
Abstract
Liver cancer remains a global health challenge, with an estimated incidence of >1 million cases by 2025. Hepatocellular carcinoma (HCC) is the most common form of liver cancer and accounts for ~90% of cases. Infection by hepatitis B virus and hepatitis C virus are the main risk factors for HCC development, although non-alcoholic steatohepatitis associated with metabolic syndrome or diabetes mellitus is becoming a more frequent risk factor in the West. Moreover, non-alcoholic steatohepatitis-associated HCC has a unique molecular pathogenesis. Approximately 25% of all HCCs present with potentially actionable mutations, which are yet to be translated into the clinical practice. Diagnosis based upon non-invasive criteria is currently challenged by the need for molecular information that requires tissue or liquid biopsies. The current major advancements have impacted the management of patients with advanced HCC. Six systemic therapies have been approved based on phase III trials (atezolizumab plus bevacizumab, sorafenib, lenvatinib, regorafenib, cabozantinib and ramucirumab) and three additional therapies have obtained accelerated FDA approval owing to evidence of efficacy. New trials are exploring combination therapies, including checkpoint inhibitors and tyrosine kinase inhibitors or anti-VEGF therapies, or even combinations of two immunotherapy regimens. The outcomes of these trials are expected to change the landscape of HCC management at all evolutionary stages.
Similar content being viewed by others
Introduction
Liver cancer remains a global health challenge and its incidence is growing worldwide1,2. It is estimated that, by 2025, >1 million individuals will be affected by liver cancer annually3. Hepatocellular carcinoma (HCC) is the most common form of liver cancer and accounts for ~90% of cases. Hepatitis B virus (HBV) infection is the most prominent risk factor for HCC development, accounting for ~50% of cases4. The risk attributed to hepatitis C virus (HCV) infection has substantially decreased owing to patients achieving sustained virological response (SVR) with antiviral drugs5. Nonetheless, patients with cirrhosis are still considered to be at high risk for HCC incidence even after HCV clearance. Non-alcoholic steatohepatitis (NASH), associated with metabolic syndrome or diabetes mellitus, is becoming the fastest growing aetiology of HCC, particularly in the West6. Additionally, reports on mutational signatures have established aristolochic acid and tobacco as potential pathogenetic cofactors in HCC7.
The molecular pathogenesis of HCC varies according to the distinct genotoxic insults and aetiologies. Although our understanding of the pathophysiology and drivers of the disease has improved, this knowledge is yet to be translated into clinical practice. Approximately 25% of HCC tumours present actionable mutations; however, the prevalence of most mutations is <10%, thereby complicating proof-of-concept studies7,8. Indeed, dominant mutational drivers in HCC, such as TERT, TP53 and CTNNB1, remain undruggable9. In addition, the translation of molecular and immune classes into biomarkers that guide therapies is still under investigation. Currently, specific advancements in our understanding of the mechanisms underlying NASH-associated HCC have provided new insights into the contributions of the tumour microenvironment, particularly the immune system and platelet activation, in the pathophysiology of this disease10,11.
The diagnosis of HCC is usually based on non-invasive criteria, although there is a growing need for molecular characterization of the tumour using tissue biopsies in clinical practice12,13. In terms of prevention, beyond vaccines preventing HBV infection and anti-viral therapies for HBV and HCV infection, cumulative data support the preventive role of coffee and aspirin14. The management of HCC has markedly improved since the early 2010s8,12,13,15. Hepatic resection and liver transplantation have been the mainstay curative treatments in HCC cases. Refinements in patient selection have resulted in enhanced surgical resection outcomes and remarkable 10-year post-liver transplantation survival rates for tumours down-staged beyond Milan criteria12,16. Local ablation with radiofrequency remains the backbone of image-guided ablation for non-surgical early-stage HCC, despite progress in other techniques15. Adjuvant therapies to preclude relapse, following these potentially curative approaches, are an unmet medical need, as randomized controlled trials (RCTs) have so far yielded negative results. For intermediate-stage HCC, transarterial chemoembolization (TACE) has been the most widely used treatment and the standard of care over the past two decades17. Transarterial radioembolization (TARE) has shown efficacy in phase II investigations18 but has not been established as a primary standard of care by guidelines. Other loco-regional devices or radiation oncology approaches are not expected to improve the intermediate treatment armamentarium in the short term.
Currently, systemic therapies, including immune-checkpoint inhibitors (ICIs), tyrosine kinase inhibitors (TKIs) and monoclonal antibodies, have challenged the use of conventional therapies for HCC. Approximately, 50–60% of patients with HCC are estimated to be exposed to systemic therapies in their lifespan, particularly in advanced stages of the disease8. The field has witnessed substantial progress in the development of systemic therapies in the past 5 years, with studies reporting a marked increase in overall survival and in the quality of life of patients8. For example, the natural history of advanced-stage HCC cases involves a median survival of ~8 months and the approved combination of atezolizumab (anti-PDL1 antibody) and bevacizumab (anti-VEGF antibody) has more than doubled this life expectancy and improved the patient-reported outcomes19. Sorafenib20 and lenvatinib21 remain as the most effective single-drug therapies. In case of progression to single-agent regimens, regorafenib22, cabozantinib23 and ramucirumab24 have also proven improved survival benefits. Single-agent ICIs provide substantial clinical benefits in 15–20% of responders but, so far, biomarkers have failed to identify this group25,26. Furthermore, phase III trials investigating the efficacy of combination therapy, that is, combining ICIs with TKIs or combining PD1/PDL1 axis inhibitors with CTLA4 inhibitors, are ongoing. The results of these trials are expected to change the landscape of HCC management at all evolutionary stages.
This Primer provides an update on the advancements in HCC pathogenetic mechanisms and its treatment since our first review1. We discuss the increasing contribution of non-alcoholic fatty liver disease (NAFLD) and NASH to the development of HCC as well as the specific key molecular mechanisms associated with this risk factor. Additionally, we summarize the current knowledge and trends in epidemiology, diagnosis, screening and management. In particular, we describe the evidence-based data generated with new therapies and the prospects of novel combination therapies in the adjuvant setting as well as in intermediate-stage and advanced-stage HCC. Finally, we discuss the role of biomarkers, liquid biopsy and patient-reported outcomes in the future management of this devastating disease.
Epidemiology
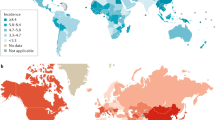
Liver cancer is the sixth most common cancer worldwide, with 841,080 new liver cancer cases in 2018, and the fourth leading cause of cancer-related death globally3 (Fig. 1). The highest incidence and mortality of HCC are observed in East Asia and Africa, although HCC incidence and mortality are increasing in different parts of Europe and in the USA27. Indeed, Surveillance Epidemiology End Results (SEER) reported HCC as the fastest increasing cause of cancer-related death in the USA since the early 2000s and HCC is projected to become the third leading cause of cancer-related death by 2030 if these trends continue28.

The incidence and major aetiological factors involved in hepatocarcinogenesis are depicted in this figure. The highest incidence of hepatocellular carcinoma (HCC) is observed in East Asia, with Mongolia demonstrating the highest incidence of HCC worldwide. Hepatitis B virus (HBV) is the major aetiological factor in most parts of Asia (except Japan), South America and Africa; Hepatitis C virus (HCV) is the predominant causative factor in Western Europe, North America and Japan, and alcohol intake is the aetiological factor in Central and Eastern Europe. Non-alcoholic steatohepatitis (NASH), the main aetiology included in the category ‘Other’, is a rapidly increasing risk factor that is expected to become the predominant cause of HCC in high income regions in the near future. ASR, age-standardized incidence rate. Data from refs3,129. Reprinted from ref.3, Global Cancer Observatory, World Health Organization, Estimated age-standardized incidence rates (World) in 2020, liver, both sexes, all ages, Copyright (2020) (https://gco.iarc.fr/today/online-analysis-map?v=2020&mode=population&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=11&type=0&statistic=5&prevalence=0&population_group=earth&color_palette=default&map_scale=quantile&map_nb_colors=5&continent=0&rotate=%255B10%252C0%255D).
Risk factors
Over 90% of HCC cases occur in the setting of chronic liver disease. Cirrhosis from any aetiology is the strongest risk factor for HCC12,13. HCC is the leading cause of death in patients with cirrhosis, with an annual HCC incidence of 1–6%29. The major risk factors for HCC include chronic alcohol consumption, diabetes or obesity-related NASH, and infection by HBV or HCV (Fig. 1). Other less prevalent risk factors for HCC include cirrhosis from primary biliary cholangitis, haemochromatosis and α1-antitrypsin deficiency. Indeed, patients developing cirrhosis from haemochromatosis are at a particularly high risk of HCC, with up to 45% developing HCC in their life span30.
Hepatitis B virus infection
HBV infection accounts for ~60% of HCC cases in Asia and Africa and 20% of cases in the West4 (Fig. 1). HBV is a DNA virus that can integrate into the host genome inducing insertional mutagenesis, leading to oncogene activation31. HBV increases the risk of HCC even in the absence of cirrhosis, although most patients with HBV-induced HCC have cirrhosis at presentation. The high prevalence of endemic HBV infection in East Asia has resulted in a risk of HCC exceeding cost-effectiveness thresholds in men (40 years of age) and in women (50 years of age), thereby justifying surveillance programmes. In Africa, patients in their early 30s or 40s present with HCC, likely because of exposure to aflatoxin B1, which acts synergistically with HBV to increase the risk of HCC32. HBV vaccination programmes have led to a decrease in HCC incidence in some parts of Asia, although many jurisdictions are yet to implement universal vaccination programmes33.
Hepatitis C virus infection
Chronic HCV infection is the most common underlying liver disease among patients with HCC in North America, Europe and Japan4 (Fig. 1). Unlike HBV, HCV is an RNA virus that does not integrate into the host genome and, therefore, the risk of HCC is primarily limited to those who develop cirrhosis or chronic liver damage with bridging fibrosis. With the use of direct-acting antiviral (DAA) therapy, an increasing proportion of patients with HCV infection have been successfully treated to achieve an SVR, resulting in a 50–80% reduction in the risk of HCC5. However, several patients, particularly, racial minorities, ethnic minorities or people from low socioeconomic regions, are yet to be tested for HCV and remain unaware of their infection34. Additionally, patients with HCV-induced cirrhosis continue to have a persistent risk of developing HCC (>2% per year) even after SVR and should therefore remain under close surveillance35,36.
Hepatitis D virus infection
Hepatitis D virus (HDV) is an RNA virus that requires the presence of HBV surface antigens for its replication and, therefore, for infectivity. HDV is estimated to affect 20–40 million people globally and is associated with a more severe course of liver disease, including increased fibrosis and risk of cirrhosis, than patients with HBV infection alone. Similarly, several cohort studies suggest that HBV/HDV co-infection is associated with an increased risk of HCC compared with HBV infection alone. In one of the largest studies to date, the risk of HCC was significantly higher among those with acute HDV infection (RR 6.1, 95% CI 2.8–11.7) or chronic HDV infection (RR 3.9, 95% CI 1.6–7.2) than among those with HBV infection alone37.
Alcohol
Excessive alcohol intake causes alcoholic liver disease, cirrhosis and HCC. Currently, an increasing number of persons have cirrhosis from chronic alcohol consumption or NASH. Alcohol-related cirrhosis has an annual incidence ranging from 1% in population-based studies to 2–3% in tertiary care referral centres and accounts for ~15–30% of HCC cases depending on the geographical region38. Chronic alcohol intake can also increase the risk of HCC from other aetiologies; for example, several studies reveal an increased risk of HCC in HBV carriers who consume alcohol compared with those who do not consume alcohol39. Although alcohol consumption shares many pathophysiological processes with other forms of cirrhosis, in particular NASH, there is evidence supporting distinct alcohol-specific pro-tumorigenic mechanisms in patients.
NASH
Another common aetiological factor for cirrhosis in people is NASH, which is the precursor step in the development of HCC in patients with diabetes mellitus or obesity. Owing to the increasing prevalence of obesity, NASH has become the most common cause of cirrhosis in most regions of the world. Since 2010, the proportion of HCC attributed to NASH has rapidly increased, currently representing 15–20% of cases in the West6. Furthermore, the population attributable fraction of metabolic syndrome and NASH is likely to be >20% owing to its co-existence in patients with other liver diseases40. Although the annual incidence of HCC is lower in NASH-related cirrhosis (1–2% per year) than in viral-mediated cirrhosis (3–5% per year), the incidence is >1.1 per 100 person-years, indicating that surveillance is cost-effective and should therefore be implemented41. Several studies have demonstrated that 25–30% of NASH-associated HCC cases occur in the absence of cirrhosis, which hampers the applicability of surveillance programmes currently targeting only patients with cirrhosis. However, a cohort study from the national Veterans Affairs health system found that the annual incidence of HCC in individuals with non-cirrhotic NASH falls below the cost-effectiveness threshold, thus advising against surveillance41,42.
Age, sex and other factors
Several sociodemographic characteristics have been associated with HCC, particularly in patients with cirrhosis. Ageing is a strong risk factor, with the highest age-specific incidence reported in individuals >70 years of age43. Furthermore, HCC also has a strong male predominance (male to female ratio of 2–3:1), likely related to a clustering of risk factors among men as well as differences in sex hormones44. Studies have reported a higher incidence of HCC among racial or ethnic minorities, in particular Hispanics, than among white individuals. This discrepancy in incidence might partly be due to the high incidence of single-nucleotide variants in PNPLA3, linked to NASH-associated HCC45. Epidemiological studies have also highlighted the increased risk of HCC associated with smoking46. However, the role of diet in moderating the risk of HCC remains unclear, with the exception of studies showing a preventive effect of coffee and aspirin47.
Mechanisms/pathophysiology
The pathophysiology of HCC is a complex multistep process. The interplay of various factors is at the origin of the early steps of hepatocyte malignant transformation and HCC development. These factors include a genetic predisposition, reciprocal interactions between viral and non-viral risk factors, the cellular microenvironment and various immune cells, and the severity of the underlying chronic liver disease. An altered microenvironment is a key enabling characteristic of cancer and is known to participate in all stages of malignant progression, from the initial transformation phases, through to invasion and, ultimately, to metastasis. In our previous Primer, we described the main oncogenic drivers and signalling pathways involved in the initiation, development and progression of HCC1. Herein, we explain in detail our current understanding of the mechanisms underlying NASH-associated HCC.
Cell of origin
The cell of origin of HCC is debated. Similar to any type of cancer, the cell of origin could be a liver stem cell, a transit amplifying population or mature hepatocytes. In general, the presence and role of stem cells in the liver is in itself debatable. Moreover, mature hepatocytes are long-lived cells and retain considerable proliferative potential in response to injury. Many mouse models support the possibility that HCC originates in transformed mature hepatocytes, although others posit that putative liver stem cells could be the source48. Paradoxically, intrahepatic cholangiocarcinomas and tumours showing mixed HCC or cholangiocarcinoma morphology often seem to arise from mature hepatocytes, emphasizing the concepts of metaplasia and cell plasticity (that is, transdifferentiation). This finding confirms the notion that the morphology and epigenetic landscape of a tumour does not necessarily reflect its cell of origin49,50.
Cancer driver gene mutations in HCC
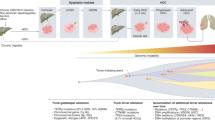
High throughput next-generation sequencing has enabled the identification of cancer driver genes with oncogenic functions or tumour suppressive functions that are recurrently altered in HCC. Telomerase activation via TERT promoter mutations, viral insertions, chromosome translocation or gene amplification are the most frequent driver gene alterations, observed in ~80% of HCC7,51. Studies have demonstrated the activation of the Wnt–β-catenin signalling pathway in 30–50% of the cases, caused by mutations in CTNNB1 (encoding β-catenin), AXIN1 or APC (inhibitors of Wnt pathway) inactivation7,51. Other frequent mutations or genetic alterations are found in TP53, RB1, CCNA2, CCNE1, PTEN, ARID1A, ARID2, RPS6KA3 or NFE2L2, all of which alter cell cycle control. Additionally, variants in genes involved in epigenetic regulation, oxidative stress, and the AKT–mTOR and MAPK pathways have been implicated in HCC (see previous Primer1). Furthermore, recurrent focal chromosome amplifications in CCND1, FGF19, VEGFA, MYC or MET leading to over-expression result in the activation of various oncogenic signalling pathways, including of receptor tyrosine kinases52. Although cancer driver gene mutations accumulate randomly, specific genes are related to precise molecular HCC subclasses, defined by transcriptomic profiles and histological phenotypes8,9,53 (Fig. 2). Overall, only ~20–25% of patients with HCC have at least one potential actionable mutation as per current standards7,8,54.

