful for patients with this specific type of tumour.
In general, the well-established cell lines described in Supplementary Table 2 are easy models to explore tumorigenesis mechanisms and achieve high experimental reproducibility, mainly due to their long-term growth ability, short replication doubling time and low maintenance costs. However, several significant weaknesses have been described, such as long-term serum-based culture conditions that favour the accumulation of new genomic alterations as seen in many other long-term cultured cell lines137,138,139,140. New mutations obviously are not wanted in studying the effects of mutations leading to malignant outgrowth. Furthermore, in vitro maintenance often supports the selection of cell clones that are not representative of the genetic heterogeneity of the original tumour123,137,141. In addition, cell cultures grown as a monolayer might lack polarization and realistic cell–cell contacts within the tumour bulk. Finally, in the absence of cancer stromal cells and cell–matrix interactions, the fundamental tissue architecture provided by cellular and molecular components of the tumour microenvironment is not recapitulated3,137.
In addition to immortalized 2D cell lines, primary cultures of human CCA tissue have been established142,143,144,145. The overall success rate for CCA cell line isolation and establishment is relatively low (approximately 10%)146, partly due to insufficient numbers of tumour cells in resected tissues. Notably, contaminating non-tumour cells, such as fibroblasts, must be removed. Primary cultures are grown under serum-free and growth factor-enhanced conditions, which better resemble the in vivo tumour condition. Also, primary CCA cultures can be used shortly after derivation, so that more of the morphological and functional characteristics of their tissue of origin are retained147. In primary cultures, cell differentiation is constrained and the stem-like component is partially preserved, and thereby these cultures reflect tumour heterogeneity. However, the short time window to reach senescence in primary cultures hampers long-term experiments and their reproducibility.
A major limitation, independent of whether cell lines or primary CCA cultures are used, is the absence of acellular and cellular components of the tumour microenvironment, the presence of which would benefit the model. To address this problem148,149, different strategies have emerged in 2D cell cultures, including conditioned medium experiments, indirect co-culture through porous membrane cell culture inserts150 and direct co-culture151. In some cases, these experiments are performed with primary cultures of tumour and stromal cells (that are cancer-associated fibroblasts (CAFs) and monocytes/macrophages)5,152. In other cases, CCA cell lines are made to interact with immortalized stromal cell lines148,150,153 (Supplementary Table 2). Although these systems do not fully recapitulate the complex tumour microenvironment, they enable study of the crosstalk between CCA cells and other cell types, deepening our understanding of the role of different stromal cell types in tumour progression and drug response mechanisms148,149,152.
Based on statements on histological assessment (Table 1) and unanimous agreement, the expert panel (Box 1) strongly suggests to state in publications the origin of any cell line (previously established or new) according to the new CCA classification (iCCA, pCCA or dCCA). In addition, information regarding cell culture conditions should be provided in publications to standardize the procedures (such as choice of plastic support and cell culture medium, level of confluence, isolation procedure for primary culture, and passaging and subculturing methods).
3D culture recapitulating tumour organization
To facilitate personalized or precision medicine, patient material is used to study treatment responses. Although 2D CCA models are a step closer to the in vivo conditions in the patient compared with the established CCA cell lines, 3D culture models, including spheroids and organoids, resemble physiological conditions even more thoroughly. Spheroids are 3D aggregates of cells grown without a predefined culture substrate to adhere to5,154, whereas organoids self-organize in a matrix-rich 3D environment with which they interact155,156,157. Although traditional organoids represent an epithelial cell culture, there is a consensus that 3D models should ideally be upgraded to include epithelial stem cells, cells from the tumour microenvironment (for example, fibroblasts and/or immune cells) and extracellular matrix components to enable the analysis of cell–cell and cell–matrix interactions.
Spheroids
Tumour spheroids, typically generated as 3D multicellular aggregates from 2D-grown adherent cells, sometimes including stromal cells such as fibroblasts and endothelial cells, are used to model tumour biology5,154. They can be grown in natural and/or synthetic hydrogels157,158, and the increased complexity of these models enhances the understanding of tumour pathobiology, including tumour homeostasis and organization. In contrast to 2D cultures, tumour spheroids inherently recapitulate the gradient of oxygen supply and drug diffusion occurring within the tumour. However, their use as high-throughput, robust platforms is still limited due to the complexity of the culture conditions.
Organoids
Robust protocols for deriving biliary organoids from both mouse and human primary tissue explants or biopsy samples have been established6,156, and are complemented by methods that enable the derivation and propagation of organoids from induced pluripotent stem cells159 or cells collected from bile160,161. In addition to organoids derived from healthy donors, the successful establishment of organoid cultures from tumour tissues6,7,9,162,163 can substantially add to the toolbox of preclinical and translational CCA research. The overall consensus in the field is that the efficiency of establishing these CCA organoids from different patient tumours should be at least 25%. Efficiency should reach over 50% to guarantee the applicability of organoids to personalized medicine. Working with CCA organoids inevitably has limitations, including the overgrowth of non-malignant cholangiocyte organoids. Using specific tumour enrichment medium164, resorting to hand-picking non-malignant or tumour organoids to clean up the culture and xenotransplantation are ways to address this challenge. It is agreed that tumorigenicity needs to be confirmed for all CCA organoid lines, preferably via mutation analysis (standalone or as part of whole-genomic profiling). Proof of organoid tumorigenicity in immunocompromised mice and histopathological analysis are additional tests that can be performed. A shortcoming of CCA organoids is that an established line does not fully reflect the polyclonal nature of the original tumour. This might hamper insights into drug sensitivity or clonal regrowth of treated CCA tumours.
