
Abstract
In animals, PIWI-interacting RNAs (piRNAs) direct PIWI proteins to silence complementary targets such as transposons. In Drosophila and other species with a maternally specified germline, piRNAs deposited in the egg initiate piRNA biogenesis in the progeny. However, Y chromosome loci cannot participate in such a chain of intergenerational inheritance. How then can the biogenesis of Y-linked piRNAs be initiated? Here, using Suppressor of Stellate (Su(Ste)), a Y-linked Drosophila melanogaster piRNA locus as a model, we show that Su(Ste) piRNAs are made in the early male germline via 5′-to-3′ phased piRNA biogenesis initiated by maternally deposited 1360/Hoppel transposon piRNAs. Notably, deposition of Su(Ste) piRNAs from XXY mothers obviates the need for phased piRNA biogenesis in sons. Together, our study uncovers a developmentally programmed, intergenerational mechanism that allows fly mothers to protect their sons using a Y-linked piRNA locus.
Similar content being viewed by others


Main
In animals, the PIWI-interacting RNA (piRNA) pathway generates small RNAs that direct silencing of transposable elements and other selfish genetic elements1. Loss of piRNAs derepresses transposons2,3,4,5, dysregulates gene expression6,7,8 and reduces fertility. At the core of piRNA-mediated silencing are 18–35-nucleotide (nt) piRNAs that bind to and guide PIWI proteins to their targets via nucleotide sequence complementarity2,9,10,11,12. The three D. melanogaster PIWI proteins have specialized functions in the germline: Piwi represses transposon transcription in the nucleus, whereas Ago3 and Aubergine (Aub) cleave piRNA precursor and transposon transcripts in the cytoplasm4,12,13,14,15,16,17,18,19,20,21,22.
Animals often use pre-existing piRNAs to direct slicing of complementary transcripts and initiate piRNA biogenesis from the resulting 5′-monophosphorylated cleavage products23. For example, in the D. melanogaster female germline, Ago3 and Aub are loaded with piRNAs derived from complementary transcripts (transposon messenger RNAs and piRNA precursors), and the 3′ cleavage product of Ago3 slicing is used to make antisense Aub-loaded piRNAs and vice versa. This positive feedback loop—the ‘ping-pong’ cycle—amplifies the transposon-targeting population of piRNAs4,24. The ping-pong pathway also initiates 5′-to-3′ fragmentation of the remainder of the cleavage product into tail-to-head, phased piRNAs loaded in Piwi19,20,25,26. Phased piRNA biogenesis requires the endonuclease Zucchini (Zuc; PLD6 in mammals) and the RNA helicase Armitage (Armi; MOV10L1 in mammals)27,28,29,30. The ping-pong pathway increases only piRNA abundance, whereas production of phased primary piRNAs adds sequence diversity to the piRNA19 pool.
The ping-pong cycle requires pre-existing piRNAs to initiate the amplification process. In D. melanogaster, maternally deposited piRNAs serve this purpose, providing a pool of piRNAs that can initiate the ping-pong cycle17,31,32,33,34. For example, the inability of naïve mothers to provide P-element-derived piRNAs when mated with P-element-infested fathers causes derepression of selfish elements and sterility in their offspring, a phenomenon called hybrid dysgenesis32,35,36,37,38,39,40,41,42,43.
Stellate (Ste) and Suppressor of Stellate (Su(Ste)) in D. melanogaster provided the founding paradigm of piRNA-directed repression44,45,46,47,48. Ste is a repetitive gene whose unchecked expression results in Ste protein crystals, amyloid-like protein aggregates that cause male sterility via unknown mechanisms49. To ensure male fertility, Ste genes on the X chromosome are normally repressed by Su(Ste) piRNAs that are antisense to Ste and are produced from Y chromosome transcripts12,50,51,52. Su(Ste) locus comprises tandem repeats nearly identical (~90%) to Ste. Ste is the major silencing target of the piRNA pathway in the D. melanogaster male germline7,51,52,53,54,55, requiring armi, zuc, krimp, spn-E, vas, aub and ago3, but not piwi or rhino (rhi), suggesting that Ste repression is primarily dependent on cytoplasmic cleavage of the Ste mRNA12,27,56,57,58,59. Because Su(Ste) is encoded on the Y chromosome, fly mothers—which lack a Y chromosome—cannot provide their sons with Su(Ste) piRNAs to initiate biogenesis. How the male germline produces Su(Ste) piRNAs in the absence of maternally deposited Su(Ste) piRNAs is unknown.
In this Article, we describe the mechanism by which the male germline represses Ste in the absence of maternally deposited Su(Ste) piRNAs. We show that Su(Ste) piRNAs are produced by Armi- and Zuc-dependent phased piRNA biogenesis in male germline stem cells (GSCs) and early spermatogonia (SGs), days before expression of Ste target RNAs in spermatocytes. Phased biogenesis of Su(Ste) piRNAs in GSCs/SGs is critical to repress Ste later in spermatocytes and thus for male fertility. Our data show that males from XX mothers use maternally deposited 1360/Hoppel piRNAs to cleave Su(Ste) precursors and initiate 5′-to-3′ phased biogenesis of Su(Ste) piRNAs in the early germline (GSCs/SGs). We show that the requirement for Armi, a protein essential for phased piRNA biogenesis, in Su(Ste) piRNA production in males is relieved when XXY females provide maternal Su(Ste) piRNAs to their sons’ germline. These data explain how maternally deposited piRNAs can direct production of non-homologous piRNA guides in the germline of the progeny. Our study reveals a mechanism for intergenerational transmission of piRNA-coded memory in the absence of direct homology and demonstrates that the phased piRNA pathway can protect offspring from selfish genetic elements not encountered by their mothers.
Results
Su(Ste) transcription starts days before Ste expression
To investigate Su(Ste) piRNA precursor expression and processing into piRNAs during D. melanogaster spermatogenesis, we used single-molecule RNA fluorescent in situ hybridization (smRNA-FISH)60,61. By leveraging nucleotide polymorphisms between Ste and Su(Ste), we used a single in situ probe to detect Su(Ste) and a collection of Stellaris in situ probes to visualize Ste (Methods). smRNA-FISH can detect Ste mRNA and Su(Ste) precursor transcripts but not mature piRNAs, because small RNAs are not retained in formaldehyde-fixed tissues62 (Methods).