Hepatocellular carcinoma (HCC) can be classified into two major molecular groups based on transcriptomic-based phenotypic classes52,67,68,69,70. The proliferation class is characterized by more aggressive tumours with poor histological differentiation, high vascular invasion and increased levels of α-fetoprotein (AFP)53. This class can be further divided into two subclasses: S1 or iCluster 3 (refs67,68), characterized by Wnt–TGFβ activation, which drives an immune-exhausted phenotype71, and S2 or iCluster 1 (refs67,68), characterized by a progenitor-like phenotype, with the expression of stem cell markers (CK19, EPCAM) and activated IGF2 and EPCAM signalling pathways53. Hepatitis B virus (HBV)-associated tumours present frequent activation of classical cell proliferation pathways such as PI3K–AKT–mTOR, RAS–MAPK, MET and IGF cascades. In addition, frequent TP53 mutations, high chromosomal instability and global DNA hypomethylation represent additional hallmarks of this class. The non-proliferation class52,67,68,69,70 is characterized by less aggressive tumours with well to moderate histological differentiation, low levels of AFP and less frequent vascular invasion53. These tumours are related to non-alcoholic steatohepatitis (NASH), alcoholic steatohepatitis and hepatitis C virus (HCV) infection. Distinct subgroups have been characterized within this class: the WNT–β-catenin CTNNB1 subclass presents frequent CTNNB1 mutations and activation of the WNT–β-catenin signalling pathway, which drives an immune-excluded phenotype with low immune infiltration52,70,71, and the interferon subclass presents a highly activated IL6–JAK–STAT signalling pathway, with a more inflamed tumour microenvironment. This class present chromosomal stability with frequent TERT promoter mutations. Data from refs1,7,8,9,52,53,66,67,68,69,70,71. FLC, fibrolamellar carcinoma; IHC, immunohistochemistry; miRNA, microRNA; TCGA, The Cancer Genome Atlas.
In addition to cancer driver mutations, the cooperation of risk factors is well described in the pathogenesis of HCC. For example, the toxic effect of aflatoxin B1 is potentiated by HBV infection, particularly in patients with a null polymorphism of GSTT1 (refs55,56). In addition, polymorphisms in PNPLA3, TM6SF2 and HSD17B13 have been identified to be associated with the severity of NASH and HCC incidence, specifically in patients with high chronic alcohol intake57,58.
Viral infection-associated molecular alterations
The most frequent site of HBV-mediated insertional mutagenesis is located within the TERT promoter, leading to an overexpression of telomerase, the enzyme responsible for the maintenance of telomere length59. The activation of telomerase prevents the erosion of the chromosomes that physiologically occur at each cell division during ageing. The ectopic activation of telomerase protects cells from senescence and promotes cell transformation60. Other recurrent insertions associated with HBV were identified to activate potent oncogenes, such as CCNA2 or CCNE1, involved in cell cycle control. These oncogenic alterations induce replicative stress and complex rearrangements throughout the genome61. In a small set of patients with HCC, adeno-associated virus 2 demonstrated a similar insertional oncogenic mutagenesis, with a common hot spot of viral insertion within the TERT promoter, CCNA2 and CCNE1 (ref.62). These observations illustrate that specific oncogenes, activated by viral infection, act as early facilitators of hepatocyte transformation. By contrast, HCV infection does not drive a strong, direct oncogenic effect and the induction of mutations results from the oxidative stress caused by chronic inflammation.
HCC-related mutational signatures
During the development of chronic liver disease and cirrhosis, which are the basis for the onset of HCC in most cases, hepatocytes progressively accumulate numerous genetic mutations and epigenetic changes. During this process, several risk factors inducing DNA mutations are associated with specific mutational signatures7,63. Exome sequencing analyses of HCC have identified mutational signature 22 and signature 24, especially in patients from Asia and Africa exposed to aristolochic acid (A>T mutations in CTG trinucleotide) and aflatoxin B1 (C>A mutations), respectively7,64. Signature 4 (C>A and dinucleotide mutations) and signature 16 (T>C mutation at TpA dinucleotide) were respectively associated with tobacco smoking and alcohol intake65. Whether this observation could be translated for preventive measures remains to be elucidated. These observations underline the role of the liver in detoxifying numerous metabolites, which can damage the hepatocyte genome by inducing passenger or driver mutations, leading to carcinogenesis.
Molecular classes of HCC
Several studies based on genomic, epigenomic, histopathological and immunological analyses have established a molecular and immune classification of HCC1,9,66 (Fig. 2). Molecular classes of HCC have been defined based on the main molecular drivers and pathways involved9,66,67,68,69,70 or depending on the immune status of the tumour8,71. These molecular classes correlate with specific genomic disturbances, histopathological fingerprints and clinical outcomes. The proliferation class accounts for ~50% of HCCs and is overall enriched in mutations in TP53 and in amplifications of FGF19 or CCND1 (ref.52); additionally, it is more common in HBV-associated HCC and has the worst prognosis. The proliferation class includes two subclasses — the proliferation-progenitor cell group and the proliferation–Wnt–TGFβ group. The proliferation-progenitor cell group, which represents 25–30% of HCC52,67, is characterized by the activation of classic cell proliferation pathways (such as PI3K–AKT–mTOR signalling, RAS–MAPK pathway and MET and IGF signalling cascades9) and by the expression of progenitor cell markers (such as EPCAM and α-fetoprotein), and corresponds to cluster 1 of The Cancer Genome Atlas (TCGA)68. The proliferation–WNT–TGFβ group, which represents 20% of HCC cases, is characterized by non-canonical activation of Wnt and correlates with cluster 3 of the TCGA. Conversely, the non-proliferation tumour class, which accounts for 50% of HCC, is more prevalent in alcohol-associated HCC and HCV-related HCC; these tumours present better outcomes and correlate with cluster 2 of the TCGA68. Within the non-proliferative class, at least two distinct subgroups have been delineated — one characterized by a dominant canonical Wnt signalling associated with mutations in CTNNB1 (ref.72) and the second characterized by the activation of IFNα signaling52.
Reports on the classification of HCC according to the immune cell status have further expanded our understanding of the molecular traits of HCC71 (Fig. 2). This classification provides complementary information based upon immune traits and divides HCC tumours into distinct subclasses — immune-active, immune-exhausted, immune-intermediate and immune-excluded. The immune class, which includes both the immune-active and immune-exhausted subclasses, is characterized by immune cell infiltrates of distinct nature. The immune-active HCC tumours (found in 20% of cases) are enriched with active helper T (CD4+) cell infiltrates and cytotoxic T (CD8+) cell infiltrates and respond to ICIs. Conversely, immune-exhausted tumours are dominated by TGFβ-driven CD8+ cell exhaustion status. Immune-excluded tumours, which represent the other end of the spectrum, are characterized by a paucity of T cell infiltrates and an increase of regulatory T (Treg) cells and are dominated by canonical Wnt signalling and other immune-dissuasive cascades. Immune-excluded tumours are proposed to be primarily resistant to ICIs73.
NASH-associated HCC
Obesity is linked to an increased risk of cancer in multiple organs74. Obesity can induce systemic changes, including altered immune function and systemic endocrine changes, which are hallmarks of multiple types of cancer. Current evidence shows that fatty liver disease is rapidly becoming the leading cause of HCC in the West6. Studies have demonstrated that liver-specific mechanisms through which NAFLD or NASH promote HCC involve metabolic and oxidative stress, altered immune function, pathological inflammatory responses, and altered endocrine or adipokine signalling10,75.
Oxidative stress
Hepatocytes overloaded by fatty acids cause oxidative stress and endoplasmic reticulum (ER) stress, which induce pathological inflammation and cell damage10,11. One study proved the causative role for ER stress in NASH-induced HCC in mice; ER stress in mouse hepatocytes led to the activation of inflammatory signalling pathways, specifically NF-κB and TNF, leading to HCC induction76. However, these pathogenic mechanisms are yet to be proven in human HCC. The deranged fatty acid metabolism in hepatocytes can cause DNA damage owing to increased reactive oxygen species (ROS), produced as a result of mitochondrial dysfunction77. In addition, the altered expression of specific metabolic enzymes can affect hepatocytes by decreasing their ability to repair DNA damage78. The metabolic dysfunction also results in altered inflammatory signalling; for example, high expression levels of IL-17 (a tumour-promoting cytokine) have been observed in human NASH79. In NASH, lipid generation might not only be increased but possibly also altered to generate more pathogenic lipids that serve as oncometabolites80,81. For example, continuous activation of mTORC2 in mouse hepatocytes increased the generation of the sphingolipid glucosylceramide, causing increased ROS production, which can ultimately lead to HCC development80. Similarly, altered cholesterol metabolism may also contribute to HCC pathogenesis81, potentially through the production of pro-tumorigenic nuclear receptor ligands. Although autophagy can have anti-tumour functions, one study demonstrated an important role for lipophagy (that is, autophagic degradation of lipid droplets) in HCC pathogenesis. The overexpression of sequestosome 1 (also known as p62), which regulates lipophagy, in hepatocytes of patients with NASH and in a mouse model was linked to HCC development82.
Studies have shown a higher risk of HCC in patients with NASH than in those with NAFLD6. One experimental study revealed that fatty acid-induced oxidative stress in hepatocytes upregulated STAT1 and STAT3, both of which are pro-inflammatory transcription factors that usually act in parallel83. Remarkably, in this mouse model, high levels of STAT1 caused progression to NASH, while high levels of STAT3 promoted HCC, independently of each other83. This suggests that similar inflammatory signals can differentially promote the progression of NAFLD to NASH or to HCC. As NAFLD is more prevalent than NASH in the general population6, this finding underscores the need for a better understanding of how NAFLD per se, irrespective of NASH, can progress to HCC.
Taken together, ER stress, pathological lipophagy, increased ROS production and diminished reducing power (low NADH or NADPH levels) could cause oncogenic genetic alterations in fatty acid-overloaded hepatocytes and promote the expansion of malignant cells.
Immune infiltration of fatty liver
Immune cell infiltration of the fatty liver is a histopathological hallmark of NASH10. The development of animal models that accurately replicates human HCC is essential for basic studies exploring pathogenesis and for translational studies84,85,86,87,88,89,90,91,92,93,94,95,96,97,98 (Fig. 3; Box 1). Several experimental models have shown that immune cells and cytokines play an important role in HCC pathogenesis. For example, prolonged NASH in mouse models induces CD8+ T cell activation, which results in hepatocyte damage, leading to HCC99. In addition, NAFLD causes a selective loss of intrahepatic CD4+ T cells, which are crucial for eliciting an effective anti-tumour adaptive immune response100. Other immune cell types, including B cells, Treg cells, natural killer cells and different types of myeloid cell, have been linked to NASH-induced HCC pathogenesis10,75. Interestingly, in line with clinical data14 (see section on prevention), platelet recruitment and activation in the liver also contribute to HCC development in mice, specifically via platelet glycoprotein Ibα (GPIbα) signalling, suggesting a therapeutic potential of this pathway101. An altered cytokine milieu was also shown to underlie the causative role of NASH in HCC11. For example, NASH was shown to overexpress hepatic IL-6 and TNF, which are drivers of HCC in other aetiologies as in NASH102.

Translational research in hepatocellular carcinoma (HCC) represents a two-way road between preclinical and clinical models. At one end, preclinical models aim at understanding the pathogenesis and mechanisms involved in disease initiation and progression and build the groundwork for the development of clinical therapies. The selection of preclinical models represents a compromise between time, complexity and clinical relevance. For instance, cell lines provide fast, relatively simple but less clinically relevant information while patient-derived xenograft (PDX) models are slow, complex but more relevant. At the other end, clinical studies are focused on drug development and biomarker discovery and their outcomes, albeit negative, often lead to new hypotheses that require preclinical investigation. Phase I studies aim at understanding the pharmacokinetics and toxicity profiles of newly developed drugs, phase II studies are meant to explore preliminary efficacy and phase III randomized controlled trials represent the highest level of evidence necessary for regulatory approval. Biomarkers allow for the selection of enriched populations that are most likely to benefit from certain treatments based on their mechanism of action. GEMM, genetically engineered mouse model.
All of the above-described mechanisms could simultaneously promote HCC on a background of fatty liver disease. However, their relative contribution in human HCC is currently unknown. The analysis of mutational signatures in NASH-associated HCC versus HCC from other aetiologies could help to delineate the relative contributions of various factors.
Chronic inflammation
HCC is a prototypical inflammation-associated cancer, with ~90% of the HCC burden being associated with prolonged inflammation owing to viral hepatitis, excessive alcohol intake, NAFLD or NASH. The immune microenvironment plays a pivotal role in the pathogenesis of HCC103. In HCC, the presence of immune infiltrates is associated with a better prognosis, likely owing to more effective anti-tumour immunity71,104. Mouse models of HCC have revealed that immune signals, such as IL-6, lymphotoxin-α and TNF, can accelerate hepatocarcinogenesis and affect tumour aggressiveness50,105; nevertheless, immune responses also limit liver cancer progression103. Importantly, the liver harbours the largest number of immune cells in the body and maintains a unique immune state, considerably more tolerant than other organs, allowing it to withstand the constant flow of inflammatory signals from the gut103. Understanding this unique hepatic immune system is likely important in the context of the complex interaction between malignant hepatocytes and the liver immune system103,106. Remarkably, studies in mice and humans suggest that VEGF secreted by malignant hepatocytes generates an immune-tolerant, pro-tumorigenic microenvironment52,107, suggesting that blocking the VEGF cascade could be effective by altering liver immune tolerance. Interestingly, combinations of an ICI with specific targeted therapies, such as VEGF inhibitors, showed more potent survival benefits than the use of single agents19,108.
In the chronically inflamed liver, multiple cell types, including macrophages, stellate cells, endothelial cells and different lymphocyte subtypes, interact with hepatocytes103,106. The role of innate immune cells and fibroblasts in HCC pathogenesis was described in detail in our previous Primer1. Understanding the role of the adaptive immune system is gaining increased attention in view of its importance in immuno-oncology therapies. Notably, insights from mouse models reveal that virtually every immune cell type can have both pro-tumour and anti-tumour roles103. The two major pro-tumorigenic mechanisms through which immune cells promote HCC include the secretion of cytokines and growth factors that favour proliferation or counteract apoptosis of tumour cells as well as, paradoxically, suppressing the anti-tumour function of neighbouring lymphocytes. Studies have demonstrated the NF-κB and JAK–STAT pathways as the key inflammatory signalling pathways involved in promoting HCC109; this finding was further supported in a transcriptome analysis of human HCC110. The major anti-tumour function of the adaptive immune system is mediated via immune surveillance and by the elimination of pre-malignant or fully transformed malignant hepatocytes104.
Adaptive immune system in HCC
Cytotoxic T (CD8+) cells are considered the key effectors of anti-tumour immunity. Accordingly, one study showed that their depletion in mice could increase HCC burden111 and another study showed that these T cells mediate the surveillance of premalignant hepatocytes112. Paradoxically, in several specific cases, CD8+ T cell depletion in mice resulted in a reduced tumour load, indicating that these cells can also have pro-tumorigenic functions99. Analyses of human HCC samples revealed the presence of functional CD8+ T cells expressing anti-tumour effector molecules, such as granzyme A, granzyme B and perforin, in some patients113. Nonetheless, single-cell RNA sequencing of T cells in human HCC suggests that, in many cases, these CD8+ T cells are dysfunctional114. The causes of CD8+ T cell dysfunction, evident by decreased proliferation and a decreased ability to produce cytotoxic effector molecules, are not sufficiently clarified to date. Treg cells are considered a major culprit in mediating T cell dysfunction in HCC and higher numbers of Treg cells within the tumour are associated with worse disease outcomes115. The immunosuppressive functions of Treg cells might be mediated via the secretion of CD10 and TGFβ116, suggesting that targeting these cytokines might sensitize HCC to ICIs. Interestingly, the hyaluronic acid receptor, layilin, was linked to the suppressive function of HCC-infiltrating Treg cells. Layilin induction caused CD8+ T cell dysfunction in human HCC and its overexpression in human lymphocytes was associated with a unique mRNA expression signature114.
Although B cells were thought to be innocent bystanders in cancer, the emerging evidence supports their active participation in the crosstalk between the adaptive immune system and cancer117. In mouse models of HCC, B cells both promoted and supressed tumour growth118. Furthermore, one study showed that IgA-expressing lymphocytes supported HCC growth by actively suppressing CD8+ T cell function111. Finally, human and mouse studies have shown that tertiary lymphoid structures, which have important roles in the adaptive immune response to cancer119, demonstrated pro-tumour and anti-tumour response capacities in HCC120,121. Thus, tertiary lymphoid structures, similar to macrophages and lymphocytes, could be either anti-tumorigenic or pro-tumorigenic in HCC.
Cirrhotic microenvironment and cancer field
Although some aetiologies are more likely to induce HCC than others (for example, HCV versus autoimmune hepatitis), once the patient reaches the cirrhotic stage, the risk of HCC is adequate to render surveillance cost effective12,13. The key cell involved in the liver response to chronic damage is the hepatic stellate cell122, which, upon activation, undergoes phenotypic changes and synthesizes extracellular matrix components, mostly collagen and growth factors, that promote the migration of endothelial cells, neoangiogenesis and fibrosis123. The subsequent distortion of the hepatic architecture and disorganized vasculature are the histological substrate for cirrhosis and portal hypertension. In response, premalignant senescent hepatocytes secrete chemokines that interfere with senescent surveillance and impair immune-mediated tumour suppression in vivo112. Furthermore, experimental models have documented the importance of CD4+ lymphocytes in NAFLD-related HCC100 as well as the interplay between the innate immune system and the intestinal microbiota to favour HCC development124,125. Thus, besides fibrosis, the immune system contributes substantially to the cancer field effect in HCC.
