
Abstract
Inflammasome signalling is an emerging pillar of innate immunity and has a central role in the regulation of gastrointestinal health and disease. Activation of the inflammasome complex mediates both the release of the pro-inflammatory cytokines IL-1β and IL-18 and the execution of a form of inflammatory cell death known as pyroptosis. In most cases, these mediators of inflammation provide protection against bacterial, viral and protozoal infections. However, unchecked inflammasome activities perpetuate chronic inflammation, which underpins the molecular and pathophysiological basis of gastritis, IBD, upper and lower gastrointestinal cancer, nonalcoholic fatty liver disease and obesity. Studies have also highlighted an inflammasome signature in the maintenance of gut microbiota and gut–brain homeostasis. Harnessing the immunomodulatory properties of the inflammasome could transform clinical practice in the treatment of acute and chronic gastrointestinal and extragastrointestinal diseases. This Review presents an overview of inflammasome biology in gastrointestinal health and disease and describes the value of experimental and pharmacological intervention in the treatment of inflammasome-associated clinical manifestations.
Key points
-
Inflammasomes are expressed in both immune and non-immune cells, contributing to their functional ties to infection, IBD, cancer, autoinflammation and autoimmune conditions.
-
Inflammasome sensors function by recognizing and responding to a pathogen (lipopolysaccharide, microbial DNA or bacterial flagellin) or to a danger signal (ion flux, self-DNA or ATP).
-
Inflammasomes control the magnitude of inflammation and cell death in response to pathogen-associated molecular patterns or danger-associated molecular patterns, which, in part, determines a protective or detrimental outcome in the host.
-
Experimental and pharmacological interventions have yielded success in the treatment of inflammasome-mediated disorders, such as autoinflammatory enterocolitis.
-
The inflammasome–gut microbiota axis and its relevance to health and disease are influenced by genetic, environmental and experimental factors.
-
The deep and complex relationship between inflammasomes, pathogens and the microbiota provides an exciting platform for basic and clinical research with which to understand health and disease.
Similar content being viewed by others


Introduction
The immune system has an extraordinary capacity to recognize and respond to a range of microbial patterns and danger signals. In the gastrointestinal tract, trillions of microorganisms colonize the mucosal surface and lumen and constantly release immunomodulatory molecules that interact with and shape the immune system1. Most of these microorganisms are symbionts or so-called commensals and do not generally evoke a detrimental inflammatory response. However, pathogens have the ability to invade the mucosal barrier and underlying tissue, inducing the production of cytokines, chemokines and antimicrobial molecules.
A central and rapid mechanism triggering an inflammatory response occurs through activation of the innate immune signalling complex called the inflammasome2,3. The inflammasome is formed in a variety of immune and non-immune cells when a subset of cytosolic pattern-recognition receptors known as inflammasome sensors recognizes pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs)4. To date, members from three families of pattern-recognition receptors are known to have an established role in the assembly of an inflammasome: NLRP1, NLRP3, NLRC4 and neuronal apoptosis inhibitory proteins (NAIPs) from the NOD-like receptor (NLR) family; absent in melanoma 2 (AIM2) from the AIM2-like receptor (ALR) family; and pyrin from the tripartite motif-containing protein (TRIM) family5 (Fig. 1). Several other sensors have been suggested to activate the inflammasome, including the NLR family members NLRP6, NLRP7, NLRP9 (NLRP9b in mice) and NLRP12, the DNA sensor IFNγ-inducible protein 16 (IFI16) and the RNA sensor retinoic acid-inducible gene I protein (RIG-I; also known as DDX58); however, the ability of these sensors to form inflammasome complexes is still uncertain6. On activation, inflammasome sensors recruit the adaptor protein apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC; also known as PYCARD) and the cysteine protease caspase 1, and form a single cytoplasmic aggregate known as the inflammasome speck6. Caspase 1 is activated in this complex, inducing proteolytic cleavage of the pro-inflammatory cytokines pro-IL-1β and pro-IL-18 and the pore-forming protein gasdermin D, which results in the release of bioactive IL-1β and IL-18 into the extracellular milieu and pyroptosis, respectively7. Bioactive IL-1β and IL-18 and certain DAMPs can be secreted through the gasdermin D pores before rupture of the host cell membrane, cell lysis and cell death8,9,10.

The inflammasome sensors NLRP1b, NLRP3, NLRC4, AIM2 and pyrin are all capable of forming a canonical inflammasome complex containing the adaptor protein ASC and the cysteine protease caspase 1. NLRP1b and NLRC4 also recruit caspase 1 without ASC owing to the presence of a CARD domain in their structure64,65,66. Activation of NLRP3 and NLRC4 requires the kinase NEK7 and the NLR family members neuronal apoptosis inhibitory proteins (NAIPs), respectively. Caspase 1 cleaves the precursor cytokines pro-IL-1β and pro-IL-18 and the pore-forming protein gasdermin D. The active fragment of gasdermin D oligomerizes and forms pores on the cell membrane, resulting in pyroptosis73,74,75,76,77,78,79,80. These pores also allow passive release of biologically active IL-1β and IL-18 from the cell. The non-canonical inflammasome is defined by a requirement for human caspase 4, human caspase 5 or mouse caspase 11 for the activation of the NLRP3 inflammasome complex16. Activation of these caspases leads to cleavage of gasdermin D and pyroptosis73,74,75. The pore-forming fragment of gasdermin D activates the NLRP3 inflammasome and caspase 1-dependent maturation of IL-1β and IL-18 (refs73,74).
Inflammation and cell death responses triggered by the inflammasome contribute to the pathogenesis of a spectrum of acute and chronic diseases in the gastrointestinal tract, including infection, gastritis, IBD, cancer, nonalcoholic fatty liver disease (NAFLD) and obesity11,12. Inflammasomes might also support a healthy gut microbiota and regulate metabolic and gut–brain homeostasis, with a majority of studies focusing on bacteria of the gut microbiota13,14. This Review describes the central roles of inflammasome signalling in the gastrointestinal tract, highlighting its major functionalities in shaping the outcome of acute and chronic gastrointestinal diseases in the context of infectious agents, genetic susceptibilities, physiological aberrations and the changing landscape of the bacterial gut microbiota. I also discuss the potential translation and efficacies of pharmacological interventions targeting the inflammasome pathways in gastrointestinal diseases.
Inflammasome biology and activation
An inflammasome complex is generally categorized into a canonical or non-canonical inflammasome. The inflammasome sensors NLRP1, NLRP3, NLRC4, NAIPs, AIM2 and pyrin are capable of assembling a canonical inflammasome complex15 (Fig. 1). The canonical inflammasome is a protein complex comprising one or more inflammasome sensors, ASC and caspase 1. The non-canonical inflammasome refers to an activation pathway of the NLRP3 inflammasome that specifically requires mouse caspase 11, human caspase 4 or human caspase 5 (ref.16). The current conceptual view is that inflammasome sensors either directly bind and interact with a ligand, such as flagellin, DNA or lipopolysaccharide (LPS), or indirectly sense a physiological aberration, such as ion fluxes and mitochondrial dysfunction17. The former case is exemplified by AIM2, caspase 4, caspase 5, caspase 11 and NAIPs. Direct binding between double-stranded DNA (dsDNA) and AIM2, independently of the DNA sequence, via electrostatic interaction initiates activation of the AIM2 inflammasome18,19,20,21. Similarly, direct binding between cytosolic LPS and caspase 4, caspase 5 or caspase 11 initiates activation of the non-canonical NLRP3 inflammasome pathway22. Activation of the NLRC4 inflammasome requires initial recognition of the ligand by NAIP23,24,25,26,27. The single NAIP encoded in the human genome is capable of recognizing the needle and inner rod components of the type 3 secretion system (T3SS) and flagellin subunits of bacteria28,29,30. Of the mouse NAIPs, NAIP5 and NAIP6 directly bind flagellin subunits, and NAIP1 and NAIP2 bind the needle and inner rod components of the T3SS, respectively28,31,32,33,34. Ligand-bound NAIPs then recruit and induce oligomerization of NLRC4 in order to catalyse the assembly of a functional NAIP–NLRC4 inflammasome complex35,36 (Fig. 1).
The inflammasome sensors NLRP1, NLRP3 and pyrin do not bind directly to a ligand but respond to a subset of cellular events triggered by PAMPs and DAMPs. Human NLRP1, rat NLRP1 and mouse NLRP1b are activated by any protease capable of mediating site-specific proteolytic cleavage at the amino terminus or function to find domain (FIIND) of NLRP1 (refs37,38,39,40), such as the lethal factor component of the anthrax lethal toxin41. NLRP3 indirectly responds to a multitude of PAMPs and DAMPs, including pathogens, toxins, crystalline substances and host and microbial metabolites14. These NLRP3 activators trigger physiological aberrations in the cell, signified by potassium efflux42,43,44, lysosomal rupture45, mitochondria disruption and release of either mitochondrial reactive oxygen species (ROS) or oxidized mitochondrial DNA46,47,48,49 or calcium influx and reduction in cAMP levels50. These signals converge on a pathway that drives activation of the NLRP3 inflammasome, which is mediated by serine/threonine-protein kinase NEK7 (Fig. 1).
The most recently described inflammasome sensor, pyrin, responds to inactivation of host small GTPases of the RHO family51 (Fig. 1). RHO-inactivating events occur as a result of infection by bacteria capable of secreting RHO-inactivating toxins, such as infection by the diarrhoeal pathogen Clostridium difficile51,52, or as a result of host genetic mutations, such as mutations S208A and S242R in MEFV (which encodes pyrin), which impair either actin dynamics or phosphorylation of pyrin53,54.
On activation, inflammasome components assemble a single speck of 1 μm in diameter55,56,57,58,59. Cryoelectron microscopy analysis revealed that ASC and caspase 1 act in a prionoid-like fashion to form filamentous structures and catalyse formation of the inflammasome speck59,60,61,62. Although the use of cryoelectron microscopy analysis of overexpressed inflammasome proteins can provide potentially ground-breaking insights into inflammasome structures and assembly, these techniques can lead to artefactual observations and would require further experimental support.
Caspase 1 contains a CARD. Inflammasome sensors that do not carry their own CARD must first recruit ASC — a bipartite protein composed of a pyrin domain and a CARD, the former can interact with the pyrin domain of CARD-less inflammasome sensors, and the latter can subsequently bind the CARD of caspase 1 (ref.63). Indeed, inflammasome sensors that have a CARD, such as NLRC4 and NLRP1, can activate inflammasome responses or induce pyroptosis without ASC, whereas inflammasome sensors that have a pyrin domain but not a CARD, such as NLRP3 and AIM2, require ASC for inflammasome activation23,51,56,64,65,66.