In wild-type testes, Ste transcripts were first detected in the nuclei of spermatocytes (Fig. 1a,b). In contrast, in XO males, which lack Su(Ste), Ste transcripts were readily detected in the spermatocyte cytoplasm (Fig. 1c), leading to production of Ste protein crystals, a known cause of subfertility. Notably, in XO males, cytoplasmic Ste mRNA was observed only in spermatocytes (Fig. 1c), suggesting that Ste is transcriptionally silent in early germ cells (that is, GSCs and SGs).

a, Early stages of D. melanogaster spermatogenesis. The stem cell niche is formed by the non-dividing somatic cells of the hub (asterisk). The GSCs are physically attached to the hub and divide asymmetrically. The gonialblasts (GBs), the differentiating daughters of GSCs, undergo four rounds of mitotic divisions with incomplete cytokinesis. Resultant 16-cell SGs then enter meiotic prophase as spermatocytes. The expression patterns of nos-gal4 and bam-gal4 drivers in the adult male germ line are also indicated. GSCs and early SGs are indicated by a yellow dotted line; cyan lines indicate the zone of spermatocytes in this and all subsequent figures. b(i), c(i) Expression of Ste mRNA (red) and antisense Su(Ste) precursor (green) in the wild-type (b) and in XO (c) testes (smRNA-FISH). Magnified view of boxed areas is shown in b(ii), b(iii), c(ii) and c(iii). Arrow points to Su(Ste) transcripts in a GSC nucleus. b and c represent z-projections that cover the depth of the testes, whereas b(ii), b(iii) c(ii) and c(iii) only cover the depth of the cells presented. Dotted white lines indicate the nuclear periphery. Red, Ste RNA; green, antisense Su(Ste) RNA; blue, DAPI. Scale bars, 20 µm (b(i), c(i)) and 5 µm (b(ii), b(iii), c(ii) and c(iii)). d, Length profile of Ste-, Su(Ste)- (Supplementary Table 3) and flamenco-derived small RNAs in control (y1w1118/Y; nos-gal4:VP16/TM2) testis. flamenco produces 21-nt siRNAs79. The data are the mean from two independent biological samples. Source numerical data are available in source data.
Our smRNA-FISH experiments readily detected Su(Ste) expression in GSCs (Fig. 1b), earlier than previously reported50. Thus, Su(Ste) expression precedes that of Ste by ~2–3 days (Fig. 1a). In GSCs and SGs, Su(Ste) transcription was detectable only from the genomic strand that produces piRNA precursors antisense to Ste mRNA (Extended Data Fig. 1a). The steady-state abundance of nuclear antisense Su(Ste) transcripts peaked in late SGs/early spermatocytes and was undetectable by the time Ste expression was first detected, in late spermatocytes (Fig. 1b).
Ping-pong amplification of Ste-targeting piRNAs should require the presence of both antisense Su(Ste) and sense Ste RNA in the same cells. Our data, however, show that antisense Su(Ste) piRNA precursors are transcribed and processed into Ste-targeting piRNAs before the first detectable accumulation of Ste mRNA. Supporting the idea that antisense Su(Ste) precursors and sense Ste mRNA are not present in the same germ cell types, we did not detect short interfering RNAs (siRNAs) production from Su(Ste) loci (Fig. 1d). (siRNAs are produced by Dicer proteins from double-stranded RNAs63). We conclude that ping-pong amplification is unlikely to explain the biogenesis of Su(Ste) piRNAs in GSCs and SGs (Extended Data Fig. 1b,c).
Su(Ste) transcripts are processed in early male germ cells
Consistent with earlier studies27,54, we found that processing of antisense Su(Ste) precursors into mature piRNAs in GSCs/SGs depends on components of the phased piRNA biogenesis pathway. In wild-type GSCs/SGs, Su(Ste) transcripts were detected as a single nuclear focus, corresponding to nascent transcripts from the Su(Ste) loci (Fig. 2a). In contrast, in armi1/72.1 or zucEY11457/− loss-of-function mutants, the nuclear foci of Su(Ste) transcripts were enlarged, and multiple cytoplasmic foci appeared, probably representing accumulation of unprocessed piRNA precursor transcripts (Fig. 2b,c). Similar Su(Ste) cytoplasmic foci were detected when armi or zuc mRNA was specifically depleted in germ cells by RNA interference (RNAi) using pVALIUM22 transgenes (armiTRIP.GL00254 and zucTRIP.GL00111; henceforth, armiRNAi and zucRNAi) driven by nanos(nos)-Gal4 (ref. 64; Figs. 1a and 2d,e). The appearance of Su(Ste) cytoplasmic foci in zuc and armi mutants (Fig. 2f) concurs with the increase in the steady-state abundance of Su(Ste) transcripts measured by quantitative reverse transcription polymerase chain reaction (qRT–PCR) in zucEY11457/− mutant testis enriched for SGs by over-expressing dpp: Su(Ste) precursors increased 1.9 ± 0.7-fold in mutants versus control testis (two-tailed, one sample t-test, P = 0.025), while act5C transcripts changed 1.1 ± 0.7-fold (two-tailed, one sample t-test, P = 0.7; Extended Data Fig. 2 and Supplementary Table 1).