The permissive microenvironment in cirrhosis that promotes tumour development is commonly referred to as the cancer field effect. Different genomic studies have characterized the dominant molecular elements deregulated in this microenvironment. Numerous gene signatures derived from cirrhotic tissue correlate with the risk of HCC development and can be used to risk-stratify patients110,126,127. These gene signatures correlate with cancer risk as well as with likelihood of patient hepatic decompensation and overall survival126,127. More studies have detailed the genomic traits of the inflammatory microenvironment in cirrhosis that contribute to HCC development128. An immune-mediated cancer field molecular subclass was detected in 50% of adjacent cirrhotic tissue from patients with HCC. This subclass can be further stratified based on lymphocyte infiltration and on the activation of either immunosuppressive or pro-inflammatory signals. The immunosuppressive subclass, which showed enrichment in TGFβ signalling, T cell exhaustion and overexpression of immune checkpoints (such as CTLA4, TIGIT, LAG3), represented 10% of patients and had a higher risk of HCC development (threefold increased risk at 5 and 10 years)128.
The crucial part played by the tumour microenvironment in the natural history of HCC is a strong rationale for modulating the dynamic cross-talk between hepatocytes and the liver immune system as a therapeutic strategy103.
Diagnosis, screening and prevention
Given that most cases of HCC occur in an identifiable patient population, that is, in those with chronic hepatitis B or cirrhosis, many patients are diagnosed through surveillance129,130. Nevertheless, given the under-implementation of screening in some clinical practices, a proportion of patients with HCC might present incidentally with a liver mass, identified on cross-sectional imaging performed for other reasons or owing to symptomatic advanced-stage HCC after developing abdominal pain, weight loss or worsening of liver dysfunction. Such incidental diagnosis has been estimated to occur in 50% of cases globally, particularly in developing jurisdictions.
Diagnosis
Imaging
Patients with an abnormal surveillance test, that is, detection of a liver nodule in abdominal ultrasonography or high serum α-fetoprotein levels (>20 ng/ml), belong to at-risk populations and require timely diagnostic evaluation. Most lesions <1 cm in diameter detected on ultrasonography are not HCC or are very difficult to diagnose. Hence, cross-sectional imaging is not required and short-term follow-up with a repeat ultrasonography after 3 months is sufficient. For lesions ≥1 cm in diameter, either quadruple-phase CT or dynamic contrast-enhanced MRI should be performed13,14. HCC lesions are brighter than the surrounding liver in the arterial phase in a CT scan or MRI and less bright than the surrounding parenchyma in the venous and delayed phases, related to the differential blood supply of the tumour compared with the background liver131. This phenomenon of ‘arterial enhancement and delayed washout’ has a sensitivity of 89% and a specificity of 96% for HCC and is regarded as the radiological hallmark of HCC, which is sufficient for a diagnosis without requiring histological confirmation132. The specificity of MRI using hepatobiliary contrast agents seems to be lower than that using extracellular agents; therefore, its role in the non-invasive diagnosis of HCC remains unclear133. Nonetheless, practice guidelines are increasingly recommending biopsies to molecularly characterize HCC13. Importantly, imaging criteria for HCC diagnosis only applies to at-risk patients, including those with cirrhosis or chronic HBV infection.
Histopathology
Although most HCCs have characteristic features in imaging, ~10% of the tumours (but up to 30% of tumours 1–2 cm in diameter) have an atypical presentation, lacking the imaging hallmarks of HCC. The International Consensus Group for Hepatocellular Neoplasia has proposed major histological features of HCC, which include stromal invasion, increased cell density, intratumoural portal tracts, unpaired arteries, pseudo-glandular pattern and diffuse fatty changes134. If there is a clinical suspicion for HCC but the appearance is atypical by imaging, a biopsy or second contrast-enhanced study should be performed13. The sensitivity of a biopsy is ~70% and is even lower in tumours <2 cm because of the potential for missed lesions as well as the difficulty in distinguishing well-differentiated HCC from dysplastic nodules. Some patients require multiple biopsies for a diagnosis, so patients with a negative biopsy should continue to be followed with serial contrast-enhanced imaging135. If the lesion enlarges but retains its atypical appearance for HCC, a repeat biopsy should be considered.
Screening
The prognosis for HCC is driven by the tumour stage, with curative options providing a 5-year survival exceeding 70% for early-stage HCC compared with a median survival of ~1–1.5 years for symptomatic advanced-stage cases treated with systemic therapies1,2. Thus, professional societies recommend HCC surveillance in high-risk individuals, including those with cirrhosis and subgroups of patients with chronic HBV infection (Table 1). The highest level of data supporting HCC surveillance comes from an RCT in China among 17,920 persons with HBV infection136. HCC-related mortality was decreased by 37% in patients randomized to surveillance compared with those who were not screened for HCC. Whether the survival benefit would have persisted if the analytic plan accounted for the use of block randomization (that is, using randomization of villages as opposed to individuals) is unclear137. A subsequent RCT among patients with cirrhosis was terminated given poor enrolment as patients did not accept the risk of being randomized to the no-surveillance arm138. Hence, surveillance recommendations in patients with cirrhosis are based on level II data, with cohort studies demonstrating an association between HCC surveillance and early tumour detection, curative treatment receipt and improved overall survival, which persists after adjusting for lead-time and length-time biases139. Decision analysis models have demonstrated that surveillance is cost effective in patients with compensated cirrhosis and, therefore, this strategy has been adopted by guidelines in this population140.
Guidelines across scientific societies concur that screening for HCC should be performed semi-annually as a 6-month interval yields improved survival compared with annual surveillance and non-inferior outcomes compared with a 3-month interval141. Nevertheless, optimal surveillance modalities are being debated. Increasing data have highlighted that abdominal ultrasonography, the most commonly recommended surveillance modality, is operator-dependent and has a poor performance in patient subgroups such as those with obesity and NASH142. These data have led to an increased interest in blood-based biomarkers and alternative imaging modalities for screening purposes. Although several biomarkers and biomarker panels (for example, GALAD score) have shown promising results in phase II (case–control) biomarker studies, most still require validation in large phase III (cohort) studies143,144. The only blood-based biomarker currently validated for HCC surveillance is α-fetoprotein139. A meta-analysis evaluating surveillance modalities found that the pooled sensitivity of ultrasonography for early HCC detection was significantly increased from 45% when used alone to 63% when combined with α-fetoprotein, albeit with a small decrease in specificity owing to false-positive results with α-fetoprotein145. Even though surveillance with CT or MRI likely has increased sensitivity for the early detection of HCC145, concerns about radiation, contrast exposure, radiologic capacity and cost limit their widespread implementation. As we await the evaluation of newer surveillance modalities, semi-annual ultrasonography with13 or without α-fetoprotein12 remains the recommended surveillance strategy. Given the higher burden of HCC in East Asia, surveillance is typically performed using more intensive protocols — with a combination of ultrasonography, cross-sectional imaging and serum biomarkers.
The ideal surveillance tool should be highly reproducible, not operator dependent (unlike abdominal ultrasonography), have a good accuracy, and easy to implement in different clinical settings. Liquid biopsy is one such tool that fulfils all these requirements. Mutation analysis of circulating tumour DNA (ctDNA) detects tissue mutations in patients at early-stage HCC after resection146. Similarly, aberrant ctDNA methylation patterns have been studied as surveillance tools in HCC146,147,148,149,150,151 (Box 2). The accuracy of these approaches as surveillance tools is currently being investigated.
Prevention
In our original Primer1, we discussed the primary prevention of HCC with vaccines, whereas here we focus on the different emerging prevention strategies. Besides treating the primary cause (for example, viral hepatitis), no intervention proven to prevent HCC development is currently available in patients at high risk. The effective suppression of HBV replication with antivirals and universal HBV vaccination have decreased HCC incidence152,153. Similarly, the high cure rate of HCV with the new DAA therapy has reduced the incidence of HCC in patients with chronic HCV infection5. However, evidence on the impact of alcohol cessation or reversion of NAFLD and the risk of HCC is lacking. Studies have tested different therapies, such as vitamin A, vitamin K and retinol analogues, for HCC chemoprevention. Currently, numerous uncontrolled, retrospective, population-based studies have suggested a role for metformin, statins, coffee and aspirin in HCC prevention154, regardless of the aetiology of liver disease. However, statins have been extensively studied for therapeutic repurposing in different indications, with disappointing results in controlled trials showing no evidence for a decreased incidence of HCC. Conversely, reports on aspirin are compelling in terms of HCC prevention, including data from nationwide Swedish registries14 showing that, after a median follow-up of 8 years, aspirin use reduced the estimated cumulative incidence of HCC from 8% to 4%. Several cohort and case–control studies demonstrated a dose-dependent relationship between coffee consumption and reduced HCC incidence in the general population as well as in patients with chronic liver disease155. Based on available data, European Association for the Study of Liver (EASL) guidelines offer a strong recommendation for coffee consumption as a chemoprevention strategy in patients with chronic liver disease12. Nevertheless, controlled studies are needed to establish the role of these interventions.
Management
HCC is a unique neoplasm as ~80–90% of cases develop in patients with cirrhosis and, therefore, the application of different therapeutic options might be limited because of the patient’s overall health status. The management of HCC has substantially improved over the past decade. The treatment is assigned according to tumour stages and the expected benefits of major interventions, following the Barcelona Clinic Liver Cancer (BCLC) staging system12,156,157 (Fig. 4). In principle, patients with early-stage HCC tumours are the preferred candidates for resection, transplantation and local ablation (Fig. 4), whereas patients at intermediate stages are first candidates for TACE and those with advanced disease will first receive systemic therapies. These therapies have substantially improved the reported natural history of untreated cases at each of the stages, with median survival times for early, intermediate and advanced HCC of ~36, ~16 and ~6 months, respectively, in patients with well-preserved liver function defined as Child-Pugh A (according to the Child-Pugh score) and compensated disease158,159. In order to prevent collateral liver dysfunction, certain therapies (such as resection and systemic therapies) are mostly applied in this patient population.

The Barcelona Clinic Liver Cancer (BCLC) staging system consists of five stages depending on disease extension, liver function and performance status. Asymptomatic patients with low tumour burden and good liver function (BCLC 0/A) should be treated with local curative treatments (resection, ablation or transplantation, depending on the presence of portal hypertension, number of nodules and liver function). Asymptomatic patients with multinodular disease and adequate liver function (BCLC B) should receive chemoembolization and patients with portal thrombosis or extrahepatic spread (BCLC C) should be treated with systemic therapies. Ongoing phase III trials in all disease stages are depicted. AFP, α-fetoprotein; DDLT, deceased-donor liver transplantation; ECOG, Eastern Cooperative Oncology Group; HCC, hepatocellular carcinoma; LDLT, living-donor liver transplantation; M1, distant metastasis; N1, lymph node metastasis; OS, overall survival; RCT, randomized controlled trial; TACE, transarterial chemoembolization. aPatients with end-stage liver disease Child-Pugh class C should first be considered for liver transplantation. bPatients with preserved hepatic function Child-Pugh class A with normal bilirubin and no portal hypertension are optimal candidates for hepatic resection. cSorafenib and lenvatinib are also considered first-line treatment in case of contraindication for atezolizumab + bevacizumab. Adapted with permission from ref.156, Wiley.
Surgical interventions
Surgical treatment, which includes both hepatic resection and liver transplantation, has long been the backbone of curative therapies for HCC, yielding the best outcomes, with a 5-year survival of ~70–80%12,13 (Table 2; Supplementary Table 1). The decision between resection and transplantation requires consideration of the patient’s liver function, the presence and extent of portal hypertension, performance status, and tumour characteristics such as size, number and involvement of the hepatic and portal veins. The local regulations governing the availability and allocation of organs must also be incorporated into the decision-making process. Western guidelines have advocated on the principal of selecting the ideal candidates leading to the best outcomes for surgical resection whilst relegating the non-ideal candidates to other therapies12,13. However, several studies now challenge this principal based on the fact that resection on suboptimal candidates for surgery might provide similar or even better outcomes than loco-regional therapies. Nonetheless, level 1 evidence supporting this approach of resection for non-ideal candidates is not yet available.
Resection
Hepatic resection is considered the treatment of choice in patients with HCC without cirrhosis in whom post-operative hepatic decompensation is not a major concern12,13,160. Of note, however, resection for HCC in non-cirrhotic NAFLD livers is associated with morbidity as high as 20%, similar to that observed in patients with cirrhosis161,162. In patients with cirrhosis, Western guidelines have restricted resection to those with a single tumour (regardless of size), with well-preserved liver function (Child-Pugh A with total bilirubin <1 mg/dl), the absence of clinically relevant portal hypertension (no varices or ascites) or a hepatic venous pressure gradient (<10 mmHg), as well as with a preserved performance status Eastern Cooperative Oncology Group score ((ECOG score) 0). Adherence to these selection criteria have resulted in a 5-year survival of ~70% and perioperative mortality of <3%163. The Child-Pugh score, model for end-stage liver disease and indocyanine green clearance are the conventionally used tests to assess liver function prior to resection12. Currently, several studies have validated the albumin–bilirubin score (ALBI score) to be able to accurately stratify patients for resection with more granularity than the Child-Pugh score164,165.
Analysis of data from a large prospective registry found that the majority (>60%) of hepatic resections were performed in patients who did not meet the criteria of Western guidelines, either in terms of liver function, performance status or tumour characteristics166 (Table 2). This study showed that the presence of one risk factor did not adversely affect overall survival, although resection in patients with both portal hypertension and elevated bilirubin resulted in a significantly lower survival than in candidates who met the criteria. Other studies have reported that resection in patients with portal hypertension or Child-Pugh B resulted in a 5-year survival of <50% with high morbidity and a perioperative mortality of 4%167,168. Overall, liver function, portal hypertension and the extent of liver resection directly impact outcome and, therefore, these variables should all be integrated into the selection process169. Whether outcomes can be improved with a minimally invasive approach still needs to be confirmed170.
Other potential indications for resection that require further study prior to being adopted in the management guidelines are the expansion of criteria to multinodular tumours or segmental vein invasion. One randomized trial and several retrospective studies suggest that expanding the criteria for resection to patients with multiple tumours and well-preserved liver function might lead to better outcomes for resection when compared with TACE171. Similarly, resection for HCC invading the segmental branches of the portal vein led to survival outcomes ranging from 29 to 49 months in Western and Japanese series172,173.
The recurrence of HCC after hepatic resection remains a major obstacle, with recurrence rates as high as 70% at 5 years, even in patients with a single tumour ≤2 cm (ref.174). Recurrences can be divided into either early (<2 years), resulting from micrometastases following resection, or late (>2 years), resulting from de novo tumours arising in a microenvironment predisposed to carcinogenesis175. Modifications of surgical technique, such as the anatomical approach, as well as non-anatomical resection with a margin of 2 cm, have shown varying degrees of success in reducing recurrence176,177. Neoadjuvant treatment with embolization as well as adjuvant administration of retinoids, adoptive immunotherapy and 131I-lipiodol embolization have all been tested unsuccessfully178,179. Additionally, the STORM trial randomizing patients to sorafenib versus placebo after resection or ablation showed no benefit in recurrence-free survival180. A decrease in late recurrence after resection has been reported with the use of antiviral agents for HBV in uncontrolled investigations181. Similarly, DAA therapy in patients with HCV cirrhosis and a history of treated HCC has been shown to be safe and likely beneficial, with improved overall survival in a meta-analyses182,183, thus not confirming preliminary discouraging results184. Current data suggest that treatment of HCC recurrence with resection, salvage transplantation, ablation, TACE and systemic therapies achieves outcomes close to those achieved in primary HCC185. Finally, pre-emptive liver transplantation has been proposed for patients with high risk of recurrence (that is, those with microvascular invasion)186.
Liver transplantation
Patients with cirrhosis and a limited tumour burden (the Milan Criteria — single tumour ≤5 cm or 2–3 tumours ≤3 cm without vascular invasion) are considered for liver transplantation187. The outcomes have been excellent, with a 5-year and a 10-year survival of 70% and 50%, respectively, and recurrence rates of 10–15% at 5 years16 (Table 2; Supplementary Table 1). Long-term outcomes of liver transplantation are considered superior than resection, which has a 70% recurrence rate and a 10-year survival of 7–15%188. However, transplantation is plagued by organ shortage with prolonged waiting times, leading to patient dropout from the waiting list because of tumour progression. The probability of a cure via resection becomes similar to liver transplantation when drop-out rates exceed 20%189,190.