Although it has been known for decades that caspase 1 induces proteolytic cleavage of the substrates pro-IL-1β and pro-IL-18 (refs67,68,69,70), how caspase 1 mediates pyroptosis was unknown71,72. Studies have now identified the pore-forming protein gasdermin D as a substrate of inflammatory caspases73,74,75. Caspase 1 and caspase 11 cleave gasdermin D between its Asp276 and Gly277 residues73,74, yielding an active 30–31 kDa amino-terminal fragment that oligomerizes and forms pores on the host cell membrane76,77,78,79,80 (Fig. 1). The pores are 10–21 nm in diameter, and biologically active and pleotropic forms of the cytokines IL-1β and IL-18 and certain DAMPs are released through them, even before lytic demise of the cell8,9,10,76,77,78,79,80 (Fig. 1). These cytokines and DAMPs collectively regulate intestinal inflammation, host defence, gut barrier functions and host–microbiota homeostasis81,82. Thus, secretion of cytokines and execution of cell death via the inflammasome are critical to the functional activity of the gastrointestinal tract.
Infectious diseases
Microbial pathogens from all domains of life are capable of colonizing and infecting the mammalian gut. These pathogens continue to pose serious concerns to public health and cause substantial morbidity and mortality worldwide83. The inflammasome operates in intestinal cells and immune cells as a cytosolic sentinel that serves to protect the host from invading microorganisms (Fig. 2).

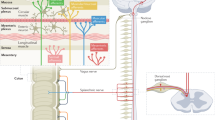
a | Expression in humans. b | Expression in mice. References for expression data are given in Supplementary table 1. AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD); NAIP, neuronal apoptosis inhibitory protein; NLRC, nucleotide-binding domain, leucine-rich repeat-containing protein (NLR) family CARD domain-containing protein; NLRP, NACHT, LRR and PYD domains-containing protein.
Bacteria
Bacteria carry an array of PAMPs, some of which are delivered into the host cytoplasm, where they activate the inflammasome (Fig. 3). A thematic overview of the mechanisms of inflammasome activities in the recognition of bacteria and host defence, with a focus on gastrointestinal pathogens, is provided herein.

Pathogens carry a plethora of pathogen-associated molecular patterns (PAMPs). These PAMPs either directly bind and activate an inflammasome sensor or induce a physiological change in the cell that is sensed by an inflammasome sensor5. Pathogens also cause substantial damage to the host cell, a process that induces the liberation of danger-associated molecular patterns (DAMPs) that activate the inflammasome15. AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD); LPS, lipopolysaccharide; NAIP, neuronal apoptosis inhibitory protein; NLRC4, nucleotide-binding domain, leucine-rich repeat-containing protein (NLR) family CARD domain-containing protein 4; NLRP3, NACHT, LRR and PYD domains-containing protein 3; ROS, reactive oxygen species; T3SS, type 3 secretion system.
NAIP–NLRC4 and caspase 11 — direct sensors of pathogen-associated molecular patterns
Flagellin, T3SSs and LPS of bacteria are some of the most potent activators of the innate immune system5. Flagellated bacteria that are of clinical importance in the gut and are recognized by the NAIP–NLRC4 inflammasome include Salmonella enterica subsp. enterica serovar Typhimurium, Escherichia coli, Shigella flexneri and Listeria monocytogenes (Fig. 4a). Flagellin subunits of bacteria delivered into the cytoplasm by T3SSs are recognized directly by NAIP sensors, triggering activation of the NLRC4 inflammasome24,25,26,27. Indeed, studies have shown that mice lacking NAIPs or NLRC4 are highly susceptible to infection by S. Typhimurium55,84,85,86,87, S. flexneri26,88,89, L. monocytogenes90, the mouse-specific enteropathogen Citrobacter rodentium91,92 and other extragastrointestinal pathogens that carry flagellin and/or the T3SS63, highlighting the physiological importance of the NAIP–NLRC4 inflammasome in the host defence against bacterial infection. The release of IL-1β as a result of NAIP–NLRC4 inflammasome activation drives IL-1 receptor (IL-1R) signalling to promote neutrophil recruitment to the intestine and prevent colonization of gastrointestinal bacteria such as S. Typhimurium85,93,94.

a | Inflammasomes mediate host protection against Gram-negative bacteria by inducing the secretion of IL-1β and IL-18 and pyroptosis55,59,84,85,91,92,254,255. NLRP6 and NLRP12 negatively regulate inflammation133,134,256,257. NLRP6 responds to Toll-like receptor (TLR)-induced autophagy in goblet cells and mediates secretion of mucus137. RNA-bound DEAH box protein 9 (DHX9) interacts with NLRP9b, inducing the assembly of an inflammasome complex161. The NLRP6–DHX15 complex binds to viral RNA and induces the production of type I and type III interferons165. b | Intestinal macrophages can discriminate pathogens from commensals85. c | Activation of caspase 1, caspase 8 or caspase 11 leads to cell death, which removes and extrudes the infected enterocyte from the epithelium93,94,104. d | The inflammasome can reduce bacterial load by inhibiting bacterial uptake, which limits macrophage movement and stiffness, and can promote the production of reactive oxygen species (ROS)105. Pyroptotic macrophages liberate either whole bacteria or bacteria entrapped within pore-induced intracellular traps103. These entities are phagocytosed by neutrophils84,106. ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD); dsRNA, double-stranded RNA; EMCV, encephalomyocarditis virus; KC, keratinocyte chemoattractant (also known as CXCL1); MAVS, mitochondrial antiviral-signalling protein; NAIP, neuronal apoptosis inhibitory protein; NF-κB, nuclear factor-κB; NLRC4, nucleotide-binding domain, leucine-rich repeat-containing protein (NLR) family CARD domain-containing protein 4; ssRNA, single-stranded RNA.
NAIPs and NLRC4 are expressed in both immune cells and enterocytes (Fig. 2). Infection of intestinal macrophages with S. Typhimurium, but not with the commensal bacteria Bacteroides fragilis, Enterococcus faecalis or Lactobacillus plantarum, triggers activation of the NAIP–NLRC4 inflammasome85, suggesting that innate immune detection of flagellin and the T3SS enables the host immune cell to, in part, discriminate between pathogenic and commensal bacteria (Fig. 4b). In addition, intestinal macrophages, unlike bone marrow-derived macrophages, are relatively unresponsive to activators of the NLRP3 inflammasome owing to rapid proteasomal degradation of NLRP3 and pro-IL-1β95, potentially representing a mechanism to restrain activation of an inflammasome sensor in a cell type that normally exposes and responds to a plethora of signals.
In addition to the NAIP–NLRC4 inflammasome, LPS from Gram-negative bacteria including S. Typhimurium, E. coli and C. rodentium have been found to be recognized by caspase 4, caspase 5 and caspase 11 on the basis of cell culture and mouse studies16. LPS from these bacteria is liberated in the cytoplasm through a host-mediated process requiring interferon-inducible GTPases96. These interferon-inducible effector proteins rupture the membrane of the pathogen-containing vacuole and/or the bacterial cell membrane96. Cytosolic Gram-negative bacteria that naturally escape the pathogen-containing vacuole, such as S. flexneri and Burkholderia thailandensis, transport their LPS into the cytoplasm of macrophages, where the hexa-acylated lipid A portion of the LPS binds and activates caspase 4, caspase 5 and caspase 11 (refs22,97,98). Indeed, the naturally cytosolic bacteria Burkholderia pseudomallei, B. thailandensis and a genetically engineered strain of S. Typhimurium that are incapable of remaining in the pathogen-containing vacuole of macrophages and thus enter the host cytoplasm aberrantly, are rapidly cleared from the host in a caspase 11-dependent manner99,100. Furthermore, outer membrane vesicles carrying LPS are shed by E. coli and most Gram-negative bacteria101. These outer membrane vesicles are phagocytosed by macrophages, leading to LPS-induced activation of caspase 11 and the non-canonical NLRP3 inflammasome even in the absence of bacterial invasion of the host cell102.
Emerging evidence indicates that pyroptosis executed by NAIP–NLRC4 or caspase 11 is an effective host-protective mechanism that either drives bacteria out of infected host cells or removes an infected cell in its entirety from the host63. In the former scenario, the expelled bacteria released from an infected macrophage could be free or trapped within a cellular corpse composed of pyroptotic debris, both of which are cleared by neutrophils84,103. In the latter scenario, enterocytes in the mouse intestine infected with bacteria, such as S. Typhimurium, can be physically extruded in their entirety from the gut epithelium into the lumen for removal93,104 (Fig. 4c). Rearrangement of the cytoskeleton in enterocytes is required for epithelial cell extrusion, and caspase 8 has been implicated in this extrusion process during S. Typhimurium infection owing to the ability of NLRC4 to engage caspase 8-dependent cell death58,93,94,104.
Activation of the NAIP–NLRC4 inflammasome can also prevent further bacterial uptake and enhance intracellular bactericidal activity in macrophages105 (Fig. 4d). Unlike macrophages and enterocytes, neutrophils are resistant to pyroptosis despite expressing all essential components of the inflammasome, including gasdermin D10,106 (Fig. 2). This cell-intrinsic resistance to pyroptosis enables neutrophils to phagocytose extracellular bacteria or bacteria entrapped in pyroptotic debris, secrete an abundant amount of IL-1β to amplify local inflammation and generate ROS to facilitate intracellular pathogen killing84,106 (Fig. 4d).
NLRP3 and pyrin — sensors of physiological aberrations
Bacteria that either evade or fail to activate the NAIP–NLRC4 inflammasome or caspase 11 can be recognized by other inflammasome complexes22,98 (Fig. 4). Of clinical importance in this bacterial category are the Gram-negative bacterium Helicobacter pylori and several toxin-producing foodborne bacteria, such as Staphylococcus aureus. H. pylori, the most common bacterial cause of gastric cancer107, encodes flagella and flagellin A (FlaA) subunits whose structures evade detection by the NAIP–NLRC4 inflammasome108. Instead, H. pylori infection induces ROS production, potassium efflux and lysosomal destabilization in the host cell, ultimately leading to activation of the canonical NLRP3 inflammasome109,110,111,112,113. Further studies have shown that the H. pylori virulence factors cag pathogenicity island and urease B subunit potentiate activation of the NLRP3 inflammasome and secretion of IL-1β109,112,113. Excessive production of IL-1β could be a vital link between H. pylori infection and tumorigenesis in the stomach. Indeed, a study found that a transgenic mouse strain engineered to overexpress human IL-1β in the stomach is prone to the development of gastric cancer, either spontaneously or after colonization with Helicobacter felis, a model organism for H. pylori114. The specific inflammasome involved in the development of gastric cancer is not known; however, an association between polymorphisms in the genes encoding NLRP3 and caspase 1 and patients with gastric cancer has been reported115. Furthermore, H. pylori infection suppresses the expression of the microRNA molecule miR-22 in gastric epithelial cells, which leads to elevated expression of NLRP3 and cellular proliferation116. These studies suggest that activation of the inflammasome by Helicobacter spp. infection contributes to the molecular basis of gastritis and gastric cancer.