In addition to fully transformed CCA organoids, non-malignant cholangiocyte organoids can be a genetically flexible platform to functionally annotate the influence of specific genetic alterations on CCA pathobiology. Thus, recurrent iCCA genetic alterations (such as BAP1, NF1, SMAD4, PTEN, KRAS, AKT and IDH1/2 mutations, and FGFR2 fusions and MYC overexpression) were engineered in vitro in both human165,166 and mouse liver organoids167. Collectively, these studies provide convincing evidence that liver organoids, in which few genetic hits were introduced to recapitulate recurrent patterns of putative iCCA driver mutations, give rise to CCA upon subcutaneous or orthotopic transplantation into mice. Therefore, this approach is suitable for modelling genetically-defined cholangiocarcinogenesis in bipotent liver precursors and generating models for precision oncology research12.
Based on the available data and unanimous agreement, the expert panel strongly suggests (Box 1): the use of a specific tumour ‘enrichment’ medium (that is, tumour initiating medium as described by Broutier et al.7) to minimize contamination in non-tumour organoids; mutation and phenotypic analyses should be done to confirm the malignant origin of established organoid lines and reported in publications; and every organoid culture should be characterized before clinical applications such as drug screening.
Complex 3D culture systems
Although a hydrogel-based extracellular matrix is used to support the 3D growth of cells for both spheroids and organoids, this is typically a mouse tumour-derived basement membrane extract (Matrigel or BME) that does not represent the full human or tumour extracellular matrix155. Moreover, additional stromal cells, such as fibroblasts and immune cells are generally lacking in these cultures. The tumour microenvironment is crucial in the initiation, progression and invasion of CCA through a complex interaction between tumour cells, stromal cells and the extracellular matrix168. Targeting this desmoplastic, stroma-rich tumour microenvironment might be essential to overcome chemoresistance169,170,171. Thus, it would seem vital to include the CCA extracellular environment in vitro to mimic tumour composition, cell–cell and cell–matrix interactions172, morphology and tumour architecture more closely.
Current efforts are focused on the generation of future complex models (assembloids) that integrate the epithelial CCA component with 3D bioprinted scaffolds that recapitulate the anatomy of the biliary system173,174. This includes immune cells that shape tumour growth and drug sensitivity through direct or paracrine interaction, and stromal cells that create a physical barrier for drug delivery in addition to a pro-tumorigenic microenvironment. The challenges reside in the co-culture of autologous cell types derived from the same patient, as each cell type will have a unique growth dynamic and timeline. The use of cryopreservation protocols and human-induced pluripotent stem cell-derived generation of cell types from the same background cell might overcome these issues.
Addressing clinical needs
The experimental models described previously will facilitate the translation from experimental and preclinical work to the clinical setting. Whereas some models have provided relevant insights into the basic mechanisms of cancer progression, unravelling and allowing the analysis of cell signalling pathways, and cell–cell or tumour–microenvironment interactions, others have provided results that can be cautiously translated into the design of more effective treatments for CCA or the development of new clinical trials in humans. A few studies have indicated that the use of genetically defined cellular and animal models can advance the discovery of actionable vulnerabilities associated with druggable iCCA oncogenic drivers. Specifically, three independent studies found that RAS–ERK signalling is necessary and sufficient to support the oncogenic activity of FGFR2 fusions in PDXs91, GEMMs175 and organoid-based iCCA models167, and that combination therapies capable of more robust and durable suppression of RAS–ERK improve the therapeutic efficacy of clinically approved FGFR tyrosine kinase inhibitors91,167,175. Likewise, IDH1/KRAS-driven models have revealed that pharmacological targeting of mutated IDH1 sensitizes iCCA to host-mediated immune responses, which can be enhanced by concomitant administration of immune checkpoint inhibitors121.
The increasing availability of novel circulating biomarkers beyond the conventional serum tumour markers warrants validation for specific uses. Additional prognostic biomarkers might enable more accurate patient risk assessment and stratification in clinical trials. Predictive biomarkers for selecting the optimal therapy, such as circulating tumour DNA-based assays for FGFR2 fusions and IDH1 mutations176,177, are already in clinical use and will push the field forward. Finally, additional pharmacodynamic biomarkers capable of tracking disease evolution more accurately than the carbohydrate antigen 19-9 (CA 19-9), which is a tumour marker used in the management of biliary and pancreatic cancer, and that can reveal the emergence of drug resistance are warranted178, as shown for resistance to FGFR2 inhibitors179. Exposing FGFR2-mutated cells to an irreversible FGFR inhibitor (TAS-120) was found to provide a clinical benefit in patients who had developed resistance to reversible FGFR inhibitors via on-target mutations, the only exception being mutations targeting the gatekeeper residue.
CCA organoids have proven helpful for elucidating fundamental mechanisms of cancer progression and biomarker discovery7. Although the successful derivation of CCA organoids has lagged behind some other tumour types, organoids have high potential as tools for improving CCA research and therapy180. With further improvement in clinical applicability through continued advances in stem cell biology, organoid cultures and single-cell sequencing, a possible golden era for CCA organoids in personalized medicine is within reach.