a–e, smRNA-FISH for antisense Su(Ste) precursor transcript (green) in control y1w1118 testis (a) and in piRNA pathway mutant testes of the indicated genotypes: armi mutant (b); zuc mutant (c); armi RNAi (d); zuc RNAi (e)). The corresponding magnified regions of the niche marked by quadrates are shown in a(ii), b(ii), c(ii), d(ii) and e(ii) GSC and early SGs are indicated by yellow dotted lines; cyan lines indicate zone of spermatocytes. Arrowheads point to nuclear transcripts; arrows point to cytoplasmic RNA foci. The asterisks indicate the hub. Blue, DAPI. Scale bars, 5 µm. f, Quantification of cytoplasmic Su(Ste) RNA foci in GCSs and SG cells. Signal intensity was measured by maximum projection of z-stacks that encompass the entire cell. Box plots show the median and interquartile range (IQR); whiskers denote 1.5× IQR (n = 90 for control; n = 54 for nos>armiTRIP.GL00254; n = 33 for armi1/72.1; n = 34 for nos>zucTRIP.GL00111; n = 31 for zucEY11457/−). P = 2.2 × 10−16 for Kruskal–Wallis test (one-way analysis of variance on ranks) comparing all genotypes and control; Benjamini–Hochberg-corrected P values for post hoc pairwise two-tailed Mann–Whitney tests: P = 2 × 10−16 for nos>armiTRIP.GL00254 versus control; P = 7.2 × 10−9 for armi1/72.1 versus control; P = 9.4 × 10−9 for nos>zucTRIP.GL00111 versus control; P = 2 × 10−16 for zucEY11457/− versus control. Source numerical data are available in source data.
By contrast, Su(Ste) piRNA precursor transcripts did not accumulate when vas—the helicase required for ping-pong piRNA processing19,65—was depleted by nos-driven RNAi (Extended Data Fig. 3a–d). Similarly, depletion of either of the endonucleases in the ping-pong pathway (Aub or Ago3) did not stabilize Su(Ste) precursor transcripts in GSCs/SGs (Extended Data Fig. 3a–d).
In the phased piRNA biogenesis pathway, the endonuclease Zuc fragments piRNA precursors into head-to-tail pre-piRNAs, and the overwhelming majority of phased pre-piRNAs bear a uridine as their 5′-terminal nucleotide (pre-piRNAs become mature piRNAs after their 3′ ends are trimmed and 2′-O-methylated). Conversely, piRNA guides produced by the ping-pong pathway frequently have an adenine at position 10, because endonucleases in the ping-pong pathway often have an intrinsic preference for targets with an adenine at the position that then becomes the tenth nucleotide of a new mature piRNA66. Transposon-derived piRNAs in testis are made by both the ping-pong and the phased biogenesis pathways54, and thus exhibit both the enrichment of uridines as the first nucleotide (67 ± 3%) and a higher frequency of adenines as the tenth nucleotide (37.2 ± 0.3%; Extended Data Fig. 3e). Supporting the idea that processing of Su(Ste) precursors into piRNAs in GSCs/SGs is catalysed by Zuc19,20, we find that, although the majority of Su(Ste)-derived piRNAs begin with a uridine (77 ± 1% at position 1 versus 28.4 ± 0.2% at all positions), they show no enrichment for adenine as the tenth nucleotide (21 ± 1% at position 10 versus 25.9 ± 0.3% at all positions; Extended Data Fig. 3e). Together, these results suggest that, in GSC/SGs, the phased piRNA biogenesis pathway dominates the production of piRNAs from Su(Ste) transcripts.
Ste silencing requires zuc and armi in early male germ cells
Repression of Ste in late spermatocytes depends on zuc and armi expression during a short window in early spermatogenesis. When armi or zuc mRNA was depleted by nos-driven RNAi (nos>armiRNAi or nos>zucRNAi) throughout the germline (Fig. 1a), we observed derepression of Ste RNA (Fig. 3a–c), Ste protein accumulation (Fig. 3i) and reduced fertility (Fig. 3j). In contrast, using bam-gal4 (Fig. 1a) to deplete armi or zuc in >4-cell SG stages (bam>armiRNAi or bam>zucRNAi) had no observable effect on Ste repression or fertility (Fig. 3d,e), suggesting that armi and zuc are dispensable for Ste repression after the four-cell spermatogonial stage.

a–h, Ste smRNA-FISH (red) in the testes in control y1w1118 testis (a) and in piRNA pathway mutant testes of the indicated genotypes: armi nos-driven RNAi (b); zuc nos-driven RNAi (c); armi bam-driven RNAi (d); zuc bam-driven RNAi (e); armi mutant (f); armi mutant, nos rescue (g); armi mutant, bam rescue(h). GSCs and early SGs are indicated by yellow dotted lines; cyan lines indicate zone of spermatocytes. The asterisks indicate the hub. Scale bars, 20 µm. i, Anti-Ste and anti-Tubulin western blots of whole testis lysates from the indicated genotypes. j, Male fertility of indicated genotypes (number of progeny/male/7 days). Box plots show the median and IQR; whiskers denote 1.5× IQR (n = 20 males per genotype). P < 10−5 for Kruskal–Wallis test (one-way analysis of variance on ranks) comparing all genotypes and controls; Benjamini–Hochberg-corrected P values for all post hoc pairwise two-tailed Mann–Whitney tests are shown. Source numerical data and unprocessed blots are available in source data.
Consistent with the idea that Ste silencing requires Armitage in early germ cells, expression of an armi-gfp transgene under the control of nos-gal4 restored Ste repression in armi1/72.1 testes (Fig. 3f,g). In contrast, expression of the same rescue construct driven by bam-gal4 failed to rescue the armi mutant phenotype (Fig. 3h). Collectively, these data suggest that Su(Ste) piRNAs are produced in early germ cells by the phased biogenesis pathway.
Ste silencing requires both Aub and Ago3
In the phased biogenesis pathway, the products of Zuc-catalysed fragmentation of piRNA precursors are loaded into PIWI Argonaute proteins and mature to become piRNAs20,23. In fly testis, >80% of Su(Ste)-derived piRNAs in Aub and Ago3 are derived from the antisense precursor transcript54, suggesting that both proteins are programmed with antisense Su(Ste) piRNAs during phased biogenesis in GSC/SGs. Both Aub and Ago3 are required for repression of Ste mRNA in spermatocytes54 (Fig. 4a–e). Antisense Su(Ste)-piRNA-guided Aub and Ago3 are thus non-redundant in silencing Ste.