Toxins secreted by bacteria can induce a state of intracellular physiological aberration that is sensed by inflammasome sensors. The Gram-positive bacterium C. difficile is a causative agent of diarrhoea associated with the use of antibiotics, toxic megacolon and colonic perforation117. The toxins toxin A (TcdA) and toxin B (TcdB) secreted by C. difficile can inactivate host transforming protein RHOA at the GTPase switch I region; this RHO-inactivating activity is sensed by pyrin, resulting in the assembly of the pyrin inflammasome51,52. The physiological function of pyrin in the gastrointestinal tract remains unexplored, and further studies are required to clarify its role. However, wild-type mice treated with the IL-1R antagonist anakinra are protected from C. difficile-toxin-induced intestinal injury118. This finding indicates that activation of the pyrin inflammasome in response to C. difficile infection might lead to excessive IL-1-mediated inflammation and pathology that is detrimental to the host. Pharmacological inhibitors of microtubules are efficacious in blocking the pyrin inflammasome in cell culture systems54,119,120, but their clinical applications against pyrin-mediated pathology have not been tested in animal models.
The haemolysin of the Enterobacteriaceae member Proteus mirabilis can activate NLRP3 and drive inflammation in the mouse intestine121. Similarly, α-haemolysins, β-haemolysins and γ-haemolysins of the Gram-positive foodborne bacterium S. aureus mediate activation of the NLRP3 inflammasome in mouse macrophages and mice122,123, and the pore-forming cytolysin listeriolysin O of the Gram-positive foodborne bacterium L. monocytogenes potentiates activation of the NLRP3, NAIP–NLRC4 and AIM2 inflammasomes90,124,125,126,127,128. Infection with either L. monocytogenes or S. aureus activates caspase 1 in human macrophages via NLRP3 and NLRP7 (ref.129). NLRP7 (encoded in the human, but not the mouse, genome) is a sensor of bacterial lipopeptides; its activation in macrophages impairs intracellular replication of L. monocytogenes or S. aureus129. Further studies have shown that, in macrophages or embryonic fibroblasts, inflammasome components accumulate on the phagosome and promote phagosome–lysosome fusion and phagosomal acidification27,105,130,131,132. The functional diversity of the inflammasome in both immune and non-immune cells ensures host protection against a range of bacteria with preferential tropisms and virulence strategies.
NLRP6 and NLRP12 — enigmatic sensors
In line with the functional versatility of other inflammasome sensors, NLRP6 and NLRP12 have multiple functions during bacterial infection. The functions of these sensors are enigmatic owing to the lack of expert consensus on their biological functions. Both NLRP6 and NLRP12 can negatively regulate activation of the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways in macrophages infected with L. monocytogenes, S. Typhimurium, E. coli or other non-gastrointestinal bacteria (Fig. 4a), resulting in a state of reduced inflammation that can be readily exploited by bacterial pathogens during infection of a cell or a mouse133,134,135,136. Others have proposed that NLRP6 and NLRP12 mediate activation of caspase 1 in response to infection by C. rodentium and Yersinia pestis, respectively137,138, and that NLRP12 functions in dampening the cell-intrinsic migratory capacity of neutrophils139. A further study suggests that the C57Bl/6J mice that are used widely in research harbour a missense mutation in the Nlrp12 gene that impairs the ability of NLRP12-expressing macrophages to draw neutrophils to the site of inflammation in response to bacterial infection140. These conflicting studies highlight that the biological functions of NLRP6 and NLRP12 should be revisited and should take into account the genetic background of the mouse strains, cell types, bacterial agents and the experimental approaches used.
Detrimental roles of the inflammasome
Overt activation of the inflammasome is detrimental, especially during systemic bacterial dissemination, which might arise from intestinal perforation. Mice lacking caspase 11 or the pyroptosis-inducing effector gasdermin D are remarkably resistant to LPS-induced or E. coli-induced endotoxaemia compared with wild-type mice16,73,97,98,141,142,143,144, suggesting that caspase 11-mediated pyroptosis drives lethality in endotoxaemia. Deletion of the Nlrp3 gene in mice offers protection against polymicrobial endotoxaemia following caecal ligation and puncture145,146. In this model of systemic inflammation, the lack of inflammasome activity impairs the production of pro-inflammatory and endogenous lipid mediators but increases the levels of pro-resolving lipid mediators145. Moreover, LPS-induced endotoxaemia promotes secretion of bile acids from hepatocytes in mice; bile acids are a form of DAMP that either synergize with ATP to activate the NLRP3 inflammasome147 or signal through the membrane receptor G protein-coupled bile acid receptor 1 (GBAR1) to inhibit the NLRP3 inflammasome148.
In other models of systemic inflammation, activation of the NAIP–NLRC4 inflammasome by systemic delivery of cytosolic flagellin to mice results in the production of inflammatory lipid mediators called eicosanoids149. Synthesis of eicosanoids initiates fluid loss into the mouse intestine and peritoneal cavity and ultimately causes rapid host death within 30 min149. The presence of a multidrug-resistant E. coli pathobiont in the gut can also trigger activation of the NAIP–NLRC4 inflammasome and increase susceptibility to sepsis150, whereas certain strains of E. coli, such as strain O21:H+, engage activation of the NAIP–NLRC4 inflammasome to inhibit the body wasting induced by S. Typhimurium infection151. Thus, the inflammasome influences the magnitude of inflammation towards bacterial infection and, in part, determines either a protective or detrimental outcome in the host.
Viruses
Norwalk and Norwalk-like viruses, rotavirus, astroviruses, adenoviruses, caliciviruses and coronaviruses are among the leading causes of acute gastroenteritis worldwide152. Evidence has emerged that inflammasomes respond to some of these gastrointestinal viruses. Earlier studies had demonstrated that the DNA virus adenovirus induces the production of IL-1β in circulation and in hepatocytes and Kupffer cells of mice153. Further functional studies have shown that adenovirus, specifically its virion component, triggers activation of the NLRP3 inflammasome and secretion of IL-1β in human THP-1 macrophages and primary mouse macrophages154. Adenovirus-induced secretion of IL-1β in human monocyte-derived macrophages and peripheral blood mononuclear cells partially requires Toll-like receptor 9 (TLR9)155,156, presumably owing to TLR9-mediated sensing of viral DNA as the virus penetrates the cell membrane and enters the endosome. Injection of an adenovirus vector into mice triggers NLRP3-dependent production of IL-1β and other pro-inflammatory cytokines, highlighting a physiological role for the NLRP3 inflammasome in licensing an innate immune response to adenovirus in vivo154. This finding is also consistent with the observation that IL-1R, which is activated downstream of inflammasomes, incites an acute and deleterious inflammatory signalling cascade in mice infected with adenovirus153. The magnitude of the inflammatory response is reduced in mice lacking IL-1R or in wild-type mice treated with an anti-IL-1 antibody153, suggesting that blockade of IL-1 signalling prevents overt inflammation that might otherwise be damaging to the host.
Similar to adenovirus, enterovirus EV71, a sporadic cause of diarrhoea and a common cause of hand, foot and mouth disease in humans157, is sensed by the NLRP3 inflammasome158,159. Inflammasome-mediated production of IL-18 is crucial for the protection against enterovirus EV71 in mice158,159. Administration of IL-18 to infected mice reduces viral loads and the production of pro-inflammatory cytokines159. The precise viral components of enterovirus EV71 that activate NLRP3 are unknown; it is possible that the single-stranded RNA (ssRNA) viral genome and/or the structural proteins of the capsid might trigger activation of the NLRP3 inflammasome.
The double-stranded RNA (dsRNA) virus rotavirus is one of the most common causes of gastroenteritis in children160. The role of inflammasomes in the host defence against this virus was revealed when the poorly characterized NLR member NLRP9b was found to mediate sensing of rotavirus and drive the production of IL-18 and pyroptosis in organoids generated from the small intestine of mice161. Furthermore, mice lacking NLRP9b are substantially more susceptible to rotavirus infection, harbour a higher viral load and develop more severe intestinal pathology than wild-type mice161. Mouse NLRP9b and its human counterpart, NLRP9, are strongly and specifically expressed in ileal epithelial cells and primary intestinal epithelial cells, respectively161 (Fig. 2). Both mouse NLRP9b and human NLRP9 cooperate with the RNA sensor DEAH box protein 9 (DHX9) to sense dsRNA161 (Fig. 4a), implicating the existence of an RNA-sensing NLRP9 inflammasome complex in the host defence against rotavirus infection. A protective role of inflammasomes was also observed in a previous study showing that administration of bacterial flagellin to mice — causing activation of NLRC4 and the membrane-bound flagellin sensor TLR5, which induced the production of IL-18 and IL-22, respectively — mediates clearance of rotavirus within 24 h (ref.162). Recapitulating the beneficial effects of flagellin, coadministration of IL-18 and IL-22 similarly drives anti-rotavirus immunity in infected mice, supporting the view that inflammasome signalling can mediate protection against rotavirus infection.
Similar to NLRP9, NLRP6 is strongly expressed in the intestinal tract of humans and mice163,164 (Fig. 2). In concert with the RNA sensor DHX15, NLRP6 interacts with viral RNA and mediates the recognition of the ssRNA viruses encephalomyocarditis virus and murine norovirus165 (Fig. 4a). In this case, NLRP6 does not assemble an inflammasome. Instead, the RNA-bound NLRP6–DHX15 complex migrates to the mitochondria, where it interacts with the signalling adaptor mitochondrial antiviral-signalling protein (MAVS) to induce the production of type I and type III interferons and viral clearance165 (Fig. 4a). These studies underscore emerging roles of the inflammasome and associated signalling molecules in antiviral defence of the gastrointestinal tract. The relative contribution of IL-1β and IL-18 secretion versus pyroptosis in driving immunity to viruses remains largely unknown. The current availability of mice lacking gasdermin D will enable exploration of this emerging research area.