The use of extended criteria for liver transplantation has been an active area of investigation. Some proposed measures, such as the University of California San Francisco (UCSF) criteria, Up-to-Seven criteria, total tumour volume and α-fetoprotein criteria, and the Milan and α-fetoprotein model have been validated in studies191,192,193. Among these, only the UCSF criteria have been adopted as an upper limit of tumour burden for down-staging to Milan criteria for liver transplantation by the American Association for the Study of Liver Diseases (AASLD) guidelines13. Other retrospective and non-validated studies involving ~2,000 patients transplanted for a single tumour ≤6 cm or 2–3 tumours ≤5 cm demonstrated a 10-year survival rate similar to the Milan criteria194,195. Overall, the ‘Metroticket’ concept clarifies that an ideal cut-off value is difficult to establish owing to a continuous spectrum of outcomes that are incrementally correlated based on tumour size, number and α-fetoprotein levels193,196. Another approach to expand the transplantation criteria is based on the biological behaviour of a tumour, determined by a combination α-fetoprotein level and 18F-fluorodeoxyglucose uptake on PET scans197. Nevertheless, transplantation of HCC beyond the Milan criteria remains experimental except in cases where the tumour has been successfully down-staged to within the criteria.
Studies have explored neoadjuvant therapies, such as TACE or ablation, to prevent tumour progression while on the waiting list (bridging therapy) or to reduce tumour burden to within the Milan criteria (down-staging). A response to neoadjuvant therapies reduces dropout from the waiting list as well as the risk of post-transplant recurrence198,199. A multicentre analysis involving ~2,500 patients demonstrated a 10-year survival of 52% in patients successfully down-staged to Milan criteria16. The response to neoadjuvant therapy assessed by the modified Response Evaluation Criteria In Solid Tumours (mRECIST) has been proposed as a criterion for selecting patients for transplantation and as predictor of death after liver transplantation200. Similarly, a response (evident by a decrease in α-fetoprotein levels) while on the waiting list has been shown to correlate with recurrence-free survival after transplantation201. However, these models need further validation before they can be incorporated into guidelines.
The scarcity of cadaveric organs has led to long waiting times for transplantation, resulting in dropout owing to tumour progression; the use of marginal donors (donors >60 years of age, those with diabetes, BMI >35 kg/m2 or severe graft steatosis) and of living donors has been advocated to expand the access to transplantation. However, some notes of caution have been raised. Although the use of living donors yields similar survival rates as using deceased donors, some studies reported higher recurrence rates with the former despite adjustment for tumour characteristics (mostly size and number)202. This high recurrence may potentially be explained by the fact that a shorter waiting time with living donors prevents the identification of molecularly aggressive HCCs, which are prone to dropout with longer waiting times203. In this regard, genomic studies have identified the proliferative–progenitor subclass of HCC, characterized by an aggressive phenotype and high α-fetoprotein levels, to be associated with high recurrence rates after liver transplantation203. As patients with HCC listed for liver transplantation often have better liver function than those listed for hepatic decompensation, the preferential use of marginal organs has been proposed for patients with HCC. Nonetheless, studies have shown that the use of marginal donors is linked with a significantly higher risk of HCC recurrence198,199.
The 10-year recurrence rate after transplantation is 10–15% for HCC tumours within Milan criteria and 20% in those down-staged to the Milan criteria16. So far, no adjuvant treatment has been shown to prevent recurrence after liver transplantation. An RCT exploring sirolimus-based immunosuppression versus standard immunosuppression revealed no difference in overall survival or recurrence-free survival204. In summary, selection based on Milan criteria leads to low recurrence rates, which so far have not been able to further decrease with molecular therapies.
Image-guided ablation
Image-guided ablation is accepted as a potentially curative therapy for small, early-stage HCC tumours12,205. In principle, there are two indications for these therapies according to guidelines, either as first choice therapy for single, very early tumours <2 cm or as an alternative to surgery in early-stage single tumours, generally up to 4 cm, or 2–3 tumours ≤3 cm (refs12,205) (Fig. 4). The latter patients are unsuitable for resection due to liver dysfunction or tumour multi-nodularity and additionally present with formal contraindications for liver transplantation. Ablation is used to direct injury to the tumour and is achieved via chemical, thermal or electrical methods15. Historically, percutaneous ethanol injection is the seminal technique for local ablation and is still recommended for tumours <2 cm, especially when located in the proximity of major vessels or bile ducts. Currently, radiofrequency ablation (RFA) is the established thermal technology along with microwave ablation (MWA), whereas other ablative techniques, such as cryoablation (CRA) and laser interstitial thermotherapy, are less used15. Irreversible electroporation (IRE) achieves tumour destruction by inducing electrical damage to the tumour cells without a significant thermal component.
Assessment of response
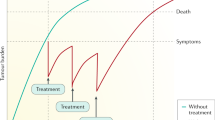
Identifying patients who respond to image-guided ablation as well as to loco-regional and systemic therapies is essential to guide appropriate management of these patients. RECIST is the standard imaging approach in oncology, but this approach has several limitations in the assessment of treatment response of HCC. Consequently, several groups have proposed the mRECIST for HCC, aimed at capturing differences in the viable tumour (that is, non-necrotic tissue) as opposed to differences in absolute tumour shrinkage206,207. These criteria have been recently refined and their performance reviewed elsewhere208. Overall, mRECIST identifies 2–3-fold more responders than standard RECIST in patients receiving loco-regional treatments as well as in those receiving systemic therapies21,209. Similarly, overall response rates (ORRs) assessed by mRECIST have been associated with better survival in patients receiving local therapies and systemic therapies208,209,210,211.
Radiofrequency ablation
RFA is the most used technique for local ablation and several RCTs have demonstrated the superiority of RFA to percutaneous ethanol injection in objective response rates and overall survival212,213,214 as well as similar survival rates when compared with surgical resection in appropriately selected patients215,216,217,218 (Table 2; Supplementary Table 1). Thus, AASLD and EASL guidelines have adopted RFA as the front-line primary treatment for single tumours <2 cm and as an alternative for surgery in early-stage single tumours 3–4 cm or 2–3 tumours <3 cm (refs12,205). The main predictor of treatment failure is tumour size. When RFA is used as first-line therapy for early-stage HCC, complete response rates by mRECIST range from 70% to 90%219,220 and is significantly associated with better overall survival220. In addition, studies have reported a median overall survival of ~60 months and a 5-year recurrence rate of 50–70% with RFA12,205,221.
Microwave ablation
MWA has the advantage of achieving a larger ablation zone than RFA as several needles can be used at simultaneously15. Several trials comparing RFA and MWA reported no differences in the primary endpoint or in local tumour progression at 2 years222. Similarly, three meta-analyses comparing percutaneous MWA and RFA showed a similar efficacy between the two percutaneous techniques223,224, with a trend towards greater efficacy but higher complication rates in tumours >3 cm treated with MWA compared with treatment with RFA225. The lack of phase III data led to the proposal of this treatment in early-stage HCC with only a low level of evidence12. Overall, MWA is easy to deliver and is widely used in clinical practice, although no evidence of superiority to RFA is available.
Other ablative techniques
Other ablative techniques have been the subject of limited research so far. Combining RFA with TACE or lyso-thermosensitive liposomal doxorubicin has not improved outcomes compared with RFA alone226,227. A multicentre randomized trial comparing RFA and CRA reported no differences in overall survival and tumour-free survival228, whereas a large retrospective study showed a significant advantage for CRA in liver cancer-specific survival compared with RFA229. IRE is a mostly non-thermal technology with the theoretical advantage that it avoids unnecessary thermal damage to critical structures; however, this advantage is counterbalanced by the complexity of multiprobe technology requiring general anaesthesia. Preliminary results from small series have shown early signals of efficacy with IRE224 and laser ablation230. Nevertheless, these techniques are not yet ready for recommendation in conventional clinical practice15.
Radiotherapy
External beam radiation therapy can achieve radiological responses in HCC tumours across a range of sizes and stages within the liver as well as palliation of extrahepatic metastases. In HCC tumours confined to the liver, prospective studies of stereotactic body radiation therapy with photons or protons show high rates of radiological responses with acceptable safety in predominantly Child-Pugh A populations, although these findings are limited by uncontrolled study designs231,232. A pooled analysis examined the outcomes of 102 patients with unresectable HCC and Child-Pugh A liver function treated with photon stereotactic body radiation therapy from 24 to 54 Gy over six fractions and reported objective responses in 54% of patients and a median overall survival of 17 months231. Other smaller uncontrolled studies have reported better outcomes in this population232 as well as in HCC lesions with tumour macrovascular invasion233,234.
Most studies comparing radiotherapy with other locoregional therapies in HCC are retrospective in nature and are limited due to selection bias and population heterogeneity235,236. The randomized phase III APROH trial comparing proton beam radiotherapy to RFA under a non-inferiority design, involving 144 patients with small HCC tumours (that is, up to 2 tumours <3 cm) and well-preserved liver function, met the pre-specified target for non-inferiority in the per-protocol population, with a hazard ratio of 0.52 (95% CI 0.26–1.05) for 2-year liver progression-free survival (PFS)237.
Collectively, these studies support a potential role for radiotherapy in selected patients, particularly those with small tumours not amenable to resection or transplantation. Additional randomized studies with longer follow-up and pooled analyses are required to confirm whether these approaches are similar to RFA and to define the optimal radiation modality. Studies combining palliative radiotherapy with immunotherapy in advanced HCC are under way.
Transarterial therapies
Two RCTs and a subsequent meta-analysis involving patients with intermediate-stage HCC have demonstrated survival benefits with TACE compared with suboptimal therapies, including tamoxifen or best supportive care (that is, management of pain and nutritional and psychological support)17,238,239. As a result of these studies, TACE has been globally adopted as standard of care for patients with intermediate-stage HCC12,205,240. Overall, the median survival ranges from 19.4 months in uncontrolled investigations241 and up to 37 months in RCTs242,243,244,245, with an estimated average of median overall survival of ~30 months15 (Table 3; Supplementary Table 1). Large case-series assessing the safety of conventional TACE reported a treatment-related mortality of 0.6%241. Over the past few years, the introduction of drug-eluting bead TACE has offered an alternative to conventional lipiodol TACE. Drug-eluting bead TACE has been associated with a reduction in systemic drug exposure and drug-related adverse events246,247, albeit with similar outcomes to conventional TACE, except for a median survival of >45 months in single-arm studies248. TACE is usually indicated by physicians on demand according to radiological response, generally assessed according to mRECIST206,208. Indeed, according to a large meta-analysis, response to mRECIST was associated with better survival (HR 0.39, 95% CI 0.26–0.61)249. However, combinations of TACE and TKIs have so far failed to provide beneficial clinical outcomes243,250,251,252,253,254.
TARE is a procedure involving the intra-arterial delivery of glass microspheres or resin microspheres embedded with yttrium. Uncontrolled studies and small RCTs in highly selected centres have reported results similar to TACE for the treatment of intermediate-stage HCC or even better outcomes in terms of time to progression18,255,256. The AASLD guidelines recommend TARE to patients with intermediate-stage HCC with a level 2 evidence12,205. TARE was explored owing to preliminary encouraging results in patients with portal vein thrombosis255,257 but three consecutive RCTs comparing TARE with sorafenib in advanced-stage HCC failed to meet the primary endpoint of superior overall survival258,259,260. Consequently, guidelines have adopted a negative recommendation for this indication12,205,240.
Systemic therapies
The benchmark for clinical trial design in HCC is the SHARP study20, which established the selection criteria and stratification factors, such as the use of the BCLC staging system, Child-Pugh A liver function and performance status (ECOG 0 or 1), all of which form the basis for future phase III trials investigating advanced-stage HCC. Several studies over the past decade have established other prognostic factors that led to modifications in study designs. These modifications include the separation of extrahepatic spread and macrovascular invasion, the importance of elevated α-fetoprotein levels, and the incorporation of mRECIST assessments156.
First-line therapies
In patients with advanced-stage HCC, the SHARP trial demonstrated the superiority of sorafenib to placebo (overall survival, 10.7 months versus 7.9 months), which represented a breakthrough in HCC management (Table 4). Sorafenib was the only available standard of care for advanced HCC for a decade. A further meta-analysis established that sorafenib was more effective in patients with HCV-associated HCC and liver-only disease (that is, without metastases) than in those with HCC from non-HCV causes or in patients with extrahepatic disease261. Since the approval of sorafenib in 2007, several new effective drugs have been established as second-line treatment after progression on sorafenib as have more effective drugs in the first-line setting (Fig. 5).

a | The mechanisms of action of targeted therapies approved based on phase III data. Green boxes indicate positive results based on phase III trials with a superiority design, in the first-line setting compared with placebo (versus sorafenib) or sorafenib (versus atezolizumab plus bevacizumab) or in the second-line setting compared with placebo (versus regorafenib, cabozantinib and ramucirumab). Yellow boxes indicate positive results based on phase III trials with a non-inferiority design (lenvatinib). Red boxes indicate other FDA-approved drugs based on non-randomized phase II trials (nivolumab, pembrolizumab and nivolumab plus ipilimumab). b | Mechanisms of action of the combination of molecular and immune targeted therapies. Checkpoint blockade monotherapies benefit a small subset of patients (~15–20%). Combining tyrosine kinase inhibitors or VEGF inhibitors with immune-checkpoint inhibitors can modulate the immune microenvironment by enhancing both dendritic cells (DCs) and cytotoxic T lymphocytes and inhibiting tumour-associated macrophages (TAMs), regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs), thereby creating a more inflamed microenvironment and favouring the development of more effective and durable responses to checkpoint inhibitors. Data from refs1,8,19,20,21,22,23,24,25,263,264. Part a adapted from ref.8, Springer Nature Limited. Part b adapted from ref.15, Springer Nature Limited.
A global open label randomized phase III study (REFLECT) demonstrated the efficacy of lenvatinib, which was the first new drug approved for advanced-stage HCC in the first-line setting in over 10 years21. The REFLECT study excluded patients with extrahepatic main portal vein invasion or in whom >50% of the liver was involved. The primary endpoint of the study was overall survival and was powered for superiority and non-inferiority, but the trial met only the latter endpoint. The final results established an improved median overall survival for lenvatinib (13.6 months) compared with sorafenib (12.3 months). In addition, lenvatinib also significantly improved PFS (7.4 months versus 3.7 months; HR 0.66, 95% CI 0.57–0.77; P < 0.001) and ORR (24.1% versus 9.2%; OR 3.13, 95% CI 2.15–4.56; P < 0.0001) compared with sorafenib according to mRECIST (Table 4). Unlike sorafenib, lenvatinib is a small molecular type V TKI, with more potent activity against VEGF receptors and the FGFR family. In this regard, the side effect profiles are different, with higher grade hypertension and proteinuria occurring with lenvatinib and increased hand–foot skin reaction occurring with sorafenib; both drugs are associated with asthenia, anorexia, diarrhoea and weight loss. Overall, both treatments are associated with grade 3–4 drug-related adverse events in ~50% of the treated patients, resulting in a ~15% withdrawal rate (Table 4).
The combination of atezolizumab (anti-PDL1 antibody) and bevacizumab (anti-VEGF antibody) was the first regimen to improve overall survival compared with sorafenib19,262. The IMbrave150 trial, an open-label study that randomized patients to sorafenib or to a combination of atezolizumab and bevacizumab as first-line therapy for advanced HCC, demonstrated an improvement in overall survival with the combination therapy. An updated analysis published as a conference abstract shows the median survival of patients receiving sorafenib was 13.4 months and the median survival of the combination arm was 19.2 months. PFS was improved from 4.3 months in the sorafenib arm to 6.8 months in the combination arm and RECIST ORR was increased from 11% in the sorafenib arm to 30% in the combination arm, and the median duration of response for the combination arm was 18.1 months by RECIST 1.1 and 16.3 months by mRECIST19,262 (Table 4). Patient-reported outcomes were also favourable for the combination arm, with the median time to deterioration of quality of life being 11.2 months compared with 3.6 months for sorafenib. Tolerability was more favourable in the combination group compared with sorafenib, with the most common side effects being hypertension, proteinuria and low-grade diarrhoea. The autoimmune events that occurred with atezolizumab were reported as manageable. Upper-gastrointestinal endoscopies were required within 6 months prior to enrolment for the treatment of varices in all patients to mitigate the risk of bleeding associated with bevacizumab. This concept represents a change in practice, especially for the screening of patients in first-line therapy, as upper gastrointestinal endoscopies will have to be performed prior to treatment initiation. As a consequence of the positive findings, atezolizumab plus bevacizumab have become the standard of care in first-line therapies for advanced HCC, except in patients with untreated varices or in those with contraindications for VEGF inhibitors or immunotherapy156.
Second-line therapies
Currently, based on positive phase III data, three regimens (regorafenib, cabozantinib and ramucirumab) are approved for the treatment of advanced HCC after progression on sorafenib according to guidelines (Table 4). In addition, based upon promising phase Ib/II studies, three additional therapies, namely nivolumab, pembrolizumab and nivolumab plus ipilimumab, have been approved by the FDA25,263,264 after first-line treatment with sorafenib.