a–i, Representative images of Ste smRNA-FISH (red) in the testes in control y1w1118 testis (a) and in piRNA pathway mutant testes of the indicated genotypes: aub nos-driven RNAi (b); aub bam-drive RNAi (c); ago3 nos-driven RNAi (d); ago3 bam-driven RNAi (e); aub mutant (f); aub mutant, bam-driven rescue (g); ago3 mutant (h), ago3 bam-driven rescue (i). GSCs and early SGs are indicated by a yellow dotted line; cyan lines indicate spermatocytes. The asterisks indicate the hub. Scale bars, 20 µm. Experiments were repeated three times with similar results. These results show that Aub and Ago3 programmed with antisense Su(Ste) piRNAs are required for efficient repression of Ste. Note that, in fly testes and ovaries, transposon-derived piRNAs partition between Aub and Ago3: most antisense, phased, 1U-enriched piRNAs are bound to Aub, while most sense, ping-pong produced, 10A-biased piRNAs are loaded in Ago3 (refs. 4,54). Yet antisense, phased, 1U-enriched Su(Ste) piRNA are loaded into both Aug and Ago3 (ref. 54). Our analyses also show that piRNAs produced from the cleavage products of slicing of Ste transcripts by Su(Ste) piRNAs (that is, responder Ste piRNAs1) are most frequently loaded in Ago3 (>51 ± 8% in Ago3 versus >7 ± 2% in Aub).
We find that efficient repression of Stellate occurs when expression of Aub and Ago3 begins no later than the spermatogonial four-cell stage, that is, before Su(Ste) precursor transcription reaches its peak (Fig. 1a,b). Expressing a gfp-aub rescue transgene using bam-gal4 driver restored Ste repression in loss-of-function aubHN2/QC42 mutants (Fig. 4f,g). Ste was also silenced when a bam-driven FLAG-Myc-ago3 rescue transgene was expressed in ago3T2/T3 mutant males (Fig. 4h,i). We conclude that both Aub and Ago3 programmed with antisense Su(Ste) piRNAs are required for efficient repression of Ste.
1360 piRNAs trigger phased biogenesis of Su(Ste) piRNAs
Efficient repression of Ste requires production of Su(Ste) piRNAs days before Ste is first expressed (Fig. 1). Production of Su(Ste) piRNAs in early male germ cells requires Zuc and Armi, components of the phased piRNA biogenesis pathway (Figs. 2 and 3). Typically, phased piRNA biogenesis is initiated by a PIWI protein-catalysed, piRNA-directed slicing event that generates a long 5′-monophosphorylated 3ʹ-cleavage product (pre-pre-piRNA). The pre-pre-piRNA is then fragmented by Zuc into phased, tail-to-head pre-piRNAs19,20,25,66. But Ste piRNAs that could trigger phased fragmentation of Su(Ste) precursors are not produced by mothers (see below).
We propose that maternally inherited 1360/Hoppel transposon-derived piRNAs initiate phased production of Su(Ste) piRNAs that direct cleavage of the 1360/Hoppel sequence residing at the 5′ end of Su(Ste) precursor RNAs (Fig. 5a). Several observations support this idea: (1) transcription of Su(Ste) starts inside a 1360/Hoppel transposon insertion upstream of the sequence complementary to Ste (ref. 52); (2) ovaries contain abundant 1360/Hoppel transposon-derived piRNAs (~18,200 ± 400 per 10 pg total RNA); and (3) mothers deliver 1360/Hoppel piRNA to their male offspring via the oocyte32.

a, Model for initiation of phased biogenesis of Su(Ste) piRNAs by maternal 1360/Hoppel piRNAs. b, Frequency of 0–20-nt overlaps between Su(Ste) 5′-monophosphorylated long RNAs and 1360/Hoppel piRNAs on opposite genomic strands in control (y1w1118/Y; nos-gal4:VP16/TM2) testis. The standard score (number of standard deviations from the mean) and the corresponding P value (two-sided Z-test) of the 10-nt overlap (Z10) is shown. Data are for all possible permutations of two small RNA datasets and two 5′-monophosphorylated long RNA datasets (n = 2 × 2 = 4). c, Change in steady-state abundance of 5′-monophosphorylated long RNA datasets in nos>armiRNAi males (n = 2 for control; n = 2 for nos>armiRNAi) and in zucEY11457/− mutants (n = 3 for control; n = 3 for zucEY11457/−); P values are shown for two-sided Mann–Whitney test. d, Left: metaplot of piRNA 5′-end density along Su(Ste) long monophosphorylated RNAs in nos>dpp testis. Data are for all possible permutations of small RNA and 5′-monophosphorylated long RNA datasets (12 permutations; n = 3 for 5′-monophosphorylated long RNA datasets; n = 2 for small RNA datasets used to identify putative cleavage products among 5′-monophosphorylated long RNAs; n = 2 for small RNA datasets used to plot piRNA density). Black line indicates the median; grey area shows IQR. Right: autocorrelation analyses of the median piRNA density data in the metaplot. In b, c and d, only 5′-monophosphorylated long RNAs that span a Su(Ste) locus and whose 5′ ends lie in the 100 nt flanking the upstream 1360/Hoppel insertion were used for analyses (Supplementary Table 3; control testis: trial 1, 57 long RNAs; trial 2, 21 long RNAs; nos>dpp testis: trial 1, 33 long RNAs; trial 2, 49 long RNAs; trial 3, 42 long RNAs). Source numerical data are available in source data.
To test this model, we sequenced ≥ 200-nt long, 5′-monophosphorylated RNAs from adult testis to identify putative pre-pre-piRNAs. Like all Argonautes, PIWI proteins cleave their targets between nucleotides t10 and t11, the target nucleotides complementary to piRNA nucleotides g10 and g11. In the piRNA producing loci 42AB and petrel, the 5′ ends of long RNAs most frequently lay between nucleotides g10 and g11 of an antisense piRNA, supporting the idea that these monophosphorylated RNAs are bona fide pre-pre-piRNAs (Z10 = 5.1, P = 6 × 10‒7 for 42AB; Z10 = 8.5, P = 2.3 × 10‒17 for petrel; Extended Data Fig. 4a). As expected, we detected no antisense piRNAs overlapping with the 5′ ends of monophosphorylated RNAs from the genic loci nos, bam and bgcn, consistent with these RNAs being mRNA turnover intermediates (Extended Data Fig. 4a).