Protozoa
Protozoal pathogens cause several neglected tropical diseases and are public health risks largely affecting the developing world166. Inflammasomes are known to recognize major protozoal pathogens, including Leishmania spp., Plasmodium spp. and Toxoplasma spp.167. However, information concerning the biological importance of inflammasomes in the context of gastrointestinal protozoa is limited. The foodborne protozoan Entamoeba histolytica causes amoebiasis, which is characterized by dysentery, amoebic colitis, amoeboma (the formation of fibrotic tissue masses in the intestinal wall) and invasive disease of the brain, lung and liver168. There is evidence to support the idea that E. histolytica infection activates caspase 1 and induces secretion of IL-1β and IL-18 in both human and mouse macrophages169. Pharmacological blockade of potassium efflux inhibits activation of caspase 1, IL-1β-release and pyroptosis in THP-1 macrophages or intestinal epithelial cells that have been infected with E. histolytica170. Further studies indicated that the parasite integrin-binding cysteine protease E. histolytica cysteine protease gene 5 (CP5) mediates activation of the host α5β1 integrin during parasite–macrophage contact, leading to the release of ATP and subsequent activation of the canonical NLRP3 inflammasome171. The importance of the inflammasome in E. histolytica infection is partially inferred by the observation that recombinant IL-1β and IL-18 are cleaved and inactivated by E. histolytica CP5 (refs172,173).
Similar to pathogenic protozoa, the commensal gut protozoan Tritrichomonas musculis in mice also triggers activation of the inflammasome in intestinal epithelial cells in mice174. In this context, T. musculis–induced activation of the inflammasome promotes IL-18 secretion, T helper 1 (TH1) cell and TH17 cell immunity and protection against S. Typhimurium infection174, highlighting a favourable outcome of this host–protozoan engagement. However, intestinal colonization by T. musculis also drives sustained inflammation that elevates the likelihood of developing T cell-driven colitis and sporadic colorectal tumours in mice174. The precise inflammasome complex triggered by T. musculis is unknown; however, it is likely that protozoa-induced activation of the inflammasome in the gastrointestinal tract is a double-edged sword that contributes to both health and disease.
Intestinal inflammation, IBD and cancer
The ability of inflammasomes to respond to danger signals broadens their clinical relevance to diseases other than infectious diseases175,176. Genetic analyses in humans have pinpointed an association between mutations in genes encoding NLRs and intestinal inflammation, autoinflammatory conditions and cancer14,177. The contribution of individual inflammasome sensors and related molecules in the pathogenesis of intestinal inflammation, IBD and cancer is discussed herein.
NLRP3
NLRP3 is a global sensor of PAMPs and DAMPs and is expressed in multiple cell types (Fig. 2), contributing to its links to a plethora of clinical manifestations. Genetic analysis identified an association between six polymorphisms located in a region 4.7 kb downstream of the NLRP3 gene and the development of Crohn’s disease in some, but not all, European populations178,179. Peripheral blood mononuclear cells derived from patients with Crohn’s disease who were homozygous for any one of these six polymorphisms have an impaired ability to express NLRP3 and secrete IL-1β in response to LPS stimulation in vitro178. A further study reported that a heterozygous NLRP3 polymorphism, resulting in a mutation at residue Q705K, is associated with poor survival in patients with advanced colorectal cancer (CRC)180. Global expression analysis of genes encoding cytosolic innate immune sensors revealed that the expression of genes encoding NLRP3 and other inflammasome sensors is substantially reduced in tumour tissues compared with non-tumour-associated tissues in patients with CRC181. Similar observations have been made in mice during colitis-associated CRC182. However, how reduced expression of these genes is initially triggered or executed mechanistically is unknown.
Several groups have found that mice lacking NLRP3, ASC or caspase 1 and administered a combination of the DNA-damaging agent azoxymethane (AOM) and the chemical colitogen dextran sodium sulfate (DSS) are highly sensitive to the development of colitis and colitis-associated CRC183,184,185,186,187,188. DSS-induced activation of the inflammasome in mice also causes a loss of enteric neurons and onset of abnormal gut motility189. The activity of the NLRP3 inflammasome during DSS-induced colitis is controlled by the microRNA molecule miR-223, which binds the miR-223-binding site within the 3′ untranslated region of NLRP3 (ref.190). However, the state of hyporesponsiveness of colonic macrophages to NLRP3 activators is lost during DSS-induced colitis95, which might fuel inflammation. Indeed, studies in mice have also reported a deleterious or non-existent role for NLRP3 or caspase 1 and caspase 11 in colitis and colitis-associated CRC191,192,193,194. The biological basis of these discrepancies is unclear; however, differences in gut microbiota, diet and/or housing conditions might affect the activity of the inflammasome (the differences in the microbiota are specifically discussed later). Indeed, a ketogenic diet or caloric restriction that increases the production of the ketone body β-hydroxybutyrate from the liver contributes to the inhibition of the NLRP3 inflammasome195. Similarly, omega-3 fatty acids signal through the metabolite-sensing receptors G protein-coupled receptor 40 (GPR40; also known as FFAR1) and GPR120 (also known as FFAR4) to inhibit activation of the NLRP3 inflammasome and prevent diet-induced insulin resistance196. By contrast, short-chain fatty acids from dietary fibres bind to the metabolite-sensing receptor GPR43 (also known as FFAR2) on enterocytes, stimulating potassium efflux that drives activation of the NLRP3 inflammasome and protection against colitis197,198. A diet rich in cholesterol or saturated fatty acids also promotes activation of the NLRP3 inflammasome in mice199,200,201,202,203, whereby a cholesterol-rich diet increases inflammation and tumour burden199, but a high-fat diet reduces glucose tolerance and insulin sensitivity200. These studies provide evidence to indicate that dietary components, in association with the gut microbiota, are critical modulators of inflammasome activity and susceptibility to the development of intestinal inflammation, cancer and metabolic syndromes.
Activation of the NLRP3 inflammasome leads to secretion of both IL-1β and IL-18. Although injection of recombinant IL-1β into mice has been shown to mediate protection against DSS-induced colitis204, studies have found variable levels of IL-1β in the colon tissue of inflammasome-deficient mice in response to DSS163,184,185,204,205,206,207,208,209,210,211. These findings would suggest that the role of IL-1β is not always coupled with inflammasome-mediated protection of intestinal inflammation and colitis-associated CRC.
The protective role of the NLRP3 inflammasome in colitis and CRC is attributed in part to the secretion of IL-18 (Fig. 5a). Mice lacking IL-18 are highly susceptible to the development of colitis and CRC183,212,213. Injection of recombinant IL-18 reduces intestinal inflammation and tumour burden in inflammasome-deficient mice that had been administered AOM and DSS; the same treatment also ameliorated colitis in inflammasome-deficient mice exposed to the chemical colitogen oxazolone183,187,204,211,214. IL-18 functionally exerts healing properties184,185,187,214 and triggers natural killer cell-mediated killing of colonic tumour cells that have metastasized to the liver215. Another study challenged the view that IL-18 is beneficial in intestinal inflammation and reported that the IL-18 and IL-18 receptor signalling pathway drives depletion of mucus-secreting goblet cells, such that a loss of the mucous layer augments sensitivity to DSS-induced colitis in mice216. Similarly, a transgenic mouse strain overexpressing IL-18 develops aggressive colitis in response to DSS owing to increased levels of infiltrating macrophages in the colon217. One possibility that might reconcile the diametrically opposing roles of IL-18 is the ability of this cytokine to downregulate the soluble IL-22 receptor called IL-22-binding protein (IL-22BP)218. IL-22BP modulates the bioavailability of IL-22, a cytokine capable of suppressing early intestinal damage but also promoting tumour development over time218 (Fig. 5a).

a | Oncogenic assaults such as azoxymethane (AOM) and dextran sodium sulfate (DSS) cause damage, leading to the release of danger-associated molecular patterns (DAMPs). Bacteria can invade enterocytes and introduce pathogen-associated molecular patterns (PAMPs) into the host cell. DAMPs and PAMPs are sensed by inflammasomes183,184,185,187,188,204. IL-18 promotes downregulation of soluble IL-22-binding protein (IL-22BP), which controls the ability of IL-22 to suppress inflammation or induce tumorigenesis in the gut218. b | Nucleotide-binding domain, leucine-rich repeat-containing protein (NLR) family CARD domain-containing protein 4 (NLRC4) and neuronal apoptosis inhibitory proteins (NAIPs) can block cellular proliferation and tumorigenesis192,226. c | DNA-dependent protein kinase (DNA-PK) induces colorectal tumorigenesis via activation of AKT and the transcription factor MYC205,206. This response is inhibited by absent in melanoma 2 (AIM2)205,206. AIM2 also triggers the production of antimicrobial peptides (AMPs) in intestinal epithelial cells to modulate the gut microbiota207,208. A similar negative regulatory role for NLRC3 has been described229. d | NLRP6 and NLRP12 contribute to the pathogenesis of gastrointestinal infection, acute colitis and colorectal cancer133,134,163,234,235,236,244,248,256,257. Question marks denote unknown mediators. GFR, growth factor receptor; mTOR, mechanistic target of rapamycin; NLRP, NACHT, LRR and PYD domains-containing protein; PI3Ks, phosphoinositide 3-kinases; ROS, reactive oxygen species; STAT3, signal transducer and activator of transcription 3; T3SS, type 3 secretion system; TLR, Toll-like receptor.
Inappropriate activation of the NLRP3 inflammasome can result in damage to the small intestine214. In mice, enteropathy induced by NSAIDs, such as indomethacin, can be attenuated through the use of neutralizing antibodies to IL-1β or inhibitors of the NLRP3 inflammasome, including the ATP scavenger apyrase, the P2X purinoceptor 7 (P2X7) antagonist brilliant blue G and the cytoskeletal inhibitor colchicine214. These studies support the view that unwarranted activation of the NLRP3 inflammasome is a catalyst for small intestinal damage. The availability of more specific inhibitors of the NLRP3 inflammasome, such as MCC950 and CY-09 (refs219,220,221), enables clinical trials to explore new options for the treatment of human diseases182.