Regorafenib, a multi-kinase inhibitor targeting VEGFR1–3 and other kinases, was the first agent to be approved in the second-line setting upon demonstrating a survival advantage over placebo (10.6 months versus 7.8 months) for patients who tolerated and had documented progression on sorafenib22 (Table 4). The median survival with regorafenib was 10.6 months versus 7.8 months with placebo (HR 0.63, 95% CI 0.50–0.79; P < 0.0001). The median PFS was 3.1 months versus 1.5 months (HR 0.46, 95% CI 0.37–0.56; P < 0.0001) and ORR was 11% and 4% for regorafenib and placebo, respectively. The most common grade 3–4 events were hypertension, hand–foot skin reaction, fatigue and diarrhoea.
Cabozantinib is a multi-kinase inhibitor with unique activity against VEGFR2, AXL and MET. The CELESTIAL trial demonstrated an improvement in the median overall survival for cabozantinib (10.2 months) compared with placebo (8 months; HR 0.76, 95% CI 0.63–0.92; P = 0.0049) and an improvement in median PFS (5.2 months with cabozantinib versus 1.9 months with placebo; HR 0.44, 95% CI 0.36–0.52; P < 0.001)23. Both treatment arms had single digit objective response rates. The most common grade 3–4 events were palmar–plantar erythrodysesthesia, hypertension, increased aspartate aminotransferase levels, fatigue and diarrhoea.
Ramucirumab is the only biomarker-guided therapy for HCC. The REACH-2 trial that investigated ramucirumab was enriched for patients with baseline α-fetoprotein levels of ≥400 ng/dl. Ramucirumab demonstrated an improvement in overall survival (8.5 months versus 7.3 months in the placebo group; HR 0.710, 95% CI 0.531–0.949; P = 0.0199)24. PFS was increased with ramucirumab compared with placebo (2.8 months versus 1.6 months; HR 0.452, 95% CI 0.339–0.603; P < 0.0001) but the proportion of patients with objective response did not differ significantly between groups. The most common grade 3–4 treatment-related adverse events were hypertension, hyponatraemia and increased aspartate aminotransferase levels. All six regimens tested in phase III trials for advanced HCC with beneficial effects in survival showed an HR of ≤0.6 for PFS in contrast to ~15 randomized negative studies assessing systemic therapies. This observation led to the proposition of this restrictive threshold as a cut-off for assessing PFS-based benefits with molecular therapies highly likely to ultimately capture overall survival differences156,265. This PFS–HR threshold has been validated in five new RCTs and, as a result, it has been proposed as a primary endpoint for the trial design of studies in advanced HCC156.
Based on phase Ib/II data, nivolumab and pembrolizumab (anti-PD1 inhibitors) were approved as single agents and ipilimumab (CTLA4 monoclonal antibody) was approved in combination with nivolumab263,266. CheckMate 040 assessed nivolumab as monotherapy in 262 patients mostly as second line, demonstrating an ORR of 14% by RECIST with a median duration of response of 17 months (95% CI 6–24)25. The median overall survival was 15.6 months and the treatment was generally well tolerated. Similarly, the KEYNOTE-224 trial showed an ORR of 17% (RECIST v1.1) with pembrolizumab and these rates were durable with a median time to progression and progression-free survival of 4.9 months and a median overall survival of 12.9 months264. The pembrolizumab-associated adverse effects were tolerable. However, two phase III studies were unable to confirm the findings of these single-arm studies. CheckMate 459, exploring nivolumab versus sorafenib in the first-line setting, reported a median overall survival of 16.4 months for nivolumab and 14.7 months for sorafenib (P = 0.07)267. Similarly, KEYNOTE-240 reported a median survival of 13.9 months for pembrolizumab compared with 10.6 months for placebo (P = 0.02); however, the results did not hit the pre-specified P value required for statistical significance26. Both drugs achieved a durable ORR of 15–18% and remain approved in the USA. An expansion arm in the CheckMate 040 study evaluated the combination of nivolumab plus ipilimumab in patients who progressed on prior sorafenib in a three-arm randomized study involving 148 patients263. The combination of nivolumab plus ipilimumab achieved an objective response of 31% with a median duration of response of 17 months and a median overall survival of 23 months. Although the combination regimen induced immune-related toxicities requiring systemic corticoid administration in 51% of cases, the efficacy of outcomes resulted in an accelerated approval by the FDA for second-line therapy. As a result, phase III trials are currently exploring this combination therapy versus either sorafenib or lenvatinib268.
Emerging combination regimens
Across tumour types in oncology, new immunotherapy combination strategies are being developed to augment tumour responsiveness to immune-checkpoint inhibition269. In HCC, ICIs have shown promising activity when paired with anti-angiogenic agents, other molecularly targeted therapies and complementary ICIs (Fig. 5). The VEGF pathway promotes local immune suppression through the inhibition of antigen-presenting cells and effector cells as well as through the activation of suppressive elements, including Treg cells, myeloid-derived suppressor cells and tumour-associated macrophages, providing the rationale for combining ICIs with anti-angiogenic agents270. A phase Ib trial of the combination of lenvatinib and pembrolizumab as first-line therapy in 100 unresectable patients with HCC demonstrated durable, objective radiographic responses by mRECIST in 46%, with a median PFS of 9.5 months and a median overall survival of 22 months108. The efficacy of this combination has prompted an ongoing phase III trial investigating this combination therapy versus lenvatinib as monotherapy271. Based upon the unique immunomodulatory and antiangiogenic profile of cabozantinib, another phase III trial to determine the efficacy of the combination of cabozantinib with atezolizumab compared with sorafenib or cabozantinib alone272, is ongoing273. A number of trials testing the combinations of a variety of other multi-kinase inhibitors plus ICIs are under way (Fig. 5).
The inhibition of complementary, non-redundant immune-checkpoint pathways may augment the proportion of patients achieving anti-tumour immune responses274 (Fig. 5). The addition of a CTLA4 inhibitor to the inhibition of PD1 or PDL1 has shown higher rates of durable responses in multiple tumour types, albeit with higher rates of immune-related toxicity275. Phase III trials testing the combination of nivolumab and ipilimumab as front-line therapy268 are ongoing. A trial tested the combination of durvalumab with tremelimumab in 75 patients with advanced HCC after failure of prior sorafenib276. Radiographic responses by RECISTv1.1 occurred in 24% of patients, with a median PFS and overall survival of 2.7 and 18.7 months, respectively. This regimen was tolerable, with a requirement of systemic corticosteroid in 24% of patients. A confirmatory phase III trial of this combination regimen compared with durvalumab or sorafenib as monotherapy277 has been completed and results are awaited.
An important question in the evaluation of the efficacy of the combination regimen is to understand whether improvements in time-to-event medians and objective response rates are due to synergy and not because of the independent additive effects of two active agents, which can also be achieved by a sequential approach. The depth and the durability of objective radiographic responses may inform this inference278. In the absence of head-to-head trials or established biomarkers to guide the choice of therapy, treatment decisions must rely upon the magnitude of benefits, the toxicity profile and drug availability.
Biomarkers of response to systemic therapy
To date, biomarker data to help decision-making and to guide treatment for advanced stages of HCC are limited. An elevated level of serum α-fetoprotein is an established biomarker of poor prognosis across all stages of HCC and is associated with tumour VEGF pathway activation279,280. Pre-treatment serum levels of α-fetoprotein became the first biomarker predictive of response, with the finding of a survival benefit of ramucirumab over placebo only in patients with α-fetoprotein levels ≥400 ng/ml (ref.24). Thus, ramucirumab is only indicated when α-fetoprotein levels are beyond this cut-off value. However, unlike ramucirumab, the treatment benefits from multi-kinase inhibitors, including sorafenib, lenvatinib, regorafenib and cabozantinib, occur across a range of baseline α-fetoprotein values, likely owing to a broader spectrum of target inhibition22,23,245,261. In patients with elevated α-fetoprotein levels at baseline, changes in α-fetoprotein levels on treatment were shown to correlate with clinical outcomes on systemic therapy, with declining α-fetoprotein levels linked to prolonged PFS and overall survival and increasing α-fetoprotein levels associated with tumour progression245,280. Nevertheless, additional studies of α-fetoprotein kinetics are required.
Few studies evaluating TKIs in HCC have reported on the biomarkers associated to response281,282. A variety of candidate biomarkers of benefit from immune-checkpoint inhibition are under investigation across different solid tumours, including HCC. A meta-analysis of outcomes from >3,500 patients showed that tumour PDL1 expression is associated with a worse prognosis in HCC, including a poorly differentiated histology, high levels of α-fetoprotein and shorter overall survival283. Non-randomized studies of nivolumab and pembrolizumab suggest higher rates of radiographic response25,264 and prolonged PFS264 in patients with positive tumour or combined tumour and non-tumour PDL1 expression scores, although interpretation is limited owing to assay heterogeneity and small sample sizes. Analyses of PDL1 expression and its association with survival endpoints are awaited from RCTs of ICIs in monotherapy as well as in combination regimens. Tumour lymphocytic infiltration53, immune class gene signature71 and CTNNB1 mutation status73 in subsets of HCC tumours also warrant examination for predictive value in patients treated with ICIs.
Proof-of-concept studies in HCC based on trial enrichment for biomarkers have shown distinct results. Early clinical trials demonstrated that enriching patients with advanced HCC with immunopositivity for FGF19 (a known oncogene in HCC) led to a significant ORR of 16% (versus 0% in patients with no immunoreactivity) when treated with FGFR4 inhibitors284. Conversely, other trials enriching patients for RAS mutations285 or MET286 immunoreactivity have resulted in negative outcomes.
COVID-19 infection and HCC
There is no definitive data on the impact of COVID-19 (a disease caused by the SARS-CoV-2 coronavirus that causes fever, shortness of breath and, in rare cases, acute respiratory distress) in patients with HCC. The known indirect impacts relate to the large numbers of patients with COVID-19 requiring hospitalization and critical care, which has diverted the resources away from patients with HCC. This diversion will likely result in a second wave of patients with other ailments requiring health-care services at an increased rate once the pandemic wave settles and social restrictions are de-escalated287. A survey from the American Cancer Society found that 50% of patients with cancer reported an impact to their cancer care, including delays in therapy. Further, over one-third of patients expressed concern about their ability to afford cancer-related care given the repercussions of the COVID-19 pandemic on the job market288. In 15–54% of patients with COVID-19, an elevation of transaminases has been observed289; however, the long-term impact of elevated transaminases in HCC outcomes is currently unknown. In addition, considering the potential role of immune derangement in the pathophysiology of COVID-19, the effect of immune-based therapies in the course or outcomes of patients with HCC with COVID-19 is unclear. To address these issues, the International Liver Cancer Association has developed a guidance document to help adapt the clinical management of patients with HCC during this unprecedented time290.
Quality of life
Quality of life is becoming a major endpoint in oncology research. Improvements in quality of life are captured by changes in patient-reported outcomes, which result from clinical benefits from treatments and from the impact of adverse events and tolerability of potentially toxic drugs. A longstanding challenge to systemic therapy in advanced HCC has been the limited tolerability to treatment, owing in part to adverse events from the treatments themselves and in part to symptomatic comorbidity from the underlying liver disease, which, in turn, is confounded by the increasing tumour burden. In the pivotal clinical trials investigating multi-kinase inhibitors such as sorafenib, lenvatinib or regorafenib, the rate of treatment-related adverse events of grade ≥3 generally exceeded 50%20,21,22,291. However, dose reductions to delay treatment-related adverse events are prevalent but may interfere with efficacy.
Treatment-related adverse events
The advent of immune-checkpoint inhibition for advanced HCC has expanded the treatment landscape to include ICIs as monotherapy as well as in combination19,25,26. Beyond the potential for deep and durable immune responses with immune-targeted therapies in subsets of patients, ICIs have also shown favourable adverse event profiles in comparison to standard therapies such as sorafenib. In phase III RCTs of ICIs as monotherapy in HCC, the rates of grade 3–4 treatment-related adverse events ranged from 18.2% to 22%26,267 for single agents and 37% for combination regimens19. Although the adverse event profiles are favourable overall, immune-related toxicity can occur in any organ system, ranging from mild and manageable events such as rash, joint aches or hypothyroidism, to severe and potentially life-threatening events such as pneumonitis, enterocolitis or myocarditis292. Immune-related adverse events of any grade occur in ~27% of patients treated with drugs targeting PD1 or PDL1, with adverse events of grade ≥3 occurring in 6% of cases292. Systemic corticosteroids for the management of immune-related toxicity were required in 8.2% of patients treated with pembrolizumab as monotherapy in an RCT but in up to 50% of patients receiving nivolumab plus ipilimumab263. The generally favourable safety profile of ICIs has enabled their combination with other agents, as discussed in prior sections, with varying degrees and types of additive toxicity19,263. The toxicity associated with various new ICI combinations may impact the choice of therapy for individual patients.
Health-related quality of life
Beyond treatment-related adverse events, comorbidity from the underlying liver disease also impacts the quality of life in patients with advanced HCC. Owing to the complex relationship between tumour burden and underlying liver function, HCC-specific health-related quality of life (HRQOL) assessments are necessary in clinical management as well as in assessing the safety and efficacy of new therapies19,20,22,293 (Table 5; Supplementary Table 2). The most common instruments used to assess HRQOL in patients with HCC are the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core-30 (EORTC QLQ-C30 and its HCC-specific module, the EORTC QLQ-HCC18 (ref.294)), the Functional Assessment of Cancer Therapy-Hepatobiliary (FACT-Hep) survey and its derivative the FACT Hepatobiliary Symptom Index (FHSI8)295. Patients with HCC report significantly lower physical well-being and lower overall HRQOL than the general population, patients with chronic liver disease and patients with other cancers296. The most commonly reported symptoms driving HRQOL were fatigue, pain, insomnia, anorexia and weight loss, and sexual dysfunction. Poor HRQOL seems to be driven by a combination of liver-related and tumour-related factors, with worse Child-Pugh class and increased tumour burden being independently associated with lower HRQOL scores. Emerging data suggest that HRQOL, particularly role functioning (involvement in life situations involving family, partner relationship, work or household chores), may be independently associated with survival in patients with advanced HCC297.
HRQOL assessments are now incorporated into most HCC RCTs as a secondary endpoint. In the phase III trial of nivolumab versus sorafenib, HRQOL assessments using the FACT-Hep survey showed substantially higher scores for HRQOL in the nivolumab arm than in the sorafenib arm, extending for >100 weeks on treatment and with at least 70% instrument completion rate at all time points267. Similarly, the RCT of atezolizumab plus bevacizumab in advanced HCC also showed substantial prolongation in time to deterioration in quality of life, physical functioning and role functioning using the EORTC QLQ-C30 instrument for the combination regimen compared with sorafenib19 (Supplementary Table 2). The HRQOL outcomes from these trials reinforce the favourable benefit-to-risk ratio for immune-checkpoint inhibition in advanced HCC and establish the utility of patient-reported outcomes in HCC clinical trials.
Outlook
HCC is one of the few malignancies where the major risk factors have been delineated. Although vaccinations and anti-viral therapies have dramatically reduced HCC occurrence, its incidence is steadily growing as a result of other aetiological factors, such as alcohol abuse and NASH, especially in the West41. Strategies aimed at decreasing the risk factors of NASH might also decrease HCC incidence in the future. Enhanced surveillance methods for patients at risk might facilitate the identification of more patients with curative stage disease. New serum biomarkers or ctDNA have the potential to replace imaging as a screening modality or even as a diagnostic modality in the future143,149. With these advancements, in the future, more patients are likely to present with early-stage disease that is more amenable to curative approaches. In addition, even those that present beyond resection or transplant criteria may have better preserved liver function, which will allow the sequential use of numerous lines of systemic anti-cancer therapies leading to improvements in survival.
The marked survival benefit of atezolizumab plus bevacizumab has validated the use of combination therapies as an approach to improving outcomes in patients. More specifically, targeting angiogenesis has proven to be a specifically important part of HCC management. The importance of targeting angiogenesis was emphasized with ramucirumab, which demonstrated survival advantages in second-line treatment for advanced HCC. Nonetheless, the effect of bevacizumab plus atezolizumab goes beyond the expected anti-VEGF effect, particularly to expand the target population to anti-PDL1 responders. The effects of such combination have been reviewed elsewere298,299. In summary, besides normalizing angiogenesis, VEGF inhibitors have been shown to suppress Treg cells, myeloid-derived suppressor cells, and tumour-associated macrophages and to increase cytotoxic T cell activity as well as the maturation of dendritic cells. Overall, these effects might enable the switching of cold tumours into hot tumours, thereby allowing an additional effect of ICIs. A similar mechanism has been identified with TKIs300, where oncogene-mediated T cell exclusion can be reverted by blocking, for instance, CDK4 or CDK6 or MAPK signalling, resulting in dendritic cell activation, T cell infiltration, increased tumour antigen presentation and increased IFNγ sensitivity.
Numerous ongoing phase III trials exploring combinations of TKIs (lenvatinib, cabozantinib and apatinib) and ICIs and combinations of CTLA4 inhibitors (ipilimumab and tremelimumab) and other ICIs are currently ongoing. In the latter of the two combination regimens, a major role in priming and peripheral activation of the immune system is expected301. The next big challenge in the field is to identify the novel combination regimens for a continued improvement in overall survival in the front-line setting. With a high bar now set with a HR 0.66 (for overall survival with bevacizumab plus atezolizumab versus sorafenib) and a median overall survival of 19.2 months19,262, there is no obvious combination partner. The next several years will incorporate the testing of new agents in clinical trials, which will be rationally designed based on basic science and will simultaneously drive future research.