A common limitation of experimental models is their inability to fully mimic all aspects of the tumour biology and personalized cancer features of individual patients. For example, the tumour microenvironment is a complex mix of cancerous and non-cancerous cells. The extracellular matrix dynamics, that is constantly remodelled by tumour cells, CAFs and tumour-associated macrophages, create a desmoplastic environment. In addition, there is considerable heterogeneity within and between tumours. It is challenging to capture this in experimental models, but it is essential in assessing drug resistance and tumour progression. Owing to the lack of the tumour microenvironment, drug screenings performed in vitro do not fully reflect in vivo efficacy, resulting in newly developed drugs failing in phase I to phase III clinical trials181. Finally, common risk factors and co-existing diseases that characterize human CCA (primary sclerosing cholangitis, liver flukes, chronic viral hepatitis, liver cirrhosis and others) are generally absent in the existing models. Thus, generating new models that combine established risk factors and concomitant morbidities for the human tumour with specific genetic alterations such as those reported earlier might recapitulate human CCA more accurately.
Study strengths and limitations
The Delphi method was applied to reach a consensus on the criteria required to establish valid preclinical models for the study of CCA. For this purpose, we built a task force of 45 renowned experts. Although we recognized that a more extensive panel could be preferred, we believe that the number of experts, their relevance in the CCA field, and the variety of backgrounds represented, including basic scientists, pathologists and clinicians, strengthened the validity of the consensus. During the process, the experts raised numerous comments, suggestions and questions, which were openly and rigorously discussed and incorporated into the study. This interactive and dynamic approach and the absence of dominant voices, which often inhibit the expression of minority viewpoints, resulted in fair and balanced contributions and the achievement of the final consensus statements and recommendations.
Experimental models are essential for a better understanding of carcinogenesis and tumour progression, for testing antitumour therapies and for deciphering therapeutic resistance mechanisms. A wide range of experimental models of CCA are available from simple, practical and inexpensive to more complex models resembling human cancer biology, albeit with a more challenging implementation process and higher costs (Table 4). The choice of model depends on what is requested of it, its accessibility, and, most importantly, its ability to answer a well-defined scientific question. 2D cultures and engrafted subcutaneous murine models are the most-used models to dissect signalling pathways, identify therapeutic targets and investigate drug resistance mechanisms. Depending on the type of research, in vivo orthotopic implantation models are preferred over ectopic CCA models. Both have advantages and limitations, as previously discussed. GEMMs seem to mimic pathobiological features of human tumorigenesis more closely than other models, despite being complex and expensive. Regarding in vitro models, tremendous progress has been made in better recapitulating the tumour 3D structure. The difficulty in employing these models includes the relatively high costs of setting up the culture and the availability of starting material (human CCA tissue).
In addition to providing an inventory, including evaluating advantages and disadvantages, of the most accurate experimental models currently available to the CCA scientific community, we present recommendations on minimal criteria for using these models. Using a Delphi-based process, a panel of experts in the field reached a consensus on these criteria as proposed herein. Obviously, disease models should ultimately lead to knowledge transfer from (basic) laboratory research to the clinic, to better understand the disease and offer innovative therapies. As the choice of model is highly dependent on the research question, it is highly recommended that results are gathered using different models to provide a comprehensive tumour mimic. This fosters the consolidation of scientific data with well-defined minimal criteria before validating them in humans by manipulating ex vivo samples or clinical trials.
Conclusions
Biomedical research relies entirely on in vitro and in vivo experimental models, a prerequisite for research in basic and applied sciences. This Consensus Statement is based on a set of recommendations on experimental models of CCA and information that should be specified in publications on these models developed and endorsed by an international group of experts to provide guidance to the scientific community. As a complement, the experts provided a brief overview of currently available models, highlighting the advantages and disadvantages that scientists should be aware of. Importantly, this Consensus Statement was prepared based on the expertise of both researchers and clinicians from different specialties (cell biologists, molecular biologists, oncologists, hepatologists and pathologists), and thus ensures the relevance of these statements and recommendations for a broad range of scientific communities, from health-care professionals to scientists directly investigating this fatal cancer.
References
Marin, J. J. G., Herraez, E., Lozano, E., Macias, R. I. R. & Briz, O. Models for understanding resistance to chemotherapy in liver cancer. Cancers 11, 1677 (2019).
Banales, J. M. et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 17, 557–588 (2020).
Zach, S., Birgin, E. & Rückert, F. Primary cholangiocellular carcinoma cell lines. J. Stem Cell Res. Transplant. 2, 1013 (2015).
Martinez-Becerra, P. et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 9, 1693–1704 (2012).
Raggi, C. et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 66, 102–115 (2017).
Marsee, A. et al. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell 28, 816–832 (2021).
Broutier, L. et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 23, 1424–1435 (2017).
Weiswald, L. B., Bellet, D. & Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 17, 1–15 (2015).
Nuciforo, S. et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 24, 1363–1376 (2018).
Loeuillard, E., Fischbach, S. R., Gores, G. J. & Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 982–992 (2019).
Leiting, J. L. et al. Biliary tract cancer patient-derived xenografts: surgeon impact on individualized medicine. JHEP Rep. 2, 100068 (2020).
Saborowski, A. et al. Murine liver organoids as a genetically flexible system to study liver cancer in vivo and in vitro. Hepatol. Commun. 3, 423–436 (2019).
Wang, J. et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J. Hepatol. 71, 742–752 (2019).
Primrose, J. N. et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 20, 663–673 (2019).
Jusakul, A. et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 7, 1116–1135 (2017).
Abou-Alfa, G. K. et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 796–807 (2020).