NLRC4
Studies in humans have revealed that gain-of-function mutations in the gene encoding NLRC4 are associated with enterocolitis and autoinflammation222,223,224. For example, a substitution mutation within the HD1 domain of the NLRC4 protein is linked to recurrent enterocolitis and autoinflammation in several members of the same family222. A newborn baby from this family and carrying this mutation died 23 days after birth owing to severe gastrointestinal complications, fever and systemic inflammation222. A further study identified that a heterozygous de novo substitution mutation in the nucleotide-binding domain of NLRC4, potentially causing destabilization and autoactivation of the protein, was linked to macrophage activation syndrome, gastrointestinal pathology, splenomegaly and systemic inflammation223. Monocytes or macrophages carrying the aforementioned disease-associated mutations undergo spontaneous activation of the inflammasome and pyroptosis and secrete increased levels of IL-1β and IL-18 (refs222,223,224), suggesting that unchecked inflammasome activation contributes to the molecular basis of these autoinflammatory conditions. Of clinical importance is that the aforementioned patient with the substitution mutation in NLRC4 responded to treatment with the recombinant IL-1R antagonist anakinra223. A newly identified patient with the heterozygous de novo mutation in NLRC4, who also experienced enterocolitis and skin rash, responded to a combined blockade therapy targeting IL-1β and IL-18 (ref.225). These studies exemplify the success of experimental targeted therapies in the treatment of inflammasome-mediated disorders.
In animal studies, deletion of the genes encoding NLRC4 or NAIPs heightens the susceptibility of mice to the development of colitis and colitis-associated CRC in response to AOM and DSS192,226. Mechanistically, NLRC4 inhibits cellular proliferation and promotes apoptosis via an undefined mechanism192, whereas NAIPs inhibit proliferation in enterocytes by blocking phosphorylation and activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) independently of NLRC4 (ref.226) (Fig. 5b). A possible scenario is that either spontaneous activation or loss of function associated with the NAIP or NLRC4 signalling axis could upset the equilibrium of the gut ecosystem and drive intestinal inflammation and tumorigenesis. Immunization of mice with tumour cell lines expressing flagellin — to trigger activation of the NAIP–NLRC4 and TLR5 pathways – induces clearance of tumour cells by innate immune cells and tumour-specific CD4+ and CD8+ T cells227. Thus, modulation of the NAIP–NLRC4 and/or the IL-1–IL-18 pathways might provide a promising avenue for cancer immunotherapy.
AIM2 and other signalling molecules
The inflammasome-associated sensor AIM2 also contributes to the pathogenesis of colitis and CRC205,206,207,208. The levels of AIM2 expression in tumour tissues often predict survival in patients with CRC228. Patients whose tumour cells lack AIM2 expression are threefold more likely to die within 5 years of diagnosis than patients whose tumour cells retain some level of expression228. Reflecting the scenario in humans, the expression of the Aim2 gene is markedly reduced in inflamed and tumour-associated tissues of mice, and mice lacking AIM2 are hypersusceptible to both colitis-associated and spontaneous colorectal tumorigenesis205,206. AIM2 has functions independent of the inflammasome that drive protection against both colitis-associated and spontaneous CRC, the latter of which arises from a mutation in the mouse homologue of the human APC gene or from aberrant β-catenin activation205,206. AIM2 suppresses the ability of the DNA-binding kinase DNA-dependent protein kinase (DNA-PK) to activate RAC serine/threonine-protein kinase (AKT) and transcription factor MYC proto-oncogene protein, ultimately preventing excessive proliferation of stem cells in the colon205,206 (Fig. 5c). A similar negative regulatory effect of the orphan receptor NLRC3 on the AKT–mechanistic target of rapamycin (mTOR) pathways activated during colitis-associated CRC or spontaneous CRC has been described229 (Fig. 5c). Pharmacological inhibitors of the AKT–mTOR pathway are efficacious against the development of colitis-associated or spontaneous CRC in mice lacking either AIM2 or NLRC3 (refs205,206,229). The precise identity or source of the ligand(s) activating these sensors in the colon is not known. It is possible that self-DNA liberated from the damaged intestinal barrier or DNA derived from the gut microbiota might activate AIM2 (ref.230) (Fig. 5c). Consistent with this hypothesis, AIM2 can localize to DNA in the nucleus of mouse intestinal epithelial cells and bone marrow cells following dsDNA breaks caused by ionizing radiation or chemotherapeutic agents231. The generation of conditional mouse models that enable deletion or re-expression of AIM2 and other related sensors in all cells or in certain cell types once tumours are established would provide additional insights into the molecular and cellular mechanisms of the operation of these sensors. These studies would also aid further assessment of the translational potential of targeting these sensors in cancer immunotherapy.
Indeed, therapeutic inhibition of the AIM2 inflammasome might, in some cases, reduce the adverse gastrointestinal effects associated with chemotherapy232. The chemotherapeutic and cytotoxic agent irinotecan (CPT-11), a topoisomerase I inhibitor used in the treatment of a variety of cancers, elicits the release of self-DNA, subsequent activation of AIM2 and secretion of IL-1β and IL-18 in enterocytes, leading to intestinal mucositis, late-onset diarrhoea and microbiota disturbance in mice232,233. Deletion of Aim2 or pharmacological inhibition of the AIM2 inflammasome in mice reduces the incidence of irinotecan-induced diarrhoea without dampening the anticancer efficacy of irinotecan232.
Further studies have shown that NLRP6 (refs137,163,234,235,236,237), NLRP1b (ref.204) and caspase 11 (refs209,210,211) are important innate immune rheostats of the gut, providing additional targets for experimental therapies. NLRP6 controls intestinal inflammation and tumorigenesis via multiple mechanisms (discussed further later), including modulation of IL-18 secretion163,234,235,237, curtailing the colonization of pro-colitogenic gut bacteria163 and promoting secretion of mucus in goblet cells137,236 (Fig. 5d). NLRP6 expression in enterocytes and inflammatory monocytes has been reported to mediate protection against DSS-induced colitis163,235,237. NLRP1b regulates activation of the inflammasome and secretion of IL-1β and IL-18 specifically in non-haematopoietic cells204, whereas caspase 11 triggers IL-1β–IL-18-dependent and IL-1β–IL-18-independent functions209,210,211, with both sensors mediating protection against DSS-induced colitis. A side-by-side comparison revealed that, under isobiotic conditions with normalized gut microbiota across genotypes, mice lacking caspase 1, but not mice lacking caspase 11, were hypersensitive to DSS-induced colitis188. Evidence from this study would seem to favour a role for canonical inflammasome complexes over the non-canonical inflammasome in this mouse model of intestinal inflammation. Although these studies point towards a protective role of inflammasomes in the context of intestinal inflammation and tumorigenesis, a study found that deletion of the genes encoding the mucous layer components core 1-derived and core 3-derived intestinal O-glycans in mice causes colonic mucous barrier breach, spontaneous colitis and the development of colorectal tumours, and further deletion of the genes encoding caspase 1 and caspase 11 reduces the propensity of colitis and colorectal tumorigenesis239. These findings illustrate that in the presence of pre-existing immunological dysregulation triggering overt inflammation, blockade of inflammasome signalling might inhibit an inflammatory circuitry that normally fuels disease progression. Development of safe and efficacious modulators of AIM2, NLRP1 and NLRP6 and investigations into the bilateral interaction between inflammasomes and the gut ecosystem would create new avenues for the treatment of intestinal inflammation and cancer.
Inflammasome–gut microbiota axis
The gut microbiota has an important role in the development and progression of a range of clinical manifestations, especially in association with the gastrointestinal tract. Evidence supporting this view includes the ability of postoperative diversion of the faecal stream to prevent recurrent IBD in humans and the use of broad spectrum antibiotics in the amelioration of chemical-induced colitis in mice177. The latest developments in the field have suggested that the inflammasome regulates gut microbiota composition, which would imply that the inflammasome–gut microbiota axis might be a centrepiece in the development of intestinal inflammation, cancer and metabolic syndromes177,238,240,,240,241.
Colitis and cancer
The concept of inflammasomes controlling the biogeography of intestinal bacteria was spearheaded by studies showing that mice lacking NLRP6 harbour a pro-colitogenic gut microbiota that renders these mice more susceptible to DSS-induced colitis than wild-type mice163. This pro-colitogenic signature is defined by an increased relative abundance of Prevotellaceae and the phylum Candidatus Saccharibacteria, reduced relative abundance of Lactobacillus spp. and the transmissible nature of the pro-colitogenic microbiota through means of cross-fostering and cohousing techniques163,242. Furthermore, NLRP6 dampens spontaneous colitis in mice lacking the anti-inflammatory cytokine IL-10 by reducing levels of the mucus-degrading bacterium Akkermansia muciniphila243. Carriage of a transmissible pro-colitogenic gut microbiota has also been described in mice lacking the inflammasome components AIM2, NLRP3, ASC, caspase 1 or caspase 11 or mice lacking the related NLR member NLRP12 (refs163,186,205,207,208,209,244,245). Similar to the findings with NLRP6, reciprocal exchange of the gut microbiota between wild-type mice and mutant mice lacking AIM2 or NLRP12 elevates the susceptibility of wild-type mice, but reduces the susceptibility of mutant mice, to colitis-associated tumorigenesis205,244, suggesting that the colitogenic potential and transmissibility of the gut microbiota influence disease outcome.
Other studies, however, challenged the idea that NLRP6 controls the biogeography of the gut microbiota246,247. Phylogenetic analyses of wild-type mice and littermate-controlled mice lacking either NLRP6 or ASC failed to reveal any difference in the composition of the gut microbiota246. Lifetime separation of wild-type mice and mice lacking NLRP6 also does not seem to promote alterations of their gut microbiota profile or differential susceptibility to DSS-induced colitis246, casting doubt over the previously suggested role of NLRP6 and ASC in driving dysbiosis163. This study also argued that any difference associated with the gut microbiota might be entirely dependent on the facilities used to house the animals246. Thus, food sources, gut microbiota and sterilization techniques used by different mouse vivarium across the globe can influence the activation status of inflammasomes. Future experimental approaches should be more tightly controlled to examine the role of the gut microbiota in any disease context, including the use of littermate controls and careful evaluation of cage-to-cage and room-to-room effects and the overall sterility of the facility.
Despite the uncertainty surrounding the nexus between NLRP6 and the gut microbiota, the mechanistic functions of NLRP6 have been further characterized against the backdrop of intestinal inflammation, cancer and metabolic syndromes. NLRP6 senses the microbial metabolite taurine and drives the secretion of IL-18 and antimicrobial peptides to maintain a healthy gut microbiota and to promote antitumour immunity in mice163,248 (Fig. 5d). By contrast, the microbial metabolites histamine and spermine suppress NLRP6-dependent IL-18 secretion in mice, indicating that components of the gut microbiota can reciprocate and fine-tune the activities of NLRP6 (ref.248) (Fig. 5d). The NLRP6–gut microbiota axis modulates the expression of the chemokine CC-chemokine ligand 5 (CCL5) and the pro-inflammatory cytokines IL-6 and TNF and, therefore, the overall inflammatory state of the intestinal tract163,237,242. In the absence of NLRP6, the dysbiotic gut microbiota developed as a result induces the expression of CCL5 in enterocytes; CCL5, in turn, mediates the secretion of IL-6 and drives proliferation of epithelial cells and tumour formation in mice163,242. Deletion of the gene encoding CCL5 in mice prevents colitis and colitis-associated tumorigenesis despite these mice carrying a dysbiotic gut microbiota163,242, placing dysbiosis upstream of CCL5-mediated immune dysregulation. Furthermore, inflammatory monocytes residing in the lamina propria promote NLRP6-dependent and IL-18-dependent secretion of TNF and feature a protective role in DSS-induced colitis and colitis-associated tumorigenesis237.