Mechanisms
Our understanding of the molecular pathogenesis and heterogeneity of the disease has also advanced, although this knowledge is yet to influence clinical practice or trial design. Developing data linking molecular subtypes with therapeutic interventions will bridge this gap. As technology evolves, the increasing ability to classify tumours using liquid biopsies or other techniques will serve as a platform for incorporating our molecular understanding of the disease into treatment decisions. These data will also help delineate the mechanisms of resistance to current therapies and lead to personalized medicine tailored to individual patient needs. Ultimately, translating tumour biology into the clinic will continue to improve patient outcomes.
Management
Currently, image-based diagnoses are being challenged because of the need for a more profound molecular understanding of the disease. In this regard, a standardized routine for collecting tumour biopsies in clinical practice is emerging and recommended8,15. The therapeutic armamentarium of HCC has been growing, including improvements in ablation techniques, loco-regional therapies and systemic therapies8,15. As is typical in cancer medicine, once agents have shown efficacy in the advanced setting, they are shifted into earlier stages of the disease, where survival benefits can be amplified. Unfortunately, sorafenib failed to improve outcomes in the adjuvant180 and intermediate-stage settings250. Now, phase III trials with newer immunotherapy agents (alone or in combination) in the adjuvant setting after curative resection or ablation and in combination with locoregional therapies are ongoing (Fig. 4). Shifting these agents into earlier lines of therapy and for patients with earlier disease stages holds the promise of curative treatment for more patients. In addition, the pursuit of biomarkers for assessing response to therapy is ongoing. Although single agent PD1 inhibitors are very active in ~15–20% of patients, this activity was not sufficient to improve survival in randomized studies. The ability to select patients most likely to benefit from a given regimen providing long-lasting responses would be of great value and ongoing translational studies will hopefully offer answers, including an elucidation of the mechanisms of resistance71,73. The capacity of combinations of ICIs with TKIs or VEGF inhibitors to switch cold tumours into hot tumours299,300 (Fig. 5) has already resulted in nearly doubled response rates and survival benefits compared with single agents108 (Fig. 4). With the advent of atezolizumab plus bevacizumab demonstrating significant survival benefits over sorafenib, two major questions emerge: whether other combination regimens will be equally efficacious or improve the survival mark currently established in advanced HCC, and whether the current combination regimen and other regimens reaching ≥30% objective responses can improve outcomes at earlier disease stages. Although vaccine-related therapeutic strategies have not yet yielded significant clinical activity, there is growing interest in cell-based strategies such as chimeric antigen receptor T cell therapy, which is now being studied in early-stage HCC based on its approval in haematologic cancers302. Novel antibody targets are being pursued with both naked antibodies and antibody–drug conjugates to novel epitopes unique to HCC. Overall, we envision major advancements in the management of all stages of the disease based on current investigations in the next 5 years.
Change history
12 February 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41572-024-00500-6
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2, 16018 (2016).
Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 380, 1450–1462 (2019).
International Agency for Research on Cancer. GLOBOCAN 2018. IARC https://gco.iarc.fr/today/online-analysis-map?v=2020&mode=population&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=11&type=0&statistic=5&prevalence=0&population_groupearth&color_palette=default&map_scale=quantile&map_nb_colors=5&continent=0&rotate=%255B10%252C0%255D (2020).
Akinyemiju, T. et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level. JAMA Oncol. 3, 1683 (2017).
Kanwal, F. et al. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology 153, 996–1005.e1 (2017).
Estes, C., Razavi, H., Loomba, R., Younossi, Z. & Sanyal, A. J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018).
Schulze, K. et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 47, 505–511 (2015). A comprehensive genomic study conducted in HCC describing the landscape of mutations and mutational signatures.
Llovet, J. M., Montal, R., Sia, D. & Finn, R. S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 15, 599–616 (2018).
Zucman-Rossi, J., Villanueva, A., Nault, J.-C. & Llovet, J. M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239.e4 (2015).
Anstee, Q. M., Reeves, H. L., Kotsiliti, E., Govaere, O. & Heikenwalder, M. From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16, 411–428 (2019).
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M. & Sanyal, A. J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 (2018).
European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatocellular carcinoma. J. Hepatol. 69, 182–236 (2018).
Marrero, J. A. et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723–750 (2018).
Simon, T. G. et al. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N. Engl. J. Med. 382, 1018–1028 (2020).
Llovet, J. M. et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. https://doi.org/10.1038/s41575-020-00395-0 (2020).
Tabrizian, P. et al. A US multicenter analysis of 2529 HCC patients undergoing liver transplantation: 10-year outcome assessing the role of down-staging to within Milan criteria [abstract 15]. Hepatology 70, 10–11 (2019).
Llovet, J. & Bruix, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 37, 429–442 (2003). This paper is a meta-analysis of randomized studies providing the rationale to use transarterial chemoembolization in intermediate HCC as standard of care.
Salem, R. et al. Y90 radioembolization significantly prolongs time to progression compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 151, 1155–1163.e2 (2016).
Finn, R. S. et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020). This paper is the first study demonstrating survival benefit for any systemic therapies compared with the standard of care sorafenib in advanced HCC.
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008). The first study demonstrating survival benefit for systemic therapies (sorafenib) in advanced HCC compared with placebo.
Kudo, M. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018). This is the first study demonstrating a survival benefit similar to the standard of care sorafenib in advanced HCC compared with placebo.
Bruix, J. et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 (2017). The first study demonstrating survival benefit in second-line treatment for patients with advanced HCC progressing to sorafenib therapy.
Abou-Alfa, G. K. et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 (2018).
Zhu, A. X. et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 282–296 (2019).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017).
Finn, R. S. et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 38, 193–202 (2020).
McGlynn, K. A., Petrick, J. L. & London, W. T. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin. Liver Dis. 19, 223–238 (2015).
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014).
Trinchet, J.-C. et al. Complications and competing risks of death in compensated viral cirrhosis (ANRS CO12 CirVir prospective cohort). Hepatology 62, 737–750 (2015).
Fracanzani, A. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non–iron-related chronic liver disease. Hepatology 33, 647–651 (2001).
Wang, J., Chenivesse, X., Henglein, B. & Bréchot, C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature 343, 555–557 (1990).
Kew, M. C. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 23, 405–409 (2003).
Chang, M. H. et al. Long-term effects of hepatitis B immunization of infants in preventing liver cancer. Gastroenterology 151, 472–480.e1 (2016).
Jain, M. K. et al. Evaluation of a multifaceted intervention to reduce health disparities in hepatitis C screening: a pre-post analysis. Hepatology 70, 40–50 (2019).
Ioannou, G. N. et al. Increased risk for hepatocellular carcinoma persists up to 10 years after HCV eradication in patients with baseline cirrhosis or high FIB-4 scores. Gastroenterology 157, 1264–1278.e4 (2019).
Llovet, J. M. & Villanueva, A. Effect of HCV clearance with direct-acting antiviral agents on HCC. Nat. Rev. Gastroenterol. Hepatol. 13, 561–562 (2016).
Puigvehí, M., Moctezuma-Velázquez, C., Villanueva, A. & Llovet, J. M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 1, 120–130 (2019).
Jepsen, P., Ott, P., Andersen, P. K., Sørensen, H. T. & Vilstrup, H. Risk for hepatocellular carcinoma in patients with alcoholic cirrhosis. Ann. Intern. Med. 156, 841 (2012).
Lin, C. W. et al. Heavy alcohol consumption increases the incidence of hepatocellular carcinoma in hepatitis B virus-related cirrhosis. J. Hepatol. 58, 730–735 (2013).
Welzel, T. M. et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 108, 1314–1321 (2013).
Kanwal, F. et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 155, 1828–1837.e2 (2018).
Mittal, S. et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 14, 124–131.e1 (2016).
Rich, N. E., Yopp, A. C., Singal, A. G. & Murphy, C. C. Hepatocellular carcinoma incidence is decreasing among younger adults in the United States. Clin. Gastroenterol. Hepatol. 18, 242–248.e5 (2020).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 68, 394–424 (2018).
Rich, N. E. et al. Racial and ethnic differences in presentation and outcomes of hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 17, 551–559.e1 (2019).
Lee, Y.-C. A. et al. Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int. J. Epidemiol. 38, 1497–1511 (2009).
Bravi, F., Bosetti, C., Tavani, A., Gallus, S. & La Vecchia, C. Coffee reduces risk for hepatocellular carcinoma: an updated meta-analysis. Clin. Gastroenterol. Hepatol. 11, 1413–1421.e1 (2013).
Sia, D., Villanueva, A., Friedman, S. L. & Llovet, J. M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152, 745–761 (2017).
Pikarsky, E. Neighbourhood deaths cause a switch in cancer subtype. Nature 562, 45–46 (2018).
Seehawer, M. et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 562, 69–75 (2018).
Guichard, C. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 44, 694–698 (2012).
Chiang, D. Y. et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 68, 6779–6788 (2008).
Calderaro, J., Ziol, M., Paradis, V. & Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 71, 616–630 (2019).
Hyman, D. M., Taylor, B. S. & Baselga, J. Implementing genome-driven oncology. Cell 168, 584–599 (2017).
Bressac, B., Kew, M., Wands, J. & Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350, 429–431 (1991).
Wang, B. et al. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: Evidence from an updated meta-analysis. J. Hepatol. 53, 508–518 (2010).
Romeo, S. et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465 (2008).
Buch, S. et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 47, 1443–1448 (2015).
Paterlini-Bréchot, P. et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 22, 3911–3916 (2003).
Nault, J.-C., Ningarhari, M., Rebouissou, S. & Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 544–558 (2019).
Bayard, Q. et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 9, 5235 (2018).
Nault, J.-C. et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 47, 1187–1193 (2015).
Letouzé, E. et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 8, 1315 (2017).
Ng, A. W. T. et al. Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Sci. Transl. Med. 9, eaan6446 (2017).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Rebouissou, S. & Nault, J.-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 72, 215–229 (2020).
Hoshida, Y. et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 69, 7385–7392 (2009).
Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341.e23 (2017).
Lee, J.-S. et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 12, 410–416 (2006).
Boyault, S. et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45, 42–52 (2007).
Sia, D. et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 153, 812–826 (2017).
Lachenmayer, A. et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 18, 4997–5007 (2012).
Ruiz de Galarreta, M. et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019).
Renehan, A. G., Tyson, M., Egger, M., Heller, R. F. & Zwahlen, M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–578 (2008).
Sutti, S. & Albano, E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 17, 81–92 (2020).
Nakagawa, H. et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014).
Nishida, N. et al. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 31, 1646–1653 (2016).
Tummala, K. S. et al. Inhibition of de novo NAD+ synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell 26, 826–839 (2014).
Gomes, A. L. et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30, 161–175 (2016).
Guri, Y. et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32, 807–823.e12 (2017).
Liu, D. et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 10, eaap9840 (2018).
Umemura, A. et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 29, 935–948 (2016).
Grohmann, M. et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 175, 1289–1306.e20 (2018).
Henderson, J. M., Zhang, H. E., Polak, N. & Gorrell, M. D. Hepatocellular carcinoma: mouse models and the potential roles of proteases. Cancer Lett. 387, 106–113 (2017).
Negro, F. Natural history of NASH and HCC. Liver Int. 40, 72–76 (2020).
Rudalska, R. et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 20, 1138–1146 (2014).
Martinez-Quetglas, I. et al. IGF2 is up-regulated by epigenetic mechanisms in hepatocellular carcinomas and is an actionable oncogene product in experimental models. Gastroenterology 151, 1192–1205 (2016).
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014).
Cook, N., Jodrell, D. I. & Tuveson, D. A. Predictive in vivo animal models and translation to clinical trials. Drug Discov. Today 17, 253–260 (2012).
Singh, M. & Ferrara, N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat. Biotechnol. 30, 648–657 (2012).
Newell, P., Villanueva, A., Friedman, S. L., Koike, K. & Llovet, J. M. Experimental models of hepatocellular carcinoma. J. Hepatol. 48, 858–879 (2008).
Bresnahan, E., Ramadori, P., Heikenwalder, M., Zender, L. & Lujambio, A. Novel patient-derived preclinical models of liver cancer. J. Hepatol. 72, 239–249 (2020).
Moriya, K. et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4, 1065–1067 (1998).
Hagel, M. et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 5, 424–437 (2015).
Day, C.-P., Merlino, G. & Van Dyke, T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53 (2015).
Jayson, G. & Harris, J. How participants in cancer trials are chosen: ethics and conflicting interests. Nat. Rev. Cancer 6, 330–336 (2006).
Febbraio, M. A. et al. Preclinical models for studying NASH-driven HCC: how useful are they? Cell Metab. 29, 18–26 (2019).
Sharpless, N. E. & DePinho, R. A. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 5, 741–754 (2006).
Wolf, M. J. et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564 (2014).
Ma, C. et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257 (2016).
Malehmir, M. et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 25, 641–655 (2019).
Park, E. J. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208 (2010).
Ringelhan, M., Pfister, D., O’Connor, T., Pikarsky, E. & Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 19, 222–232 (2018).
Wada, Y., Nakashima, O., Kutami, R., Yamamoto, O. & Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 27, 407–414 (1998).
Yuan, D. et al. Kupffer cell-derived TNF triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell 31, 771–789.e6 (2017).
Crispe, I. N. The liver as a lymphoid organ. Annu. Rev. Immunol. 27, 147–163 (2009).
Horwitz, E. et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to Sorafenib treatment. Cancer Discov. 4, 730–743 (2014).
Finn, R. S. et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 38, 2960–2970 (2020).
Hou, J., Zhang, H., Sun, B. & Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J. Hepatol. 72, 167–182 (2020).
Hoshida, Y. et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 359, 1995–2004 (2008). This study is the first molecular indication of the importance of the cancer field effect in the outcome of patients with HCC.
Shalapour, S. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345 (2017).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Flecken, T. et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 59, 1415–1426 (2014).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017).
Langhans, B. et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol. Immunother. 68, 2055–2066 (2019).
Garnelo, M. et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 66, 342–351 (2017).
Bruno, T. C. New predictors for immunotherapy responses sharpen our view of the tumour microenvironment. Nature 577, 474–476 (2020).
Schneider, C. et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 61, 1733–1743 (2012).
Sautès-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Calderaro, J. et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 70, 58–65 (2019).
Finkin, S. et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 16, 1235–1244 (2015).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Higashi, T., Friedman, S. L. & Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 121, 27–42 (2017).
Dapito, D. H. et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 (2012).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018).
Hoshida, Y. et al. Prognostic gene expression signature for patients with hepatitis C–related early-stage cirrhosis. Gastroenterology 144, 1024–1030 (2013).
Budhu, A. et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 10, 99–111 (2006).
Moeini, A. et al. An immune gene expression signature associated with development of human hepatocellular carcinoma identifies mice that respond to chemopreventive agents. Gastroenterology 157, 1383–1397.e11 (2019).
Singal, A. G., Lampertico, P. & Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J. Hepatol. 72, 250–261 (2020).
Papatheodoridis, G. et al. PAGE-B predicts the risk of developing hepatocellular carcinoma in Caucasians with chronic hepatitis B on 5-year antiviral therapy. J. Hepatol. 64, 800–806 (2016).
Shinmura, R. et al. Cirrhotic nodules: association between MR imaging signal intensity and intranodular blood supply. Radiology 237, 512–519 (2005).
van der Pol, C. B. et al. Accuracy of the liver imaging reporting and data system in computed tomography and magnetic resonance image analysis of hepatocellular carcinoma or overall malignancy — a systematic review. Gastroenterology 156, 976–986 (2019).
Paisant, A. et al. Comparison of extracellular and hepatobiliary MR contrast agents for the diagnosis of small HCCs. J. Hepatol. 72, 937–945 (2020).
Kojiro, M. et al. Pathologic diagnosis of early hepatocellular carcinoma: a report of the international consensus group for hepatocellular neoplasia. Hepatology 49, 658–664 (2009).
Forner, A. et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: Prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 47, 97–104 (2007).
Zhang, B.-H., Yang, B.-H. & Tang, Z.-Y. Randomized controlled trial of screening for hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 130, 417–422 (2004).
Lederle, F. A. Screening for liver cancer: the rush to judgment. Ann. Intern. Med. 156, 387 (2012).
Poustchi, H. et al. Feasibility of conducting a randomized control trial for liver cancer screening: is a randomized controlled trial for liver cancer screening feasible or still needed? Hepatology 54, 1998–2004 (2011).
Singal, A. G., Pillai, A. & Tiro, J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta-analysis. PLoS Med. 11, e1001624 (2014). This paper is a meta-analysis defining the accuracy of distinct surveillance strategies for the early detection of HCC.
