
Abstract
In recent years, exceptional technological advances have enabled the identification and interrogation of rare circulating tumour cells (CTCs) from blood samples of patients, leading to new fields of research and fostering the promise for paradigm-changing, liquid biopsy-based clinical applications. Analysis of CTCs has revealed distinct biological phenotypes, including the presence of CTC clusters and the interaction between CTCs and immune or stromal cells, impacting metastasis formation and providing new insights into cancer vulnerabilities. Here we review the progress made in understanding biological features of CTCs and provide insight into exploiting these developments to design future clinical tools for improving the diagnosis and treatment of cancer.
Similar content being viewed by others

Introduction
Despite advances in cancer research and treatment, metastasis remains the leading cause of cancer-related death worldwide1. The limitation of current clinical diagnostic tools to predict metastatic progression and detect minimal residual disease is a major hurdle to improve outcomes. Standard-of-care (SOC) approaches such as tissue biopsies, imaging modalities and tumour markers are often limited in their ability to capture all relevant aspects of a patient’s cancer due to insufficient diagnostic thresholds, sampling bias related to temporal and spatial tumour heterogeneity, and limited access to metastatic lesions2,3.
Blood-based tumour-derived materials provide an alternative, real-time and minimally invasive method for early detection, prognosis and prediction of response to anticancer agents4,5. Among the most intriguing liquid analytes are circulating tumour cells (CTCs), which are shed from primary and/or metastatic tumours into the bloodstream to eventually seed metastases at distant sites. As pioneers of the metastatic process, CTCs are extraordinary cells that offer an opportunity to capture and interrogate the most aggressive cancer clones, providing a privileged insight into the biology and vulnerabilities of blood-borne metastasis5,6.
An entire research field dedicated to the investigation of CTCs as a liquid biopsy analyte has produced remarkable progress and led to the translational implementation of CTCs as biomarkers in the setting of clinical trials7,8. However, despite scientific advances and technological innovations, the rare nature of CTCs poses considerable challenges, and as a consequence, their use in routine clinical practice has been slow5,9,10,11,12. Moving use of CTCs forward to realize their full potential as a highly accurate and predictive tool for antimetastatic therapies has both opportunities and challenges.
Here we provide a current and comprehensive overview of state-of-the-art developments in this dynamic field, with a focus on the role of CTCs in blood-borne metastasis. We describe various aspects influencing the metastatic potential of CTCs, including the formation of homotypic (cancer cells only) and heterotypic (cancer cells together with immune or stromal cells) clusters. We further review current technologies for CTC detection, capture and downstream analysis. Clinical translational aspects are discussed in reference to current and future trials aimed at targeting CTCs. Finally, we highlight key challenges and necessary research priorities to foster translational applications.
Progression of CTCs to metastasis
Patterns of metastatic cancer dissemination
Metastasis occurs via direct intravasation of tumour cells into blood vessels or the lymphatic system and, less frequently, by their direct spreading into neighbouring tissues. Although the tumour, node and metastasis (TNM) staging system uses positive lymph node status as a marker of advanced cancer13, to date there is limited experimental evidence that cancer cells must traverse the lymphatic system before seeding distant metastasis. In contrast, a larger body of literature supports the haematogenous route as a main metastasis conduit14,15. We therefore focus this Review on blood-borne dissemination of cancer.
While decades of scientific research has revealed detailed pathogenetic mechanisms leading to primary tumour formation, the biological foundations of metastatic disease remain inadequately understood. Nevertheless, over the past 20 years important progress has been made in elucidating key aspects in this regard. Metastasis is a multistep process contingent on invasive, metastasis-competent cell clones in the primary tumour (or metastatic lesions) that escape via intravasation and transit the bloodstream as CTCs. When they reach a point of extravasation, CTCs can home to distant niches and grow into secondary lesions14,15.
Two mechanistic models of cancer cell dissemination have been proposed: (1) the linear or late dissemination model and (2) the parallel progression or early dissemination model. The former postulates that natural selection within the primary tumour will lead to the dissemination of the fittest tumour cell clones over time to give rise to metastasis, where only small degrees of divergence should be present between metastatic and primary lesions16. Along these lines, primary tumour and metastatic tissue typically demonstrate high level of genetic similarities, including in metachronous lesions17,18. However, contradictory observations of genetic discrepancies between metastases and primary tumours favour the notion that tumour cell escape may occur early during tumour evolution19,20, perhaps even at preneoplastic stages21,22, following the logic of a parallel progression model19,21,23,24. Opponents of this theory argue that extensive intratumour heterogeneity might obscure shared but rare metastasis-competent subclones, which are not detected by current sequencing technologies.
These models of metastatic spread were enriched by phylogeny studies applying evolutionary principles, which revealed additional, and more complex than originally anticipated, patterns of metastasis-to-metastasis dissemination and tumour self-seeding25,26,27,28. Although plausible and repeatedly confirmed in both mouse models and patient samples of various cancer types29,30,31,32, this concept has been challenged too, given the knowledge that complete phylogeny is difficult to achieve with current methods and their limitations (for example, insufficient sequencing depth, sampling bias and heterogeneity, and different bioinformatics approaches)25,28. Nevertheless, the analysis of unique subclonal somatic mutations that occur during branching and progression of cancer cell lineages has revealed that metastatic spread follows intricate patterns, with monoclonal or polyclonal seeding between metastatic lesions29,31,33,34. Consequently, cancer cell dissemination is likely not only a solitary endeavour but may also occur via polyclonal events that play a central role in diversifying the metastatic process, far away in both space and time from the primary tumour35,36,37.
Step-by-step sequence of metastatic events
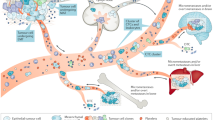
The first critical step in metastatic dissemination is cancer cell invasion, followed by intravasation into the proximal blood vessels (Fig. 1a,b). Access to the circulation can be achieved via passive shedding or active cell invasion38. In the case of passive shedding, tumour fluid dynamics and reduced barrier function of immature tumour neovasculature facilitate physical expulsion of tumour cells into the periphery38,39,40. However, the frequency and exact conditions leading to passive tumour cell shedding remain poorly understood.

a, Invasion of cancer cells. The formation of invasive features (for example, invadopodia) and hypoxic conditions (indicated by a blue haze) favour release of cancer cells away from the primary tumour site via upregulation of hypoxia inducible factor 1α (HIF1α), NMYC downstream-regulated gene 1 protein (NDRG1) and vascular endothelial growth factor A (VEGFA). This is further enhanced by metastasis-promoting features, including the expression of CXC-chemokine receptor 4 (CXCR4) and angiopoietin-like protein 4 (ANGPTL-4), the secretion of matrix metalloproteinases (MMPs), decreased expression of phosphoglycerate dehydrogenase (PHGDH) and cytoskeletal rearrangements. External conditions conducive to spreading are further provided by physical factors (for example, fluid pressure and stiffness) and surrounding cells (for example, cancer-associated fibroblasts and endothelial cells) in the tumour microenvironment. Mechanical stimuli from the tumour microenvironment promote pro-metastatic conditions by activating Yes-associated protein 1 (YAP)–transcriptional co-activator with PDZ-binding motif (TAZ) in cancer associated fibroblasts, favouring cancer cell invasion. Further, paracrine factors secreted by endothelial cells may reduce PHGDH levels in cancer cells, potentiating cell migration and invasion. Intravasation (part b) and circulation (part c). Circulating tumour cells (CTCs) and their clusters have a short half-life in circulation, due to hostile conditions, including physical forces (that is, shear stress) and anoikis. CTCs can escape the immune system via downregulation of major histocompatibility complex class I (MHC-I), expression of immune checkpoint molecules (for example, programmed cell death protein 1 (PD1) ligand 1 (PDL1) and CD47) or through support from platelets and neutrophils. Cell-intrinsic factors (for example, expression of anti-apoptotic factors) enhance CTC survival and successful transit, while circadian rhythmicity (and related hormone fluctuations) dictates the timing of CTC intravasation events, reaching a peak during the rest phase. d, Extravasation. The efficiency of extravasation relies upon the expression of adhesion molecules (for example, CD44, mucin 1 (MUC1) and sialyl-Lewis A (sLeA)/sialyl-Lewis X (sLeX), chemokine release, physical properties (for example, CTC cluster size and deformability) and supporting cells (for example, neutrophils via the formation of neutrophil extracellular traps (NETs)). Signalling via transforming growth factor-β (TGFβ)/SMAD family member 3 (SMAD3) leads to the upregulation of various adhesion-related molecules and facilitates CTC vascular adhesion. e, Successful homing into a new environment is dependent on niche factors (that is, various organ-specific cell types), but is also influenced by the preset metastatic potential of CTCs. f, CTCs may spread from the primary tumour or from metastatic lesions to either seed new metastasis (metastasis-to-metastasis dissemination) or return to the primary tumour site (tumour self-seeding). DTC, disseminated tumour cell; HIFPH2, hypoxia-inducible factor prolyl hydroxylase 2; NK natural killer; N-WASP, neural Wiskott–Aldrich syndrome protein.
Active cancer cell invasion can be triggered by hypoxia41. Hypoxia-inducible factor 1α (HIF1α) increases expression of the adhesion molecules L1 cell adhesion molecule (L1CAM; via angiopoietin-like protein 4 (ANGPTL4) signalling)42 and CXC-chemokine receptor 4 (CXCR4)43, resulting in enhanced CTC–endothelium binding and intravasation. Hypoxia has further been linked to the formation and intravasation of CTC clusters, via upregulation of cell–cell adhesion molecules41 (Fig. 1a). Expression of HIF prolyl hydroxylase 2 (HIFPH2) by endothelial cells promotes escape of tumour cells into blood vessels by impeding endothelial integrity and vessel maturation44. Transient binding of tumour cells to perivascular macrophages further increases endothelial permeability through the production of vascular endothelial growth factor A (VEGFA), in a HIF1α-independent manner45. Vascular endothelial cells can induce metabolic changes in cancer cells, such as the downregulation of phosphoglycerate dehydrogenase (PHGDH), to further facilitate cancer cell invasion via activation of the hexosamine–sialic acid pathway and increased sialylation of integrin αVβ3 (ref.46) (Fig. 1b). Tumour cell-intrinsic factors contributing to active invasion include the loss of function of nuclear receptor subfamily 2 group F member 1 (NR2F1), accompanied by increased canonical Wnt signalling, the expression of epithelial-to-mesenchymal transition (EMT)-related transcription factors (for example, TWIST1 and zinc-finger E-box-binding homeobox (ZEB1))47 and the formation of invadopodia via neural Wiskott–Aldrich syndrome protein (N-WASP)-directed cytoskeletal reorganization48 (Fig. 1a).
