
Abstract
Liver sinusoidal endothelial cells (LSECs) line the low shear, sinusoidal capillary channels of the liver and are the most abundant non-parenchymal hepatic cell population. LSECs do not simply form a barrier within the hepatic sinusoids but have vital physiological and immunological functions, including filtration, endocytosis, antigen presentation and leukocyte recruitment. Reflecting these multifunctional properties, LSECs display unique structural and phenotypic features that differentiate them from the capillary endothelium present within other organs. It is now clear that LSECs have a critical role in maintaining immune homeostasis within the liver and in mediating the immune response during acute and chronic liver injury. In this Review, we outline how LSECs influence the immune microenvironment within the liver and discuss their contribution to immune-mediated liver diseases and the complications of fibrosis and carcinogenesis.
Key points
-
Liver sinusoidal endothelial cells (LSECs) that line the hepatic sinusoids have important physiological roles and mediate the filtration and scavenger functions of the liver.
-
LSECs also have innate and adaptive immunological functions, including antigen presentation and maintenance of the balance between tolerance and effector immune responses.
-
In inflammatory liver diseases, LSECs influence the composition of hepatic immune populations by mediating diapedesis of leukocyte subsets via distinct combinations of adhesion molecules and chemokines.
-
LSECs play a crucial part in the cellular crosstalk that regulates progressive chronic liver disease, which leads to fibrosis and carcinogenesis.
-
The role of LSECs in initiating immune responses and contributing to progressive liver disease makes them a potential therapeutic target for treating inflammatory liver diseases.
Similar content being viewed by others
Introduction
Sinusoidal endothelial cells line what constitutes a unique vascular bed in the liver, which receives blood from both the hepatic artery and the portal veins into the hepatic parenchyma (Fig. 1). Studies of these cells isolated from animals usually refer to them as liver sinusoidal endothelial cells (LSECs), whereas isolated human cells have also been referred to as human hepatic sinusoidal endothelial cells (HSECs). For the purpose of this Review, we use the term LSEC. The exposure of these sinusoidal endothelial cells to blood originating from both the gut and the systemic circulation means they are ideally situated to remove and recycle blood-borne proteins and lipids. In combination with Kupffer cells (KCs; liver-resident macrophages), LSECs constitute the most powerful scavenger system in the body1. This activity is facilitated by the presence of fenestrae in LSECs, their lack of a classical basement membrane and their expression of promiscuous scavenger receptors combined with the most potent endocytic capacity in the body2. Thus, virus particles3, advanced glycation end products4 and modified LDL cholesterol5 can be cleared from the circulation within minutes by this route.

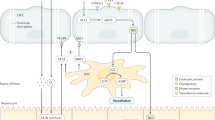
a | Low-power image of human liver tissue (stained with haematoxylin and eosin) illustrating the lobular organization of the liver, with zonal architecture indicated relative to the position of the portal tract. b | Expanded periportal section of the same image to illustrate the different vascular compartments within the parenchyma. c | Immunohistochemical staining of stabilin 1, which highlights liver endothelial cell distribution within hepatic tissue in a normal liver section. d | A comparison of the structure of liver sinusoidal endothelium and glomerular endothelium.
Endothelial cells in different vascular beds are generated from common early embryological precursors and have broadly similar histological appearance and functional roles throughout the body. However, extensive variations in phenotype and function arise as a consequence of local microenvironmental signals dependent on anatomical localization6. The vascular architecture in the human liver is acquired by 17–25 weeks of gestation, but different vessels within the liver have distinct embryonic origins. Thus, portal vessels are derived from vitelline veins, whereas sinusoids develop from capillary vessels of the septum transversum and acquire their distinctive fenestrated phenotype by week 20 of gestation7 under the control of transcription factor GATA4 (ref.8). From this point onward, sinusoidal endothelial cells remain functionally and phenotypically distinct from the other vascular endothelial cells in the liver microenvironment and assume a phenotype that has many similarities with lymphatic endothelial cells9. The unique characteristics of LSECs are presented in Box 1. Both lymphatic and sinusoidal endothelial cells have minimal basement membranes and loosely organized cell junctions10 and share a complement of receptors such as lymphatic vessel endothelial hyaluronic acid receptor (LYVE1)11, prospero homeobox protein 1 (PROX1)12, podoplanin13 and liver/lymph node-specific ICAM3-grabbing non-integrin (LSIGN; also known as CLEC4M)14. It has been shown that the phenotype of sinusoidal endothelial cells varies across the liver acinus; a study of human liver tissue published in 2017 demonstrated that zone 1 LSECs are CD36hi and LYVE1low, whereas zone 2 and zone 3 LSECs are CD36low, LYVE1hi and CD32hi (ref.15). The presence of fenestrations or membranous pores organized into sieve plates is a feature that also distinguishes LSECs from the other hepatic endothelial populations2.
Fenestrations are not unique to hepatic endothelial cells and are also found in endothelium in endocrine glands such as the pancreas16, kidney17, spleen18 and bone marrow19 and are sometimes observed in tumour vasculature20. However, unlike other fenestrated endothelial populations such as those in the kidney, hepatic fenestrations lack a diaphragm or basal lamina and are grouped into organized sieve plates, rendering LSECs highly permeable (Fig. 1d). Many studies have implicated vascular endothelial growth factor (VEGF) as an essential factor for regulation of fenestrations21, but dynamic changes in hepatic fenestration number and size can occur rapidly in response to agents such as alcohol22, dietary constituents23 and fasting24 or calorie restriction25. The fenestrations act as a ‘dynamic filter’ (ref.26) to permit the access of macromolecules to parenchymal cells, and in addition, these pores might allow circulating viruses to gain access to hepatocytes27. Evidence from animal studies suggests that fenestrations can constitute up to 40% of the cell and that the size, distribution and clustering of the pores in sieve plates varies with the zonal distribution of the endothelium28 and across the endothelial surface. Up to one-third of these pores are organized into complex labyrinths, and many are associated with components of microtubules29, caveoli and coated pits to form a transport network that could impose additional regulation on the traffic of material into the cells30, enabling them to govern the movement of materials to and from the liver parenchyma.
Balancing tolerance and immune response
The permissive nature of sinusoidal endothelium probably evolved to handle the constant exposure of the liver to microbial and food antigens derived from the gastrointestinal tract via the portal vein. The liver needs to ensure that damaging immune responses are not precipitated against harmless antigens while at the same time being able to eliminate invading pathogens. The first site of exposure to these antigens occurs within the hepatic sinusoids, and both KCs and LSECs are important players in taking up and eliminating soluble antigens entering via the portal vein and in determining the nature of any immune response such antigens trigger.
The initial critical step in an immune response is the innate pathway of antigen uptake by pattern recognition receptors31. Pattern recognition receptors are highly evolutionarily conserved and include the Toll-like receptor (TLR) family and the scavenger receptors31. An example of how the liver regulates inflammatory and immune responses is seen in the recognition of the TLR4 ligand lipopolysaccharide (LPS) by KCs and LSECs. Chronic exposure of both KCs and LSECs to LPS leads to an LPS-refractory state, and in LSECs specifically, LPS exposure is associated with reduced nuclear translocation of nuclear factor-κB (NF-κB) and subsequent reduced leukocyte adhesion32. This mechanism prevents the liver from being in a constantly activated inflamed state in response to the constant exposure to bacterial products from the gut. Studies of other TLRs demonstrate that LSECs can respond to signals mediated via TLR1–TLR4, TLR6, TLR8 and TLR9, but their activation has cell-specific responses that are restricted compared with classical antigen-presenting cells, thereby contributing to an organ-specific response to antigens and the tolerogenic environment of the liver33.