Abou-Alfa, G. K. et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684 (2020).
Roskoski, R. Jr Properties of FDA-approved small molecule protein kinase inhibitors: a 2022 update. Pharmacol. Res. 175, 106037 (2022).
Izquierdo-Sanchez, L. et al. Cholangiocarcinoma landscape in Europe: diagnostic, prognostic and therapeutic insights from the ENSCCA registry. J. Hepatol. 76, 1109–1121 (2022).
WHO Classification of Tumours Editorial Board. Digestive System Tumours: WHO Classification of Tumours 5th edn Vol. 1 (WHO, 2019).
Goyal, L. et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 7, 252–263 (2017).
Lowery, M. A. et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a phase 1 study. Lancet Gastroenterol. Hepatol. 4, 711–720 (2019).
Saborowski, A., Vogel, A. & Segatto, O. Combination therapies for targeting FGFR2 fusions in cholangiocarcinoma. Trends Cancer 8, 83–86 (2022).
Wu, M. J., Shi, L., Merritt, J., Zhu, A. X. & Bardeesy, N. Biology of IDH mutant cholangiocarcinoma. Hepatology 75, 1322–1337 (2022).
Brierley, J. D., Gospodarowicz, M. K. & Wittekind, C. TNM Classification of Malignant Tumours 8th edn (Union for International Cancer Control, 2017).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Guglielmi, A. et al. Intrahepatic cholangiocarcinoma: prognostic factors after surgical resection. World J. Surg. 33, 1247–1254 (2009).
Kendall, T. et al. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 39 (Suppl. 1), 7–18 (2019).
Zen, Y., Quaglia, A., Heaton, N., Rela, M. & Portmann, B. Two distinct pathways of carcinogenesis in primary sclerosing cholangitis. Histopathology 59, 1100–1110 (2011).
Radwan, N. A. & Ahmed, N. S. The diagnostic value of arginase-1 immunostaining in differentiating hepatocellular carcinoma from metastatic carcinoma and cholangiocarcinoma as compared to HepPar-1. Diagn. Pathol. 7, 149 (2012).
Shirakawa, H. et al. Glypican-3 is a useful diagnostic marker for a component of hepatocellular carcinoma in human liver cancer. Int. J. Oncol. 34, 649–656 (2009).
Lei, J. Y., Bourne, P. A., diSant’Agnese, P. A. & Huang, J. Cytoplasmic staining of TTF-1 in the differential diagnosis of hepatocellular carcinoma vs cholangiocarcinoma and metastatic carcinoma of the liver. Am. J. Clin. Pathol. 125, 519–525 (2006).
Zong, Y., Xiong, Y., Dresser, K., Yang, M. & Bledsoe, J. R. Polyclonal PAX8 expression in carcinomas of the biliary tract – frequent non-specific staining represents a potential diagnostic pitfall. Ann. Diagn. Pathol. 53, 151762 (2021).
Clark, B. Z., Beriwal, S., Dabbs, D. J. & Bhargava, R. Semiquantitative GATA-3 immunoreactivity in breast, bladder, gynecologic tract, and other cytokeratin 7-positive carcinomas. Am. J. Clin. Pathol. 142, 64–71 (2014).
Zen, Y. et al. Biliary intraepithelial neoplasia: an international interobserver agreement study and proposal for diagnostic criteria. Mod. Pathol. 20, 701–709 (2007).
Fujikura, K. et al. Comparative clinicopathological study of biliary intraductal papillary neoplasms and papillary cholangiocarcinomas. Histopathology 69, 950–961 (2016).
Komori, T. et al. CT imaging comparison between intraductal papillary neoplasms of the bile duct and papillary cholangiocarcinomas. Eur. Radiol. 29, 3132–3140 (2019).
Schlitter, A. M. et al. Intraductal papillary neoplasms of the bile duct: stepwise progression to carcinoma involves common molecular pathways. Mod. Pathol. 27, 73–86 (2014).
Goeppert, B. et al. Integrative analysis defines distinct prognostic subgroups of intrahepatic cholangiocarcinoma. Hepatology 69, 2091–2106 (2019).
Quigley, B. et al. Hepatobiliary mucinous cystic neoplasms with ovarian type stroma (so-called “hepatobiliary cystadenoma/cystadenocarcinoma”): clinicopathologic analysis of 36 cases illustrates rarity of carcinomatous change. Am. J. Surg. Pathol. 42, 95–102 (2018).
Zen, Y. et al. Mucinous cystic neoplasms of the liver: a clinicopathological study and comparison with intraductal papillary neoplasms of the bile duct. Mod. Pathol. 24, 1079–1089 (2011).
Chan-On, W. et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 45, 1474–1478 (2013).
Farshidfar, F. et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 19, 2878–2880 (2017).
Fujimoto, A. et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 6, 6120 (2015).
Gao, Q. et al. Activating mutations in PTPN3 promote cholangiocarcinoma cell proliferation and migration and are associated with tumor recurrence in patients. Gastroenterology 146, 1397–1407 (2014).
Nepal, C. et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 68, 949–963 (2018).
Nakamura, H. et al. Genomic spectra of biliary tract cancer. Nat. Genet. 47, 1003–1010 (2015).
Ong, C. K. et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 44, 690–693 (2012).
Zou, S. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696 (2014).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Andersen, J. B. et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 142, 1021–1031.e15 (2012).
Sia, D. et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 144, 829–840 (2013).
Montal, R. et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 73, 315–327 (2020).