Although as many as 1 × 106 cancer cells per gram of tumour tissue are estimated to be shed into the circulation49, metastatic efficiency of CTCs appears to be low, with estimates around 0.01%50,51. CTCs generally exhibit short circulation times (25–30 min for single CTCs and 6–10 min for CTC clusters), where the shorter circulation time of clusters may be ascribed to a rapid arrest and homing at distant sites. A rapid escape and less exposure to hostile conditions in blood may ultimately contribute to CTC survival. These hostile circulatory conditions include high shear stress causing deformation, anoikis, fragmentation and cell death52. Remarkably, the process of fragmentation can trigger the priming and accumulation of pro-metastatic immune cells (for example, myeloid cells), promoting successful metastatic colonization of surviving CTCs52 (Fig. 1c). Moderate levels of shear stress can also lead to transcriptional changes, favouring motility and migration53. CTCs in circulation can enter cell cycle arrest or increase the expression of anti-apoptotic proteins such as BCL-2 (refs.54,55). To evade clearance by immune cells, CTCs can express programmed cell death protein 1 (PD1) ligand 1 (PDL1) or CD47 (refs.56,57,58) (Fig. 1c). Neutrophils support CTCs in circulation by enhancing their proliferation and survival59, as well as suppressing the host’s adaptive immune system (CD8+ T cells)60 and innate immune system (natural killer (NK) cells)61 (Fig. 1c).
Extravasation, in analogy to intravasation, may be promoted passively or actively. Both mechanical trapping (with capillaries as small as a few micrometres in diameter) and active adhesion may depend on CTC configuration (for example, single CTC versus CTC cluster)62, cluster composition (homotypic versus heterotypic)63,64 and geometrical shape65. Active CTC extravasation mimics diapedesis of leukocytes in terms of cytoskeletal and signalling changes66, including the expression of ligands and receptors for direct endothelial cell wall interaction67 (Fig. 1c). For example, expression of sialylated carbohydrate ligands (sialyl-Lewis A and sialyl-Lewis X)68, mucin 1 (MUC1)69 or CD44 (ref.70) by cancer cells and their ligation to selectins, intercellular adhesion molecule 1 (ICAM1) or β1 integrin on endothelial cells triggers CTC rolling and subsequent adhesion, enabling transendothelial migration (Fig. 1d). Endothelial adhesion is further enhanced via inflammatory mediators such as CXC-chemokine 12 (CXCL12)71. Reactive oxygen species (ROS) and SMAD family member 3 (SMAD3) enhance CTC adhesion via transforming growth factor-β (TGFβ) signalling72. The secretion of ANGPTL4, VEGF, matrix metalloproteinases and CC-chemokine ligand 2 (CCL2) by cancer cells or a disintegrin and metalloproteinase 12 (ADAM12) by endothelial cells increases endothelial permeability to facilitate extravasation73,74,75. CTC-induced damage to endothelial cells presents another means of escape, mediated by programmed endothelial cell necrosis via receptor-interacting serine/threonine-protein kinase 1 (RIPK1)76. Furthermore, various other cell types besides endothelial cells extrinsically support CTC extravasation. Platelets, for example, facilitate extravasation via ATP–P2Y purinoceptor 2 (P2Y2) interaction, causing endothelial retraction64. Neutrophils immobilized via endothelial glycocalyx binding create a pro-inflammatory local milieu through secretion of interleukin-8 (IL-8) and CXCL1, resulting in vascular leakiness61,77, release of neutrophil extracellular traps78, macrophage 1 antigen (MAC-1)-mediated ‘docking’ platforms79 or matrix metalloproteinases61 that promote dissemination (Fig. 1c,d). Experimental mouse and zebrafish models show that endothelial cells themselves may act as accomplices by wrapping around and expulsing arrested CTCs40.
After exiting the bloodstream, CTCs may home into their new metastatic site as disseminated tumour cells (DTCs) (Fig. 1e). While a comprehensive review of metastatic colonization upon extravasation is beyond the scope of this Review and has been provided well elsewhere80,81,82, we briefly discuss several key factors dictating DTC fate. Homing may be influenced by anatomical features, as well as by biological characteristics, of CTCs. For instance, in the case of colorectal cancer, a ‘direct’ vascular connection between the primary site and the metastatic site enables rapid entrapment of CTCs in the liver83. On the molecular side, CTCs may undergo chemotaxis to the bone marrow via expression of CXCR4 (ref.84). When travelling as heterotypic clusters with platelets, CTCs attract granulocytes that help establish neoplastic lesions at secondary sites85.
In the metastatic niche, the associated microenvironment exerts a critical influence on CTCs as they stop and possibly enter a stage of dormancy82. Organ-specific cell types have been suggested to provide DTC sanctuaries and promote dormancy, including perivascular osteoblasts in bone86,87, NK cells and hepatic stellate cells in the liver88, astrocytes in the brain89 and haematopoietic stem cells in the bone marrow90 (Fig. 1e). Activation and proliferation of DTCs can be subsequently prompted by inflammatory stimuli or osteoclast activity via calcium-associated signalling91,92. Whether physical factors differ between organs and influence metastasis remains poorly understood.
Physical and metabolic hallmarks of CTC biology
Tumour mechanics and physical properties are intricately linked with the state and function of cancer cells, tumour progression and metastasis93. Typically, tumours are stiffer than healthy tissue, often due to changes in their extracellular matrix94. Rigidity can lead to gene expression changes promoting tumour cell motility, while perpendicular alignment facilitates spread (sometimes referred to as ‘highways’ for tumour cell invasion)39,95. Leaky tumour vasculature can cause fluid accumulation, which, in combination with space constraints present in a rapidly growing tumour mass, results in high interstitial pressure and flow gradients, creating permissive avenues for tumour cell escape39,95. Cancer cells can actively migrate by repurposing their nucleus to create hydrostatic pressure, pulling and releasing the nucleus, akin to an engine piston, via actomyosin contractility96. Mechanical stimuli from the tumour microenvironment promotes pro-metastatic conditions via Yes-associated protein 1 (YAP)–transcriptional co-activator with PDZ-binding motif (TAZ)-dependent cancer-associated fibroblast (CAF) activation and enhanced cell invasion and migration97,98. Once in the bloodstream, CTCs experience various shear stress levels, where high levels lead to cellular fragmentation52 and intermediate levels may promote endothelial adhesion and extravasation40.
Homeostatic and metabolic stimuli affect CTCs throughout the metastatic passage. ROS from myeloid-derived suppressor cells lead to the upregulation of NOTCH1 expression on CTCs through the ROS–nuclear factor erythroid 2-related factor 2 (NRF2)–antioxidant response element axis, and NOTCH1 relays proliferative signals via its ligand JAGGED1, promoting proliferation and survival99. Studies using the 4T1 syngeneic mouse model of breast cancer reveal that CTCs, compared with the primary tumour, have elevated mitochondrial biogenesis and respiration via peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1A) upregulation, as well as increased mitochondrial DNA and ATP production100,101. In human epidermal growth factor receptor 2 (HER2)-positive breast cancer, oxidative stress transitions CTCs to a HER2-negative status, providing one possible explanation for the observed phenotypic discrepancy between primary lesions and metastatic lesions102. In the study cited, both HER2-positive cells and HER2-negative cells exhibited comparable tumour-initiating capacity, but HER2-negative CTCs were less proliferative. Oxidative stress experienced by metastasizing cancer cells can be counteracted via fatty acid oxidation driven by nuclear receptor subfamily 4 group A member 1 (NUR77) and mitochondrial trifunctional enzyme subunit-β signalling to maintain NADPH and ATP levels103. CTC clustering enhances metastatic potential by ROS clearance via mitophagy and HIF1α activity104.
Metabolic features of CTCs can also influence the metastatic process. CTCs can increase their survival and metastatic potential by dynamically adapting to organ-specific requirements via metabolic changes that counteract oxidative stress in the oxygen-rich lung environment105,106. Mice receiving a fat-rich diet show increased release of CTCs with excess free fatty acids incorporated into the cellular membrane, promoting tissue invasion and increased lung metastasis107. Palmitic acid has been shown to increase metastatic capability of cancer cells via CD36-mediated signalling108. Lipid rafts and raft-specific proteins, such as caveolin and flotillin, are also emerging as important regulators of metastatic potential, CTC survival and, therefore, patient prognosis109. The metastatic potential of CTCs is additionally enhanced by extracellular glucose via the exchange factor directly activated by cAMP 1 (EPAC)–RAP1 and O-linked β-N-acetylglucosamine pathways110, pyruvate metabolism via α-ketoglutarate-induced collagen remodelling111 or the production of superoxide via a mitochondrial overload through aerobic glycolysis112. Interestingly, heterogeneity in glucose metabolism affects metastatic potential and growth capabilities differently; while the downregulation of PHGDH increases invasive and migratory potential, high PHGDH expression is required for tumour growth at the primary or metastatic sites46. Protein catabolism113 via proline dehydrogenase, iron metabolism via stabilization of pro-metastatic collagen lysyl hydroxylase dimers114 or acidic conditions via cathepsin B-mediated degradation of extracellular matrix115 further contribute to increasing metastasis. Collectively, these data allude to the extraordinary physical and metabolic plasticity of CTCs, allowing them to surmount adverse conditions to successfully spread.
Biological features of CTCs
CTC phenotypic heterogeneity and CTC clusters
Epithelial cells typically undergo anoikis upon losing touch with their surrounding environment; thus, metastatic seeding is generally inefficient50,51,116. Therefore, this raises the following question: what properties do CTCs have that sanction their successful metastatic dissemination? Enhanced survival and tumour-seeding capacity seems to be contained within a small fraction of tumour-initiating cells or metastasis-initiating cells with stem-like properties15,117,118,119. EMT has been proposed as a crucial requirement for metastasis, because it increases both contact-independent survival (that is, resistance to anoikis) and invasive potential15,118,120. Preclinical studies have demonstrated that EMT transcription factors, such as SNAIL and TWIST, suppress cell–cell contact and increase motility and invasiveness in vitro121,122. However, silencing of SNAIL and TWIST could reduce but not completely inhibit metastasis in vivo, challenging the necessity of EMT-related transcriptional regulators for the metastatic process123,124. Remarkably, in various models based on EMT lineage tracing, loss of epithelial cadherin (E-cadherin) or modulation of TWIST expression demonstrated that EMT prevents the successful seeding of metastatic tumours123,124,125. Observations of epithelial-to-mesenchymal plasticity suggest that this transformation need not be binary and irreversible but rather may be fluent and transitional, functioning as an influencer rather than a driver of metastasis126,127,128. Accordingly, an intermediate level of EMT has been described in CTCs, and correlates with plasticity, stem cell-like characteristics, poor treatment response and disease progression118,119,128. Such hybrid states have been observed at the invasive edge of xenografts and patient tumour tissues of common carcinoma types22,128,129,130, yet their functional contribution to invasion, dissemination and metastatic colonization requires further investigation.