Among the Su(Ste)-derived, long, 5′-monophosphorylated RNAs overlapping the upstream 1360/Hoppel transposon insertion, their 5′ ends most often corresponded to the scissile phosphate predicted from a complementary antisense 1360/Hoppel piRNA (Z10 = 8, P = 10‒15; Fig. 5b). Our data therefore support the hypothesis that the majority of these monophosphorylated RNAs are pre-pre-piRNAs whose 5′ ends are made by 1360/Hoppel piRNA-directed cleavage. Consistent with the idea that long RNAs from 42AB, petrel and Su(Ste) are pre-pre-piRNAs processed by the phased biogenesis pathway, their steady-state abundance increased 1.7–5.4-fold when phased biogenesis in males was blocked in zucEY11457/− mutants or using nos-driven armiRNAi (Fig. 5c and Extended Data Fig. 4b). By contrast, the abundance of 5′-monophosphorylated RNAs from nos, bam and bgcn did not change in zucEY11457/− or nos>armiRNAi males (Extended Data Fig. 4b).
To examine Su(Ste) piRNA biogenesis in early male germ cells in more detail, we used nos>dpp males, in which SG overproliferate67,68,69. Among the ≥200-nt long, 5′-monophosphorylated RNAs from nos>dpp testis, we identified putative Su(Ste) pre-pre-piRNAs spanning both the 1360/Hoppel and Ste-derived sequences that could have been produced by 1360/Hoppel piRNA-guided slicing (Fig. 5d). The 5′ ends of Su(Ste) piRNAs concentrated in periodic peaks starting from Su(Ste) pre-pre-piRNA 5′ termini (Fig. 5d). Consistent with Zuc-catalysed fragmentation of pre-pre-piRNAs into tail-to-head pre-piRNAs, autocorrelation analyses showed that most piRNA 5′ ends lay at regular intervals, ~25–26 nt apart from each other (Fig. 5d). For Su(Ste)-derived pre-pre-piRNAs whose 5′ ends were in the last 100 nt of the 1360/Hoppel sequence, most Su(Ste) piRNA 5′ ends occurred at ~25–27-nt intervals extending as far as ≥100 nt into the region of the Su(Ste) transcript that is antisense to Ste (Fig. 5d). Together, these data suggest that 1360/Hoppel piRNAs slice Su(Ste) precursors to initiate 5′-to-3′ phased production of Su(Ste) piRNAs capable of silencing Ste mRNA.
Su(Ste) piRNAs made in XXY females silence Ste in progeny
The remarkable stability of Argonaute-protected small RNAs70,71 probably underlies the intergenerational inheritance of transposon-targeting piRNAs in animals with maternally deposited germ plasm. Similarly, our model assumes that piRNA•PIWI complexes deposited by mothers can cleave complementary RNAs in the germline of their sons. To experimentally test this assumption, we used XXY female flies to artificially deposit Su(Ste) piRNAs in oocytes. Y chromosome-encoded Su(Ste) piRNA precursors and Su(Ste) piRNAs were detected in XXY (2,700 ± 80 piRNAs per 10 pg total RNA) but not XX ovaries (30 ± 30 piRNAs per 10 pg total RNA; Fig. 6a, Extended Data Fig. 5 and Supplementary Table 2). These maternally produced Su(Ste) piRNAs were able to repress a gfp-Ste transgene in XXY females (Extended Data Fig. 6).

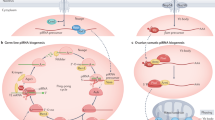
a, Length profile of Su(Ste)-derived (Supplementary Table 3) small RNAs in XX and XXY ovaries. The data are the mean from three independent biological samples. b–e, Representative images of smRNA-FISH for Ste (red) and antisense Su(Ste) (green) in the testes of control (b(i) y1w1118/Y; nos-gal4:VP16/TM2) and nos>armiRNAi (c(i)) sons from XX mothers, and in testes of control (d(i)) and nos>armiRNAi (e(i)) sons from XXY mothers. The asterisks indicate the hub. Red, Ste RNA; green, antisense Su(Ste) piRNA precursor; blue, DAPI. Magnified view of Su(Ste) piRNA precursor in situ hybridization signal at the apical tip of the testis is shown in b(ii), c(ii), d(ii) and e(ii). Arrowheads point to nuclear Su(Ste) transcripts; arrow points to cytoplasmic Su(Ste) RNA. Scale bars, 20 µm (b(i)–e(i)) and 5 µm (b(ii)–e(ii)). Experiments were repeated three times with similar results. Source numerical data are available in source data. f, Top: model of developmental regulation of Su(Ste) piRNA biogenesis and Ste repression in males. Su(Ste) piRNA precursors are transcribed in early germ cells (GSCs and SGs), where they are processed to produce antisense Su(Ste) piRNAs by Armi- and Zuc-dependent, phased fragmentation. These Su(Ste) piRNAs are loaded into Aub and Ago3, which are later used in spermatocytes to cleave Ste transcripts. Phased fragmentation of Su(Ste) piRNA precursor is initiated by 1360/Hoppel piRNAs deposited by XX mothers. Bottom: Su(Ste) piRNAs deposited by XXY mother can replace the need for Armi- and Zuc-dependent phased piRNA production.
Strikingly, when Su(Ste) piRNA biogenesis was blocked in sons, maternal Su(Ste) piRNAs from XXY mother were sufficient to silence Ste in the testis: unlike nos>armiRNAi males from XX mothers, nos>armiRNAi sons derived from XXY females effectively repressed Ste (Fig. 6b–e and Extended Data Figs. 7 and 8). We conclude that maternal deposition of Su(Ste) piRNAs by XXY mothers suffices to silence Ste mRNA and bypasses the requirement for phased piRNA production pathway in early male germ cells.