Marquardt, J. U., Andersen, J. B. & Thorgeirsson, S. S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 15, 653–667 (2015).
Moeini, A., Haber, P. K. & Sia, D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 3, 100226 (2021).
Holczbauer, A. et al. Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterology 145, 221–231 (2013).
Guest, R. V. et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 74, 1005–1010 (2014).
Lee, J. S. et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 36, 1306–1311 (2004).
Lee, J. S. et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 12, 410–416 (2006).
Massa, A. et al. Evolution of the experimental models of cholangiocarcinoma. Cancers 12, 2308 (2020).
Fava, G. et al. γ-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 65, 11437–11446 (2005).
Mohr, R. et al. In vivo models for cholangiocarcinoma–what can we learn for human disease? Int. J. Mol. Sci. 21, 4993 (2020).
Cadamuro, M. et al. Animal models of cholangiocarcinoma: what they teach us about the human disease. Clin. Res. Hepatol. Gastroenterol. 42, 403–415 (2018).
Hu, M. H. et al. Targeting SHP-1-STAT3 signaling: a promising therapeutic approach for the treatment of cholangiocarcinoma. Oncotarget 8, 65077–65089 (2017).
Samukawa, E. et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 51, 1674–1684 (2017).
Pawar, P. et al. Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma: role of calmodulin. Clin. Cancer Res. 15, 1288–1296 (2009).
Colyn, L. et al. Dual targeting of G9a and DNA methyltransferase-1 for the treatment of experimental cholangiocarcinoma. Hepatology 73, 2380–2396 (2021).
Hou, Y. J. et al. Inhibition of active autophagy induces apoptosis and increases chemosensitivity in cholangiocarcinoma. Lab. Invest. 91, 1146–1157 (2011).
Merino-Azpitarte, M. et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 67, 72–83 (2017).
Meng, F., Yamagiwa, Y., Ueno, Y. & Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 44, 1055–1065 (2006).
Lobe, C. et al. Zinc finger E-box binding homeobox 1 promotes cholangiocarcinoma progression through tumor dedifferentiation and tumor-stroma paracrine signaling. Hepatology 74, 3194–3212 (2021).
Gentilini, A. et al. Extracellular signal-regulated kinase 5 regulates the malignant phenotype of cholangiocarcinoma cells. Hepatology 74, 2007–2020 (2021).
Vallejo, A. et al. FOSL1 promotes cholangiocarcinoma via transcriptional effectors that could be therapeutically targeted. J. Hepatol. 75, 363–376 (2021).
Olaru, A. V. et al. MicroRNA down-regulated in human cholangiocarcinoma control cell cycle through multiple targets involved in the G1/S checkpoint. Hepatology 54, 2089–2098 (2011).
Zhang, J., Han, C. & Wu, T. MicroRNA-26a promotes cholangiocarcinoma growth by activating β-catenin. Gastroenterology 143, 246–256.e8 (2012).
Zhu, H., Han, C., Lu, D. & Wu, T. miR-17-92 cluster promotes cholangiocarcinoma growth: evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am. J. Pathol. 184, 2828–2839 (2014).
Zhu, H. et al. Neuropilin-1 regulated by miR-320 contributes to the growth and metastasis of cholangiocarcinoma cells. Liver Int. 38, 125–135 (2018).
Han, S. et al. Suppression of miR-16 promotes tumor growth and metastasis through reversely regulating YAP1 in human cholangiocarcinoma. Oncotarget 8, 56635–56650 (2017).
Razumilava, N. et al. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 60, 599–605 (2014).
McVeigh, L. E. et al. Development of orthotopic tumour models using ultrasound-guided intrahepatic injection. Sci. Rep. 9, 9904 (2019).
Erice, O. et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1335–1344 (2018).
Cardinale, V. et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 185, 1724–1739 (2015).
Wu, Z. et al. Significance of S100P as a biomarker in diagnosis, prognosis and therapy of opisthorchiasis-associated cholangiocarcinoma. Int. J. Cancer 138, 396–408 (2016).
Cadamuro, M. et al. Low-dose paclitaxel reduces S100A4 nuclear import to inhibit invasion and hematogenous metastasis of cholangiocarcinoma. Cancer Res. 76, 4775–4784 (2016).
Peraldo Neia, C. et al. Gene and microRNA modulation upon trabectedin treatment in a human intrahepatic cholangiocarcinoma paired patient derived xenograft and cell line. Oncotarget 7, 86766–86780 (2016).
Peraldo-Neia, C. et al. Anti-cancer effect and gene modulation of ET-743 in human biliary tract carcinoma preclinical models. BMC Cancer 14, 918 (2014).
Wang, Y. et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 380, 163–173 (2016).
Saha, S. K. et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 6, 727–739 (2016).
Kabashima, A. et al. Fibroblast growth factor receptor inhibition induces loss of matrix MCL1 and necrosis in cholangiocarcinoma. J. Hepatol. 68, 1228–1238 (2018).
Lidsky, M. E. et al. Leveraging patient derived models of FGFR2 fusion positive intrahepatic cholangiocarcinoma to identify synergistic therapies. NPJ Precis. Oncol. 6, 75 (2022).
Wu, Q. et al. EGFR inhibition potentiates FGFR inhibitor therapy and overcomes resistance in FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 12, 1378–1395 (2022).
Sirica, A. E. et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology 47, 1178–1190 (2008).
Fingas, C. D. et al. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology 52, 550–561 (2010).