A unique characteristic of LSECs is their expression of high levels of several scavenger receptors compared with conventional endothelium. Scavenger receptors are a diverse family of pattern recognition receptors that, like TLRs, are highly evolutionarily conserved34. In contrast to TLRs, they were believed to be functionally redundant and to perform silent uptake of ligands. However, gathering evidence suggests that this is not the case and that scavenger receptors have an important cell-specific role in immune responses34. They have been shown to promote potent pro-inflammatory and anti-inflammatory signalling as well as to directly interact with TLRs. Membrane-bound scavenger receptors recognize their extracellular ligands, which leads to internalization of the ligand, termed endocytosis, and trafficking from the cell membrane to intracellular compartments such as endosomes. The high levels of scavenger receptors on LSECs give them a high endocytic capacity. One of the most extensively studied scavenger receptors on LSECs is the mannose receptor (MR)1,35,36. Others include the homologous scavenger receptors stabilin 1 and stabilin 2 (ref.37) and related molecules such as C-type lectins, including the type 2 receptor subclass dendritic cell-specific ICAM3-grabbing non-integrin (DCSIGN; also known as CD209) and LSIGN14,38. The members of the C-type lectin group are involved in varied functions ranging from cell–cell interaction to uptake of serum glycoproteins. A third lectin with a similar structure to DCSIGN and LSIGN has been identified and designated the liver and lymph node sinusoidal endothelial cell C-type lectin (LSECTIN; also known as CLEC4G)39. This lectin has been shown to be co-expressed with LSIGN and is encoded in the same cluster of lectin-encoding genes as DCSIGN and LSIGN.
Innate immunity
Several of the C-type lectin receptor family members have been directly implicated in viral uptake. Both DCSIGN and LSIGN have been shown to interact with the Ebola virus and HIV, as well as the coronavirus40,41. Both these receptors have also been shown to be expressed on LSECs and bind the E2 glycoprotein of the hepatitis C virus (HCV) and facilitate hepatocyte infection14. LSECTIN has also been implicated in the uptake of severe acute respiratory syndrome coronavirus and HCV42,43. The ability of LSECs to bind multiple viruses through their diverse endocytic receptors gives them a crucial role in the response to viral infections and a specific role in mediating rapid clearance of blood-borne viruses44. In a mouse model of adenovirus infection, 90% of virus is found in LSECs and 10% is found in KCs within a minute of intravenous viral infusion44. A study published in 2017 reported that HIV-like particles are taken up by mouse LSECs at a rate of 100 million particles per minute3. After receptor-mediated endocytosis of circulating matrix breakdown products, the subsequent transit from early endosomes to late endosomes takes several hours45. The transit of viruses internalized by LSECs is less well understood. LSECs enable direct entry of certain viruses such as Ebola, whereas with other viruses, such as HCV and HBV, LSECs promote hepatotropism by facilitating parenchymal cell infection46. Rapid uptake of virus can also lead to redistribution to other cells, for instance, in animal models of HBV, viral particles are preferentially taken up by LSECs and subsequently passed on to infect underlying hepatocytes47. In the case of HCV, innate sensing of viral infection by LSECs leads to downstream signalling and release of paracrine signals such as the pro-viral molecule bone morphogenetic protein 4 (BMP4), which increases viral infection of hepatocytes48. On the other hand, direct sensing of HCV RNA in LSECs also leads to the release of type I and type III interferon-rich exosomes that inhibit HCV replication49. The balance of such responses will determine whether virus infection is established or prevented, thereby emphasizing the critical role that LSECs play in hepatotropic viral infections.
Adaptive immunity
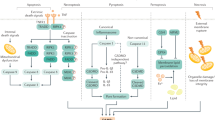
LSECs not only regulate innate immune responses but also directly regulate adaptive immune responses through antigen presentation to T cells (Fig. 2). Knolle's group demonstrated that LSECs can cross-present antigen to CD8+ T cells50 by using scavenger receptors, notably the mannose receptor, to take up, process and transfer antigen to major histocompatibility complex (MHC) class I (ref.51). The presentation of antigen, including oral antigens, by LSECs drives a tolerogenic response in naive CD8+ T cells that is mediated by upregulation on LSECs of the co-inhibitory molecule programmed cell death 1 ligand 1 (PDL1), which can activate its receptor programmed cell death protein 1 (PD1) on naive T cells50,52,53. Endocytosis of antigens by the mannose receptor on LSECs has been shown to promote CD8+ T cell tolerance54, including tolerance to tumour antigens55. However, it is crucial that rapid effector responses can be generated locally to harmful pathogens; consistently, LSEC-driven T cell activation changes in response to antigen load and local inflammatory factors. For example, in a culture model with mouse LSECs in which antigens at varying concentrations were delivered to LSECs for cross presentation to CD8+ T cells, high antigen concentrations led to a shift from tolerogenic to effector T cell differentiation56 as a consequence of enhanced T cell receptor (TCR) signalling that overcame PD1-mediated tolerogenic responses. This response is also affected by local levels of IL-2. Furthermore, rapid activation of CD8+ T cells by LSECs occurs in the presence of IL-6 trans-signalling, and this activation not only drives rapid effector T cell differentiation but also primes T cells to respond to other inflammatory signals and leads to sustained effector responses57.

a | Liver sinusoidal endothelial cells (LSECs) express major histocompatibility complex class I (MHC-I) receptors and can cross-present antigen to CD8+ cytotoxic T cells. At low antigen concentrations, this presentation leads to tolerance and deletion of CD8+ T cells. b | If antigen concentrations are high, then antigen cross presentation to CD8+ T cells leads to a memory effector T cell phenotype. c | In the context of hepatotrophic infections such as hepatitis B, CD8+ T cells adhere to the sinusoids in a platelet-dependent process and then probe for infected hepatocytes through LSEC fenestrae. Detection of an infected hepatocyte leads to diapedesis (the process of cells actively crossing capillaries)-independent killing. d | LSECs can also present antigen to CD4+ T cells via expression of MHC class II, which leads to the induction of suppressor T cells (CD25hi regulatory T cells). TCR, T cell receptor.
LSECs also express MHC class II molecules that enable them to present antigens to CD4+ T cells58. However, the low levels of co-stimulatory molecules on LSECs means that rather than driving naive CD4+ T cell differentiation to T helper cells59, they promote the development of regulatory T cells60. In vivo studies have shown that these tolerogenic properties of LSECs can control autoimmunity. Circulating inflammatory CD4+ T cells (T helper 1 (TH1) cells and TH17 cells) were shown to interact repeatedly with liver sinusoidal endothelium, and this interaction successfully suppressed inflammatory cytokine release in mice61. The induction of autoantigen-specific T regulatory cells by LSECs was also shown to have important systemic effects by ameliorating damage in mouse models of autoimmune central nervous system disease62,63. This finding has therapeutic implications for systemic as well as local immunity and has led to development of nanotechnology-based strategies to deliver autoantigen to LSECs as part of tolerance induction protocols63. C-type lectins also contribute to the unique ability of LSECs to control T cell differentiation. Thus, LSECTIN on LSECs inhibits T cell activation and effector functions through its interaction with CD44 on activated T cells64.
LSECs in inflammatory liver disease
In addition to their roles as pathogen recognition and antigen-presenting cells, LSECs also have a critical role in regulating the recruitment of leukocytes into liver tissue (Box 2). A key step in the progression of liver injury or infection, regardless of aetiology, is the development of hepatitis as a consequence of the recruitment of leukocytes from the circulation. The balance and retention of immune subsets within the liver determines whether injury resolves, persists or progresses to either liver failure or chronic hepatitis and cirrhosis65. Leukocyte recruitment from the blood occurs as a consequence of a multistep adhesion cascade that enables leukocytes flowing in the circulation to be captured by activated endothelial cells and then to migrate through the endothelium towards sites of infection or injury66. The cascade consists of sequential steps mediated by interactions between receptors on the surface of leukocytes and endothelial cells. The general paradigm applies to all vascular beds, but tissue-specific and inflammation-specific interactions provide powerful local regulation of where, when and which leukocytes are recruited. The steps in the cascade are broadly described as rolling or tethering, in which the leukocyte is captured from the circulation and induced to roll on the endothelial surface. In most vascular beds, this step is mediated by a family of receptors termed selectins, but other receptors are involved under specific circumstances, such as in the hepatic sinusoids67. Leukocyte rolling is followed by activation of leukocyte integrins in response to tissue-derived chemoattractant cytokines (chemokines) sequestered in the endothelial glycocalyx68,69, which leads to firm adhesion mediated by integrins binding to immunoglobulin superfamily members on the endothelial surface. This adhesion is followed by intravascular crawling of the adherent leukocyte on the endothelium, which occurs before the final step of transmigration, in which the leukocyte migrates across the endothelium, through the post-endothelial tissue and into the liver parenchyma. The transmigration step is mediated by a complex series of receptor–ligand interactions, with cytoskeletal changes in both the endothelial cells and the leukocytes, and which enable the cell to cross the endothelium without disrupting the vascular barrier70.