Discussion
The piRNA pathway is required for production of functional germ cells in animals. In species like Drosophila, whose germline is specified by maternally inherited determinants, the oocyte germ plasm contains piRNA•PIWI complexes that instruct their progeny to silence transposons antisense to the inherited piRNAs. Intergenerational continuity of the piRNA pathway in these species therefore relies on the continued passage of information through the germline. Such maternal inheritance is not possible for Y chromosome-encoded piRNAs, as females lack a Y chromosome. How can mothers instruct their sons to make piRNAs from precursors on the Y chromosome? Our data suggest that the D. melanogaster male germline relies on maternally deposited, transposon-derived piRNAs to trigger production of Su(Ste) piRNAs antisense to Ste (Fig. 6f). The production of such Ste-silencing piRNAs is possible because piRNA-directed cleavage of an RNA triggers the production of tail-to-head strings of piRNA via the phased piRNA biogenesis pathway. This model explains how fly males make piRNAs for which no homologous piRNA guides can be deposited by mothers. Our study also reveals that abundant Su(Ste) piRNAs are produced before the onset of transcription of their target, Ste. Such spatiotemporal separation may be required for effective repression of Ste mRNA.
In the fly germline, the proteins Rhino and Kipferl bind heterochromatic piRNA-producing loci and initiate transcription of precursor transcripts from both genomic strands57,72,73,74. Promoter-independent, RNA polymerase II transcription of these dual-strand piRNA clusters occurs throughout each locus, ignoring splice sites and polyadenylation sequences75,76,77,78. This atypical transcription strategy maximizes production of transposon-targeting piRNAs. Su(Ste) piRNA biogenesis in the male germline is unlikely to involve such non-canonical transcription of Su(Ste). First, our smFISH experiments detected Su(Ste) transcripts from only one genomic strand. Second, loss of rhi in fly males has no effect on Ste silencing56.
Taken together, our data suggest that the fly male germline has evolved a strategy that uses maternally supplied, transposon-derived piRNAs to generate Y chromosome-derived, Su(Ste) piRNAs that silence the selfish genetic element Ste. This strategy allows fly females to instruct their sons to produce piRNAs from sequences absent from the maternal genome. We speculate that this same mechanism may be used by mothers to protect their sons from selfish DNA in other animal species that deposit germline determinants in oocytes.
Methods
Statistics and reproducibility
No statistical method was used to determine the sample size. For all biological samples, the maximum possible sample size (n = 3–90) was chosen for each type of data ensuring that variability arising from all accountable sources was incorporated in the analyses (day of data collection, reagent lots, and experimenter). No data were excluded from the analyses. The experiments were not randomized, because this study did not involve treatment or exposure of animals to any agent. Instead, the goal of this work was to compare untreated wild-type/control flies and untreated mutant flies: all wild-type animals were compared with all mutant animals. The Investigators were not blinded to allocation during experiments and outcome assessment. Blinding was not performed during data collection, because methods used for data acquisition (smFISH, western blotting, qRT–PCR and high-throughput sequencing) are not influenced by the experimenter’s knowledge of the fly genotype. Blinding was not performed during data analyses, because analyses were performed with the same automated algorithms and programming code. During analyses, wild-type control and mutant datasets are also easily identified and are directly compared with another.
Fly husbandry and strains used
Flies (D. melanogaster strain w1118; 0–7 days old) were raised in standard Bloomington medium at 25 °C. The following stocks were obtained from the Bloomington Stock Center: C(1)RM/C(X:Y)y1f1w1, armi1, armi72.1, aubHN2, aubQC42, zucEY11457, Df(2L)BSC323, nos-gal4:VP16, bam-gal4:VP16, UAS-flag3-myc6-ago3 (ref. 80), UAS-gfp-aub, UAS-armi-gfp, UAS-dpp, and RNAi lines for armi: TRIP.GL00254, aub: TRIP.GL00076, ago3: TRIP.HMC02938, vasa: TRIP.HMS00373, zuc: TRIP.GL00111. To generate UAS-gfp-Ste (SteXh:CG42398), cDNAs was synthetized (Invitrogen, sequence is provided in Supplementary Table 4), and inserted into UAST-gfp vector, after the gfp cDNA cassette, between BglII and XbaI sites. Transgenic lines carrying these transgenes were generated at BestGene.
To assay male fertility, a single male of indicated genotype (0–1 days old) was crossed to three y1w1118 virgin females (0–2 days old) at room temperature. Flies were removed after 7 days, and the number of progeny was scored.
Western blots
Testes (20 pairs per sample) were dissected and rinsed twice with 0.1 M phosphate buffer saline pH 7.2 (PBS), snap frozen and kept at −80 °C until use. Testes were homogenized in 100 µl (PBS), supplied with c0mplete protease inhibitor + ethylenediaminetetraacetic acid (Roche), and mixed with 100 µl of 2× Laemmli Sample Buffer (Bio-Rad). Cleared lysates were separated on a 12% Tris-glycine gel (Thermo Scientific) and transferred onto polyvinylidene fluoride membrane (Immobilon-P, Millipore). Mouse anti-α-Tubulin (clone 4.3; 1:3,000) (Walsh 1984) was obtained from the Developmental Studies Hybridoma Bank. The generation of polyclonal anti-Ste antibody (used at 1:10,000) was outsourced to Covance and was produced by immunizing guinea pigs with KLH-conjugated Ac-KPVIDSSSGLLYGDEKKWC (53–70 amino acids of Ste). Horseradish peroxidase-conjugated goat anti-mouse IgG (115-035-003; 1:10,000; Jackson ImmunoResearch Laboratories) and anti-guinea pig IgG (106-035-003; 1:10,000; Jackson ImmunoResearch Laboratories) secondary antibodies were used. The signals were detected by Pierce ECL Western Blotting Substrate enhanced chemiluminescence system (Thermo Scientific).