Both plasticity-dependent and plasticity-independent routes of metastasis may exist in parallel127. An essential counterargument to the necessity of EMT was provided by observations of cell contact-dependent, collectively migrating and highly metastatic CTC clusters, which were first described several decades ago131,132. The polyclonal nature of metastatic colonies29,31,33,34 and the mutually beneficial or synergistic interactions between subclones133,134 indicate that cancer spread occurs not only via single CTCs but also via heterogeneous CTC clusters35,36,37. Several studies show that CTC clusters, comprising a minority of the total CTC events in the peripheral circulation, display up to a 100-fold increase in metastatic potential compared with individual CTCs35,36,37,125,135. Evidence from patient samples and mouse models demonstrate that intratumour hypoxia triggers the upregulation of genes encoding proteins involved in cell adhesion to enable collective CTC cluster shedding41. We and others have demonstrated that homotypic clustering enhances various cellular properties, including upregulating stem-like features37,136,137,138 via methylation of metastasis suppressor genes136 and hypomethylation of binding sites for the transcription factors OCT4, SOX2 and NANOG137 (Fig. 2a). Clustering may further be enhanced by circulating galectin 3 or cancer-associated MUC1 (ref.139), homotypic ICAM interactions138 or CD44 interacting with p21 protein–activated kinase 2 (PAK2)37. It can also increase survival and self-renewal capacity via CDK6 or via increased size or number of desmosome and hemidesmosomes35,36. CD44 was among the first markers to identify breast cancer cells with increased tumour-initiating capacity in solid tumours117, and was later shown, together with MET, epithelial cell adhesion molecule (EpCAM) and CD47, to characterize highly metastatic subpopulations118. Importantly, CD44 expression is ubiquitous in the haematopoietic cell compartment, and therefore should be cautiously used as a CTC marker on its own140. Expression of CK14, previously ascribed to stem-like cells, is enriched in CTC clusters compared with single CTCs and is required for distant metastasis36. A phenotypic signature in CTCs with stem-like features (EpCAM–, HER2+, EGFR+, heparanase (HPSE)+, NOTCH1+) was shown to confer competency for brain and lung metastasis141.

Clustering of circulating tumour cells (CTCs) may occur exclusively between tumour cells (homotypic CTC clusters), as well as between tumour cells and other cell types (heterotypic CTC clusters). This results in enhanced proliferation and survival in the circulation, enabling superior metastatic proficiency. a, Homotypic clustering of CTCs leads to the creation of typically oligoclonal clusters, kept together by cell adhesion molecules (for example, plakoglobin, claudins and CD44). Expression of these molecules and cluster formation are promoted by hypoxic conditions. CTC clustering triggers epigenetic changes (for example, hypomethylation of binding sites for OCT4, NANOG and SOX2), leading to stem-like cell behaviour, which facilitates metastasis seeding. b, Heterotypic CTC clusters (for example, between tumour cells and neutrophils, cancer-associated fibroblasts or platelets) display increased proliferation, invasion and homing at the metastatic site, as well as protection against immune surveillance. E-cadherin, epithelial cadherin; GPIb-IX-V, glycoprotein Ib–IX–V; GPVI, glycoprotein VI; N-cadherin, neural cadherin; VCAM1, vascular cell adhesion molecule 1.
Clustering occurs not only between CTCs (homotypic) but also between CTCs and other cell types (heterotypic clusters), including platelets, myeloid cells and CAFs59,85,99,142,143,144,145,146,147 (Fig. 2b). The interaction of CTCs with platelets occurs rapidly in the circulation143, promoting plasticity and metastasis-initiating capacity142, for example via RhoA–MYPT1–PP1-mediated YAP1 signalling63 and increased vascular permeability via platelet-derived ATP–P2Y2 interaction64. Platelets provide protection against T cell-mediated clearance via the glycoprotein A repetitions predominant (GARP)–TGFβ axis145, as well as NK cell-mediated clearance via platelet-derived major histocompatibility complex class I (ref.144). We demonstrated for the first time that neutrophils are another accomplice in forming heterotypic CTC clusters59. Neutrophils are recruited by CXCL5- and CXCL7-dependent chemotaxis85, and establish cell contact with CTCs via vascular cell adhesion molecule 1 (VCAM1)-mediated adhesion, increasing their proliferative and metastatic potential59 (Fig. 2b). They also facilitate adhesion and extravasation via the formation of neutrophil extracellular traps78 or IL-1β and matrix metalloproteinase secretion61 (Fig. 1c). Neutrophils also offer CTCs protection from immune surveillance60,61, a benefit similarly observed for CTC clustering with myeloid-derived suppressor cells and macrophages99,148. Heterotypic adherens junctions between invasive cancer cells and stromal CAFs mediated by E-cadherin and neural cadherin (N-cadherin), respectively, have been shown to promote collective invasion, where CAFs function as leader cells with migratory–invasive features146,147 and support metastasis (tumours bring their own soil)149 (Fig. 2b). Beyond the biological implications of clustering, mathematical models propose that cluster shape additionally affects CTC behaviour in the circulation. Compact clusters flow closer to the endothelial wall than linear ones150, yet passage of clusters through narrow capillaries occurs in ‘single chains’151. Together, the phenotypic plasticity and variations in clustering profoundly impact the ability of CTCs to metastasize, highlighting possible strategies to interfere with the metastatic process.
Molecular heterogeneity of CTCs
CTCs are dynamic cell populations that are constantly replenished from multiple tumour regions and anatomical locations, enabling a snapshot assessment of tumour heterogeneity. By studying the genomic, transcriptomic and epigenetic profile of CTCs, we can further explore their biology. The development of single-cell technologies and sophisticated bioinformatics tools has allowed the dissection of rare CTC populations at single-cell resolution152. For example, single-CTC analysis has uncovered copy number variants, therapeutic targets (for example, HER2) and resistance mutations (for example, in PIK3CA) that only partially overlap with primary tumours or metastatic lesions from the same patient102,153,154. Dynamically regulated HER2 expression in CTCs differs from HER2 expression in primary tumours: HER2-positive CTCs may activate multiple redundant pathways (for example, insulin receptor, MET and IGF1), while HER2-negative cells show activation of Notch and DNA damage components102. Mutationally defined metastasis-prone CTC subclones exhibit similarities but also private alterations compared with primary tumours155, including mutations in genes encoding proteins regulating motility (for example, dynein axonemal heavy chain 8 (DNAH8), ephrin B receptor 1 (EPHB1)156, microtubule–actin crosslinking factor 1 (MACF1) and neural precursor cell-expressed developmentally downregulated protein 9 (NEDD9)157). Activation of stem cell-like signalling pathways such as Wnt and Notch158,159 and dedifferentiation via loss of NK2 homeobox 1 (NKX2-1)160 further enhance the metastatic potential of CTCs. An in vivo loss-of-function CRISPR screen identified specific molecular dependencies required by CTCs for successful completion of the metastatic cascade161. For example, knockout of the gene encoding PLK1 led to a remarkable reduction in CTC intravasation, as well as metastasis formation161. Other regulatory elements, including long non-coding RNAs such as colon cancer-associated transcript 1 (CCAT1) and HOX transcript antisense RNA (HOTAIR), have been shown to function as key pro-metastatic contributors by inducing microenvironmental changes that promote invasion, migration and organotropic colonization via the TGFβ–ZEB1/ZEB2 axis or the nuclear factor-κB (NF-κB) pathway162. CTCs from different primary tumours exhibit ‘preset’ organotropism, and CTCs captured from different vascular sites exhibit distinct molecular features163. For instance, a series of seminal studies identified gene expression features that mediate metastasis of breast cancer to the lungs, brain or bone164,165,166. Tropism to bone has also been linked to SMAD signalling167 and ZEB1 activity in breast cancer cells with an epithelial phenotype168. MYC was identified as a crucial regulator for tropism and adaptation to the brain microenvironment169. Differences at the molecular level between CTC clusters and single CTCs include the higher expression of the cell–cell junction molecules plakoglobin and claudins35,137, hypomethylation of binding sites for stem cell-like transcription factors (that is, NANOG, OCT4 and SOX2) and increased proliferation in clusters137. In summary, CTC phenotypic and molecular heterogeneity fuels the adaptive processes required for metastasis.
Timing of CTC release
In addition to the aforementioned dimensions of heterogeneity, propagation dynamics of CTCs is emerging as an equally important element for tumour cell dissemination170,171. The role of the circadian rhythm for tumour onset172,173,174,175 and growth dynamics176,177 has been investigated and explored clinically via the concept of chronotherapy. This aims to increase the efficacy of antineoplastic drugs by administering treatment at optimized times178,179,180. However, the effect of the circadian rhythm on CTC release and metastatic dissemination was determined only recently170,171. Current practice for detecting CTCs assumes that peripheral blood counts do not change significantly throughout the day. This has potentially caused inconsistent results, limiting the clinical implementation of CTCs as a liquid biopsy analyte. Observations from use of fluorescence in vivo flow cytometry in orthotopic mouse models of human prostate cancer suggested that CTCs are subject to circadian rhythmicity171. Our laboratory recently elucidated those temporal dynamics of CTC intravasation, which vary dramatically based on circadian rhythm, both in mouse models and in patients with breast cancer, and are dictated by rhythmic variations in hormone levels (for example, melatonin) that result in the highest CTC counts during sleep170 (Fig. 1c). That study also demonstrated marked differences in Ki67 expression in both the primary tumour and CTCs, reaching a peak during the rest phase, suggesting the need to standardize the timing of tissue biopsies used for prognostic and predictive information (that is, directly influencing clinical decision-making). These findings argue for a re-evaluation of current biopsy standards and suggest innovative, time-controlled clinical trials exploring their translational value.
Detection and analysis of CTCs
Implementing CTCs as a liquid biopsy analyte in the clinic will require unbiased, effective, rapid and affordable capture technologies to reliably isolate sufficient numbers of CTCs. These capture technologies must also be compatible with advanced sequencing tools and functional assays, to generate data for accurate patient stratification and therapeutic decision-making (Fig. 3a). When isolated in a viable form, CTCs are also amenable to an exceptional range of molecular and functional investigations of the biology and vulnerabilities of metastatic cancer (Fig. 3a). Since they may represent aggressive subclones with high metastatic propensity, CTCs fulfil a unique role as a liquid biopsy analyte. We speculate that their phenotypic and molecular analysis might reveal more relevant information than classical tissue biopsies (isolation of random subclones)181,182 or analysis of other circulating analytes such as circulating tumour DNA (detection of dying subclones)183. However, this will need to be further investigated. Access through minimally invasive blood draws could allow frequent, longitudinal assessment of the effect of clinical interventions and may enable early detection of cancer or recurrence184. This renders CTCs as an ideal source of biomarkers for real-time clinical applications and personalized medicine. Due to their rare nature, however, capturing CTCs remains challenging, and efficient CTC enrichment is critical for reproducible downstream analysis and applications.

Available circulating tumour cell (CTC) detection technologies and examples of how CTCs can be included in clinical trial designs. a, CTC capture tools include antigen-dependent technologies (for example, immune capture via immobilized antibodies, antibody coated beads or coated intravascular guidewires) and antigen-independent technologies (for example, density gradient centrifugation, microfluidic systems based on deformability and size, size-based filtration systems, electrical charge-based technologies or cytapheresis). The latter do not require a priori knowledge of phenotypic profiles and are thought to capture more heterogeneous CTC populations compared with antigen-dependent methods. Downstream analysis of CTCs includes direct drug phenotyping, the creation of CTC-derived xenograft ‘avatar’ models and multi-omics interrogations at the single cell-level: epigenomic, proteomic, genomic and transcriptomic. b, Validation of the use of CTCs in the setting of innovative clinical trials includes randomizing and benchmarking CTCs against standard-of-care (SOC) diagnostic and therapeutic approaches or other liquid biopsy analytes (for example, circulating tumour DNA (ctDNA)) and testing of CTC-based treatment strategies. The figure shows various possibilities for CTC-based clinical trial design: randomization of CTC-based liquid biopsies versus tissue biopsies to guide the choice of therapy; randomization based on the presence of CTCs (positive versus negative) to guide the choice of therapy (that is, experimental or targeted versus SOC); randomization of patients who are positive for CTCs to treatment with different targeted or experimental drugs; randomization according to longitudinal or repeated CTC assessment to guide subsequent lines of therapy (for example, targeted or experimental treatment versus SOC); randomized trials which compare either use of CTCs alone with SOC diagnostic approaches (for example, medical imaging) or use of other liquid biopsy analytes (for example, ctDNA) or each individual modality with combined use of modalities.