The relationship between NLRP6 and intestinal microorganisms is further exemplified by the ability of NLRP6 to respond to Toll-like receptor (TLR)-induced autophagy in goblet cells137. This NLRP6 response mediates secretion of mucus via granule exocytosis and prevents colonization of the mouse gut by C. rodentium137. Further studies have shown that sentinel goblet cells located at the colonic crypt entrance execute NLRP6-dependent secretion of mucin 2 (MUC2) to expel intruding bacteria found in the inner mucous layer236 (Fig. 5d). In the small intestine of mice, a loss of endogenous NLRP6 expression owing to stress is linked to the development of intestinal pathology and changes to the relative abundance of Bacteroidetes, Clostridiales, Firmicutes, Lachnospiraceae, Lactobacillaceae, Ruminococcaceae and Streptococcaceae249. The peroxisome proliferator-activated receptor-γ (PPARγ) agonist rosiglitazone, among other functions, elevates the expression of NLRP6 in the small intestine and prevents stress-related intestinal pathology in mice164,249. Whether this therapeutic effect is directly or specifically dependent on the ability of rosiglitazone to modulate the expression of NLRP6 remains unclear.
An additional function of NLRP6 is its ability to suppress activation of the canonical NF-κB and MAPK pathways in response to infection by the gastrointestinal pathogens L. monocytogenes, S. Typhimurium and E. coli133 (Fig. 5d). This immunomodulatory pathway operates in the presence or absence of an altered gut microbiota composition133, highlighting that certain biological functions of NLRP6 can clearly be uncoupled from the gut microbiota and that previous studies identifying a connection with the gut microbiota should be interpreted with care and revisited.
Nonalcoholic steatohepatitis, obesity and diet
The influence of the inflammasome–gut microbiota axis extends to the development of nonalcoholic steatohepatitis (NASH) and obesity. Mice lacking NLRP3, NLRP6, ASC, caspase 1 or IL-18 have a gut microbiota profile that is associated with exacerbated hepatic steatosis and inflammation and NASH progression, phenotypes of which are transmissible to wild-type mice via gut microbiota transfer245. A further study found that mice lacking ASC are prone to hepatosteatosis and obesity induction when fed a high-fat diet245. These abnormalities are abrogated following treatment with the antibiotics ciprofloxacin and metronidazole245, suggesting a connection with the gut microbiota in the context of inflammasome signalling and obesity.
A role for inflammasome sensors and diet in regulating microbial diversity and disease is further demonstrated in mice lacking NLRP3, which when fed a high-fat and high-carbohydrate diet develop microbial dysbiosis compared with mice lacking NLRP3 fed with standard chow; the degree of dysbiosis between the two groups seems to be more substantial than the changes observed between wild-type mice that had been fed a high-fat, high-carbohydrate diet and wild-type mice that had been fed with chow250. Compared with wild-type mice on a high-fat and high-carbohydrate diet, the dysbiotic change in mice lacking NLRP3 on the same diet was associated with elevated levels of triglyceride in the liver and faeces and increased intestinal permeability, liver injury and inflammation of the adipose tissue250. Moreover, a diet composed of high fat and high cholesterol induces a reduction in the relative abundance of Prevotella spp., which is associated with reduced expression of pro-IL-1β in the intestine of mice carrying a mutation that predisposes them to spontaneous inflammatory bone disease251.
Gut–brain axis
The gut microbiota also operates at the interface between inflammasome signalling and the gut–brain axis. An example of this relationship is the observation that inhibition of the inflammasome-mediated IL-1β signalling pathway in the small intestine in mice by S. Typhimurium-encoded E3 ubiquitin-protein ligase SlrP apparently prevents infection-induced anorexia owing to a blockade of signal transduction to the hypothalamus via the vagus nerve252. Furthermore, it was reported that mice lacking caspase 1 or treatment of wild-type mice with a semisynthetic antibiotic minocycline, which potentially functions to inhibit caspase 1 and modulates the gut microbiota, are protected from developing depressive-like and anxiety-like behaviours, even when experiencing chronic stress253. The increased resistance to stress in these mice is associated with an increase in the relative abundance of species of Akkermansia, Blautia and Lachnospiraceae and a reduced abundance of Allobaculum spp., Bifidobacterium spp., Turicibacter spp. and Clostridium spp.253. These studies highlight a complex and potentially emerging link between inflammasomes, microorganisms and the gut–brain axis.
Conclusions
Inflammasome signalling is a central pillar of innate immunity that triggers inflammation and cell death. The antimicrobial function of the inflammasome is largely attributed to IL-1β and IL-18 and pyroptosis: IL-1β and IL-18 activate an inflammatory circuitry, whereas pyroptosis expels the entire infected cell from the body or liberates concealed pathogen from the infected host cell to facilitate pathogen elimination by other antimicrobial mechanisms. These diverse and multifaceted immune mechanisms are operated in both intestinal epithelial cells and immune cells. Although microorganisms across all kingdoms of life are detected by the inflammasome pathways, the mechanisms regulating activation of the inflammasome in response to many pathogens remain unclear, especially in the context of gastrointestinal viruses and protozoa.
Uncontrolled inflammation and cell death underpin the molecular basis of immunopathology and fuel the progression of chronic inflammatory conditions and cancer. Inflammasome activities largely prevent the development of colitis and gastrointestinal cancer owing to their functions in tumour immunosurveillance, mucus production, cell-renewal, suppression of proliferation of intestinal epithelial stem cells and antitumorigenic responses. The specific PAMPs and DAMPs that trigger activation of the inflammasome in the gut in non-infectious diseases remain largely unknown. What is clear is that the development of intestinal inflammation and cancer is intimately linked to dysregulated inflammasome signalling and/or dysbiosis. This connection is in part owing to the ability of the inflammasome and the gut microbiota to regulate one another. Although advanced sequencing technologies have been informative in providing a snapshot of the global changes of the gut microbiota in the presence of inflammasome dysregulation, the causative microbial species or population responsible for a specific clinical manifestation has not been identified. Furthermore, spatial and temporal changes to the gut microbiota over the length of the intestine and over time with respect to inflammasome dysregulation have not been investigated. Nevertheless, further studies are needed in light of new evidence suggesting that the role of the inflammasome in regulating the composition of the gut microbiota is not always apparent and is, in fact, heavily influenced by a multitude of genetic, environmental and experimental factors associated with animal models246,247.
Given that experimental therapies have yielded success in the treatment of inflammasome-mediated disorders53,182,219,221,223,225, strategies targeting distinct aspects of the inflammasome pathways will become an emerging area of research and development. Discovery of novel small-molecule compounds for use in pharmacological activation or inhibition of the inflammasome and in the prevention and treatment of disease in humans will probably take priority in the next decade of inflammasome research. The deep and complex relationship between inflammasomes, pathogens and the microbiota provides an exciting platform for basic and clinical research with which to understand health and disease.
References
Belkaid, Y. & Hand, T. W. Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Man, S. M. & Kanneganti, T. D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 16, 7–21 (2016).
Rathinam, V. A. & Fitzgerald, K. A. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165, 792–800 (2016).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Conos, S. A., Lawlor, K. E., Vaux, D. L., Vince, J. E. & Lindqvist, L. M. Cell death is not essential for caspase-1-mediated interleukin-1beta activation and secretion. Cell Death Differ. 23, 1827–1838 (2016).
Evavold, C. L. et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44 (2018).
Heilig, R. et al. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 48, 584–592 (2018).
Szabo, G. & Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 12, 387–400 (2015).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Levy, M., Kolodziejczyk, A. A., Thaiss, C. A. & Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 17, 219–232 (2017).
Man, S. M. & Kanneganti, T. D. Regulation of inflammasome activation. Immunol. Rev. 265, 6–21 (2015).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 (2013).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). This study describes the existence of a non-canonical inflammasome.
Liston, A. & Masters, S. L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17, 208–214 (2017).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009). References 18–21 show that AIM2 is a cytosolic inflammasome sensor of double-stranded DNA.
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004). This study provides the first genetic evidence for the existence of an NLRC4 inflammasome (previously known as the ICE-protease activating factor (IPAF) inflammasome).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006).
Miao, E. A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575 (2006).
Miao, E. A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl Acad. Sci. USA 107, 3076–3080 (2010).
Amer, A. et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223 (2006).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Kortmann, J., Brubaker, S. W. & Monack, D. M. Cutting edge: inflammasome activation in primary human macrophages is dependent on flagellin. J. Immunol. 195, 815–819 (2015).
Reyes Ruiz, V. M. et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl Acad. Sci. USA 114, 13242–13247 (2017).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Rayamajhi, M., Zak, D. E., Chavarria-Smith, J., Vance, R. E. & Miao, E. A. Cutting edge: mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 191, 3986–3989 (2013).
Rauch, I. et al. NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J. Exp. Med. 213, 657–665 (2016).
Zhao, Y. et al. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J. Exp. Med. 213, 647–656 (2016).
Halff, E. F. et al. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N− and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 (2012).
Hu, Z. et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 (2015).
D’Osualdo, A. et al. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PloS one 6, e27396 (2011).
Frew, B. C., Joag, V. R. & Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659 (2012).
Finger, J. N. et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 287, 25030–25037 (2012).
Chavarria-Smith, J., Mitchell, P. S., Ho, A. M., Daugherty, M. D. & Vance, R. E. Functional and evolutionary analyses identify proteolysis as a general mechanism for NLRP1 inflammasome activation. PLoS Pathog. 12, e1006052 (2016).
Levinsohn, J. L. et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638 (2012).
Perregaux, D. & Gabel, C. A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 269, 15195–15203 (1994).
Muñoz-Planillo, R. et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Petrilli, V. et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 (2007).
Hornung, V. et al. Silica crystals and aluminum salts mediate NALP-3 inflammasome activation via phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Iyer, S. S. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
Groß, C. J. et al. K+ efflux-independent NLRP3 Inflammasome activation by small molecules targeting mitochondria. Immunity 45, 761–773 (2016).