Cell recruitment to the liver has several features that are distinct from the general adhesion cascade (Fig. 3). Recruitment of the majority of leukocytes occurs within the sinusoidal channels of the liver, in contrast to most other organs in which recruitment occurs within the post-capillary venules71. Furthermore, the recruitment of leukocytes subsets to the liver is regulated by specific combinations of typical and atypical adhesion molecules, reflecting the unique phenotype and structure of LSECs and the anatomy and rheology of the sinusoids (Box 1). The sinusoids are narrow, in some places, no wider than a flowing leukocyte, and characterized by low shear stress. These properties mean that the initial recruitment step does not require rolling and in most circumstances is selectin-independent. As a consequence, sinusoidal endothelium expresses minimal levels of selectins in vivo71,72. A summary of the key adhesion factors is outlined in Table 1.

Lymphocyte recruitment involves an adhesion cascade within the hepatic sinusoids that is influenced by the low shear environment and cellular crosstalk between parenchymal and non-parenchymal cells. Chronic parenchymal cell damage leads to the release of danger-associated molecular patterns (DAMPs) and pro-inflammatory mediators by Kupffer cells, which increase adhesion molecule expression by liver sinusoidal endothelial cells (LSECs) (step 1). Lymphocyte recruitment across activated LSECs involves a selectin-independent tethering step (step 2), followed by integrin activation and firm adhesion to immunoglobulin superfamily members on the LSEC surface (step 3). This process is influenced by paracrine factors released from hepatocytes. Lymphocytes then crawl along the luminal endothelium (step 4) until they receive a signal to transmigrate across LSECs through either a paracellular or a transcellular route (step 5). A third route of lymphocyte migration involves intracellular migration directly into the LSEC body and then migration to the adjacent LSEC, termed intracellular crawling (step 6). Release of chemotactic factors from activated hepatic stellate cells promotes subsequent migration and positioning in liver tissue (step 7). CXCR3, CXC-chemokine receptor 3; ECM, extracellular matrix; ICAM1, intercellular adhesion molecule 1; MADCAM1, mucosal addressin cell adhesion molecule 1; VAP1, vascular adhesion protein1; VCAM1, vascular cell adhesion molecule 1.
Immunoglobulin superfamily
The conventional endothelial adhesion molecules that mediate firm adhesion of leukocytes, intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1)69,73, are expressed at high levels on inflamed LSECs74. Their role in lymphocyte recruitment to the liver has been confirmed in both in vitro and in vivo assays. VCAM1, which binds the α4β1 integrin expressed on lymphocytes, has an important role in capturing lymphocytes from blood flow and mediating stabilization65,75. ICAM1 binds to αLβ2 integrin to support firm adhesion within the hepatic sinusoids71. Another family member is mucosal vascular addressin cell adhesion molecule 1 (MADCAM1), which binds to the α4β7 integrin and plays a major part in lymphocyte homing to the gut via mucosal vessels76. Our group demonstrated that this receptor was also upregulated in the liver in some chronic liver diseases, in which it promotes the recruitment of T cells activated in the gut that express high levels of α4β7 integrin, thereby contributing to the link between IBD and inflammatory liver disease77,78. Although many of these immunoglobulin superfamily members are regulated by pro-inflammatory cytokines, their adhesive function is also dependent on the formation of cell surface platforms regulated by the tetraspanin family of receptors, which form microdomains and associate laterally with ICAM1 and VCAM1 (refs79,80). For example, our group confirmed that CD151 associates with VCAM1 on LSECs and regulates lymphocyte adhesion under shear stress81.
Atypical adhesive and migratory routes
In addition to conventional adhesion molecules, our group and others have demonstrated that LSECs use atypical adhesion molecules to regulate leukocyte recruitment. Vascular adhesion protein 1 (VAP1; also known as AOC3) is a membrane-bound amine oxidase that was originally shown to mediate lymphocyte binding to high endothelial venules, the specialized post-capillary venules found in lymph nodes82. Further studies confirmed that VAP1 was expressed at high levels in chronic liver disease and mediated adhesion and transmigration across LSECs83. Models of in vitro and in vivo inflammatory liver injury corroborated its role in mediating recruitment during liver inflammation. VAP1 has unique properties generated by its enzyme activity that can upregulate expression of adhesion molecules and chemokines in LSECs, thereby amplifying leukocyte recruitment84,85,86,87. The scavenger receptor family of endothelial receptors also contributes to leukocyte recruitment to the liver. Stabilin 2 was shown to regulate lymphocyte adhesion to LSECs via the αMβ2 integrin (ref.88), and its homologue, stabilin 1, was originally shown to mediate recruitment across lymphatic endothelium89. Similarly, expression of stabilin 1 is upregulated in chronic liver disease and hepatocellular carcinoma, in which it mediates transmigration of lymphocytes across LSECs under shear stress90.
Following adherence, leukocytes crawl across the endothelial surface before undergoing transmigration, which usually occurs via endothelial junctions, termed the paracellular route91,92. Several studies have demonstrated that lymphocyte interactions with LSECs within the sinusoids trigger important immune effector mechanisms93,94, which might influence the infiltration and positioning of cells within the liver in inflammatory liver diseaes74. Thus, it is important to understand how the process of transendothelial migration through LSECs is regulated. Visualization of this process using confocal imaging of lymphocytes migrating across LSECs under shear stress demonstrated that ~50% of cells took a transcellular route of migration and migrated directly through the endothelial body90 as opposed to the conventional paracellular route. This transcellular migration route involved the formation of ICAM1-rich channels to facilitate lymphocyte migration. Although transcellular migration has been noted in some other specialized endothelial beds, its function and molecular basis remain poorly understood95. Transendothelial migration is a multistep process involving different combinations of receptors that enables preferential recruitment of particular leukocyte subsets, as described in the following sections. An additional step in migration was described in 2016 in which lymphocytes migrate into LSECs and then crawl within the endothelial cell to the cell junction, through which they enter the adjacent endothelial cell96. This process, which we term intracellular crawling, is dependent on IFNγ and could not be detected when LSECs were stimulated by other interferon family cytokines. IFNγ treatment did alter the cytoskeleton of LSECs, which might promote intracellular crawling. This process was also facilitated by the unique junctional complexes between LSECs. The functional consequences of intracellular crawling are yet to be elucidated but could have an important role in lymphocyte positioning in liver tissue.
Chemokines
Chemokines are a family of small secreted proteins ranging from 67 to 127 amino acids in size that bind to heparin sulfate on proteoglycans97,98. They play central parts in leukocyte migration during homeostasis (in development and localization in secondary lymphoid organs) as well as within tissues during inflammatory responses by binding to G protein-coupled receptors on the surface of leukocytes. Upregulation of several chemokines on liver vasculature has been demonstrated in a range of chronic inflammatory liver diseases, including alcoholic liver disease, primary sclerosing cholangitis and chronic graft rejection99,100,101,102,103. In these conditions, chemokines seem to be compartmentalized to the sinusoidal vasculature and portal vessels and have a substantial influence on immune cell localization and subsequent disease progression100,101,104. T cell migration across sinusoidal endothelium is mediated by the interferon-inducible chemokines CXC-chemokine ligand 9 (CXCL9) and particularly CXCL10, which bind the receptor CXC-chemokine receptor 3 (CXCR3)105,106. In other diseases, including chronic HCV infection, the chemokine CXCL16, which exists in a transmembrane form, is expressed on sinusoidal endothelium, hepatocytes and bile ducts, enabling it to regulate the recruitment and retention of CXCR6+ effector T cells within the liver107,108. Subsets of natural killer (NK) and NK T cells express high levels of CXCR6 that enable them to interact with CXCL16 on sinusoidal endothelium; this interaction promotes active migration along the sinusoids as part of a process of ongoing immune surveillance and patrolling109. Studies in mouse liver endothelial cells have shown that a vital property of chemokine-mediated recruitment is the transcytosis of chemokines from the basolateral side to the luminal side of sinusoidal endothelial cells110. This process is clathrin-dependent and promotes the transendothelial migration of lymphocytes across LSECs, and inhibition of this pathway reduces CD4+ T cell recruitment during liver injury111.