The past decade resulted in several technological advances to improve CTC detection and analysis3,185, exploiting distinct characteristics and phenotypes of CTCs. Broadly speaking, these can be divided into antigen-dependent and antigen-independent methods (Fig. 3a). To date, the most widely used approaches use antigens expressed on CTCs and minimally expressed on other cells in circulation to enable positive selection. To improve discrimination, this approach is often combined with depletion of haematopoietic cells via CD45-based negative selection. Both the US Food and Drug Administration (FDA)-approved CellSearch system (Menarini Silicon Biosystems, Italy) and AdnaTest CTC Select (QIAGEN, Germany) use immunomagnetic selection based on EpCAM expression186,187. Further markers, including pan-CK and CD45, are applied to increase sensitivity and specificity, respectively. The magnetic-activated cell separation (MACS) technology (Miltenyi Biotec, Germany) applies antibody-coated magnetic beads for CTC capture119. The geometrically enhanced differential immunocapture (GEDI) method combines microfluidics with different antibodies depending on the tumour type (for example, HER2 for breast cancer, and PMSA for prostate cancer) and cytokeratin positivity for enumeration188.
Antigen-agnostic detection technologies exploit physical properties such as size, charge, density or elasticity for CTC enrichment. Filter-based devices, density gradient centrifugation, capture surfaces and microfluidic systems such as ISET (Rarecells Diagnostics, France), CTC-iChip (TellBio, USA), Smart Biosurface Slides and the FDA-cleared Parsortix (ANGLE, UK) all enable detection of CTCs based on physical properties189,190,191,192,193. Multimodality approaches are being developed to further increase sensitivity and specificity. For instance, Isoflux (Fluxion Biosciences, USA) combines flow control and immunomagnetic beads194, while the Cyttel system (CYTTEL Biosciences, China) is an image-based detection tool that sequentially combines centrifugation, immunohistochemistry and fluorescence in situ hybridization to identify CTCs195. The microfluidic platforms Parsortix and CTC-iChip can also be combined with marker-based positive and negative selection (for example, EpCAM, EGFR, HER2 and CD45), imaging and micromanipulation to isolate pure CTC subsets41,59,137,189.
To tackle low CTC numbers in peripheral blood samples, innovative in vivo CTC detection technologies have been developed. For instance, direct intravascular CTC-catching guidewires coated with EpCAM-directed antibodies (CellCollector (GILUPI, Germany)) allow direct extraction of CTCs from the circulation system196. Cytapheresis enables cell fraction enrichment from large blood volumes, and combined with antigen-dependent selection, it appears to be promising for CTC isolation197. However, implementation of this approach in routine clinical practice may be difficult due to the length and invasiveness of the procedure, as well as the poor vascular health of heavily treated patients with cancer. Studies comparing different approaches to gain access to the vasculature demonstrate higher CTC numbers in tumour-draining vessels than in peripheral locations in patients with early-stage non-small-cell lung cancer184,198. This principle offers an attractive liquid biopsy scenario for patients with early-stage cancer treated with surgery. Although important, implementation of these findings remains impractical for routine CTC assessment or for patients with advanced-stage disease who do not undergo surgery.
The advances in capture technologies enabled CTC research to move far beyond mere enumeration. Detailed molecular and functional analysis has enabled the dissection of genomic, epigenomic, transcriptomic, proteomic and functional properties of CTCs at the bulk and single-cell levels. Comprehensive reviews have extensively covered these developments152,185,199,200, and therefore we mention only aspects of cell multi-omics and functional assessment of CTCs. For instance, single-cell interrogation of individual CTCs and CTC clusters combined with drug phenotyping can identify biological dependencies and potential therapeutic targets137. Single-cell resolution mass spectrometry201 and bead-based immunoassays on microfluidic platforms have been developed to characterize CTC proteins and secreted factors202,203. Although demonstrated in experimental proof-of-principle studies204,205,206, the low success rates of ex vivo CTC cultures represent a considerable challenge for their clinical translation.
Generally, while CTC capture and downstream analysis seems feasible and clinically relevant, many of the techniques outlined are far from being routinely applied today. Available CTC technologies have limitations that must be addressed to enable a robust entry into the clinical setting. This includes a better understanding of epitope expression and plasticity, as well as addressing issues relating to cell loss due to size and deformability differences, low CTC purity, device clogging, the large amounts of blood needed, the time required and difficulties with automation. Additional challenges relate to the improvement of functional assays (for example, more efficient culture methods and CTC-derived xenografts), as well as rigorous validation of molecular analysis (for example, accounting for stochastic variations, low sequencing coverage, amplification bias and high error rates, as well as variation in bioinformatics approaches152). Overcoming these challenges could eventually propel CTCs into the limelight of personalized medicine as minimally invasive, yet comprehensively informative sources of biomarkers.
CTC-based clinical applications
Vulnerabilities and targeting of CTCs
Current strategies for eliminating metastasis are identical to those applied to primary tumours: targeting growth and tumorigenesis instead of the metastatic process itself207,208. Surgery or systemic therapy for the primary tumour will not necessarily remove the source of metastasis, as dissemination may have already occurred19,21,24,32. Most antineoplastic drugs are initially tested in the metastatic setting and then reused in the adjuvant setting to prevent metastatic disease, with only partial success209. This paucity of truly metastasis-targeting agents is being challenged by several preclinical studies and thoughts for future clinical trial designs207,210. Because metastatic cancers represent the progeny of CTCs, which may be derived from selected subpopulations in tumours, targeting CTCs at various steps of the metastatic cascade would, in principle, directly interrupt the metastatic progression (Fig. 4).

Outlined here are various potential circulating tumour cell (CTC)-targeting strategies that are proposed on the basis of recent experimental work. a, Inhibition of cancer cell intravasation via normalization of the hypoxic tumour microenvironment; for example, by ephrin B2 Fc chimaera protein (EpB2) treatment leading to modulation of vascular endothelial growth factor receptor (VEGFR) signalling and vascular normalization, blockade of intravasation-relevant proteins (for example, Polo-like kinase (PLK1) inhibition) or blocking of cellular interactions between cancer and endothelial cells (for example, integrin-targeted antibodies). b, Dissociation of CTC clusters or prevention of their formation, for example via Na+/K+-ATPase inhibition, heparanase (HPSE) inhibition, stimulation with the urokinase-type plasminogen activator (urokinase) or inhibition of platelet receptors on CTCs. c, Targeting CTC survival via metabolic interference by increasing oxidative stress and inhibition of pyruvate or proline metabolism, or by use of E-selectin/tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-coated nanoliposomes that mimic the activity of natural killer (NK) cells. d, ‘Demasking’ CTCs for immune clearance by targeting immune evasion via immune checkpoint inhibitors against programmed cell death protein 1 (PD1) and PD1 ligand 1 (PDL1), cytotoxic T lymphocyte-associated antigen 4 (CTLA4) or CD47. e, Use of engineered CTCs as therapeutic vehicles (for example, prodrug conjugates) or CTCs for tumour vaccine development. f, CTC-based chronotherapy (that is, delivering treatments to be maximally effective at the times of greatest CTC production). VCAM1, vascular cell adhesion molecule 1.
Targeting hypoxia-induced cluster release using vascular normalization-inducing agents (for example, ephrin B2 Fc chimaera protein, which fine-tunes VEGF receptor (VEGFR) signalling211) has been suggested as a strategy to prevent metastasis in a preclinical models41 (Fig. 4a). The PLK1 inhibitor BI 2536 also prevents CTC intravasation161, and its clinical use could therefore provide a method to curb metastatic spread (Fig. 4a). Intravasation and extravasation could also be prevented by targeting integrins, cadherins and cell-surface glycoproteins212, targeting of invadopodia (for example, via N-WASP inhibition)48,66, or antibody targeting of CD36 and P-selectin or αIIbβ3 and α6β1 integrin antagonists212 (Fig. 4a). One class of drug currently under development aims to inhibit HPSE, which induces ICAM1-mediated cell adhesion in CTC clusters213. Urokinase, a thrombolytic agent that dissolves fibrin, also suppresses clustering in vitro and decreases the number of CTC clusters in mice214. Further, Na+/K+-ATPase inhibitors (for example, digoxin) show great promise due to their cluster-dissociation capabilities in vivo, ultimately leading to metastasis suppression in mice137 (Fig. 4b). Currently, a single-arm, proof-of-mechanism, therapeutic exploratory phase I study of digoxin in patients with advanced or metastatic breast cancer is investigating whether cardiac glycosides are able to disrupt CTC clusters in patients with breast cancer (NCT03928210)215. Heterotypic clustering can also be disrupted through cell–cell dissociation. Disrupting platelet–cancer cell interactions by blocking key platelet receptors on CTCs, such as glycoprotein Ib–IX–V and glycoprotein VI, reduces metastatic potential85,216,217 (Fig. 4b). Disrupting cell–cell interactions in CTC–neutrophil clusters via VCAM1 targeting curbs proliferation and metastatic efficiency59. On the other hand, VCAM1-mediated affinity of CTCs for neutrophils could be exploited for immune-based targeting by arming the latter with nanoscale liposomes carrying tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and the E-selectin adhesion receptor to functionalize those neutrophils by mimicking the cytotoxic activity of NK cells218 (Fig. 4c). Metabolic and homeostatic vulnerabilities could also be targeted by inhibiting pyruvate metabolism by preventing α-ketoglutarate-induced activity of prolyl 4-hydroxylase (P4HA), hampering proline metabolism or increasing oxidative stress100,111,113 (Fig. 4c). Immune checkpoint inhibitors could mark CTCs for killing by T cells57 (Fig. 4d), and dual targeting of EpCAM or HER2 in combination with use of an immune checkpoint inhibitor (that is, PD1, PDL1 or cytotoxic T lymphocyte-associated protein 4 (CTLA4)) produces greater cancer cell killing compared with single-agent therapy219,220. CTCs can also be used for cancer vaccine production using mechanically disrupted CTCs as nanolysates221 (Fig. 4e). The capacity of CTCs to home to existing tumour microenviroments26 could be exploited therapeutically through the identification of homing signals and the delivery of therapeutic payloads (Fig. 4e). As a proof of principle, systemically administered CTCs engineered to express the prodrug-converting enzyme cytosine deaminase–uracil phosphoribosyl transferase were able to convert non-toxic 5′-fluorocytosine into the cytotoxic compound 5′-fluoruridine monophosphate, resulting in CTC suicide upon homing to neoplastic tissues and killing of surrounding cancer cells as a bystander effect222. Lastly, given recent findings regarding the rhythmicity of CTC release into the bloodstream170, currently available therapeutic opportunities could be optimized, through chronotherapy-based designs, to achieve maximal effects during peaks of CTC production (Fig. 4f). These approaches offer exciting opportunities for targeting CTCs in future studies; however, given the complexity of the metastatic process, and the limited history of approaches purposely designed to target metastatic cells, their implementation in the clinical setting and demonstration of clinical value will require highly innovative and ambitious trial designs.