Andersson, K. L., Salomon, J. A., Goldie, S. J. & Chung, R. T. Cost effectiveness of alternative surveillance strategies for hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 6, 1418–1424 (2008).
Trinchet, J.-C. et al. Ultrasonographic surveillance of hepatocellular carcinoma in cirrhosis: a randomized trial comparing 3- and 6-month periodicities. Hepatology 54, 1987–1997 (2011).
Atiq, O. et al. An assessment of benefits and harms of hepatocellular carcinoma surveillance in patients with cirrhosis. Hepatology 65, 1196–1205 (2017).
Marrero, J. A. et al. α-Fetoprotein, DES-γ carboxyprothrombin, and lectin-bound α-fetoprotein in early hepatocellular carcinoma. Gastroenterology 137, 110–118 (2009).
Pepe, M. S. et al. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 93, 1054–1061 (2001).
Tzartzeva, K. et al. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta-analysis. Gastroenterology 154, 1706–1718.e1 (2018).
Labgaa, I. et al. A pilot study of ultra-deep targeted sequencing of plasma DNA identifies driver mutations in hepatocellular carcinoma. Oncogene 37, 3740–3752 (2018).
Kisiel, J. B. et al. Hepatocellular carcinoma detection by plasma methylated DNA: discovery, phase I pilot, and phase II clinical validation. Hepatology 69, 1180–1192 (2019).
Xu, R. et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 16, 1155–1161 (2017).
Qu, C. et al. Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy. Proc. Natl Acad. Sci. USA 116, 6308–6312 (2019).
Oh, C. R. et al. Genome-wide copy number alteration and VEGFA amplification of circulating cell-free DNA as a biomarker in advanced hepatocellular carcinoma patients treated with Sorafenib. BMC Cancer 19, 292 (2019).
Torga, G. & Pienta, K. J. Patient-paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol. 4, 868 (2018).
Papatheodoridis, G. V., Chan, H. L.-Y., Hansen, B. E., Janssen, H. L. A. & Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: assessment and modification with current antiviral therapy. J. Hepatol. 62, 956–967 (2015).
Chang, M.-H. et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. N. Engl. J. Med. 336, 1855–1859 (1997).
Singh, S., Singh, P. P., Singh, A. G., Murad, M. H. & Sanchez, W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology 144, 323–332 (2013).
Kennedy, O. J. et al. Coffee, including caffeinated and decaffeinated coffee, and the risk of hepatocellular carcinoma: a systematic review and dose–response meta-analysis. BMJ Open 7, e013739 (2017).
Llovet, J. M. et al. Trial design and endpoints in hepatocellular carcinoma: AASLD consensus conference. Hepatology https://doi.org/10.1002/hep.31327 (2020).
Llovet, J., Brú, C. & Bruix, J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin. Liver Dis. 19, 329–338 (1999). The first description of the BCLC classification widely used in guidelines of management of HCC.
D’Amico, G. et al. Clinical states of cirrhosis and competing risks. J. Hepatol. 68, 563–576 (2018).
Llovet, J. M. et al. Natural history of untreated nonsurgical hepatocellular carcinoma: rationale for the design and evaluation of therapeutic trials. Hepatology 29, 62–67 (1999).
Shrager, B., Jibara, G., Schwartz, M. & Roayaie, S. Resection of hepatocellular carcinoma without cirrhosis. Ann. Surg. 255, 1135–1143 (2012).
Viganò, L. et al. Liver resection for hepatocellular carcinoma in patients with metabolic syndrome: a multicenter matched analysis with HCV-related HCC. J. Hepatol. 63, 93–101 (2015).
Piscaglia, F. et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 63, 827–838 (2016).
Zhou, X.-D. et al. Experience of 1000 patients who underwent hepatectomy for small hepatocellular carcinoma. Cancer 91, 1479–1486 (2001).
Johnson, P. J. et al. Assessment of liver function in patients with hepatocellular carcinoma: a new evidence-based approach — the ALBI grade. J. Clin. Oncol. 33, 550–558 (2015).
Pinato, D. J. et al. The ALBI grade provides objective hepatic reserve estimation across each BCLC stage of hepatocellular carcinoma. J. Hepatol. 66, 338–346 (2017).
Roayaie, S. et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology 62, 440–451 (2015).
Berardi, G. et al. Development of a nomogram to predict outcome after liver resection for hepatocellular carcinoma in Child-Pugh B cirrhosis. J. Hepatol. 72, 75–84 (2020).
Ishizawa, T. et al. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology 134, 1908–1916 (2008).
Citterio, D. et al. Hierarchic interaction of factors associated with liver decompensation after resection for hepatocellular carcinoma. JAMA Surg. 151, 846–853 (2016).
Vitale, A. et al. Survival benefit of liver resection for patients with hepatocellular carcinoma across different Barcelona clinic liver cancer stages: a multicentre study. J. Hepatol. 62, 617–624 (2015).
Yin, L. et al. Partial hepatectomy vs. transcatheter arterial chemoembolization for resectable multiple hepatocellular carcinoma beyond Milan criteria: a RCT. J. Hepatol. 61, 82–88 (2014).
Kokudo, T. et al. Survival benefit of liver resection for hepatocellular carcinoma associated with portal vein invasion. J. Hepatol. 65, 938–943 (2016).
Roayaie, S., Jibara, G., Taouli, B. & Schwartz, M. Resection of hepatocellular carcinoma with macroscopic vascular invasion. Ann. Surg. Oncol. 20, 3754–3760 (2013).
Roayaie, S. et al. Resection of hepatocellular cancer ≤2 cm: results from two Western centers. Hepatology 57, 1426–1435 (2013).
Imamura, H. et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J. Hepatol. 38, 200–207 (2003).
Shi, M. et al. Partial hepatectomy with wide versus narrow resection margin for solitary hepatocellular carcinoma: a prospective randomized trial. Ann. Surg. 245, 36–43 (2007).
Hidaka, M. et al. Impact of anatomical resection for hepatocellular carcinoma with microportal invasion (vp1): a multi-institutional study by the Kyushu study group of liver surgery. Ann. Surg. 271, 339–346 (2020).
Samuel, M., Chow, P. K. H., Shih-Yen, E. C., Machin, D. & Soo, K. C. Neoadjuvant and adjuvant therapy for surgical resection of hepatocellular carcinoma. Cochrane Database Syst. Rev. 2009, CD001199 (2009).
Lee, J. H. et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 148, 1383–1391.e6 (2015).
Bruix, J. et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 16, 1344–1354 (2015).
Yin, J. et al. Effect of antiviral treatment with nucleotide/nucleoside analogs on postoperative prognosis of hepatitis B virus-related hepatocellular carcinoma: a two-stage longitudinal clinical study. J. Clin. Oncol. 31, 3647–3655 (2013).
Singal, A. G. et al. Direct-acting antiviral therapy for hepatitis C virus infection is associated with increased survival in patients with a history of hepatocellular carcinoma. Gastroenterology 157, 1253–1263.e2 (2019).
Waziry, R. et al. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: a systematic review, meta-analyses, and meta-regression. J. Hepatol. 67, 1204–1212 (2017).
Reig, M. et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 65, 719–726 (2016).
Tabrizian, P., Jibara, G., Shrager, B., Schwartz, M. & Roayaie, S. Recurrence of hepatocellular cancer after resection: patterns, treatments, and prognosis. Ann. Surg. 261, 947–955 (2015).
Ferrer-Fàbrega, J. et al. Prospective validation of ab initio liver transplantation in hepatocellular carcinoma upon detection of risk factors for recurrence after resection. Hepatology 63, 839–849 (2016).
Mazzaferro, V. et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 334, 693–700 (1996). This is a landmark study establishing the criteria for liver transplantation in HCC.
Franssen, B., Jibara, G., Tabrizian, P., Schwartz, M. E. & Roayaie, S. Actual 10-year survival following hepatectomy for hepatocellular carcinoma. HPB 16, 830–835 (2014).
Cucchetti, A. et al. The chances of hepatic resection curing hepatocellular carcinoma. J. Hepatol. 72, 711–717 (2020).
Llovet, J. M., Fuster, J. & Bruix, J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 30, 1434–1440 (1999).
Yao, F. Y. et al. Liver transplantation for hepatocellular carcinoma: Expansion of the tumor size limits does not adversely impact survival. Hepatology 33, 1394–1403 (2001).
Yao, F. Y. et al. Liver transplantation for hepatocellular carcinoma: validation of the UCSF-expanded criteria based on preoperative imaging. Am. J. Transplant. 7, 2587–2596 (2007).
Mazzaferro, V. et al. Predicting survival after liver transplantation in patients with hepatocellular carcinoma beyond the Milan criteria: a retrospective, exploratory analysis. Lancet Oncol. 10, 35–43 (2009).
Commander, S. J. et al. A long-term experience with expansion of Milan criteria for liver transplant recipients. Clin. Transplant. 32, e13254 (2018).
Ravaioli, M. et al. Liver transplantation for hepatocellular carcinoma: results of down-staging in patients initially outside the Milan selection criteria. Am. J. Transpl. 8, 2547–2557 (2008).
Mazzaferro, V. et al. Metroticket 2.0 model for analysis of competing risks of death after liver transplantation for hepatocellular carcinoma. Gastroenterology 154, 128–139 (2018).
Hong, G. et al. Alpha-fetoprotein and 18F-FDG positron emission tomography predict tumor recurrence better than Milan criteria in living donor liver transplantation. J. Hepatol. 64, 852–859 (2016).
Kulik, L. et al. Therapies for patients with hepatocellular carcinoma awaiting liver transplantation: a systematic review and meta-analysis. Hepatology 67, 381–400 (2018).
Yao, F. Y. et al. Downstaging of hepatocellular cancer before liver transplant: long-term outcome compared to tumors within Milan criteria. Hepatology 61, 1968–1977 (2015).
Cucchetti, A. et al. Including mRECIST in the Metroticket 2.0 criteria improves prediction of hepatocellular carcinoma-related death after liver transplant. J. Hepatol. 73, 342–348 (2020).
Halazun, K. J. et al. Is it time to abandon the Milan criteria? Results of a bicoastal US collaboration to redefine hepatocellular carcinoma liver transplantation selection policies. Ann. Surg. 268, 690–699 (2018).
Kulik, L. M. et al. Outcomes of living and deceased donor liver transplant recipients with hepatocellular carcinoma: results of the A2ALL cohort. Am. J. Transpl. 12, 2997–3007 (2012).
Miltiadous, O. et al. Progenitor cell markers predict outcome of patients with hepatocellular carcinoma beyond Milan criteria undergoing liver transplantation. J. Hepatol. 63, 1368–1377 (2015).
Geissler, E. K. et al. Sirolimus use in liver transplant recipients with hepatocellular carcinoma. Transplantation 100, 116–125 (2016).
Heimbach, J. K. et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 67, 358–380 (2018).
Lencioni, R. & Llovet, J. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin. Liver Dis. 30, 52–60 (2010).
Lencioni, R. New data supporting modified RECIST (mRECIST) for hepatocellular carcinoma. Clin. Cancer Res. 19, 1312–1314 (2013).
Llovet, J. M. & Lencioni, R. mRECIST for HCC: performance and novel refinements. J. Hepatol. 72, 288–306 (2020).
Meyer, T. et al. mRECIST to predict survival in advanced hepatocellular carcinoma: analysis of two randomised phase II trials comparing nintedanib vs sorafenib. Liver Int. 37, 1047–1055 (2017).
Lencioni, R. et al. Objective response by mRECIST as a predictor and potential surrogate end-point of overall survival in advanced HCC. J. Hepatol. 66, 1166–1172 (2017).
Kudo, M. et al. Analysis of survival and objective response (OR) in patients with hepatocellular carcinoma in a phase III study of lenvatinib (REFLECT). J. Clin. Oncol. 37, 186–186 (2019).
Lencioni, R. A. et al. Small hepatocellular carcinoma in cirrhosis: randomized comparison of radio-frequency thermal ablation versus percutaneous ethanol injection. Radiology 228, 235–240 (2003).
Lin, S.-M., Lin, C.-J., Lin, C.-C., Hsu, C.-W. & Chen, Y.-C. Radiofrequency ablation improves prognosis compared with ethanol injection for hepatocellular carcinoma ≤4 cm. Gastroenterology 127, 1714–1723 (2004).
Shiina, S. et al. A randomized controlled trial of radiofrequency ablation with ethanol injection for small hepatocellular carcinoma. Gastroenterology 129, 122–130 (2005).
Ng, K. K. C. et al. Randomized clinical trial of hepatic resection versus radiofrequency ablation for early-stage hepatocellular carcinoma. Br. J. Surg. 104, 1775–1784 (2017).
Xu, X.-L., Liu, X.-D., Liang, M. & Luo, B.-M. Radiofrequency ablation versus hepatic resection for small hepatocellular carcinoma: systematic review of randomized controlled trials with meta-analysis and trial sequential analysis. Radiology 287, 461–472 (2018).
Izumi, N. et al. A multicenter randomized controlled trial to evaluate the efficacy of surgery vs. radiofrequency ablation for small hepatocellular carcinoma (SURF trial). J. Clin. Oncol. 37, 4002–4002 (2019).
Xia, Y. et al. Long-term effects of repeat hepatectomy vs percutaneous radiofrequency ablation among patients with recurrent hepatocellular carcinoma. JAMA Oncol. 6, 255–263 (2020).
Lencioni, R. et al. Early-stage hepatocellular carcinoma in patients with cirrhosis: long-term results of percutaneous image-guided radiofrequency ablation. Radiology 234, 961–967 (2005).
Sala, M. et al. Initial response to percutaneous ablation predicts survival in patients with hepatocellular carcinoma. Hepatology 40, 1352–1360 (2004).
Breen, D. J. & Lencioni, R. Image-guided ablation of primary liver and renal tumours. Nat. Rev. Clin. Oncol. 12, 175–186 (2015).
Yu, J. et al. Percutaneous cooled-probe microwave versus radiofrequency ablation in early-stage hepatocellular carcinoma: a phase III randomised controlled trial. Gut 66, 1172–1173 (2017).
Hu, J. et al. Image-guided percutaneous microwave ablation versus cryoablation for hepatocellular carcinoma in high-risk locations: intermediate-term results. Cancer Manag. Res. 11, 9801–9811 (2019).
Cheng, R. G., Bhattacharya, R., Yeh, M. M. & Padia, S. A. Irreversible electroporation can effectively ablate hepatocellular carcinoma to complete pathologic necrosis. J. Vasc. Interv. Radiol. 26, 1184–1188 (2015).
Sutter, O. et al. Safety and efficacy of irreversible electroporation for the treatment of hepatocellular carcinoma not amenable to thermal ablation techniques: a retrospective single-center case series. Radiology 284, 877–886 (2017).
Peng, Z.-W. et al. Radiofrequency ablation with or without transcatheter arterial chemoembolization in the treatment of hepatocellular carcinoma: a prospective randomized trial. J. Clin. Oncol. 31, 426–432 (2013).
Tak, W. Y. et al. Phase III HEAT study adding lyso-thermosensitive liposomal doxorubicin to radiofrequency ablation in patients with unresectable hepatocellular carcinoma lesions. Clin. Cancer Res. 24, 73–83 (2018).
Wang, C. et al. Multicenter randomized controlled trial of percutaneous cryoablation versus radiofrequency ablation in hepatocellular carcinoma. Hepatology 61, 1579–1590 (2015).
Xu, J. et al. Radiofrequency ablation vs. cryoablation for localized hepatocellular carcinoma: a propensity-matched population study. Anticancer. Res. 38, 6381–6386 (2018).
Di Costanzo, G. G. et al. Radiofrequency ablation versus laser ablation for the treatment of small hepatocellular carcinoma in cirrhosis: a randomized trial. J. Gastroenterol. Hepatol. 30, 559–565 (2015).
Bujold, A. et al. Sequential phase I and II trials of stereotactic body radiotherapy for locally advanced hepatocellular carcinoma. J. Clin. Oncol. 31, 1631–1639 (2013).
Hong, T. S. et al. Multi-institutional phase II study of high-dose hypofractionated proton beam therapy in patients with localized, unresectable hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Clin. Oncol. 34, 460–468 (2016).
Tse, R. V. et al. Phase I study of individualized stereotactic body radiotherapy for hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Clin. Oncol. 26, 657–664 (2008).
Yang, J.-F. et al. Stereotactic ablative radiotherapy versus conventionally fractionated radiotherapy in the treatment of hepatocellular carcinoma with portal vein invasion: a retrospective analysis. Radiat. Oncol. 14, 180 (2019).
Wahl, D. R. et al. Outcomes after stereotactic body radiotherapy or radiofrequency ablation for hepatocellular carcinoma. J. Clin. Oncol. 34, 452–459 (2016).
Shen, P. C. et al. Comparison of stereotactic body radiation therapy and transarterial chemoembolization for unresectable medium-sized hepatocellular carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 105, 307–318 (2019).
Kim, T. H. et al. Proton beam radiotherapy vs. radiofrequency ablation for recurrent hepatocellular carcinoma: a randomized phase III trial. J. Hepatol. https://doi.org/10.1016/j.jhep.2020.09.026 (2020)
Llovet, J. M. et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 359, 1734–1739 (2002).