Blechacz, B. R. et al. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology 50, 1861–1870 (2009).
Smoot, R. L. et al. A Bax-mediated mechanism for obatoclax-induced apoptosis of cholangiocarcinoma cells. Cancer Res. 70, 1960–1969 (2010).
Fingas, C. D. et al. Targeting PDGFR-β in cholangiocarcinoma. Liver Int. 32, 400–409 (2012).
Mertens, J. C. et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 73, 897–907 (2013).
Yamada, D. et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 61, 1627–1642 (2015).
Rizvi, S. et al. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 9, 5892–5905 (2018).
Saborowski, A. et al. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc. Natl Acad. Sci. USA 110, 19513–19518 (2013).
Loeuillard, E. et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Invest. 130, 5380–5396 (2020).
Fabris, L., Sato, K., Alpini, G. & Strazzabosco, M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology 73 (Suppl. 1), 75–85 (2021).
Fitzhugh, O. G. & Nelson, A. A. Liver tumors in rats fed thiourea or thioacetamide. Science 108, 626–628 (1948).
Al-Bader, A. et al. Cholangiocarcinoma and liver cirrhosis in relation to changes due to thioacetamide. Mol. Cell Biochem. 208, 1–10 (2000).
Kamp, E. J. et al. Genetic alterations during the neoplastic cascade towards cholangiocarcinoma in primary sclerosing cholangitis. J. Pathol. 258, 227–235 (2022).
Yeh, C. N., Maitra, A., Lee, K. F., Jan, Y. Y. & Chen, M. F. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis 25, 631–636 (2004).
Lozano, E. et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 12, 91–100 (2014).
Verna, L., Whysner, J. & Williams, G. M. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 71, 57–81 (1996).
Yang, H. et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 141, 378–388.e4 (2011).
Thamavit, W. et al. Promotion of cholangiocarcinogenesis in the hamster liver by bile duct ligation after dimethylnitrosamine initiation. Carcinogenesis 14, 2415–2417 (1993).
Thamavit, W., Pairojkul, C., Tiwawech, D., Shirai, T. & Ito, N. Strong promoting effect of Opisthorchis viverrini infection on dimethylnitrosamine-initiated hamster liver. Cancer Lett. 78, 121–125 (1994).
Maronpot, R. R., Giles, H. D., Dykes, D. J. & Irwin, R. D. Furan-induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicol. Pathol. 19, 561–570 (1991).
Erice, O. et al. Genetic mouse models as in vivo tools for cholangiocarcinoma research. Cancers 11, 1868 (2019).
Xu, X. et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J. Clin. Invest. 116, 1843–1852 (2006).
O’Dell, M. R. et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 72, 1557–1567 (2012).
Hill, M. A. et al. Kras and Tp53 mutations cause cholangiocyte- and hepatocyte-derived cholangiocarcinoma. Cancer Res. 78, 4445–4451 (2018).
Ikenoue, T. et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 6, 23899 (2016).
Lin, Y. K. et al. Combination of Kras activation and PTEN deletion contributes to murine hepatopancreatic ductal malignancy. Cancer Lett. 421, 161–169 (2018).
Saha, S. K. et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 (2014).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465, 966 (2010).
Wu, M. J. et al. Mutant IDH inhibits IFNγ-TET2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov. 12, 812–835 (2022).
Zender, S. et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 23, 784–795 (2013).
Yuan, D. et al. Kupffer cell-derived TNF triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell 31, 771–789.e6 (2017).
Nakagawa, H. et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl Acad. Sci. USA 114, E3806–E3815 (2017).
Guest, R. V. et al. Notch3 drives development and progression of cholangiocarcinoma. Proc. Natl Acad. Sci. USA 113, 12250–12255 (2016).
Farazi, P. A. et al. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 66, 6622–6627 (2006).
Younger, N. T. et al. In vivo modeling of patient genetic heterogeneity identifies new ways to target cholangiocarcinoma. Cancer Res. 82, 1548–1559 (2022).
Fan, B. et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 122, 2911–2915 (2012).
Yamaguchi, N., Morioka, H., Ohkura, H., Hirohashi, S. & Kawai, K. Establishment and characterization of the human cholangiocarcinoma cell line HChol-Y1 in a serum-free, chemically defined medium. J. Natl Cancer Inst. 75, 29–35 (1985).
Valle, J. W., Lamarca, A., Goyal, L., Barriuso, J. & Zhu, A. X. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 7, 943–962 (2017).
Lau, D. K. et al. Genomic profiling of biliary tract cancer cell lines reveals molecular subtypes and actionable drug targets. iScience 21, 624–637 (2019).
Scherer, D. et al. RNA sequencing of hepatobiliary cancer cell lines: data and applications to mutational and transcriptomic profiling. Cancers 12, 2510 (2020).
Ghandi, M. et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019).
Nagtegaal, I. D. et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 76, 182–188 (2020).
Knuth, A. et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J. Hepatol. 1, 579–596 (1985).
Fausther, M. et al. Establishment and characterization of rat portal myofibroblast cell lines. PLoS ONE 10, e0121161 (2015).
Kaur, G. & Dufour, J. M. Cell lines: valuable tools or useless artifacts. Spermatogenesis 2, 1–5 (2012).
Domcke, S., Sinha, R., Levine, D. A., Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Ertel, A., Verghese, A., Byers, S. W., Ochs, M. & Tozeren, A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol. Cancer 5, 55 (2006).