Prognostic and predictive value of CTCs
As of 10 October 2022, the search term ‘circulating tumour cells’ yielded 366 studies at ClinicalTrials.gov, of which 218 studies were in progress, reflecting the growing interest in CTCs as biomarkers for precision oncology. In the setting of such trials, CTCs have been detected in the peripheral blood of all major carcinomas, and their prognostic value has been demonstrated in breast, prostate and colorectal cancer, as well as in small cell and non-small-cell lung cancers223,224,225,226,227. In patients newly diagnosed with metastatic breast cancer, elevated CTC counts before therapy are predictive of shorter disease-free and overall survival225,228,229. Negative correlation between pretreatment CTC numbers and clinical prognosis has also been reported for patients with colorectal230 and prostate231,232 cancers. Importantly, several studies have demonstrated that changes in CTC numbers in response to therapy provide superior prognostic information compared with baseline CTC status, with persistence of CTCs after therapy conferring a worse prognosis9,233,234. Evaluation of CTC cluster abundance, in addition to single-CTC counts, significantly improves the prognostic value in patients undergoing therapy235. However, given that most of these studies were performed with antigen-dependent CTC technologies, enumeration in this context poses the risk of generating false-negative results.
CTCs counts are detectable 7–9 weeks before clinical manifestation of the disease, suggesting that CTC analysis in patients can aid in the prognostication of minimal residual disease and relapse in late stages of disease236, as well as providing a tool for early cancer detection193,237. CTCs collected at surgery revealed high mutational overlap with metastasis detected 10 months later (91%) in non-small-cell lung cancer184. Despite the value of CTCs for risk stratification, therapeutic patient stratification using CTCs has been explored in several clinical trials so far with limited success, including longitudinal monitoring of response and occurrence of therapy resistance9,10,238. While the interventional SWOG-S0500 trial failed to show a benefit of CTC count-guided intervention versus physician’s choice at disease progression9, the METABREAST STIC CTC trial demonstrated instances whereby CTC count can be helpful in guiding therapeutic decisions238.
Several interventional studies have explored the benefit of therapy choice based on molecular characteristics of CTCs239,240,241,242. Two proof-of-principle studies targeted HER2-positive CTCs in HER2-negative metastatic breast cancer with trastuzumab–emtansine or lapatinib (HER2-targeted therapies)240,241. Thus far, the studies have revealed only a marginal benefit, although one study (DETECT III) still awaits completion242. In metastatic prostate cancer, androgen receptor splice variant 7 (AR-V7) expression in CTCs predicts outcome in patients treated with endocrine therapy (PROPHECY trial)243,244. This led to a phase II trial focused on the response of patients with metastatic castration-resistant prostate cancer and AR-V7-positive CTCs to the microtubule inhibitor cabazitaxel11. Nevertheless, given the recent negative result in that trial, the European Society for Medical Oncology guidelines do not endorse AR-V7 testing in this setting, as there is no benefit over current decision algorithms11,12,245.
In summary, CTCs have been incorporated into the fifth edition of the WHO Classification of Tumours: Breast Tumours246 and the seventh edition of the AJCC Cancer Staging Manual247. The term ‘cM0 (i+)’ indicates that there is no overt metastasis but tumour cells have been detected in blood, bone marrow or lymph nodes. However, CTCs have yet to be included in clinical practice guidelines of major cancer societies, including the European Society for Medical Oncology or the American Society for Clinical Oncology. Arguably, the true power of CTCs lies in their potential to represent highly metastatic tumour subclones and their richness as up-to-date sources of biomarkers for molecular and functional studies. CTCs as living cells are, in principle, amenable to ex vivo cell culture and drug phenotyping, potentially in a timely manner and suitable to inform treatment decisions204,205,248, although such workflows will have to be significantly improved to reach the clinical side. Innovative, randomized and prospective interventional trials will reveal whether there are clear benefits from using CTCs as diagnostic tools against SOC techniques in specific cancer types (Fig. 3b). The predicted strengths of CTCs should be prioritized in future validation efforts, specifically the detection of minimal residual disease, expression of clinically actionable targets for therapy selection and longitudinal follow-up (Fig. 3b).
Future challenges and priorities for CTC research
Outlining key challenges in CTC biology on one hand and promoting their clinical implementation on the other is intended to help define research priorities that address the development of CTCs as superior sources of biomarkers for improved personalized cancer care (Box 1). A formidable task lies in the better understanding of tumour clonality and its relationship with CTCs. Are CTCs part of the most aggressive clones, and are they relevant to treatment decisions? How many CTCs need to be analysed to provide such information? In vivo models of spontaneous metastasis using barcoding to track tumour clones and subsequently analyse their molecular characteristics will be instrumental in this regard. Elucidating mechanisms that dictate intravasation and extravasation will provide alternative therapeutic targets for antimetastatic therapies. Although drug phenotyping of CTCs in patients with advanced disease is currently hampered by insufficient material and limited tools, the development of technologies that allow efficient capture and improved culture conditions for CTCs may enable real-time drug testing. Timing of CTC interrogation will be critical to enable meaningful and representative application of liquid biopsies. The current standard of clinical diagnostic procedures relying on traditional work schedules will require re-evaluation in light of the circadian dependency of tumour biology. Incorporating this knowledge into diagnostic algorithms could dramatically influence the clinical value of CTCs as a liquid biopsy analyte and even change treatment schedules to maximize clinical outcome. The timing of CTC dissemination also closely relates to the presence of DTCs249,250,251. Evidence for the early dissemination of CTCs19 and parallel progression of cancer23,252 argues for a detailed consideration of potential CTC reservoirs (primary tumour, macrometastatic and micrometastatic lesions, as well as DTCs) to unlock the full prognostic and predictive potential of CTCs. While single CTCs and CTC clusters are responsible for haematogenous seeding of proliferating macrometastatic tumours, logically they are by extension also the source of dormant and micrometastatic lesions. When and how CTCs and their clusters enter dormancy remains elusive, and this aspect warrants further investigation. This knowledge could lead to novel biomarkers and therapeutic strategies to target minimal residual disease and prevent further disease spread.
Priorities addressing the clinical application of CTCs should focus on improving the detection of cancer via liquid biopsies over SOC technologies. In the setting of early-stage cancers this is of particular importance but faces serious challenges regarding sensitivity and specificity. This could be addressed by benchmarking CTCs side-by-side with other circulating analytes (for example, circulating tumour DNA and exosomes) to increase the positive and negative predictive value, or by comparing and combining liquid biopsies with SOC modalities (Fig. 3b). CTCs have already been validated as independent prognostic markers, and have therefore extended the traditional TNM system. Beyond enumeration, we expect CTCs to unleash their potential as predictive biomarkers for patient stratification based on the detection of therapeutic targets or resistance mechanisms, including longitudinal surveillance. Whether treatment decisions based on CTC readouts will complement or even surpass treatment decisions based on standard tissue biopsies needs testing and validation in interventional and time-controlled clinical trials.
Conclusion
Metastasis remains a formidable obstacle to improve outcomes for patients with cancer as it corresponds to most cases of cancer-related death1,253. A deeper understanding of the biological processes underlying the capacity of tumour cells to seed tumours at distant sites is beginning to materialize, driven partly by rigorous investigations into CTCs as precursors of blood-borne metastasis. Early observations of CTC clusters190,254,255,256 and more recent dissection of their biological traits have highlighted their complexity but have also exposed vulnerabilities and, therefore, targetable opportunities. The intrapatient heterogeneity of CTCs offers an intriguing alternative to a comprehensive biopsy of a patient’s cancer, especially considering that CTCs may be enriched in metastasis-forming cells compared with the bulk of the tumour. A plethora of observational studies and a handful of interventional trials validating the use of CTCs for prognostication and therapy stratification have emerged over the past decade. Realizing the full potential of CTCs will, however, require an evolution from mere enumeration and prognostication towards highly controlled molecular characterization to develop precise predictive biomarkers. Technology and innovation are likely to pave the way for enhanced detection and detailed characterization of rare, yet precious CTC populations, thereby overcoming current diagnostic and therapeutic limitations.
Even now, major efforts are under way to move metastasis into the focus of therapeutic strategies (with the likes of the Metastasis Working Group)207. Successful translation of our knowledge of metastasis biology and implementation of efficient CTC capture methods in innovative clinical trial design and clinical–translational frameworks will substantially strengthen these efforts. We envision a future where CTCs might have a key role inpatient stratification and therapeutic decision-making by enabling the timely identification of aggressive tumour subclones, which is relevant for designing highly effective cancer therapies. Ultimately, targeting CTCs and their clusters may prevent further metastatic spread and favour survival.
Change history
16 December 2022
In the version of this article initially published, the Peer review information section, thanking Francoise Farace, Evi Lianidou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work, was omitted and has now been amended in the HTML and PDF versions of the article.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 (2012). This large-scale next-generation sequencing study demonstrates a high degree of tumour heterogeneity not captured by single tumour biopsies, with consequences for therapy failure.
Pantel, K. & Alix-Panabieres, C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424 (2019).
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Alix-Panabieres, C. & Pantel, K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 11, 858–873 (2021).
Mohme, M., Riethdorf, S. & Pantel, K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 14, 155–167 (2017).
Lin, D. et al. Circulating tumor cells: biology and clinical significance. Signal. Transduct. Target. Ther. 6, 404 (2021).
Bidard, F. C. et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 15, 406–414 (2014).
Smerage, J. B. et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. 32, 3483–3489 (2014).
Cabel, L. et al. Clinical utility of circulating tumour cell-based monitoring of late-line chemotherapy for metastatic breast cancer: the randomised CirCe01 trial. Br. J. Cancer 124, 1207–1213 (2021).
Belderbos, B. P. S. et al. Associations between AR-V7 status in circulating tumour cells, circulating tumour cell count and survival in men with metastatic castration-resistant prostate cancer. Eur. J. Cancer 121, 48–54 (2019).
Isebia, K. T. et al. CABA-V7: a prospective biomarker selected trial of cabazitaxel treatment in AR-V7 positive prostate cancer patients. Eur. J. Cancer https://doi.org/10.1016/j.ejca.2022.09.032 (2022).
Sobin, L. H., Gospodarowicz, M. K. & Wittekind, C. TNM Classification of Malignant Tumours (John Wiley & Sons, 2011).
Nguyen, D. X., Bos, P. D. & Massague, J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284 (2009).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging biological principles of metastasis. Cell 168, 670–691 (2017).
Yachida, S. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010).
Ding, L. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010).
Haffner, M. C. et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Invest. 123, 4918–4922 (2013).
Hosseini, H. et al. Early dissemination seeds metastasis in breast cancer. Nature 540, 552–558 (2016). This study demonstrates early dissemination and seeding of metastasis using mouse models, indicating that CTC circulation is likely to be a very early event in cancer progression.
Schmidt-Kittler, O. et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc. Natl Acad. Sci. USA 100, 7737–7742 (2003).