Immune subset recruitment
The balance of immune subsets determines the progression and outcome of immune responses within the liver: persistent effector responses will drive chronic inflammatory conditions, whereas excessive amounts of immunosuppressive immune subset populations promote pathogen escape and tumour formation112,113,114. In addition to the key mediators of immune cell recruitment discussed earlier, there is now evidence that immune cell subsets utilize distinct combinations of these factors to migrate through the hepatic sinusoids under specific circumstances.
T cells
T helper cells are divided into multiple functional subsets on the basis of the cytokines they secrete and that are dependent on the microenvironment in which they are activated by antigens. In a concanavalin A liver mouse inflammation model, TH1 cell recruitment through the sinusoids was mediated by α4β1 integrin interactions, whereas TH2 cells used VAP1 (ref.84). Both effector TH17 cells and regulatory T (Treg) cells found in the liver express high levels of the chemokine receptor CXCR3 and use it to migrate across LSECs115,116. Subsequent signals determine where these cells localize within the liver, with CC-chemokine receptor 6 (CCR6)+ TH17 cells migrating towards their ligand, CC-chemokine ligand 20 (CCL20), which is secreted by bile ducts, whereas Treg cells respond to different chemokines as a consequence of their expression of CCR5, CCR4 and in some cases CCR10 (refs116,117). Treg cells were also shown to use a distinct combination of adhesion receptors, involving stabilin 1, ICAM1 and VAP1, to migrate across human LSECs under flow90, whereas recruitment of CD8+ T cells to the mouse liver is primarily dependent on ICAM1 expression by LSECs with a lesser contribution from VCAM1 (refs118,119). In autoimmune hepatitis and primary sclerosing cholangitis (PSC) associated with IBD, LSECs present the chemokine CCL25, which can trigger CCR9+ gut-homing lymphocyte interactions with MADCAM1 to promote recruitment of mucosal T cells103,120.
These distinct mechanisms of migration across LSECs are probably influenced by epithelial responses to tissue injury115,121, stromal signals122 and cooperative interactions between several cell types in the sinusoid. For instance, in a model of HBV infection, effector CD8+ T cells were shown to arrest in the sinusoids by interacting with platelets adherent to the sinusoidal surface via hyaluronan-dependent mechanisms94. Subsequently, the CD8+ T cells crawled along the sinusoids, probing through the LSEC fenestrae for viral antigens presented by underlying hepatocytes. Antigen recognition as a consequence of this probing behaviour led to effector functions by a diapedesis-independent process. A human model of cytomegalovirus (CMV) infection of LSECs led to the recruitment of effector T cells and activated Treg cells in a lymphocyte function-associated antigen 3 (LFA3)-dependent mechanism123. In this study, CMV-infected human LSECs upregulated LFA3 at intercellular junctions, and during effector T cell recruitment, the interaction of LFA3 with its ligand, CD2, on T cells contributed to TH1 cell activation.
B cells
Although B cells are present in substantial numbers in chronically inflamed liver tissue, the molecular mechanism regulating their recruitment from blood into hepatic tissue is poorly understood. Our group demonstrated that B cell recruitment across human LSECs under flow was initially mediated by VCAM1-dependent capture followed by limited intravascular crawling, which is distinct from the method of T cell recruitment124. Interestingly, the receptors involved in transmigration of B cells included ICAM1, VAP1 and stabilin 1, all of which are also involved in Treg cell transmigration across LSECs.
Neutrophils
Neutrophils are one of the earliest immune cells to be recruited to a site of tissue injury, and they are also recruited into the liver via the hepatic sinusoids125. It was originally thought that their migration was mediated by simple physical trapping within the narrow sinusoidal channels, but work from McDonald et al.126 implicated a complex multistep recruitment process involving interactions between sinusoidal hyaluronan and CD44 on the neutrophil surface. Whereas neutrophil interactions in post-sinusoidal venules followed a conventional rolling mediated by selectins and integrin-mediated adhesion, this was found not to be the case in the sinusoids, where the majority of neutrophil extravasation occurred. They found that hyaluronan was highly expressed in liver sinusoids and mediated the recruitment of neutrophils in response to LPS challenge. This interaction was dependent on hyaluronan binding to CD44 rather than to the other hyaluronan receptor, receptor for hyaluronan-mediated motility (RHAMM; also known as HMMR). A study published in 2014 also highlighted the importance of TLRs for neutrophil recruitment. TLR2–protein S100A9 signalling in particular promoted the production of the chemokines CXCL1 and CXCL2, which are known to mediate neutrophil migration, by liver macrophages in acute and chronic mouse models of liver injury127.
Monocytes
In addition to the activation of resident KCs, monocytes and macrophages are also recruited to the liver from the circulation during inflammation or in response to injury. KCs are yolk sac-derived tissue macrophages found within the hepatic sinusoids; they are immobile and probe the environment with pseudopods128. The response to liver injury also includes an influx of monocytes, which have a major role in regulating inflammation, regeneration, repair and fibrosis129. Furthermore, acute liver injury is associated with an initial influx of transcription factor GATA6+ peritoneal macrophages that enter directly through the mesothelium in a process dependent on CD44 and the danger-associated molecular pattern (DAMP) molecule ATP128. This entry is followed by the recruitment of CCR2+ monocytes from the circulation130. The subsequent recruitment signals governing monocyte migration through the sinusoids are less well characterized, but several key factors have been determined. The dominant chemokine receptor–ligand interaction that mediates migration of CD16+ monocytes across LSECs is CX3C-chemokine receptor 1 (CX3CR1) binding to its ligand CX3C-chemokine ligand 1 (CX3CL1), one of the few transmembrane chemokines. Expression of CX3CL1 is restricted to bile ducts in the normal liver, yet this protein is expressed at high levels on inflamed sinusoidal endothelium131. In this study, VAP1 also contributed to adhesion and transendothelial migration of CD16+ monocytes across LSECs. The accumulation of CD14+CD16+ monocytes has been reported in inflammatory liver disease, which is due in part to the preferential migration of this subset across LSECs compared with CD14+CD16– cells132. Monocytes are known to undergo a phenomenon of bidirectional movement across endothelium that involves a reverse migration step133,134. This migratory behaviour has been confirmed in LSECs and might have a marked effect on the fate of monocytes and the outcome of liver injury because monocyte subsets which undergo reverse transmigration are predominantly pro-inflammatory CD16+ monocytes. By contrast, those remaining in the subendothelial space are anti-inflammatory monocytes that suppress T cells and promote endotoxin tolerance135.
Interaction with other liver cells
Although we have focused on leukocyte interactions with LSECs, the crosstalk between LSECs and other liver cell populations will also influence the progression of chronic inflammatory liver diseases. KCs are found within the hepatic sinusoids in close association with LSECs and are also equipped to sense tissue injury from infection and toxins. The release of DAMPs and pathogen-associated molecular patterns triggers the inflammasome pathway in KCs136. Inflammasome activation is a key step in the progression of parenchymal liver injury, such as alcoholic liver disease, in which the release of danger signals from damaged hepatocytes stimulates the release of pro-inflammatory mediators from KCs137. Despite poor understanding of the crosstalk between LSECs and KCs, the release of these mediators probably influences LSEC phenotype and activation and leads to subsequent leukocyte recruitment138,139. Furthermore, KCs can promote LSEC capillarization, whereby LSEC morphology becomes more vascular or capillary like with a loss of fenestrations, and the formation of a characteristic basement membrane140,141.
The other cell type that populates the sinusoids is the hepatic stellate cell (HSC), which is positioned within the space of Disse. The central role of HSCs in extracellular matrix production in chronic liver disease is well established142. It is now known that LSECs play an important role in maintaining the quiescence of HSCs and that this ability is lost during capillarization of LSECs, which permits HSC activation and fibrogenesis21,143. Activated liver myofibroblasts, derived predominantly from HSCs, also have a role in the subsequent migration and positioning of lymphocytes following their recruitment through LSECs. This process is mediated by distinct combinations of cytokines including IL-6, VEGF and chemokines released by myofibroblasts122.