Gillet, J. P. et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl Acad. Sci. USA 108, 18708–18713 (2011).
Chow, M. & Rubin, H. Clonal selection versus genetic instability as the driving force in neoplastic transformation. Cancer Res. 60, 6510–6518 (2000).
Massani, M. et al. Isolation and characterization of biliary epithelial and stromal cells from resected human cholangiocarcinoma: a novel in vitro model to study tumor-stroma interactions. Oncol. Rep. 30, 1143–1148 (2013).
Fraveto, A. et al. Sensitivity of human intrahepatic cholangiocarcinoma subtypes to chemotherapeutics and molecular targeted agents: a study on primary cell cultures. PLoS ONE 10, e0142124 (2015).
Carnevale, G. et al. Activation of Fas/FasL pathway and the role of c-FLIP in primary culture of human cholangiocarcinoma cells. Sci. Rep. 7, 14419 (2017).
Lustri, A. M. et al. TGF-β signaling is an effective target to impair survival and induce apoptosis of human cholangiocarcinoma cells: a study on human primary cell cultures. PLoS ONE 12, e0183932 (2017).
Ku, J. L. et al. Establishment and characterisation of six human biliary tract cancer cell lines. Br. J. Cancer 87, 187–193 (2002).
Pastor, D. M. et al. Primary cell lines: false representation or model system? A comparison of four human colorectal tumors and their coordinately established cell lines. Int. J. Clin. Exp. Med. 3, 69–83 (2010).
Vaquero, J. et al. The IGF2/IR/IGF1R pathway in tumor cells and myofibroblasts mediates resistance to EGFR inhibition in cholangiocarcinoma. Clin. Cancer Res. 24, 4282–4296 (2018).
Zhou, Z. et al. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 9, e001946 (2021).
Okabe, H. et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 16, 2555–2564 (2009).
Nicolas-Boluda, A. et al. Photothermal depletion of cancer-associated fibroblasts normalizes tumor stiffness in desmoplastic cholangiocarcinoma. ACS Nano 14, 5738–5753 (2020).
Cadamuro, M. et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol. 70, 700–709 (2019).
Li, L. et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology 65, 501–514 (2017).
Fennema, E., Rivron, N., Rouwkema, J., van Blitterswijk, C. & de Boer, J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 31, 108–115 (2013).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Huch, M., Boj, S. F. & Clevers, H. Lgr5(+) liver stem cells, hepatic organoids and regenerative medicine. Regen. Med. 8, 385–387 (2013).
Sato, K. et al. Organoids and spheroids as models for studying cholestatic liver injury and cholangiocarcinoma. Hepatology 74, 491–502 (2021).
Girard, Y. K. et al. A 3D fibrous scaffold inducing tumoroids: a platform for anticancer drug development. PLoS ONE 8, e75345 (2013).
Wu, F. et al. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 70, 1145–1158 (2019).
Soroka, C. J., Assis, D. N. & Boyer, J. L. Patient-derived organoids from human bile: an in vitro method to study cholangiopathies. Methods Mol. Biol. 1981, 363–372 (2019).
Roos, F. J. M. et al. Human bile contains cholangiocyte organoid-initiating cells which expand as functional cholangiocytes in non-canonical Wnt stimulating conditions. Front. Cell Dev. Biol. 8, 630492 (2020).
Lampis, A. et al. MIR21 drives resistance to heat shock protein 90 inhibition in cholangiocarcinoma. Gastroenterology 154, 1066–1079.e5 (2018).
Li, L. et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 4, e121490 (2019).
Broutier, L. et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 11, 1724–1743 (2016).
Artegiani, B. et al. Probing the tumor suppressor function of BAP1 in CRISPR-engineered human liver organoids. Cell Stem Cell 24, 927–943.e6 (2019).
Sun, L. et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 21, 1015–1026 (2019).
Cristinziano, G. et al. FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma. J. Hepatol. 75, 351–362 (2021).
Leyva-Illades, D., McMillin, M., Quinn, M. & Demorrow, S. Cholangiocarcinoma pathogenesis: role of the tumor microenvironment. Transl. Gastrointest. Cancer 1, 71–80 (2012).
Hogdall, D., Lewinska, M. & Andersen, J. B. Desmoplastic tumor microenvironment and immunotherapy in cholangiocarcinoma. Trends Cancer 4, 239–255 (2018).
Cadamuro, M. et al. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1435–1443 (2018).
Mertens, J. C., Rizvi, S. & Gores, G. J. Targeting cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1454–1460 (2018).
Tanimizu, N. et al. Generation of functional liver organoids on combining hepatocytes and cholangiocytes with hepatobiliary connections ex vivo. Nat. Commun. 12, 3390 (2021).
Lee, H. et al. Cell-printed 3D liver-on-a-chip possessing a liver microenvironment and biliary system. Biofabrication 11, 025001 (2019).
Allan, A. et al. Patient-specific 3D printed model of biliary ducts with congenital cyst. Quant. Imaging Med. Surg. 9, 86–93 (2019).
Kendre, G. et al. The co-mutational spectrum determines the therapeutic response in murine FGFR2 fusion-driven cholangiocarcinoma. Hepatology 74, 1357–1370 (2021).
Wintachai, P. et al. Diagnostic and prognostic value of circulating cell-free DNA for cholangiocarcinoma. Diagnostics 11, 999 (2021).
Csoma, S. L. et al. Circulating cell-free DNA-based comprehensive molecular analysis of biliary tract cancers using next-generation sequencing. Cancers 14, 233 (2022).