Hüsemann, Y. et al. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68 (2008).
Rhim, A. D. et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012).
Klein, C. A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 9, 302–312 (2009).
Harper, K. L. et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540, 588–592 (2016).
Hong, W. S., Shpak, M. & Townsend, J. P. Inferring the origin of metastases from cancer phylogenies. Cancer Res. 75, 4021–4025 (2015).
Kim, M.-Y. et al. Tumor self-seeding by circulating cancer cells. Cell 139, 1315–1326 (2009). This is an elegant study demonstrating that cancer dissemination is not unidirectional and that CTCs home back to the lesions from where they came, with important prognostic ramifications.
Naxerova, K. & Jain, R. K. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat. Rev. Clin. Oncol. 12, 258–272 (2015).
Schwartz, R. & Schaffer, A. A. The evolution of tumour phylogenetics: principles and practice. Nat. Rev. Genet. 18, 213–229 (2017).
Cleary, A. S., Leonard, T. L., Gestl, S. A. & Gunther, E. J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 508, 113–117 (2014).
Hong, M. K. et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 6, 6605 (2015).
McFadden, D. G. et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 156, 1298–1311 (2014).
Zhao, Z. M. et al. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl Acad. Sci. USA 113, 2140–2145 (2016).
Gundem, G. et al. The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353–357 (2015). This seminal study demonstrates that tumour metastasis occurs not only from primary tumours but also from metastasis.
Maddipati, R. & Stanger, B. Z. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 5, 1086–1097 (2015).
Aceto, N. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014). We demonstrate for the first time that CTC clusters derive from oligoclonal primary tumour cell groupings and that they possess both superior survival and metastatic potential compared with that of individual CTCs.
Cheung, K. J. et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl Acad. Sci. USA 113, E854–E863 (2016).
Liu, X. et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. 9, 96–113 (2019).
Bockhorn, M., Jain, R. K. & Munn, L. L. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed. Lancet Oncol. 8, 444–448 (2007).
Follain, G. et al. Fluids and their mechanics in tumour transit: shaping metastasis. Nat. Rev. Cancer 20, 107–124 (2020).
Follain, G. et al. Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev. Cell 45, 33–52 e12 (2018).
Donato, C. et al. Hypoxia triggers the intravasation of clustered circulating tumor cells. Cell Rep. 32, 108105 (2020). Our laboratory demonstrates that hypoxic conditions favour the formation and intravasation of CTC clusters with enhanced metastatic potential, and that this can be targeted with proangiogenic agents.
Zhang, H. et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012).
Jin, F., Brockmeier, U., Otterbach, F. & Metzen, E. New insight into the SDF-1/CXCR4 axis in a breast carcinoma model: hypoxia-induced endothelial SDF-1 and tumor cell CXCR4 are required for tumor cell intravasation. Mol. Cancer Res. 10, 1021–1031 (2012).
Mazzone, M. et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136, 839–851 (2009).
Harney, A. S. et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 5, 932–943 (2015).
Rossi, M. et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747–753 (2022).
Rodriguez-Tirado, C. et al. NR2F1 is a barrier to dissemination of early stage breast cancer cells. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-21-4145 (2022).
Gligorijevic, B. et al. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 125, 724–734 (2012).
Butler, T. P. & Gullino, P. M. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 35, 512–516 (1975).
Chambers, A. F., Groom, A. C. & MacDonald, I. C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 (2002).
Merino, D. et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 10, 766 (2019).
Headley, M. B. et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531, 513–517 (2016). The authors show that myeloid cells ingesting CTC-derived material subsequently accumulate in the lung interstitium to promote the development of successful metastases from surviving tumour cells.
Lee, H. J. et al. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat. Commun. 8, 14122 (2017).
Chang, S. F. et al. Tumor cell cycle arrest induced by shear stress: roles of integrins and Smad. Proc. Natl Acad. Sci. USA 105, 3927–3932 (2008).
Smerage, J. B. et al. Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol. Oncol. 7, 680–692 (2013).
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009).
Mazel, M. et al. Frequent expression of PD-L1 on circulating breast cancer cells. Mol. Oncol. 9, 1773–1782 (2015).
Steinert, G. et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 74, 1694–1704 (2014).
Szczerba, B. M. et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557 (2019). In this study, our laboratory shows how CTCs form heterotypic clusters with neutrophils, enhancing CTC proliferative and metastatic capacity.
Coffelt, S. B. et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015).
Spiegel, A. et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 6, 630–649 (2016).
King, M. R. et al. A physical sciences network characterization of circulating tumor cell aggregate transport. Am. J. Physiol. Cell Physiol. 308, C792–C802 (2015).
Haemmerle, M. et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 8, 310 (2017).
Schumacher, D., Strilic, B., Sivaraj, K. K., Wettschureck, N. & Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24, 130–137 (2013).
Anderson, K. J., de Guillebon, A., Hughes, A. D., Wang, W. & King, M. R. Effect of circulating tumor cell aggregate configuration on hemodynamic transport and wall contact. Math. Biosci. 294, 181–194 (2017).
Leong, H. S. et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8, 1558–1570 (2014).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
McDonald, B. et al. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int. J. Cancer 125, 1298–1305 (2009).
Rahn, J. J. et al. MUC1 mediates transendothelial migration in vitro by ligating endothelial cell ICAM-1. Clin. Exp. Metastasis 22, 475–483 (2005).
Osmani, N. et al. Metastatic tumor cells exploit their adhesion repertoire to counteract shear forces during intravascular arrest. Cell Rep. 28, 2491–2500 e2495 (2019).
Teicher, B. A. & Fricker, S. P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 16, 2927–2931 (2010).
Fu, Q. et al. Primary tumor-derived exosomes facilitate metastasis by regulating adhesion of circulating tumor cells via SMAD3 in liver cancer. Oncogene 37, 6105–6118 (2018).
Padua, D. et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008).
Reymond, N., d’Agua, B. B. & Ridley, A. J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 13, 858–870 (2013).
Wolf, M. J. et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 22, 91–105 (2012).
Strilic, B. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016).
Chen, M. B. et al. Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl Acad. Sci. USA 115, 7022–7027 (2018).
Cools-Lartigue, J. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Invest. https://doi.org/10.1172/JCI67484 (2013).
Spicer, J. D. et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 72, 3919–3927 (2012).
Massague, J. & Obenauf, A. C. Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016).
Klein, C. A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 20, 681–694 (2020).
Sosa, M. S., Bragado, P. & Aguirre-Ghiso, J. A. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622 (2014).
Deneve, E. et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin. Chem. 59, 1384–1392 (2013).
Muller, A. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001).
Labelle, M., Begum, S. & Hynes, R. O. Platelets guide the formation of early metastatic niches. Proc. Natl Acad. Sci. USA 111, E3053–E3061 (2014).
Croucher, P. I., McDonald, M. M. & Martin, T. J. Bone metastasis: the importance of the neighbourhood. Nat. Rev. Cancer 16, 373–386 (2016).
Lawson, M. A. et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 6, 8983 (2015).
Correia, A. L. et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 594, 566–571 (2021).
Dai, J. et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat. Cancer 3, 25–42 (2022).
Zhang, X. H. et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 (2009).
Albrengues, J. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science https://doi.org/10.1126/science.aao4227 (2018).
Wang, H. et al. The osteogenic niche is a calcium reservoir of bone micrometastases and confers unexpected therapeutic vulnerability. Cancer Cell 34, 823–839 e827 (2018).
Pickup, M. W., Mouw, J. K. & Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15, 1243–1253 (2014).
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K. J. & Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020).
Mohammadi, H. & Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 20, 766–774 (2018).
Petrie, R. J., Koo, H. & Yamada, K. M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345, 1062–1065 (2014).
Calvo, F. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Pathak, A. & Kumar, S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc. Natl Acad. Sci. USA 109, 10334–10339 (2012).
Sprouse, M. L. et al. PMN-MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20081916 (2019).
Elia, I., Doglioni, G. & Fendt, S. M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 28, 673–684 (2018).
LeBleu, V. S. et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003 (2014).
Jordan, N. V. et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 537, 102–106 (2016).
Li, X. X. et al. Nuclear receptor Nur77 facilitates melanoma cell survival under metabolic stress by protecting fatty acid oxidation. Mol. Cell 69, 480–492 e487 (2018).
Labuschagne, C. F., Cheung, E. C., Blagih, J., Domart, M. C. & Vousden, K. H. Cell clustering promotes a metabolic switch that supports metastatic colonization. Cell Metab. 30, 720–734 e725 (2019).
Basnet, H. et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. eLife https://doi.org/10.7554/eLife.43627 (2019).
Piskounova, E. et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015).
Le, T. T., Huff, T. B. & Cheng, J. X. Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer 9, 42 (2009).
Pascual, G. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017).
Greenlee, J. D., Subramanian, T., Liu, K. & King, M. R. Rafting down the metastatic cascade: the role of lipid rafts in cancer metastasis, cell death, and clinical outcomes. Cancer Res. 81, 5–17 (2021).
Onodera, Y., Nam, J. M. & Bissell, M. J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Invest. 124, 367–384 (2014).
Elia, I. et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019).
Porporato, P. E. et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 (2014).
Elia, I. et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 8, 15267 (2017).
Guo, H. F. et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat. Commun. 9, 512 (2018).
Gatenby, R. A., Gawlinski, E. T., Gmitro, A. F., Kaylor, B. & Gillies, R. J. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 66, 5216–5223 (2006).
Mehlen, P. & Puisieux, A. Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458 (2006).
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J. & Clarke, M. F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. Usa. 100, 3983–3988 (2003).
Baccelli, I. et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 31, 539–544 (2013).
Giordano, A. et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol. Cancer Ther. 11, 2526–2534 (2012).
Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Batlle, E. et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 (2000).
Yang, J. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004).
Fischer, K. R. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015).
Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Padmanaban, V. et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444 (2019). This study demonstrates that, contrary to a wide-held belief of metastasis initiation via the loss of E-cadherin, E-cadherin loss instead reduces CTC survival and successful metastasis.
Tsai, J. H., Donaher, J. L., Murphy, D. A., Chau, S. & Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012).
Brabletz, T. To differentiate or not–routes towards metastasis. Nat. Rev. Cancer 12, 425–436 (2012).
Yu, M. et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013).
Ruscetti, M., Quach, B., Dadashian, E. L., Mulholland, D. J. & Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 75, 2749–2759 (2015).
Strauss, R. et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 6, e16186 (2011).
Friedl, P., Locker, J., Sahai, E. & Segall, J. E. Classifying collective cancer cell invasion. Nat. Cell Biol. 14, 777–783 (2012).
Liotta, L. A., Saidel, M. G. & Kleinerman, J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 36, 889–894 (1976).
Eirew, P. et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 (2015).
Miller, B. E., Miller, F. R. & Heppner, G. H. Interactions between tumor subpopulations affecting their sensitivity to the antineoplastic agents cyclophosphamide and methotrexate. Cancer Res. 41, 4378–4381 (1981).
Aceto, N., Toner, M., Maheswaran, S. & Haber, D. A. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 1, 44–52 (2015).
Chimonidou, M. et al. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 57, 1169–1177 (2011).