LSECs also have a key role in maintaining hepatocyte homeostasis. LSEC fenestrations enable the bidirectional transport of metabolites between the circulation and the liver parenchyma1. LSECs also facilitate the interaction of circulating T cells with hepatocytes by allowing T cells to extend cell surface protrusions through LSEC fenestrations144. In chronic liver injury, microparticles are released from hepatocytes, leukocytes and LSECs and provide another route for cell–cell communication145. Paracrine factors released from hepatocytes influence the expression of adhesion molecules on overlying LSECs and can promote the recruitment of flowing lymphocytes from the sinusoids121. This mechanism might be particularly important in liver cancer because malignant transformation of hepatocytes enhances their ability to secrete chemokines CXCL10, CCL2 and CCL3 and to upregulate expression of ICAM1 and VAP1 on co-cultured LSECs102,146. Work from our group has demonstrated that factors secreted by hepatoma cells upregulate the expression of CD151 in LSECs, which promotes VCAM1-mediated recruitment of lymphocytes81.
Therapeutic opportunities
The evidence presented here highlights the crucial role played by LSECs in regulating the inflammatory response to liver injury. This importance makes them and the molecules they express attractive therapeutic targets in inflammatory liver disease147,148. VAP1 is a good example149, with studies confirming that both inhibition of its enzymatic activity or antibody blockade of its adhesive function reduce hepatic inflammation and fibrosis in mouse liver injury models86, and this work has led to a clinical trial of a humanized antibody against VAP1 that is currently underway (BUTEO; NCT02239211) in patients with PSC. Chemokines and adhesion molecules expressed by inflamed LSECs are also potential targets for anti-inflammatory therapy in liver disease. For example, patients with PBC have been treated with NI-0801, a humanized monoclonal antibody against CXCL10. Interestingly, the high production rate of CXCL10 by the inflamed liver made it difficult to achieve sustained neutralization of the chemokine in vivo despite evidence that the antibody could remove chemokine from the sinusoidal endothelial bed. Although the drug was well-tolerated and demonstrated immunological changes, the overall results were negative150. Thus, therapies directed at the chemokine receptors themselves might have merit, and evidence from early trials using the dual CCR2–CCR5 antagonist cenicriviroc in patients with NASH suggests that such treatment can induce a persistent blockade151.
There is also a strong rationale to target gut-tropic chemokines in patients with liver diseases associated with IBD. Of particular relevance is PSC, a progressive biliary disease that is associated with IBD in 80% of patients and that affects ~8% of patients with IBD, particularly those with colitis152. Under physiological conditions, expression of CCL25 and MADCAM1 is absent from the liver, but in PSC both proteins are detectable on hepatic endothelium and support the aberrant recruitment of α4β7+ CCR9+ effector lymphocytes from the gut. Clinical trials are currently being considered to target the α4β7 integrin–MADCAM1 pathways in PSC using antibodies developed for treating IBD.
The tolerogenic capabilities of LSECs have also been targeted therapeutically. Nanoparticles loaded with autoantigen can be targeted to LSECs as a consequence of their potent scavenging capability; the ability of LSECs to take up molecules using their scavenger receptors is an excellent way of potentially targeting a range of therapies to the liver. Presentation of delivered autoantigens by LSECs to naive T cells results in the generation of autoantigen-specific regulatory T cells that can suppress systemic as well as local autoimmune responses. This strategy could be applied to a wide range of autoimmune and allergic conditions63. Targeting LSEC stabilin 1 and stabilin 2 with nanoparticle-based drugs153 has been suggested as a way to deliver local treatment to manage a range of conditions including ischaemia–reperfusion injury (a specific type of injury that follows liver surgery and transplantation, which is a biphasic process involving hypoxia followed by restoration of blood flow and reoxygenation) and NAFLD. Similarly, blockade of LSECTIN or the related molecule LSIGN has been shown to reduce the metastasis of colon cancer cells to the liver via impairment of interactions with LSECs in mouse models154,155.
During cirrhosis and chronic hepatitis, LSECs can undergo capillarization156. This process is associated with loss of GATA4-dependent signals8, upregulation of CD31 and VCAM1 and loss of fenestrations156,157,158. The number of fenestrations per endothelial cell not only decreases with disease159,160,161 but also with ageing162, and this phenotypic change is governed by p19ARF and p53-dependent signalling163. These changes might impede the transfer of materials to or from the parenchyma and contribute to regional hepatocyte hypoxia. Capillarization is mechanistically linked to the development of chronic inflammatory disease. In rodent models, it is associated with enhanced antigen presentation and cytotoxic T cell priming during fibrosis148, and in NASH, capillarization precedes and contributes to the transition from simple steatosis to steatohepatitis157. The changes that occur in LSECs in response to chronic inflammation also affect angiogenic pathways. Neo-angiogenesis is a key feature of chronic liver disease, and the majority of neo-vessels arise from portal vein branches and are closely associated with areas of fibrogenesis164,165. A key initiating step is the capillarization of LSECs, which leads to increased hepatocyte hypoxia and subsequent release of pro-angiogenic factors166,167. The LSEC response is context-specific; for example, acute injury can induce CXCR7 expression and a regenerative response, whereas chronic injury leads to CXCR4 induction, HSC proliferation and fibrogenesis168. During ischaemia–reperfusion injury, LSECs develop a pro-inflammatory, prothrombotic phenotype associated with vasoconstriction169. These changes have been directly linked to neutrophils because IL-33 released by LSECs during ischaemia–reperfusion injury triggers the release of neutrophil extracellular traps, which exacerbate acute hepatic injury170. In chronic injury, the changes in endothelial phenotype that accompany capillarization and precede fibrosis have been linked to alterations in signalling via the Hedgehog gene family171 and lead to vasoconstriction and increased intrahepatic vascular resistance owing to reduced nitric oxide production by LSECs172. Tumour progression in hepatocellular carcinoma is associated with changes in the phenotype of peritumoural LSECs and increased production of angiogenic factors including IL-6 (refs173,174).
These changes in LSECs therefore present opportunities for therapeutic intervention. For example, pharmacological therapy in the form of a soluble guanylate cyclase activator, which restores fenestrations, has been linked to fibrosis regression in rodent models21 and it might also be possible to use GATA4-mediated cellular reprogramming to restore the differentiated phenotype of LSECs and promote fibrosis resolution8. Similarly, therapies that restore normal Hedgehog signalling promote regression of capillarization and the reappearance of fenestrations, which suggests a potential pathway for reversal of fibrosis and the restoration of lipid transport171. However, studies testing cessation of VEGF-based cancer therapies also highlight how important the development of fenestrations can be in the context of metastasis and go some way in explaining the poor performance of some strategies using anti-VEGF drugs as cancer treatments. Withdrawal of anti-VEGF antibody therapy is associated with the development of hyperpolarized LSECs and promotion of hepatic metastasis175. Thus, low-dose, non-stop antiangiogenic therapy might present a future solution to minimize these effects.
Conclusions
Sinusoidal endothelial cells have complex interrelated roles in the maintenance of liver homeostasis and are implicated as drivers of inflammation and fibrogenesis in liver disease. Their unique positioning, phenotype and function make them attractive candidates for organ-specific therapy, and it is likely that more therapies targeting these cells will be tested in the future as new treatments to reduce liver injury and inflammation and to prevent or reverse fibrogenesis. In the absence of licenced antifibrotic therapies, strategies to maintain the differentiation of LSECs and to inhibit their ability to recruit harmful pro-inflammatory leukocytes through the selective orchestration of immune cell traffic might provide vital tools to halt the increase in mortality linked to chronic liver failure.
References
Sorensen, K. K., Simon-Santamaria, J., McCuskey, R. S. & Smedsrod, B. Liver sinusoidal endothelial cells. Compr. Physiol. 5, 1751–1774 (2015).
Poisson, J. et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017).
Mates, J. M. et al. Mouse liver sinusoidal endothelium eliminates HIV-like particles from blood at a rate of 100 million per minute by a second-order kinetic process. Front. Immunol. 8, 35 (2017).