smRNA-FISH
smRNA-FISH was conducted as described61. Testes from 2–3-day-old flies were dissected in 1× PBS, fixed in 4% formaldehyde in 1× PBS for 30 min, washed in PBS and permeabilized in 70% ethanol overnight at 4 °C. The following day, testes were rinsed with wash buffer (2× saline-sodium citrate and 10% formamide) and hybridized overnight at 37 °C in hybridization buffer (2× saline-sodium citrate, 10% dextran sulfate (Sigma, D8906), 1 mg ml−1 Escherichia coli tRNA (Sigma, R8759), 2 mM vanadyl ribonucleoside complex (NEB, S142), 0.5% bovine serum albumin (Ambion, AM2618) and 10% formamide). Following hybridization, samples were washed three times in wash buffer for 20 min each at 37 °C and mounted in VECTASHIELD with 4′,6-diamidino-2-phenylindole (DAPI, Vector Labs). Fluorescently labelled probes were added to the hybridization buffer to a final concentration of 100 nM. DNA oligo probes to detect Ste and Su(Ste) RNA were conjugated with Quasar 570, Cy3 or Cy5 fluorophores (Biosearch Technologies and IDT; for probe information, see Supplementary Table 5). Images were acquired using an upright Leica TCS SP8 confocal microscope with a 63× oil immersion objective lens (numerical aperture 1.4) and processed using ImageJ.
qRT–PCR
Total RNA was isolated by Direct-zol RNA miniprep kit (Zymo Research) from biological triplicates of XY (100 testes per sample), XX or XXY gonads (60 ovaries per sample). Complementary DNA was generated by SuperScript III Reverse Transcriptase (Invitrogen) with random hexamer primers. qPCR of technical triplicates was performed using Power SYBR Green reagent (Applied Biosystems) and the following primer pairs. Gapdh: TAA ATT CGA CTC GAC TCA CGG T and CTC CAC CAC ATA CTC GGC TC, act5C: AAG TTG CTG CTC TGG TTG TCG and GCC ACA CGC AGC TCA TTG AG, Su(Ste): TTC CGA AGT CAA GCG CTT CAA TG and GGA ATC TGT TTA ATT GCA ACA AC. Ct values were normalized to Gapdh by the \({2}^{-\Delta \Delta {C}_{{\rm{t}}}}\) method81. When calculating ∆Ct and ∆∆Ct, standard deviations (σ) were propagated in Microsoft Excel 2013 using the formula \({\sigma }_{x}=\sqrt{{\sigma }_{y}^{2}+{\sigma }_{z}^{2}}\).
TaqMan small RNA analysis
The abundance of the following piRNAs were quantified by TaqMan small RNA custom assays (Thermo Fisher Scientific): Su(Ste)-4 piRNA (target sequence: UCU CAU CGU CGU AGA ACA AGC CCG A), the most abundant Su(Ste) piRNA54: piR-dme-1643 piRNA (piRBase nomenclature), target sequence: (TAA AGC GTT GTT TTG TGC TAT ACC C), a piRNA we found to be highly abundant in the ovary based on analysis of earlier small RNA sequencing data, and 2S ribosomal RNA (rRNA) (target sequence: UGC UUG GAC UAC AUA UGG UUG AGG GUU GUA), which we utilized in this study as control. Total RNA was isolated from biological triplicates of XX and XXY ovaries (60 per sample) by Direct-zol miniprep kit (Zymo Research). Reverse transcription and qPCR were performed following the manufacturer’s protocol using TaqMan MicroRNA Reverse Transcription Kit, and TaqMan Universal PCR Master Mix II, No UNG (Thermo Fisher Scientific). qPCRs were performed in technical triplicates with the appropriate controls. Ct values were normalized to 2S rRNA levels by the \({2}^{-\Delta \Delta {C}_{{\rm{t}}}}\) method81. When calculating ∆Ct and ∆∆Ct, standard deviations (σ) were propagated in Microsoft Excel 2013 using the formula \({\sigma }_{x}=\sqrt{{\sigma }_{y}^{2}+{\sigma }_{z}^{2}}\).
Small RNA-seq library preparation and analyses
Total RNA from fly ovaries or testis was extracted using the mirVana miRNA isolation kit (Thermo Fisher, AM1560). Small RNA libraries were constructed as described82 with modifications. Briefly, before library preparation, a spike-in RNA mix, an equimolar mix of six synthetic 5′-phosphorylated RNA oligonucleotides (/phos/UGC UAG UCU UAU CGA CCU CCU CAU AG, /phos/UGC UAG UCU UCG AUA CCU CCU CAU AG, /phos/UGC UAG UCU UGU CAC GAA CCU CAU AG, /phos/UGC UAG UUA UCG ACC UUC AUA G, /phos/UGC UAG UUC GAU ACC UUC AUA G, /phos/UGC UAG UUG UCA CGA AUC AUA G), was added to each RNA sample to enable absolute quantification of small RNAs (Supplementary Table 6). To reduce ligation bias and eliminate PCR duplicates, the 3′ and 5′ adaptors both contained nine random nucleotides at their 5′ or 3′ ends, respectively (see below) and 3′ adaptor ligation reactions contained 25% (w/v) PEG-8000 final concentration (f.c.). Total RNA was run through a 15% denaturing urea–polyacrylamide gel (National Diagnostics) to isolate 15–29-nt small RNAs and remove the 30-nt 2S rRNA. After overnight elution in 0.4 M NaCl followed by ethanol precipitation, small RNAs were oxidized (to clone only 2′-O-methylated siRNAs and piRNAs) in 40 µl 200 mM sodium periodate, 30 mM borax, 30 mM boric acid (pH 8.6) at 25 °C for 30 min. After ethanol precipitation, small RNAs were ligated to 25 pmol 3′ DNA adapter with adenylated 5′ and dideoxycytosine-blocked 3′ ends (/rApp/NNN GTC NNN TAG NNN TGG AAT TCT CGG GTG CCA AGG/ddC/) in 30 µl 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM dithiothreitol (DTT) and 25% (w/v) PEG-8000 (NEB) with 600 U homemade T4 Rnl2tr K227Q at 16 °C overnight. After ethanol precipitation, the 50–90-nt (14–54-nt small RNA + 36-nt 3′ unique molecular identifier adapter) 3′-ligated product was