Gkountela, S. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112.e114 (2019). Our laboratory demonstrates that CTC clustering confers stem-like features and can be targeted with FDA-approved drugs (Na+/K+-ATPase inhibitors).
Taftaf, R. et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 12, 4867 (2021).
Zhao, Q. et al. Interaction between circulating galectin-3 and cancer-associated MUC1 enhances tumour cell homotypic aggregation and prevents anoikis. Mol. Cancer 9, 154 (2010).
Haynes, B. F., Telen, M. J., Hale, L. P. & Denning, S. M. CD44 — a molecule involved in leukocyte adherence and T-cell activation. Immunol. Today 10, 423–428 (1989).
Zhang, L. et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 5, 180ra148 (2013).
Labelle, M., Begum, S. & Hynes, R. O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590 (2011). This study demonstrates that platelet–cancer cell interactions provide important signals that enhance invasiveness, priming cancer cells for subsequent efficient metastasis.
Labelle, M. & Hynes, R. O. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2, 1091–1099 (2012).
Placke, T. et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 72, 440–448 (2012).
Rachidi, S. et al. Platelets subvert T cell immunity against cancer via GARP–TGFβ axis. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aai7911 (2017).
Gaggioli, C. et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 9, 1392–1400 (2007).
Labernadie, A. et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 19, 224–237 (2017). This study demonstrates the cooperation of heterotypic CAF–cancer cell interactions for collective cell migration and invasion to promote tumour invasion and metastasis.
Schuster, E. et al. Better together: circulating tumor cell clustering in metastatic cancer. Trends Cancer 7, 1020–1032 (2021).
Duda, D. G. et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl Acad. Sci. USA 107, 21677–21682 (2010).
Rejniak, K. A. Circulating tumor cells: when a solid tumor meets a fluid microenvironment. Adv. Exp. Med. Biol. 936, 93–106 (2016).
Au, S. H. et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl Acad. Sci. USA 113, 4947–4952 (2016).
Castro-Giner, F. & Aceto, N. Tracking cancer progression: from circulating tumor cells to metastasis. Genome Med. 12, 31 (2020).
Gasch, C. et al. Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2-negative metastatic breast cancer. Mol. Oncol. 10, 1330–1343 (2016).
Ni, X. et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl Acad. Sci. USA 110, 21083–21088 (2013).
Heitzer, E. et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 73, 2965–2975 (2013).
Lohr, J. G. et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 32, 479–484 (2014).
Faugeroux, V. et al. An accessible and unique insight into metastasis mutational content through whole-exome sequencing of circulating tumor cells in metastatic prostate cancer. Eur. Urol. Oncol. 3, 498–508 (2020).
Oskarsson, T. et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 (2011).
Yu, M. et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487, 510–513 (2012).
Winslow, M. M. et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473, 101–104 (2011).
Scheidmann, M. C. et al. An in vivo CRISPR screen identifies stepwise genetic dependencies of metastatic progression. Cancer Res. 82, 681–694 (2022).
Ming, H., Li, B., Zhou, L., Goel, A. & Huang, C. Long non-coding RNAs and cancer metastasis: molecular basis and therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 1875, 188519 (2021).
Sun, Y. F. et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin. Cancer Res. 24, 547–559 (2018).
Bos, P. D. et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). Bos et al. (2009), Kang et al. (2003) and Minn et al. (2005) constitute a series of articles demonstrating organotropism-related gene expression signatures as a mechanism for non-random metastasis to specific organs.
Kang, Y. et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003).
Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005).
Kang, Y. et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl Acad. Sci. USA 102, 13909–13914 (2005).
Mohammadi Ghahhari, N. et al. Cooperative interaction between ERalpha and the EMT-inducer ZEB1 reprograms breast cancer cells for bone metastasis. Nat. Commun. 13, 2104 (2022).
Klotz, R. et al. Circulating tumor cells exhibit metastatic tropism and reveal brain metastasis drivers. Cancer Discov. 10, 86–103 (2020). The authors demonstrate that CTC cell lines established from patients with breast cancer recapitulate patient-specific metastatic patterns in mouse models and show distinct molecular characteristics.
Diamantopoulou, Z. et al. The metastatic spread of breast cancer accelerates during sleep. Nature 607, 156–162 (2022). Our laboratory demonstrates that temporal dynamics of CTC intravasation vary dramatically dependent on circadian rhythm and are dictated by rhythmic variations in hormone levels (for example, melatonin), resulting in the highest CTC counts during the rest phase. This study also demonstrates a peak in Ki67 expression in both the primary tumour and CTCs during the rest phase.
Zhu, X. et al. In vivo flow cytometry reveals a circadian rhythm of circulating tumor cells. Light Sci. Appl. 10, 110 (2021).
Schernhammer, E. S. et al. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl Cancer Inst. 93, 1563–1568 (2001).
Schernhammer, E. S. et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl Cancer Inst. 95, 825–828 (2003).
Straif, K. et al. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 8, 1065–1066 (2007).
Travis, R. C. et al. Night shift work and breast cancer incidence: three prospective studies and meta-analysis of published studies. J. Natl Cancer Inst. https://doi.org/10.1093/jnci/djw169 (2016).
Lauriola, M. et al. Diurnal suppression of EGFR signalling by glucocorticoids and implications for tumour progression and treatment. Nat. Commun. 5, 5073 (2014).
Sephton, S. E. et al. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain Behav. Immun. 30, S163–S170 (2013).
Giacchetti, S. et al. Phase III trial comparing 4-day chronomodulated therapy versus 2-day conventional delivery of fluorouracil, leucovorin, and oxaliplatin as first-line chemotherapy of metastatic colorectal cancer: the European Organisation for Research and Treatment of Cancer Chronotherapy Group. J. Clin. Oncol. 24, 3562–3569 (2006).
Levi, F. Circadian chronotherapy for human cancers. Lancet Oncol. 2, 307–315 (2001).
Levi, F., Zidani, R. & Misset, J. L. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. Lancet 350, 681–686 (1997).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
Turajlic, S. et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173, 595–610 e511 (2018).
Schwarzenbach, H., Hoon, D. S. & Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011).
Chemi, F. et al. Pulmonary venous circulating tumor cell dissemination before tumor resection and disease relapse. Nat. Med. 25, 1534–1539 (2019). This study demonstrates that CellSearch-detected pulmonary venous CTCs obtained at surgical resection of early-stage non–small-cell lung cancer revealed greater mutational overlap with metastasis detected 10 months later than with the primary tumour. This suggested that pulmonary venous CTCs represent subclones responsible for subsequent disease relapse.
Visal, T. H., den Hollander, P., Cristofanilli, M. & Mani, S. A. Circulating tumour cells in the -omics era: how far are we from achieving the ‘singularity’? Br. J. Cancer https://doi.org/10.1038/s41416-022-01768-9 (2022).
Muller, V. et al. Prognostic impact of circulating tumor cells assessed with the CellSearch System and AdnaTest Breast in metastatic breast cancer patients: the DETECT study. Breast Cancer Res. 14, R118 (2012).
Wang, L. et al. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 43, 464–475 (2016).
Gleghorn, J. P. et al. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip 10, 27–29 (2010).
Karabacak, N. M. et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 9, 694–710 (2014).
Krebs, M. G. et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J. Thorac. Oncol. 7, 306–315 (2012).
Miller, M. C., Robinson, P. S., Wagner, C. & O’Shannessy, D. J. The Parsortix cell separation system — a versatile liquid biopsy platform. Cytom. A 93, 1234–1239 (2018).
Rosenberg, R. et al. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry 49, 150–158 (2002).
Krol, I. et al. Detection of clustered circulating tumour cells in early breast cancer. Br. J. Cancer 125, 23–27 (2021).
Harb, W. et al. Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl. Oncol. 6, 528–538 (2013).
Ning, N. et al. Improvement of specific detection of circulating tumor cells using combined CD45 staining and fluorescence in situ hybridization. Clin. Chim. Acta 433, 69–75 (2014).
Saucedo-Zeni, N. et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 41, 1241–1250 (2012).
Eifler, R. L. et al. Enrichment of circulating tumor cells from a large blood volume using leukapheresis and elutriation: proof of concept. Cytom. B Clin. Cytom. 80, 100–111 (2011).
Crosbie, P. A. et al. Circulating tumor cells detected in the tumor-draining pulmonary vein are associated with disease recurrence after surgical resection of NSCLC. J. Thorac. Oncol. 11, 1793–1797 (2016).
Gawad, C., Koh, W. & Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188 (2016).
Wang, Y. & Navin, N. E. Advances and applications of single-cell sequencing technologies. Mol. Cell 58, 598–609 (2015).
Abouleila, Y. et al. Live single cell mass spectrometry reveals cancer-specific metabolic profiles of circulating tumor cells. Cancer Sci. 110, 697–706 (2019).
Armbrecht, L. et al. Quantification of protein secretion from circulating tumor cells in microfluidic chambers. Adv. Sci. 7, 1903237 (2020).
Donato, C., Buczak, K., Schmidt, A. & Aceto, N. Mass spectrometry analysis of circulating breast cancer cells from a xenograft mouse model. STAR Protoc. 2, 100480 (2021).
Khoo, B. L. et al. Expansion of patient-derived circulating tumor cells from liquid biopsies using a CTC microfluidic culture device. Nat. Protoc. 13, 34–58 (2018).
Khoo, B. L. et al. Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget 6, 15578–15593 (2015).
Yu, M. et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220 (2014).
Anderson, R. L. et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 16, 185–204 (2019).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Parsons, S., Maldonado, E. B. & Prasad, V. Comparison of drugs used for adjuvant and metastatic therapy of colon, breast, and non-small cell lung cancers. JAMA Netw. Open 3, e202488 (2020).
Ganesh, K. & Massague, J. Targeting metastatic cancer. Nat. Med. 27, 34–44 (2021).
Groppa, E. et al. EphrinB2/EphB4 signaling regulates non-sprouting angiogenesis by VEGF. EMBO Rep. https://doi.org/10.15252/embr.201745054 (2018).
Xu, X. R., Yousef, G. M. & Ni, H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood 131, 1777–1789 (2018).
Wei, R. R. et al. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol. Sin. 39, 1326–1337 (2018).
Choi, J. W. et al. Urokinase exerts antimetastatic effects by dissociating clusters of circulating tumor cells. Cancer Res. 75, 4474–4482 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03928210 (2019).
Bambace, N. M. & Holmes, C. E. The platelet contribution to cancer progression. J. Thromb. Haemost. 9, 237–249 (2011).
Cooke, N. M., Spillane, C. D., Sheils, O., O’Leary, J. & Kenny, D. Aspirin and P2Y12 inhibition attenuate platelet-induced ovarian cancer cell invasion. BMC Cancer 15, 627 (2015).
Mitchell, M. J., Wayne, E., Rana, K., Schaffer, C. B. & King, M. R. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc. Natl Acad. Sci. Usa. 111, 930–935 (2014).
Chen, H. N. et al. EpCAM signaling promotes tumor progression and protein stability of PD-L1 through the EGFR pathway. Cancer Res. 80, 5035–5050 (2020).
Muller, P. et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci. Transl. Med. 7, 315ra188 (2015).