Smedsrod, B. Clearance function of scavenger endothelial cells. Comp. Hepatol. 3 (Suppl. 1), S22 (2004).
Li, R. et al. Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G71–G81 (2011).
Potente, M. & Makinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 18, 477–494 (2017).
Gouysse, G. et al. Relationship between vascular development and vascular differentiation during liver organogenesis in humans. J. Hepatol. 37, 730–740 (2002).
Geraud, C. et al. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J. Clin. Invest. 127, 1099–1114 (2017).
Lalor, P. F., Lai, W. K., Curbishley, S. M., Shetty, S. & Adams, D. H. Human hepatic sinusoidal endothelial cells can be distinguished by expression of phenotypic markers related to their specialised functions in vivo. World J. Gastroenterol. 12, 5429–5439 (2006).
Geraud, C. et al. Unique cell type-specific junctional complexes in vascular endothelium of human and rat liver sinusoids. PLoS ONE 7, e34206 (2012).
Choi, Y. K., Fallert Junecko, B. A., Klamar, C. R. & Reinhart, T. A. Characterization of cells expressing lymphatic marker LYVE-1 in macaque large intestine during simian immunodeficiency virus infection identifies a large population of nonvascular LYVE-1(+)/DC-SIGN(+) cells. Lymphat Res. Biol. 11, 26–34 (2013).
Tanaka, M. & Iwakiri, Y. The hepatic lymphatic vascular system: structure, function, markers, and lymphangiogenesis. Cell. Mol. Gastroenterol. Hepatol. 2, 733–749 (2016).
Yokomori, H. et al. Lymphatic marker podoplanin/D2-40 in human advanced cirrhotic liver — re-evaluations of microlymphatic abnormalities. BMC Gastroenterol. 10, 131 (2010).
Lai, W. K. et al. Expression of DC-SIGN and DC-SIGNR on human sinusoidal endothelium: a role for capturing hepatitis C virus particles. Am. J. Pathol. 169, 200–208 (2006).
Strauss, O., Phillips, A., Ruggiero, K., Bartlett, A. & Dunbar, P. R. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 7, 44356 (2017).
Milici, A. J., L’Hernault, N. & Palade, G. E. Surface densities of diaphragmed fenestrae and transendothelial channels in different murine capillary beds. Circ. Res. 56, 709–717 (1985).
Satchell, S. C. & Braet, F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. Am. J. Physiol. Renal Physiol. 296, F947–F956 (2009).
Steiniger, B. S. Human spleen microanatomy: why mice do not suffice. Immunology 145, 334–346 (2015).
Bautz, F., Rafii, S., Kanz, L. & Mohle, R. Expression and secretion of vascular endothelial growth factor-A by cytokine-stimulated hematopoietic progenitor cells. Possible role in the hematopoietic microenvironment. Exp. Hematol. 28, 700–706 (2000).
Hashizume, H. et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 156, 1363–1380 (2000).
DeLeve, L. D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 61, 1740–1746 (2015).
Mak, K. M. & Lieber, C. S. Alterations in endothelial fenestrations in liver sinusoids of baboons fed alcohol: a scanning electron microscopic study. Hepatology 4, 386–391 (1984).
Cogger, V. C. et al. Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am. J. Physiol. Heart Circ. Physiol. 310, H1064–H1070 (2016).
O’Reilly, J. N., Cogger, V. C., Fraser, R. & Le Couteur, D. G. The effect of feeding and fasting on fenestrations in the liver sinusoidal endothelial cell. Pathology 42, 255–258 (2010).
Jamieson, H. A. et al. Caloric restriction reduces age-related pseudocapillarization of the hepatic sinusoid. Exp. Gerontol. 42, 374–378 (2007).
Svistounov, D. et al. The relationship between fenestrations, sieve plates and rafts in liver sinusoidal endothelial cells. PLoS ONE 7, e46134 (2012).
Protzer, U., Maini, M. K. & Knolle, P. A. Living in the liver: hepatic infections. Nat. Rev. Immunol. 12, 201–213 (2012).
Braet, F. & Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp. Hepatol. 1, 1 (2002).
Monkemoller, V., Oie, C., Hubner, W., Huser, T. & McCourt, P. Multimodal super-resolution optical microscopy visualizes the close connection between membrane and the cytoskeleton in liver sinusoidal endothelial cell fenestrations. Sci. Rep. 5, 16279 (2015).
Braet, F. et al. Three-dimensional organization of fenestrae labyrinths in liver sinusoidal endothelial cells. Liver Int. 29, 603–613 (2009).
Chen, G. Y. & Nunez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Uhrig, A. et al. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol. 77, 626–633 (2005).
Wu, J. et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 129, 363–374 (2010).
Canton, J., Neculai, D. & Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13, 621–634 (2013).
Elvevold, K. et al. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology 48, 2007–2015 (2008).
Malovic, I. et al. The mannose receptor on murine liver sinusoidal endothelial cells is the main denatured collagen clearance receptor. Hepatology 45, 1454–1461 (2007).
Politz, O. et al. Stabilin-1 and -2 constitute a novel family of fasciclin-like hyaluronan receptor homologues. Biochem. J. 362, 155–164 (2002).
Bashirova, A. A. et al. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Exp. Med. 193, 671–678 (2001).
Liu, W. et al. Characterization of a novel C-type lectin-like gene, LSECtin: demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J. Biol. Chem. 279, 18748–18758 (2004).
Lin, G. et al. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 77, 1337–1346 (2003).
Marzi, A. et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 78, 12090–12095 (2004).
Gramberg, T. et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 340, 224–236 (2005).
Li, Y. et al. C-Type lectin LSECtin interacts with DC-SIGNR and is involved in hepatitis C virus binding. Mol. Cell Biochem. 327, 183–190 (2009).
Ganesan, L. P. et al. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 7, e1002281 (2011).
Hellevik, T. et al. Transport of residual endocytosed products into terminal lysosomes occurs slowly in rat liver endothelial cells. Hepatology 28, 1378–1389 (1998).
Cormier, E. G. et al. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl Acad. Sci. USA 101, 7270–7274 (2004).
Breiner, K. M., Schaller, H. & Knolle, P. A. Endothelial cell-mediated uptake of a hepatitis B virus: a new concept of liver targeting of hepatotropic microorganisms. Hepatology 34, 803–808 (2001).
Rowe, I. A. et al. Paracrine signals from liver sinusoidal endothelium regulate hepatitis C virus replication. Hepatology 59, 375–384 (2014).
Giugliano, S. et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 148, 392–402 e313 (2015).
Limmer, A. et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T cell tolerance. Nat. Med. 6, 1348–1354 (2000).
Burgdorf, S., Kautz, A., Bohnert, V., Knolle, P. A. & Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316, 612–616 (2007).
Limmer, A. et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur. J. Immunol. 35, 2970–2981 (2005).
Diehl, L. et al. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 47, 296–305 (2008).
Schurich, A. et al. Distinct kinetics and dynamics of cross-presentation in liver sinusoidal endothelial cells compared to dendritic cells. Hepatology 50, 909–919 (2009).
Hochst, B. et al. Liver sinusoidal endothelial cells contribute to CD8 T cell tolerance toward circulating carcinoembryonic antigen in mice. Hepatology 56, 1924–1933 (2012).
Schurich, A. et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J. Immunol. 184, 4107–4114 (2010).
Bottcher, J. P. et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 8, 1318–1327 (2014).
Lohse, A. W. et al. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology 110, 1175–1181 (1996).
Knolle, P. A. et al. Induction of cytokine production in naive CD4(+) T cells by antigen- presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology 116, 1428–1440 (1999).
Carambia, A. et al. TGF-beta-dependent induction of CD4(+)CD25(+)Foxp3(+) Tregs by liver sinusoidal endothelial cells. J. Hepatol. 61, 594–599 (2014).
Carambia, A. et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J. Hepatol. 58, 112–118 (2013).
Luth, S. et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J. Clin. Invest. 118, 3403–3410 (2008).
Carambia, A. et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J. Hepatol. 62, 1349–1356 (2015).