Murakami, T. et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl Acad. Sci. USA 109, 11282–11287 (2012).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014).
Gavrilin, M. A. et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J. Immunol. 188, 3469–3477 (2012).
Masters, S. L. et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl Med. 8, 332ra345 (2016).
Park, Y. H., Wood, G., Kastner, D. L. & Chae, J. J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 17, 914–921 (2016).
Broz, P. et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755 (2010).
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E. & Monack, D. M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Fernandes-Alnemri, T. et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 (2007).
Man, S. M. et al. Salmonella infection induces recruitment of caspase-8 to the inflammasome to modulate IL-1beta production. J. Immunol. 191, 5239–5246 (2013).
Franklin, B. S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737 (2014).
Lu, A. et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014).
Cai, X. et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222 (2014).
Baroja-Mazo, A. et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 15, 738–748 (2014).
Man, S. M., Karki, R. & Kanneganti, T. D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75 (2017).
Poyet, J. L. et al. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313 (2001).
Van Opdenbosch, N. et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 5, 3209 (2014).
Guey, B., Bodnar, M., Manie, S. N., Tardivel, A. & Petrilli, V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc. Natl Acad. Sci. USA 111, 17254–17259 (2014).
Kostura, M. J. et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc. Natl Acad. Sci. USA 86, 5227–5231 (1989).
Black, R. A., Kronheim, S. R. & Sleath, P. R. Activation of interleukin-1 beta by a co-induced protease. FEBS Lett. 247, 386–390 (1989).
Thornberry, N. A. et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774 (1992).
Ghayur, T. et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623 (1997).
Vande Walle, L. & Lamkanfi, M. Pyroptosis. Curr. Biol. 26, R568–R572 (2016).
Aglietti, R. A. & Dueber, E. C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 38, 261–271 (2017).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). References 73 and 74 demonstrate that gasdermin D is a substrate of inflammatory caspases that upon cleavage converts to a pro-pyroptotic executor mediating cell lysis.
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 25, 1285–1298 (2015).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Aglietti, R. A. et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl Acad. Sci. USA 113, 7858–7863 (2016).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Sborgi, L. et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 (2016).
Chen, X. et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020 (2016).
Joosten, L. A., Netea, M. G. & Dinarello, C. A. Interleukin-1beta in innate inflammation, autophagy and immunity. Semin. Immunol. 25, 416–424 (2013).
Dinarello, C. A., Novick, D., Kim, S. & Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 4, 289 (2013).
Navaneethan, U. & Giannella, R. A. Mechanisms of infectious diarrhea. Nat. Clin. Pract. Gastroenterol. Hepatol. 5, 637–647 (2008).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Franchi, L. et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 13, 449–456 (2012).
Kupz, A. et al. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nat. Immunol. 13, 162–169 (2012).
Man S. M. et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 111, 7403–7408 (2014).
Suzuki, S. et al. Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcdelta. PLoS Pathog. 10, e1003926 (2014).
Suzuki, S. et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl Acad. Sci. USA 111, E4254–E4263 (2014).
Wu, J., Fernandes-Alnemri, T. & Alnemri, E. S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702 (2010).
Liu, Z. et al. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287, 16955–16964 (2012).
Nordlander, S., Pott, J. & Maloy, K. J. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 7, 775–785 (2014).
Sellin, M. E. et al. Epithelium-Intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 16, 237–248 (2014).
Rauch, I. et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity 46, 649–659 (2017).
Filardy, A. A., He, J., Bennink, J., Yewdell, J. & Kelsall, B. L. Posttranscriptional control of NLRP3 inflammasome activation in colonic macrophages. Mucosal Immunol. 9, 850–858 (2016).
Ngo, C. C. & Man, S. M. Mechanisms and functions of guanylate-binding proteins and related interferon-inducible GTPases: roles in intracellular lysis of pathogens. Cell. Microbiol. 19, e12791 (2017).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Aachoui, Y. et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978 (2013).
Aachoui, Y. et al. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe 18, 320–332 (2015).
Kaparakis-Liaskos, M. & Ferrero, R. L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 15, 375–387 (2015).
Vanaja, S. K. et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and Caspase-11 activation. Cell 165, 1106–1119 (2016).
Jorgensen, I., Zhang, Y., Krantz, B. A. & Miao, E. A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128 (2016).
Knodler, L. A. et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16, 249–256 (2014). References 93, 94 and 104 highlight an expulsion mechanism induced by the inflammasome to remove an entire infected host cell from the intestinal epithelium.
Man, S. M. et al. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc. Natl Acad. Sci. USA 111, 17588–17593 (2014).
Chen, K. W. et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8, 570–582 (2014).
O’Connor, A., O’Morain, C. A. & Ford, A. C. Population screening and treatment of Helicobacter pylori infection. Nat. Rev. Gastroenterol. Hepatol. 14, 230–240 (2017).
Matusiak, M. et al. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc. Natl Acad. Sci. USA 112, 1541–1546 (2015).
Koch, K. N. et al. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Invest. 125, 3297–3302 (2015).
Perez-Figueroa, E. et al. Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immun. 22, 103–112 (2016).
Li, X. et al. Helicobacter pylori induces IL-1beta and IL-18 production in human monocytic cell line through activation of NLRP3 inflammasome via ROS signaling pathway. Pathog. Dis. 73, ftu024 (2015).
Semper, R. P. et al. Helicobacter pylori-induced IL-1beta secretion in innate immune cells is regulated by the NLRP3 inflammasome and requires the cag pathogenicity island. J. Immunol. 193, 3566–3576 (2014).
Kim, D. J., Park, J. H., Franchi, L., Backert, S. & Nunez, G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori infected dendritic cells. Eur. J. Immunol. 43, 2650–2658 (2013).
Tu, S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
Castano-Rodriguez, N., Kaakoush, N. O., Goh, K. L., Fock, K. M. & Mitchell, H. M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLOS ONE 9, e98899 (2014).
Li, S. et al. MiR-22 sustains NLRP3 expression and attenuates H. pylori-induced gastric carcinogenesis. Oncogene 37, 884–896 (2018).
Martin, J. S., Monaghan, T. M. & Wilcox, M. H. Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 13, 206–216 (2016).
Ng, J. et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139, 542–552 (2010).
Van Gorp, H. et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, 14384–14389 (2016).
Gao, W., Yang, J., Liu, W., Wang, Y. & Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, E4857–E4866 (2016).
Seo, S. U. et al. Distinct commensals induce interleukin-1beta via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 42, 744–755 (2015).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Munoz-Planillo, R., Franchi, L., Miller, L. S. & Nunez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 183, 3942–3948 (2009).
Kim, S. et al. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551 (2010).
Sauer, J. D. et al. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419 (2010).
Warren, S. E. et al. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185, 818–821 (2010).
Tsuchiya, K. et al. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185, 1186–1195 (2010).
Meixenberger, K. et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 184, 922–930 (2010).
Khare, S. et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36, 464–476 (2012).
Akhter, A. et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37, 35–47 (2012).
Thurston, T. L. et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nat. Commun. 7, 13292 (2016).
Sokolovska, A. et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat. Immunol. 14, 543–553 (2013).
Anand, P. K. et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393 (2012).
Zaki, M. H., Man, S. M., Vogel, P., Lamkanfi, M. & Kanneganti, T. D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl Acad. Sci. USA 111, 385–390 (2014).
Allen, I. C. et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PLOS ONE 8, e60842 (2013).
Silveira, T. N. et al. NLRP12 negatively regulates proinflammatory cytokine production and host defense against Brucella abortus. Eur. J. Immunol. 47, 51–59 (2017).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Vladimer, G. I. et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107 (2012).
Zamoshnikova, A. et al. NLRP12 is a neutrophil-specific, negative regulator of in vitro cell migration but does not modulate LPS- or infection-induced NF-kappaB or ERK signalling. Immunobiology 221, 341–346 (2016).
Ulland, T. K. et al. Nlrp12 mutation causes C57BL/6J strain-specific defect in neutrophil recruitment. Nat. Commun. 7, 13180 (2016).
Sarkar, A. et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am. J. Respir. Crit. Care Med. 174, 1003–1010 (2006).
Wang, S. et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509 (1998).
Li, P. et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80, 401–411 (1995).
Man, S. M. et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci. Rep. 7, 45126 (2017).
Lee, S. et al. NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am. J. Respir. Crit. Care Med. 196, 713–726 (2017).
Jin, L., Batra, S. & Jeyaseelan, S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J. Immunol. 198, 1253–1262 (2017).
Hao, H. et al. Farnesoid X receptor regulation of the NLRP3 Inflammasome underlies cholestasis-associated sepsis. Cell. Metab. 25, 856–867 (2017).
Guo, C. et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 802–816 (2016).
von Moltke, J. et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111 (2012).
Ayres, J. S., Trinidad, N. J. & Vance, R. E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 18, 799–806 (2012).
Schieber, A. M. et al. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science 350, 558–563 (2015).
Christensen, M. L. Human viral gastroenteritis. Clin. Microbiol. Rev. 2, 51–89 (1989).
Shayakhmetov, D. M., Li, Z. Y., Ni, S. & Lieber, A. Interference with the IL-1-signaling pathway improves the toxicity profile of systemically applied adenovirus vectors. J. Immunol. 174, 7310–7319 (2005).
Muruve, D. A. et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008).
Barlan, A. U., Griffin, T. M., McGuire, K. A. & Wiethoff, C. M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 85, 146–155 (2011).
Teigler, J. E., Kagan, J. C. & Barouch, D. H. Late endosomal trafficking of alternative serotype adenovirus vaccine vectors augments antiviral innate immunity. J. Virol. 88, 10354–10363 (2014).
Solomon, T. et al. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 10, 778–790 (2010).
Wang, H. et al. Reciprocal regulation between Enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 12, 42–48 (2015).
Li, Z. et al. Interleukin-18 protects mice from Enterovirus 71 infection. Cytokine 96, 132–137 (2017).
Operario, D. J. et al. Etiology of severe acute watery diarrhea in children in the global Rotavirus Surveillance Network using quantitative polymerase chain reaction. J. Infect. Dis. 216, 220–227 (2017).
Zhu, S. et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017). This study reports the existence of an NLRP9b inflammasome complex, which is expressed specifically in mouse intestinal cells and mediates cytosolic recognition of rotavirus.
Zhang, B. et al. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science 346, 861–865 (2014).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Kempster, S. L. et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G253–G263 (2011).
Wang, P. et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830 (2015).