Dombroski, J. A., Jyotsana, N., Crews, D. W., Zhang, Z. & King, M. R. Fabrication and characterization of tumor nano-lysate as a preventative vaccine for breast cancer. Langmuir 36, 6531–6539 (2020).
Parkins, K. M. et al. Engineering circulating tumor cells as novel cancer theranostics. Theranostics 10, 7925–7937 (2020).
Aggarwal, C. et al. Relationship among circulating tumor cells, CEA and overall survival in patients with metastatic colorectal cancer. Ann. Oncol. 24, 420–428 (2013).
Allard, W. J. et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 10, 6897–6904 (2004).
Cristofanilli, M. et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 351, 781–791 (2004).
Hou, J. M. et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 30, 525–532 (2012).
Scher, H. I. et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. Lancet Oncol. 10, 233–239 (2009).
Rack, B. et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl Cancer Inst. https://doi.org/10.1093/jnci/dju066 (2014).
Sparano, J. et al. Association of circulating tumor cells with late recurrence of estrogen receptor-positive breast cancer: a secondary analysis of a randomized clinical trial. JAMA Oncol. 4, 1700–1706 (2018).
Cohen, S. J. et al. Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 6, 125–132 (2006).
Danila, D. C. et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin. Cancer Res. 13, 7053–7058 (2007).
de Bono, J. S. et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 14, 6302–6309 (2008).
Liu, M. C. et al. First-line doublet chemotherapy for metastatic triple-negative breast cancer: circulating tumor cell analysis of the tnAcity trial. Cancer Manag. Res. 11, 10427–10433 (2019).
Wallwiener, M. et al. Serial enumeration of circulating tumor cells predicts treatment response and prognosis in metastatic breast cancer: a prospective study in 393 patients. BMC Cancer 14, 512 (2014).
Larsson, A. M. et al. Longitudinal enumeration and cluster evaluation of circulating tumor cells improve prognostication for patients with newly diagnosed metastatic breast cancer in a prospective observational trial. Breast Cancer Res. 20, 48 (2018).
Liu, M. C. et al. Circulating tumor cells: a useful predictor of treatment efficacy in metastatic breast cancer. J. Clin. Oncol. 27, 5153–5159 (2009).
Thery, L. et al. Circulating tumor cells in early breast cancer. JNCI Cancer Spectr. 3, pkz026 (2019).
Bidard, F. C. et al. Efficacy of circulating tumor cell count-driven vs clinician-driven first-line therapy choice in hormone receptor-positive, ERBB2-negative metastatic breast cancer: the STIC CTC randomized clinical trial. JAMA Oncol. 7, 34–41 (2021).
Georgoulias, V. et al. Trastuzumab decreases the incidence of clinical relapses in patients with early breast cancer presenting chemotherapy-resistant CK-19mRNA-positive circulating tumor cells: results of a randomized phase II study. Ann. Oncol. 23, 1744–1750 (2012).
Jacot, W. et al. Actionability of HER2-amplified circulating tumor cells in HER2-negative metastatic breast cancer: the CirCe T-DM1 trial. Breast Cancer Res. 21, 121 (2019).
Pestrin, M. et al. Final results of a multicenter phase II clinical trial evaluating the activity of single-agent lapatinib in patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cells. A proof-of-concept study. Breast Cancer Res. Treat. 134, 283–289 (2012).
Fehm, T. et al. Efficacy of the tyrosine kinase inhibitor lapatinib in the treatment of patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cells - results from the randomized phase III DETECT III trial. Cancer Res. https://doi.org/10.1158/1538-7445.SABCS20-PD3-12 (2021).
Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 371, 1028–1038 (2014). The study demonstrates that detection of AR-V7 in patients with castration-resistant prostate cancer could be used as a predictive biomarker for resistance to anti-androgen therapy.
Armstrong, A. J. et al. Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: the PROPHECY study. J. Clin. Oncol. 37, 1120–1129 (2019).
Parker, C. et al. Prostate cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 31, 1119–1134 (2020).
WHO Classification of Tumours Editorial Board (eds). WHO Classification of Tumours: Breast Tumours 5th edn, Vol. 2 (International Agency for Research on Cancer, 2019).
Edge, S. et al. (eds). AJCC Cancer Staging Manual 7th end (Springer, 2010).
Khoo, B. L. et al. Liquid biopsy and therapeutic response: circulating tumor cell cultures for evaluation of anticancer treatment. Sci. Adv. 2, e1600274 (2016).
Bidard, F. C. et al. Prognosis of women with stage IV breast cancer depends on detection of circulating tumor cells rather than disseminated tumor cells. Ann. Oncol. 19, 496–500 (2008).
Slade, M. J. et al. Comparison of bone marrow, disseminated tumour cells and blood-circulating tumour cells in breast cancer patients after primary treatment. Br. J. Cancer 100, 160–166 (2009).
Wiedswang, G. et al. Comparison of the clinical significance of occult tumor cells in blood and bone marrow in breast cancer. Int. J. Cancer 118, 2013–2019 (2006).
Hu, Z., Li, Z., Ma, Z. & Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 52, 701–708 (2020).
Gupta, G. P. & Massague, J. Cancer metastasis: building a framework. Cell 127, 679–695 (2006).
Hofman, V. et al. Detection of circulating tumor cells as a prognostic factor in patients undergoing radical surgery for non-small-cell lung carcinoma: comparison of the efficacy of the CellSearch assay and the isolation by size of epithelial tumor cell method. Int. J. Cancer 129, 1651–1660 (2011).
Lecharpentier, A. et al. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 105, 1338–1341 (2011).
Vona, G. et al. Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 156, 57–63 (2000).
Acknowledgements
The authors thank members of and collaborators with the Aceto laboratory for scientific feedback and discussions. A.R. is supported by University Hospital Zurich, the Kurt and Senta Herrmann Foundation, the Walter and Gertrud Siegenthaler Foundation and the Swiss Cancer Foundation. B.D.N.-S. is supported by University Hospital Zurich, the Sassella Foundation and the Iten Kohaut Foundation in collaboration with the USZ Foundation and the Theodor and Ida Herzog-Egli Foundation. The Aceto laboratory is supported by the European Research Council (101001652), the strategic focus area of personalized health and related technologies at ETH Zurich (PHRT-541), the Future and Emerging Technologies programme of the European Commission (801159-B2B), the Swiss National Science Foundation (212183), the Swiss Cancer League (KLS-4834-08-2019), the Basel Cancer League (KLbB-4763-02-2019) and ETH Zurich.
Author information
Authors and Affiliations
Contributions
A.R., B.D.N.-S. and A.W. researched data and wrote the article. N.A. reviewed and edited the manuscript before submission. All authors contributed substantially to discussion and writing of the article.
Corresponding author
Ethics declarations
Competing interests
N.A. co-founded and is a member of the board of PAGE Therapeutics AG, Switzerland, is listed as an inventor on patent applications related to CTCs, is a paid consultant for Swiss Re Group, Bracco Group, Tethis S.p.A., Thermo Fisher and ANGLE PLC, and is a Novartis shareholder.
Peer review
Peer review information
Nature Reviews Cancer thanks Francoise Farace, Evi Lianidou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Adjuvant setting
-
A clinical setting in which a therapy is given in addition to the primary therapy to maximize its effectiveness, in an attempt to reduce the risk of cancer relapse.
- Anoikis
-
The induction of programmed cell death (apoptosis) in anchorage-dependent cells (for example, epithelial cells) after their detachment from the extracellular matrix or neighbouring cells.
- Antigen-agnostic
-
Without a priori knowledge of antigenic epitope expression.
- Biomarkers
-
Measurable parameters of biological processes that have diagnostic, prognostic or predictive significance; they can be used to monitor pathological processes or therapeutic interventions.
- Cancer vaccine
-
A form of immunotherapy that uses tumour-associated antigens to activate and condition the patient’s immune system against the patient’s cancer.
- Circadian rhythm
-
Endogenous (internal) rhythms driven by biochemical oscillators with a period of approximately 24 hours that have major influences on the functions of an organism.
- Cytapheresis
-
A procedure in which various cells are separated from withdrawn blood and retained so that large amounts of plasma and cellular components can be harvested for transfusions and various other purposes.
- Diapedesis
-
The final step of the leukocyte extravasation cascade when leukocytes transmigrate across the blood endothelial cell barrier in response to acute inflammation.
- Glycocalyx
-
The protein or lipid-bound carbohydrate portion of the extracellular side of the cell membrane of eukaryotic or prokaryotic cells.
- Immunomagnetic selection
-
Isolation of cells via antibody-conjugated magnets, based on cell surface antigen expression.
- Lipid rafts
-
Specialized microdomains in the membrane of eukaryotic cells containing dynamic assemblies of cholesterol, sphingolipids and glycosphingolipids; they are involved in the activation of signalling cascades by favouring specific protein–protein interactions.
- Metachronous lesions
-
Two (or more) independent primary malignancies, of which the second (or third, fourth, fifth and so on) arises more than 6 months after the diagnosis of the first.
- Minimal residual disease
-
Malignant cells that remain in patients after therapy without symptoms or overt signs of disease.
- Mitophagy
-
The selective degradation of mitochondria by autophagy.
- Necrosis
-
Passive, uncontrolled cell death due to damaging insults (for example, the result of mechanical injury, exposure to toxins, hypoxia, hypothermia or infection), resulting in an uncontrolled release of cellular contents into the surrounding environment, promoting inflammation.
- Organotropism
-
The affinity for cancerous growth in particular organs, organ systems or somatic tissues leading to a nonrandom distribution of metastatic cancer cells.
- Personalized medicine
-
The tailoring of treatment to the individual patient based on the patient’s predicted response or risk of disease based on the fundamental understanding of the molecular basis of disease.
- Phylogeny
-
The study of relationships among or within cancer cells that focuses on observed heritable traits, such as DNA or amino acid sequences or morphology.
- Private alterations
-
Rare gene mutations that are usually found only in a single cell or in a small population of cells, as opposed to public mutations, which occur in founder clones and therefore all tumour cells.
- Shear stress
-
Tangential or frictional force tending to cause mechanical stress and deformation of a material by slippage along a plane or planes parallel to the imposed stress.
- Stem-like
-
A characteristic of cancer cells with traits akin to haematopoietic stem cells, including the ability to self-renew, differentiate, and generate progeny cells.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ring, A., Nguyen-Sträuli, B.D., Wicki, A. et al. Biology, vulnerabilities and clinical applications of circulating tumour cells. Nat Rev Cancer 23, 95–111 (2023). https://doi.org/10.1038/s41568-022-00536-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-022-00536-4
This article is cited by
-
tRNA-derived small RNAs in human cancers: roles, mechanisms, and clinical application
Molecular Cancer (2024)
-
Beyond the barrier: the immune-inspired pathways of tumor extravasation
Cell Communication and Signaling (2024)
-
In Vivo Labeling and Detection of Circulating Tumor Cells in Mice Using OTL38
Molecular Imaging and Biology (2024)
-
Phenotypic Transitions the Processes Involved in Regulation of Growth and Proangiogenic Properties of Stem Cells, Cancer Stem Cells and Circulating Tumor Cells
Stem Cell Reviews and Reports (2024)
-
Clinical value of folate receptor-positive circulating tumor cells in patients with esophageal squamous cell carcinomas: a retrospective study
BMC Cancer (2023)