Lo, C. et al. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology 35, 1164–1171 (2002).
Vogel, A. et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 29 (Suppl. 4), iv238–iv255 (2018).
Lencioni, R., de Baere, T., Soulen, M. C., Rilling, W. S. & Geschwind, J.-F. H. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: a systematic review of efficacy and safety data. Hepatology 64, 106–116 (2016).
Meyer, T. et al. Sorafenib in combination with transarterial chemoembolisation in patients with unresectable hepatocellular carcinoma (TACE 2): a randomised placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol. Hepatol. 2, 565–575 (2017).
Kudo, M. et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: a randomized phase III trial. Hepatology 60, 1697–1707 (2014).
Okusaka, T. et al. Transarterial chemotherapy alone versus transarterial chemoembolization for hepatocellular carcinoma: a randomized phase III trial. J. Hepatol. 51, 1030–1036 (2009).
Chau, I. et al. Alpha-fetoprotein kinetics in patients with hepatocellular carcinoma receiving ramucirumab or placebo: an analysis of the phase 3 REACH study. Br. J. Cancer 119, 19–26 (2018).
Lammer, J. et al. Prospective randomized study of doxorubicin-eluting-bead embolization in the treatment of hepatocellular carcinoma: results of the PRECISION V study. Cardiovasc. Intervent. Radiol. 33, 41–52 (2010).
Varela, M. et al. Chemoembolization of hepatocellular carcinoma with drug eluting beads: efficacy and doxorubicin pharmacokinetics. J. Hepatol. 46, 474–481 (2007).
Burrel, M. et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using drug eluting beads. Implications for clinical practice and trial design. J. Hepatol. 56, 1330–1335 (2012).
Vincenzi, B. et al. Prognostic relevance of objective response according to EASL criteria and mRECIST criteria in hepatocellular carcinoma patients treated with loco-regional therapies: a literature-based meta-analysis. PLoS ONE 10, e0133488 (2015).
Lencioni, R. et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: the SPACE trial. J. Hepatol. 64, 1090–1098 (2016).
Kudo, M. et al. Orantinib versus placebo combined with transcatheter arterial chemoembolisation in patients with unresectable hepatocellular carcinoma (ORIENTAL): a randomised, double-blind, placebo-controlled, multicentre, phase 3 study. Lancet Gastroenterol. Hepatol. 3, 37–46 (2018).
Kudo, M. et al. Sorafenib plus low-dose cisplatin and fluorouracil hepatic arterial infusion chemotherapy versus sorafenib alone in patients with advanced hepatocellular carcinoma (SILIUS): a randomised, open label, phase 3 trial. Lancet Gastroenterol. Hepatol. 3, 424–432 (2018).
Park, J. W. et al. Sorafenib with or without concurrent transarterial chemoembolization in patients with advanced hepatocellular carcinoma: the phase III STAH trial. J. Hepatol. 70, 684–691 (2019).
Kudo, M. et al. Phase III study of sorafenib after transarterial chemoembolisation in Japanese and Korean patients with unresectable hepatocellular carcinoma. Eur. J. Cancer 47, 2117–2127 (2011).
Hilgard, P. et al. Radioembolization with yttrium-90 glass microspheres in hepatocellular carcinoma: European experience on safety and long-term survival. Hepatology 52, 1741–1749 (2010).
Salem, R. et al. Radioembolization for hepatocellular carcinoma using Yttrium-90 microspheres: a comprehensive report of long-term outcomes. Gastroenterology 138, 52–64 (2010).
Mazzaferro, V. et al. Yttrium-90 radioembolization for intermediate-advanced hepatocellular carcinoma: a phase 2 study. Hepatology 57, 1826–1837 (2013).
Vilgrain, V. et al. Efficacy and safety of selective internal radiotherapy with yttrium-90 resin microspheres compared with sorafenib in locally advanced and inoperable hepatocellular carcinoma (SARAH): an open-label randomised controlled phase 3 trial. Lancet Oncol. 18, 1624–1636 (2017).
Chow, P. K. H. et al. SIRveNIB: selective internal radiation therapy versus sorafenib in Asia-Pacific patients with hepatocellular carcinoma. J. Clin. Oncol. 36, 1913–1921 (2018).
Ricke, J. et al. Impact of combined selective internal radiation therapy and sorafenib on survival in advanced hepatocellular carcinoma. J. Hepatol. 71, 1164–1174 (2019).
Bruix, J. et al. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: analysis of two phase III studies. J. Hepatol. 67, 999–1008 (2017).
Finn R. S. et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib(sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 39, (suppl 3; abstr 267) (2021).
Yau, T. et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 6, e204564 (2020).
Zhu, A. X. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952 (2018).
Llovet, J. M., Montal, R. & Villanueva, A. Randomized trials and endpoints in advanced HCC: role of PFS as a surrogate of survival. J. Hepatol. 70, 1262–1277 (2019).
Office of the Commissioner. FDA grants accelerated approval to nivolumab and ipilimumab combination for hepatocellular carcinoma. FDA https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-and-ipilimumab-combination-hepatocellular-carcinoma (2020).
Yau, T. et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 30, v874–v875 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04039607 (2019).
Zappasodi, R., Merghoub, T. & Wolchok, J. D. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598 (2018).
Rahma, O. E. & Hodi, F. S. The intersection between tumor angiogenesis and immune suppression. Clin. Cancer Res. 25, 5449–5457 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03713593 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03755791 (2018).
Bergerot, P., Lamb, P., Wang, E. & Pal, S. K. Cabozantinib in combination with immunotherapy for advanced renal cell carcinoma and urothelial carcinoma: rationale and clinical evidence. Mol. Cancer Ther. 18, 2185–2193 (2019).
Ott, P. A., Hodi, F. S., Kaufman, H. L., Wigginton, J. M. & Wolchok, J. D. Combination immunotherapy: a road map. J. Immunother. Cancer 5, 16 (2017).
Hellmann, M. D. et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018).
Kelley, R. K. et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 38, 4508–4508 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03298451 (2017).
Schmidt, E. V. et al. Assessment of clinical activity of PD-1 checkpoint inhibitor combination therapies reported in clinical trials. JAMA Netw. Open 3, e1920833 (2020).
Montal, R. et al. Molecular portrait of high alpha-fetoprotein in hepatocellular carcinoma: implications for biomarker-driven clinical trials. Br. J. Cancer 121, 340–343 (2019).
Galle, P. R. et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 39, 2214–2229 (2019).
Teufel, M. et al. Biomarkers associated with response to regorafenib in patients with hepatocellular carcinoma. Gastroenterology 156, 1731–1741 (2019).
Pinyol, R. et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 68, 1065–1075 (2019).
Li, X.-S., Li, J.-W., Li, H. & Jiang, T. Prognostic value of programmed cell death ligand 1 (PD-L1) for hepatocellular carcinoma: a meta-analysis. Biosci. Rep. 40, BSR20200459 (2020).
Kim, R. D. et al. First-in-human phase I study of Fisogatinib (BLU-554) validates aberrant fibroblast growth factor 19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 9, 1696–1707 (2019).
Lim, H. Y. et al. Phase II studies with refametinib or refametinib plus sorafenib in patients with RAS-mutated hepatocellular carcinoma. Clin. Cancer Res. 24, 4650–4661 (2018).
Rimassa, L. et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 19, 682–693 (2018).
Tapper, E. B. & Asrani, S. K. The COVID-19 pandemic will have a long-lasting impact on the quality of cirrhosis care. J. Hepatol. https://doi.org/10.1016/j.jhep.2020.04.005 (2020).
US Census Bureau. Coronavirus (COVID-19) pandemic. US Census Bureau https://www.census.gov/coronavirus (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
International Liver Cancer Association. Management of HCC during COVID-19: ILCA guidance. ILCA https://ilca-online.org/news/management-of-hcc-during-covid-19-ilca-guidance/ (2020).
Cheng, A. L. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34 (2009).
Sangro, B. et al. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 72, 320–341 (2020).
Zhu, A. X. et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 312, 57–67 (2014).
Blazeby, J. M. et al. Development of a questionnaire module to supplement the EORTC QLQ-C30 to assess quality of life in patients with hepatocellular carcinoma, the EORTC QLQ-HCC18. Eur. J. Cancer 40, 2439–2444 (2004).
Heffernan, N. et al. Measuring health-related quality of life in patients with hepatobiliary cancers: the functional assessment of Cancer Therapy-Hepatobiliary Questionnaire. J. Clin. Oncol. 20, 2229–2239 (2002).
Fan, S. Y., Eiser, C. & Ho, M. C. Health-related quality of life in patients with hepatocellular carcinoma: a systematic review. Clin. Gastroenterol. Hepatol. 8, 559–564 (2010).
Diouf, M. et al. The added value of quality of life (QoL) for prognosis of overall survival in patients with palliative hepatocellular carcinoma. J. Hepatol. 58, 509–521 (2013).
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G. & Jain, R. K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Kalbasi, A. & Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020).
Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 38, 255 (2019).
Rafiq, S., Hackett, C. S. & Brentjens, R. J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 17, 147–167 (2020).
Acknowledgements
The authors thank Florian Castet for his invaluable support in the production of this manuscript. J.M.L. acknowledges his research funding from the Accelerator Award (HUNTER, Ref. C9380/A26813, partnership between the CRUK, AECC and AIRC), National Cancer Institute (P30-CA196521), U.S. Department of Defense (CA150272P3), Samuel Waxman Cancer Research Foundation, Spanish National Health Institute (PID2019-105378RB-100) and the Generalitat de Catalunya/AGAUR (SGR-1358). E.P. acknowledges his grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Israel Science Foundation.
Author information
Authors and Affiliations
Contributions
Introduction (J.M.L.); Epidemiology (A.G.S.); Mechanisms/pathophysiology (J.Z-R., E.P., K.K. and A.V.); Diagnosis, screening and prevention (A.G.S. and A.V.); Management (J.M.L., R.K.K., S.R., R.S.F., A.V. and R.L.); Quality of life (R.K.K., A.G.S. and A.V.); Outlook (J.M.L. and R.S.F.); Overview of Primer (J.M.L.).
Corresponding author
Ethics declarations
Competing interests
J.M.L. receives research support from Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Boehringer-Ingelheim, Eisai Inc., and Ipsen and received consulting fees from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Can-Fite Biopharma, Celsion Corporation, Eli Lilly, Eisai Inc., Exelixis, Genentech, Glycotest, Merck, Nucleix and Roche. R.K.K. receives research support to the institution from Adaptimmune, Agios Inc., AstraZeneca, Bayer, Bristol-Myers Squibb, Eli Lilly, EMD Serono, Exelixis, Merck, Novartis, Partner Therapeutics, QED and Taiho; R.K.K. has received consulting for Independent Data Monitoring Committee fees from Genentech or Roche and Gilead, and travel support from Ipsen. A.V. has received consulting fees from Guidepoint and Fujifilm and advisory board fees from Exact Sciences, Gilead, Nucleix and NGM Pharmaceuticals. A.G.S. has received consulting fees from AstraZeneca, Bayer, Bristol-Myers Squibb, Eisai, Exact Sciences, Exelixis, Fujifilm, Genentech, Glycotest, GRAIL and Roche. S.R. is a consulting director and a course director for Medtronic. R.L. reports advisory fees from AstraZeneca, Celsion and Guerbet. K.K. is receiving support from AbbVie GK, Asuka Pharmaceutical, Astellas, Bristol-Myers Squibb, Dainippon-Sumitomo Pharma, EA Pharma, Eisai Inc., Gilead Sciences, Merck, Otsuka Pharmaceuticals, Shionogi and Takeda Pharmaceuticals. R.S.F. reports consulting fees from AstraZeneca, Bayer, Bristol-Myers Squibb, CStone, Eisai, Eli Lilly, Merck, Novartis, Pfizer and Roche/Genentech. All other authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Disease Primers thanks Hellmut Augustin, Massimo Colombo, Chih-Che Lin, Michael Peter Manns, Timothy Pawlik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Sustained virological response
-
(SVR). An undetectable hepatitis C virus RNA in the serum with the use of a sensitive polymerase chain assay at 6 months after completion of antiviral therapy.
- Non-alcoholic steatohepatitis
-
(NASH). Inflammation of the liver caused by excess accumulation of fat in hepatocytes.
- Metabolic syndrome
-
A multifactorial metabolic disorder, characterized by a cluster of risk factors, including abdominal obesity, insulin resistance, dyslipidaemia and elevated blood pressure, that promote the development of type 2 diabetes mellitus and cardiovascular disease.
- Mutational signatures
-
Mutational fingerprints of specific genotoxins.
- Immune-checkpoint inhibitors
-
(ICIs). Drugs that stimulate anti-tumour immune response by blocking checkpoint proteins and promoting immune-mediated elimination of tumour cells.
- Patient-reported outcomes
-
A measurement of any aspect of a patient’s health status that comes directly from the patient, without interpretation of the patient’s responses by a health-care professional or anyone else.
- Aflatoxin B1
-
A mycotoxin produced by Aspergillus species in a variety of food commodities; consumption of the toxin can cause adverse health effects, including liver cancer.
- Bridging fibrosis
-
A histological finding in the advanced stage of liver fibrosis characterized by thin and long fibrous septa that extend across lobules and connect portal spaces and central veins.
- Population attributable fraction
-
The proportion of hepatocellular carcinoma cases that would be prevented if a risk factor were eliminated.
- Transdifferentiation
-
The process by which one somatic cell is transformed into another mature somatic cell without undergoing a pluripotent state.
- Exhaustion status
-
A progressive loss of effector function due to prolonged exposition to inflammatory signals and antigen stimulation, characteristic of chronic infections and cancer.
- Tertiary lymphoid structures
-
Lymphoid micro-organs that develop at sites of chronic inflammation, including the liver.
- Hepatic stellate cell
-
Liver-specific mesenchymal cells that play a vital role in liver physiology and in wound healing, in particular fibrogenesis.
- Senescent hepatocytes
-
A hepatocyte that is metabolically active but with permanently arrested growth and resistant to apoptosis.
- Radiological hallmark of HCC
-
A pathognomonic radiological finding of hepatocellular carcinoma (HCC) characterized by hyperenhancement in the arterial phase (wash-in) and hypoenhancement in the portal venous and/or delayed phases of acquisition (wash-out) in the setting of liver cirrhosis.
- Child-Pugh score
-
A scoring system that assesses the prognosis of chronic liver disease by integrating three analytical and two clinical items, including blood levels of bilirubin, albumin, prothrombin time, presence of encephalopathy and ascites.
- Varices
-
Abnormal and enlarged veins that develop as a result of portal hypertension and may leak or rupture, causing potentially life-threatening upper gastrointestinal bleeding.
- ECOG score
-
A standardized scale for measuring a patient’s performance status and the impact of disease on their daily living abilities and level of daily functioning.
- Micrometastases
-
A small collection of tumour cells (<2 mm in size) shed from the primary tumour that spread to another organ through blood or lymph nodes.
- Anatomical approach
-
The removal of the entire neoplasm together with the segment of the liver where the tumour is located.
- Adoptive immunotherapy
-
Therapies that transfer immune cells with anti-tumour activity into a patient to mediate tumour regression.
- Salvage transplantation
-
Transplantation of liver specimens in patients with hepatocellular carcinoma recurrence after initial tumour resection.
- ‘Metroticket’ concept
-
A mathematical model estimating the exact outcomes after transplantation based upon size and number of nodules at pathological explant.
- Marginal organs
-
Organs recovered from elderly donors or with comorbidities that convey a higher risk of technical complications and/or post-transplantation dysfunction.
- Objective response
-
A measure of treatment efficacy used in clinical trials and defined as a reduction in tumour size on radiological evaluation.
- Palmar–plantar erythrodysesthesia
-
A dermatological adverse reaction to certain drugs, mainly tyrosine kinase inhibitors, causing a painful erythematous rash localized in palms, fingers and feet.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Llovet, J.M., Kelley, R.K., Villanueva, A. et al. Hepatocellular carcinoma. Nat Rev Dis Primers 7, 6 (2021). https://doi.org/10.1038/s41572-020-00240-3
Accepted:
Published:
DOI: https://doi.org/10.1038/s41572-020-00240-3
This article is cited by
-
RARRES1 inhibits hepatocellular carcinoma progression and increases its sensitivity to lenvatinib through interaction with SPINK2
Biology Direct (2024)
-
Utility and predictive value of the CRAFITY score in advanced hepatocellular carcinoma treated with transarterial chemoembolization plus tyrosine kinase inhibitors and PD-1 inhibitor
BMC Cancer (2024)
-
A hypoxia–glycolysis–lactate-related gene signature for prognosis prediction in hepatocellular carcinoma
BMC Medical Genomics (2024)
-
Recent advances in the potential role of RNA N4-acetylcytidine in cancer progression
Cell Communication and Signaling (2024)
-
Construction of a prognostic model for hepatocellular carcinoma patients receiving transarterial chemoembolization treatment based on the Tumor Burden Score
BMC Cancer (2024)