Tang, L. et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T cell immune response. Gastroenterology 137, 1498–1508.e5 (2009).
Lalor, P. F., Shields, P., Grant, A. & Adams, D. H. Recruitment of lymphocytes to the human liver. Immunol. Cell Biol. 80, 52–64 (2002).
Nourshargh, S. & Alon, R. Leukocyte migration into inflamed tissues. Immunity 41, 694–707 (2014).
McEver, R. P. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 107, 331–339 (2015).
Tanaka, Y. et al. T cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature 361, 79–82 (1993).
Campbell, J. J. et al. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 279, 381–384 (1998).
Muller, W. A. Transendothelial migration: unifying principles from the endothelial perspective. Immunol. Rev. 273, 61–75 (2016).
Wong, J. et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Invest. 99, 2782–2790 (1997).
Adams, D. H., Hubscher, S. G., Fisher, N. C., Williams, A. & Robinson, M. Expression of E-selectin and E-selectin ligands in human liver inflammation. Hepatology 24, 533–538 (1996).
Campbell, J. J., Qin, S., Bacon, K. B., Mackay, C. R. & Butcher, E. C. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J. Cell Biol. 134, 255–266 (1996).
Lalor, P. F. & Adams, D. H. The liver: a model of organ-specific lymphocyte recruitment. Expert Rev. Mol. Med. 4, 1–16 (2002).
Lalor, P. F. et al. Association between receptor density, cellular activation, and transformation of adhesive behavior of flowing lymphocytes binding to VCAM-1. Eur. J. Immunol. 27, 1422–1426 (1997).
Habtezion, A., Nguyen, L. P., Hadeiba, H. & Butcher, E. C. Leukocyte trafficking to the small intestine and colon. Gastroenterology 150, 340–354 (2016).
Grant, A. J., Lalor, P. F., Hubscher, S. G., Briskin, M. & Adams, D. H. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology 33, 1065–1072 (2001).
Grant, A. J., Lalor, P. F., Salmi, M., Jalkanen, S. & Adams, D. H. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 359, 150–157 (2002).
Barreiro, O. et al. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood 105, 2852–2861 (2005).
Barreiro, O. et al. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J. Cell Biol. 183, 527–542 (2008).
Wadkin, J. C. R. et al. CD151 supports VCAM-1 mediated lymphocyte adhesion to liver endothelium and is upregulated in chronic liver disease and hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 02016, G138–G149 (2017).
Salmi, M., Tohka, S., Berg, E. L., Butcher, E. C. & Jalkanen, S. Vascular adhesion protein 1 (VAP-1) mediates lymphocyte subtype-specific, selectin-independent recognition of vascular endothelium in human lymph nodes. J. Exp. Med. 186, 589–600 (1997).
Lalor, P. F. et al. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J. Immunol. 169, 983–992 (2002).
Bonder, C. S. et al. Rules of recruitment for Th1 and th2 lymphocytes in inflamed liver: a role for alpha-4 integrin and vascular adhesion protein-1. Immunity 23, 153–163 (2005).
Lalor, P. F. et al. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-kappaB-dependent increase in lymphocyte adhesion. Hepatology 45, 465–474 (2007).
Weston, C. J. et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Invest. 125, 501–520 (2015).
Liaskou, E. et al. Regulation of mucosal addressin cell adhesion molecule 1 expression in human and mice by vascular adhesion protein 1 amine oxidase activity. Hepatology 53, 661–672 (2011).
Jung, M. Y., Park, S. Y. & Kim, I. S. Stabilin-2 is involved in lymphocyte adhesion to the hepatic sinusoidal endothelium via the interaction with alphaMbeta2 integrin. J. Leukoc. Biol. 82, 1156–1165 (2007).
Salmi, M., Koskinen, K., Henttinen, T., Elima, K. & Jalkanen, S. CLEVER-1 mediates lymphocyte transmigration through vascular and lymphatic endothelium. Blood 104, 3849–3857 (2004).
Shetty, S. et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J. Immunol. 186, 4147–4155 (2011).
Phillipson, M. et al. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 203, 2569–2575 (2006).
Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 15, 692–704 (2015).
Wohlleber, D. et al. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep. 2, 478–487 (2012).
Guidotti, L. G. et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 161, 486–500 (2015).
Carman, C. V. et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity 26, 784–797 (2007).
Patten, D. A. et al. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment- a new step in migration. Hepatology 65, 294–309 (2016).
Moser, B. & Willimann, K. Chemokines: role in inflammation and immune surveillance. Ann Rheum. Dis. 63 (Suppl 2), ii84–ii89 (2004).
Rot, A. Chemokine patterning by glycosaminoglycans and interceptors. Front. Biosci. 15, 645–660 (2010).
Adams, D. H. et al. Hepatic expression of macrophage inflammatory protein-1 alpha and macrophage inflammatory protein-1 beta after liver transplantation. Transplantation 61, 817–825 (1996).
Afford, S. C. et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J. Pathol. 186, 82–89 (1998).
Shields, P. L. et al. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol. 163, 6236–6243 (1999).
Yoong, K. F. et al. Expression and function of CXC and CC chemokines in human malignant liver tumors: a role for human monokine induced by gamma-interferon in lymphocyte recruitment to hepatocellular carcinoma. Hepatology 30, 100–111 (1999).
Eksteen, B. et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J. Exp. Med. 200, 1511–1517 (2004).
Goddard, S. et al. Differential expression of chemokines and chemokine receptors shapes the inflammatory response in rejecting human liver transplants. Transplantation 72, 1957–1967 (2001).
Curbishley, S. M., Eksteen, B., Gladue, R. P., Lalor, P. & Adams, D. H. CXCR3 activation promotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am. J. Pathol. 167, 887–899 (2005).
Hokeness, K. L. et al. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J. Virol. 81, 1241–1250 (2007).
Heydtmann, M. et al. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J. Immunol. 174, 1055–1062 (2005).
Heydtmann, M. & Adams, D. H. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology 49, 676–688 (2009).
Geissmann, F. et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 3, e113 (2005).
Schrage, A. et al. Enhanced T cell transmigration across the murine liver sinusoidal endothelium is mediated by transcytosis and surface presentation of chemokines. Hepatology 48, 1262–1272 (2008).
Neumann, K. et al. Chemokine transfer by liver sinusoidal endothelial cells contributes to the recruitment of CD4+ T cells into the murine liver. PLoS ONE 10, e0123867 (2015).
Eksteen, B., Afford, S. C., Wigmore, S. J., Holt, A. P. & Adams, D. H. Immune-mediated liver injury. Semin. Liver Dis. 27, 351–366 (2007).
Knolle, P. A. & Thimme, R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 146, 1193–1207 (2014).
Makarova-Rusher, O. V., Medina-Echeverz, J., Duffy, A. G. & Greten, T. F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 62, 1420–1429 (2015).
Oo, Y. H. et al. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J. Hepatol. 57, 1044–1051 (2012).
Oo, Y. H. et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J. Immunol. 184, 2886–2898 (2010).
Eksteen, B. et al. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J. Immunol. 177, 593–603 (2006).
Bertolino, P. et al. Early intrahepatic antigen-specific retention of naive CD8+ T cells is predominantly ICAM-1/LFA-1 dependent in mice. Hepatology 42, 1063–1071 (2005).
John, B. & Crispe, I. N. Passive and active mechanisms trap activated CD8+ T cells in the liver. J. Immunol. 172, 5222–5229 (2004).
Miles, A., Liaskou, E., Eksteen, B., Lalor, P. F. & Adams, D. H. CCL25 and CCL28 promote alpha4 beta7-integrin-dependent adhesion of lymphocytes to MAdCAM-1 under shear flow. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1257–G1267 (2008).
Edwards, S., Lalor, P. F., Nash, G. B., Rainger, G. E. & Adams, D. H. Lymphocyte traffic through sinusoidal endothelial cells is regulated by hepatocytes. Hepatology 41, 451–459 (2005).
Holt, A. P. et al. Liver myofibroblasts regulate infiltration and positioning of lymphocytes in human liver. Gastroenterology 136, 705–714 (2009).
Bruns, T. et al. CMV infection of human sinusoidal endothelium regulates hepatic T cell recruitment and activation. J. Hepatol. 63, 38–49 (2015).