Fletcher, S. M., Stark, D., Harkness, J. & Ellis, J. Enteric protozoa in the developed world: a public health perspective. Clin. Microbiol. Rev. 25, 420–449 (2012).
Zamboni, D. S. & Lima-Junior, D. S. Inflammasomes in host response to protozoan parasites. Immunol. Rev. 265, 156–171 (2015).
Tanyuksel, M. & Petri, W. A. Jr. Laboratory diagnosis of amebiasis. Clin. Microbiol. Rev. 16, 713–729 (2003).
Mortimer, L., Moreau, F., Cornick, S. & Chadee, K. Gal-lectin-dependent contact activates the inflammasome by invasive Entamoeba histolytica. Mucosal Immunol. 7, 829–841 (2014).
Marie, C., Verkerke, H. P., Theodorescu, D. & Petri, W. A. A whole-genome RNAi screen uncovers a novel role for human potassium channels in cell killing by the parasite Entamoeba histolytica. Sci. Rep. 5, 13613 (2015).
Mortimer, L., Moreau, F., Cornick, S. & Chadee, K. The NLRP3 Inflammasome is a pathogen sensor for invasive Entamoeba histolytica via activation of alpha5beta1 integrin at the macrophage-amebae intercellular junction. PLoS Pathog. 11, e1004887 (2015).
Zhang, Z. et al. Entamoeba histolytica cysteine proteinases with interleukin-1 beta converting enzyme (ICE) activity cause intestinal inflammation and tissue damage in amoebiasis. Mol. Microbiol. 37, 542–548 (2000).
Que, X. et al. A surface amebic cysteine proteinase inactivates interleukin-18. Infect. Immun. 71, 1274–1280 (2003).
Chudnovskiy, A. et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–456 (2016).
Mathur, A., Hayward, J. A. & Man, S. M. Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 103, 233–257 (2018).
Zmora, N., Levy, M., Pevsner-Fishcer, M. & Elinav, E. Inflammasomes and intestinal inflammation. Mucosal Immunol. 10, 865–883 (2017).
Man, S. M., Kaakoush, N. O. & Mitchell, H. M. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 8, 152–168 (2011).
Villani, A. C. et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Lewis, G. J. et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm. Bowel Dis. 17, 1387–1391 (2011).
Ungerback, J. et al. Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 33, 2126–2134 (2012).
Liu, R. et al. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6, 33456–33469 (2015).
Karki, R., Man, S. M. & Kanneganti, T. D. Inflammasomes and cancer. Cancer Immunol. Res. 5, 94–99 (2017).
Zaki, M. H., Vogel, P., Body-Malapel, M., Lamkanfi, M. & Kanneganti, T. D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920 (2010).
Allen, I. C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Zaki, M. H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Hirota, S. A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Blazejewski, A. J. et al. Microbiota normalization reveals that canonical caspase-1 activation exacerbates chemically induced intestinal inflammation. Cell Rep. 19, 2319–2330 (2017).
Gulbransen, B. D. et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 18, 600–604 (2012).
Neudecker, V. et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 214, 1737–1752 (2017).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Siegmund, B., Lehr, H. A., Fantuzzi, G. & Dinarello, C. A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl Acad. Sci. USA 98, 13249–13254 (2001).
Bauer, C., Duewell, P., Lehr, H. A., Endres, S. & Schnurr, M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig. Dis. 30 (Suppl. 1), 82–90 (2012).
Youm, Y. H. et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269 (2015).
Yan, Y. et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38, 1154–1163 (2013).
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
Macia, L. et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6, 6734 (2015).
Du, Q. et al. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 105, 42–54 (2016).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Robblee, M. M. et al. Saturated fatty acids engage an IRE1alpha-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 14, 2611–2623 (2016).
Progatzky, F. et al. Dietary cholesterol directly induces acute inflammasome-dependent intestinal inflammation. Nat. Commun. 5, 5864 (2014).
Zhao, S. et al. Deoxycholic acid triggers NLRP3 inflammasome activation and aggravates DSS-induced colitis in mice. Front. Immunol. 7, 536 (2016).
Williams, T. M. et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 194, 3369–3380 (2015).
Man, S. M. et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 162, 45–58 (2015).
Wilson, J. E. et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913 (2015). References 205 and 206 identify a role for the DNA sensor AIM2 in preventing colorectal cancer in mice.
Hu, S. et al. The DNA Sensor AIM2 Maintains Intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep. 13, 1922–1936 (2015).
Ratsimandresy, R. A., Indramohan, M., Dorfleutner, A. & Stehlik, C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell. Mol. Immunol. 14, 127–142 (2017).
Demon, D. et al. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 7, 1480–1491 (2014).
Williams, T. M. et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G139–G150 (2014).
Oficjalska, K. et al. Protective role for caspase-11 during acute experimental murine colitis. J. Immunol. 194, 1252–1260 (2014).
Takagi, H. et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 38, 837–844 (2003).
Salcedo, R. et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 207, 1625–1636 (2010).
Otani, K. et al. Colchicine prevents NSAID-induced small intestinal injury by inhibiting activation of the NLRP3 inflammasome. Sci. Rep. 6, 32587 (2016).
Dupaul-Chicoine, J. et al. The Nlrp3 Inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763 (2015).
Nowarski, R. et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015).
Ishikura, T. et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 18, 960–969 (2003).
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015). This study reports a potent and selective inhibitor of the NLRP3 inflammasome and its use in multiple mouse models of inflammatory diseases.
Guo, W. et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 10, 972–985 (2014).
Jiang, H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219 (2017).
Romberg, N. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 46, 1135–1139 (2014).
Canna, S. W. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 46, 1140–1146 (2014).
Kitamura, A., Sasaki, Y., Abe, T., Kano, H. & Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 211, 2385–2396 (2014). References 222, 223 and 224 collectively report an association between mutations in NLRC4 and inflammatory conditions in humans. They also highlight the therapeutic relevance of inflammasome blockade in the treatment of these clinical manifestations.
Canna, S. W. et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 139, 1698–1701 (2017).
Allam, R. et al. Epithelial NAIPs protect against colonic tumorigenesis. J. Exp. Med. 212, 369–383 (2015).
Garaude, J., Kent, A., van Rooijen, N. & Blander, J. M. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Sci. Transl Med. 4, 120ra116 (2012).
Dihlmann, S. et al. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int. J. Cancer 135, 2387–2396 (2014).
Karki et al. NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature 540, 583–587 (2016).
Man, S. M., Karki, R. & Kanneganti, T. D. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280 (2016).
Hu, B. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016).
Lian, Q. et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. 27, 784–800 (2017).
Alexander, J. L. et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 14, 356–365 (2017).
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl Acad. Sci. USA 108, 9601–9606 (2011).
Chen, G. Y., Liu, M., Wang, F., Bertin, J. & Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Birchenough, G. M., Nystrom, E. E., Johansson, M. E. & Hansson, G. C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542 (2016).
Seregin, S. S. et al. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol. 10, 434–445 (2017).
O’Keefe, S. J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 13, 691–706 (2016).
Bergstrom, K. et al. Defective intestinal mucin-type O-glycosylation causes spontaneous colitis-associated cancer in mice. Gastroenterology 151, 152–164 (2016).
Powell, N., Walker, M. M. & Talley, N. J. The mucosal immune system: master regulator of bidirectional gut-brain communications. Nat. Rev. Gastroenterol. Hepatol. 14, 143–159 (2017).
Man, S. M., Karki, R. & Kanneganti, T. D. DNA-sensing inflammasomes: regulation of bacterial host defense and the gut microbiota. Pathog. Dis. 74, ftw028 (2016).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Seregin, S. S. et al. NLRP6 protects Il10−/− mice from colitis by limiting colonization of Akkermansia muciniphila. Cell Rep. 19, 733–745 (2017).
Chen, L. et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 18, 541–551 (2017).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Mamantopoulos, M. et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 47, 339–348 (2017).
Lemire, P. et al. The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep. 21, 3653–3661 (2017). References 246 and 247 demonstrate, using littermate-controlled experiments, that certain inflammasomes do not influence the composition of the gut microbiota in mice.
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015).
Sun, Y. et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology 144, 1478–1487 (2013).
Pierantonelli, I. et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 7, 12200 (2017).
Lukens, J. R. et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516, 246–249 (2014).
Rao, S. et al. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell 168, 503–516 (2017).
Wong, M. L. et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 21, 797–805 (2016).
Lara-Tejero, M. et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412 (2006).
Song-Zhao, G. X. et al. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 7, 763–774 (2014).
Allen, I. C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36, 742–754 (2012).
Zaki, M. H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011). References 256 and 257 reveal an important role for NLRP12 in colitis and colitis-associated colorectal cancer in mice.
Acknowledgements
S.M.M. is supported by the Australian National University Futures Award, The Gretel and Gordon Bootes Medical Research Foundation and the National Health and Medical Research Council of Australia under project grants (APP1141504 and APP1146864) and the R.G. Menzies Early Career Fellowship (APP1091544). The author apologizes to researchers whose work was not cited or was cited through reviews owing to space limitations.
Reviewer information
Nature Reviews Gastroenterology & Hepatology thanks R. Flavell, T. Monie and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Review criteria
A detailed literature review was performed using the PubMed database using a combination of the following search terms: “inflammasome”, “NLRP1”, “NLRP3”, “NLRC4”, “AIM2”, “pyrin”, “caspase-1”, “caspase-4”, “caspase-5”, “caspase-11”, “pyroptosis”, “IL-1”, “IL-18”, “infection”, “bacteria”, “viruses”, “protozoa”, “colitis”, “IBD”, “cancer” and “microbiota”. Relevant English-language papers were evaluated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Man, S.M. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol 15, 721–737 (2018). https://doi.org/10.1038/s41575-018-0054-1
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-018-0054-1
This article is cited by
-
Modified Gexia-Zhuyu Tang inhibits gastric cancer progression by restoring gut microbiota and regulating pyroptosis
Cancer Cell International (2024)
-
The interaction of inflammasomes and gut microbiota: novel therapeutic insights
Cell Communication and Signaling (2024)
-
NFκB and NLRP3/NLRC4 inflammasomes regulate differentiation, activation and functional properties of monocytes in response to distinct SARS-CoV-2 proteins
Nature Communications (2024)
-
The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation in Helicobacter pylori-associated gastritis by regulating ROS and autophagy
Cell Communication and Signaling (2023)
-
Quantitative proteomic analysis of local and systemic extracellular vesicles during Eimeria falciformis infectious cycle in the host
Parasites & Vectors (2023)