Varghese, A. M. et al. Noninvasive detection of polyclonal acquired resistance to FGFR inhibition in patients with cholangiocarcinoma harboring FGFR2 alterations. JCO Precis Oncol. 5, 44–50 (2021).
Goyal, L. et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 9, 1064–1079 (2019).
van Tienderen, G. S. et al. Hepatobiliary tumor organoids for personalized medicine: a multicenter view on establishment, limitations, and future directions. Cancer Cell 40, 226–230 (2022).
Wong, C. H., Siah, K. W. & Lo, A. W. Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 (2019).
Lozano, E. et al. Enhanced antitumour drug delivery to cholangiocarcinoma through the apical sodium-dependent bile acid transporter (ASBT). J. Control. Release 216, 93–102 (2015).
Katz, S. F. et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 142, 1229–1239.e3 (2012).
Tschaharganeh, D. F. et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592 (2014).
El Khatib, M. et al. Activation of Notch signaling is required for cholangiocarcinoma progression and is enhanced by inactivation of p53 in vivo. PLoS ONE 8, e77433 (2013).
Ikenoue, T. et al. Establishment and analysis of a novel mouse line carrying a conditional knockin allele of a cancer-specific FBXW7 mutation. Sci. Rep. 8, 2021 (2018).
Manieri, E. et al. JNK-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc. Natl Acad. Sci. USA 117, 16492–16499 (2020).
Cubero, F. J. et al. Loss of c-Jun N-terminal kinase 1 and 2 function in liver epithelial cells triggers biliary hyperproliferation resembling cholangiocarcinoma. Hepatol. Commun. 4, 834–851 (2020).
Marsh, V., Davies, E. J., Williams, G. T. & Clarke, A. R. PTEN loss and KRAS activation cooperate in murine biliary tract malignancies. J. Pathol. 230, 165–173 (2013).
Falcomata, C. et al. Genetic screens identify a context-specific PI3K/p27(Kip1) node driving extrahepatic biliary cancer. Cancer Discov. 11, 3158–3177 (2021).
Sekiya, S. & Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Invest. 122, 3914–3918 (2012).
Crawford, D. R. et al. Characterization of liver injury, oval cell proliferation and cholangiocarcinogenesis in glutathione S-transferase A3 knockout mice. Carcinogenesis 38, 717–727 (2017).
Acknowledgements
The authors thank the European Network for the Study of Cholangiocarcinoma (ENS-CCA) and the European H2020 COST Action CA18122. The authors also acknowledge the valuable contributions of the external advisory panel. C.C. is supported by Inserm, Université de Rennes 1 and by a grant from the French Ministry of Health and the French National Cancer Institute (PRT-K20-136), CHU Rennes, CLCC Eugène Marquis, Rennes. M.M.A.V. and L.J.W.v.d.L. are supported by Medical Delta Regenerative Medicine 4D (Generating complex tissues with stem cells and printing technology) and TKI-LSH grant EMC-LSH19002. S.R. is supported by the German Research Foundation (DFG, project no. 314905040 and no. 493697503) and by German Cancer Aid (Deutsche Krebshilfe, project no. 70113922). S.V. is supported by Ministerio de Ciencia, Innovación y Universidades, Convocatoria 2019 para incentivar la Incorporación Estable de Doctores (IED2019-001007-I), by FEDER/Ministerio de Ciencia, Innovación y Universidades – Agencia Estatal de Investigación (PID2020‐116344‐RB‐100) and by the Government of Navarra-Health Research Department (58; 2018). J.V. is funded by Ministerio de Ciencia e Innovación, part of Agencia Estatal de Investigación (AEI), through the Retos Investigación grant number PID2019-108651RJ-I00/DOI 10.13039/501100011033. The authors thank CERCA Programme/Generalitat de Catalunya for institutional support. R.I.R.M. and J.J.G.M. are supported by Instituto de Salud Carlos III, Spain (PI20/00189, PI19/00819) co-funded by the European Union. L. Fouassier belongs to a team supported by the Fondation pour la Recherche Médicale (Equipe FRM 2020 no. EQU202003010517) and is supported by Inserm and Sorbonne Université, INCa and ITMO Cancer of Aviesan within the framework of the 2021–2030 Cancer Control Strategy, on funds administered by Inserm.
Author information
Authors and Affiliations
Consortia
Contributions
L. Fouassier coordinated the workgroups and the process of generating and editing the manuscript. M.M.A.V. and R.E.C. coordinated the Delphi questionnaire, and all authors contributed equally to the writing and final revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.B. receives honoraria from Incyte, Servier, BI and AstraZeneca, and research funding from Avacta and Medannex. A.F. received consultancy fees from Bayer, AstraZeneca, Roche, Boston, Exact Science and Guerbert. J.W.V. reports honoraria from Agios, AstraZeneca, Genoscience Pharma, Incyte, Mundipharma EDO, Mylan, QED, Servier, SIRTex and Zymeworks, and honoraria and non-financial support from NuCana, all outside the submitted work. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Gastroenterology & Hepatology thanks Heather Francis, Marco Marzioni and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ICD-11: https://icd.who.int
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Calvisi, D.F., Boulter, L., Vaquero, J. et al. Criteria for preclinical models of cholangiocarcinoma: scientific and medical relevance. Nat Rev Gastroenterol Hepatol 20, 462–480 (2023). https://doi.org/10.1038/s41575-022-00739-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-022-00739-y