Shetty, S. et al. Recruitment mechanisms of primary and malignant B cells to the human liver. Hepatology 56, 1521–1531 (2012).
Wang, J. et al. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 358, 111–116 (2017).
McDonald, B. et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 205, 915–927 (2008).
Moles, A. et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 60, 782–791 (2014).
Wang, J. & Kubes, P. A. Reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 165, 668–678 (2016).
Tacke, F. & Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60, 1090–1096 (2014).
Dal-Secco, D. et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 212, 447–456 (2015).
Aspinall, A. I. et al. CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of CD16(+) monocytes across human liver sinusoidal endothelium. Hepatology 51, 2030–2039 (2010).
Liaskou, E. et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 57, 385–398 (2013).
Bradfield, P. F. et al. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood 110, 2545–2555 (2007).
Randolph, G. J., Sanchez-Schmitz, G., Liebman, R. M. & Schakel, K. The CD16(+) (FcgammaRIII(+)) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J. Exp. Med. 196, 517–527 (2002).
Zimmermann, H. W. et al. Bidirectional transendothelial migration of monocytes across hepatic sinusoidal endothelium shapes monocyte differentiation and regulates the balance between immunity and tolerance in liver. Hepatology 63, 233–246 (2016).
Zannetti, C. et al. Characterization of the inflammasome in human kupffer cells in response to synthetic agonists and pathogens. J. Immunol. 197, 356–367 (2016).
Tilg, H., Moschen, A. R. & Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 64, 955–965 (2016).
Knolle, P. A. et al. Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 24, 824–829 (1996).
Xu, B. et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am. J. Pathol. 163, 1275–1289 (2003).
Ford, A. J., Jain, G. & Rajagopalan, P. Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater. 24, 220–227 (2015).
Arii, S. & Imamura, M. Physiological role of sinusoidal endothelial cells and kupffer cells and their implication in the pathogenesis of liver injury. J. Hepatobiliary Pancreat Surg. 7, 40–48 (2000).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Xie, G. et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 142, 918–927.e6 (2012).
Warren, A. et al. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 44, 1182–1190 (2006).
Rautou, P. E. et al. Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology 143, 166–176.e6 (2012).
Yoong, K. F., McNab, G., Hubscher, S. G. & Adams, D. H. Vascular adhesion protein-1 and ICAM-1 support the adhesion of tumor-infiltrating lymphocytes to tumor endothelium in human hepatocellular carcinoma. J. Immunol. 160, 3978–3988 (1998).
McMahan, R. H., Porsche, C. E., Edwards, M. G. & Rosen, H. R. Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: insights into non-alcoholic fatty liver disease. PLoS ONE 11, e0159217 (2016).
Connolly, M. K. et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J. Immunol. 185, 2200–2208 (2010).
Lalor, P. F. et al. Vascular adhesion protein-1 as a potential therapeutic target in liver disease. Ann. N Y Acad. Sci. 1110, 485–496 (2007).
de Graaf, K. L. et al. (2018) NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody, in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatology Communications 2, 492–503 (2018).
Lefebvre, E. et al. Pharmacokinetics, safety, and CCR2/CCR5 antagonist activity of cenicriviroc in participants with mild or moderate hepatic impairment. Clin. Transl Sci. 9, 139–148 (2016).
Hirschfield, G. M., Karlsen, T. H., Lindor, K. D. & Adams, D. H. Primary sclerosing cholangitis. Lancet 382, 1587–1599 (2013).
Alidori, S. et al. Deconvoluting hepatic processing of carbon nanotubes. Nat. Commun. 7, 12343 (2016).
Zuo, Y. et al. Novel roles of liver sinusoidal endothelial cell lectin in colon carcinoma cell adhesion, migration and in-vivo metastasis to the liver. Gut 62, 1169–1178 (2013).
Na, H. et al. Novel roles of DC-SIGNR in colon cancer cell adhesion, migration, invasion, and liver metastasis. J. Hematol. Oncol. 10, 28 (2017).
Couvelard, A., Scoazec, J. Y. & Feldmann, G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am. J. Pathol. 143, 738–752 (1993).
Miyao, M. et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Invest. 95, 1130–1144 (2015).
Wang, B. Y., Ju, X. H., Fu, B. Y., Zhang, J. & Cao, Y. X. Effects of ethanol on liver sinusoidal endothelial cells-fenestrae of rats. Hepatobiliary Pancreat. Dis. Int. 4, 422–426 (2005).
Horn, T., Christoffersen, P. & Henriksen, J. H. Alcoholic liver injury: defenestration in noncirrhotic livers — a scanning electron microscopic study. Hepatology 7, 77–82 (1987).
Clark, S. A. et al. Defenestration of hepatic sinusoids as a cause of hyperlipoproteinaemia in alcoholics. Lancet 2, 1225–1227 (1988).
Steffan, A. M. et al. Mouse hepatitis virus type 3 infection provokes a decrease in the number of sinusoidal endothelial cell fenestrae both in vivo and in vitro. Hepatology 22, 395–401 (1995).
Ito, Y. et al. Age-related changes in the hepatic microcirculation in mice. Exp. Gerontol. 42, 789–797 (2007).
Koudelkova, P., Weber, G. & Mikulits, W. Liver sinusoidal endothelial cells escape senescence by loss of p19ARF. PLoS ONE 10, e0142134 (2015).
Onori, P. et al. Hepatic microvascular features in experimental cirrhosis: a structural and morphometrical study in CCl4-treated rats. J. Hepatol. 33, 555–563 (2000).
Fernandez, M. et al. Angiogenesis in liver disease. J. Hepatol. 50, 604–620 (2009).
Corpechot, C. et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 35, 1010–1021 (2002).
Rosmorduc, O. et al. Hepatocellular hypoxia-induced vascular endothelial growth factor expression and angiogenesis in experimental biliary cirrhosis. Am. J. Pathol. 155, 1065–1073 (1999).
Ding, B. S. et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 505, 97–102 (2014).
Peralta, C., Jimenez-Castro, M. B. & Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J. Hepatol. 59, 1094–1106 (2013).
Yazdani, H. O. et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J. Hepatol. 68, 130–139 (2017).
Xie, G. et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 62, 299–309 (2013).
Rockey, D. C. & Chung, J. J. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology 114, 344–351 (1998).
Zhuang, P. Y. et al. Higher proliferation of peritumoral endothelial cells to IL-6/sIL-6R than tumoral endothelial cells in hepatocellular carcinoma. BMC Cancer 15, 830 (2015).
Geraud, C. et al. Endothelial transdifferentiation in hepatocellular carcinoma: loss of Stabilin-2 expression in peri-tumourous liver correlates with increased survival. Liver Int. 33, 1428–1440 (2013).
Yang, Y. et al. Discontinuation of anti-VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat. Commun. 7, 12680 (2016).
Acknowledgements
The work of the authors is funded by the National Institute for Health Research (NIHR) Birmingham Biomedical Research Centre at the University Hospitals Birmingham National Health Service (NHS) Foundation Trust and the University of Birmingham. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the UK Department of Health.
Author information
Authors and Affiliations
Contributions
S.S. and P.F.L. researched data for the article, made substantial contributions to discussion of content and wrote the manuscript. D.H.A. reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing financial interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shetty, S., Lalor, P.F. & Adams, D.H. Liver sinusoidal endothelial cells — gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol 15, 555–567 (2018). https://doi.org/10.1038/s41575-018-0020-y
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-018-0020-y
This article is cited by
-
Construction of in vitro liver-on-a-chip models and application progress
BioMedical Engineering OnLine (2024)
-
Single-cell transcriptome reveals a novel mechanism of C-Kit+-liver sinusoidal endothelial cells in NASH
Cell & Bioscience (2024)
-
Heterogeneity of hepatocyte dynamics restores liver architecture after chemical, physical or viral damage
Nature Communications (2024)
-
Extra-corporeal non-liver transplant therapies for acute liver failure: Focus on plasma exchange and continuous renal replacement therapy
Indian Journal of Gastroenterology (2024)
-
A spatiotemporal atlas of cholestatic injury and repair in mice
Nature Genetics (2024)