purified from a 15% denaturing urea–polyacrylamide gel (National Diagnostics). After overnight elution in 0.4 M NaCl followed by ethanol precipitation, the 3′-ligated product was denatured in 13 µl water at 90 °C for 60 s, 1 µl 10 µM anti-2S oligo (TAC AAC CCT CAA CCA TAT GTA GTC CAA GCA-/3′ C3 Spacer/; to suppress the ligation of 2S rRNA) and 1 µl 50 µM RT primer (CCT TGG CAC CCG AGA ATT CCA; to suppress the formation of 5′-adapter:3′-adapter dimers) were added and annealed at 65 °C for 5 min. The resulting mix was then ligated to a mixed pool of equimolar amount of two 5′ RNA adapters (to increase nucleotide diversity at the 5′ end of the sequencing read: GUU CAG AGU UCU ACA GUC CGA CGA UCN NNC GAN NNU CAN NN and GUU CAG AGU UCU ACA GUC CGA CGA UCN NNA UCN NNA GUN NN) in 20 µl 50 mM Tris–HCl (pH 7.8), 10 mM MgCl2, 10 mM DTT, 1 mM ATP with 20 U of T4 RNA ligase (Thermo Fisher, EL0021) at 25 °C for 2 h. The ligated product was precipitated with ethanol, and cDNA synthesis was performed in 20 µl at 42 °C for 1 h using AMV reverse transcriptase (NEB, M0277) and 5 µl RT reaction was amplified in 25 µl using AccuPrime Pfx DNA polymerase (Thermo Fisher, 12344024; 95 °C for 2 min, 15 cycles of: 95 °C for 15 s, 65 °C for 30 s, 68 °C for 15 s; forward primer: AAT GAT ACG GCG ACC ACC GAG ATC TAC ACG TTC AGA GTT CTA CAG TCC GA; reverse primer: CAA GCA GAA GAC GGC ATA CGA GAT XXX XXX GTG ACT GGA GTT CCT TGG CAC CCG AGA ATT CCA, where XXXXXX represents the 6-nt sequencing barcode). Finally, the PCR product was purified in a 2% agarose gel. Small RNA-seq libraries samples were sequenced using a NextSeq 550 (Illumina) to obtain 79 nt, single-end reads.
The 3′ adapter (TGG AAT TCT CGG GTG CCA AGG) was removed with fastx toolkit (v0.0.14), PCR duplicates were eliminated as described83, and rRNA matching reads were removed with bowtie (parameter -v 1; v1.0.0) against D. melanogaster set in SILVA database84. Deduplicated and filtered data were analysed with Tailor85 to account for non-templated tailing of small RNAs. Sequences of synthetic RNA spike-in oligonucleotides were identified allowing no mismatches with using bowtie (parameter -v 0; v1.0.0), and the absolute abundance of small RNAs calculated. The background for Z10 calculation was all displayed data except position 10.
RNA-seq library preparation and analyses
Total RNA from sorted germ cells was extracted using the mirVana miRNA isolation kit (ThermoFisher, AM1560). Before library preparation, to remove rRNA, 1 µg total RNA was hybridized in 10 µl to a pool of 186 rRNA antisense oligos (0.05 µm f.c. each) in 10 mM Tris–HCl (pH 7.4), 20 mM NaCl by heating the mixture to 95 °C, cooling at −0.1 °C s−1 to 22 °C, and incubating at 22 °C for 5 min. RNase H (10 U; Lucigen, H39500) was added and the mixture incubated at 45 °C for 30 min in 20 µl containing 50 mM Tris–HCl (pH 7.4), 100 mM NaCl and 20 mM MgCl2. The reaction volume was adjusted to 50 µl with 1× TURBO DNase buffer (ThermoFisher, AM2238) and then incubated with 4 U TURBO DNase (ThermoFisher, AM2238) for 20 min at 37 °C. Next, RNA was purified using RNA Clean & Concentrator-5 (Zymo Research, R1016) to retain ≥200-nt RNAs, followed by the stranded, dUTP-based RNA-seq protocol described in ref. 86 using adapters with unique molecular identifiers from ref. 83. RNA-seq libraries were sequenced using a NextSeq 550 (Illumina) to obtain 79 + 79 nt, paired-end reads.
RNA-seq analysis was performed using piPipes for genomic alignment87. Briefly, before starting piPipes, sequences were reformatted to extract unique molecular identifiers83. The reformatted reads were then aligned to rRNA using bowtie2 (v2.2.0). Unaligned reads were mapped to the dm6 assembly using STAR (v2.3.1), and PCR duplicates removed83. Transcript abundance was calculated using StringTie (v1.3.4). Differential expression analysis was performed using DESeq2 (v1.18.1).
Cloning and sequencing of 5′-monophosphorylated long RNAs
Total RNA from fly ovaries or testis was extracted using mirVana miRNA isolation kit (ThermoFisher, AM1560) and used to prepare a library of 5′-monophosphorylated long RNAs as described82 with modifications. Briefly, to deplete rRNA, 1 µg total RNA was hybridized in 10 µl to a pool of rRNA antisense oligos (0.05 µm f.c. each) in 10 mM Tris–HCl (pH 7.4), 20 mM NaCl by heating the mixture to 95 °C, cooling it at −0.1 °C s−1 to 22 °C, and incubating at 22 °C for 5 min. RNase H (10 U; Lucigen, H39500) was added and the mixture incubated at 45 °C for 30 min in 20 µl containing 50 mM Tris–HCl (pH 7.4), 100 mM NaCl and 20 mM MgCl2. The reaction volume was adjusted to 50 µl with 1× TURBO DNase buffer (ThermoFisher, AM2238) and then incubated with 4 U TURBO DNase (ThermoFisher, AM2238) for 20 min at 37 °C. Next, RNA was purified using RNA Clean & Concentrator-5 (Zymo Research, R1016) to retain ≥200-nt fragments. RNA was then ligated to a mixed pool of equimolar amounts of two 5′ RNA adapters (to increase nucleotide diversity at the 5′ end of the sequencing read: GUU CAG AGU UCU ACA GUC CGA CGA UCN NNC GAN NNU CAN NN and GUU CAG AGU UCU ACA GUC CGA CGA UCN NNA UCN NNA GUN NN) in 20 µl of 50 mM Tris–HCl (pH 7.8), 10 mM MgCl2, 10 mM DTT and 1 mM ATP with 60 U of High Concentration T4 RNA ligase (NEB, M0437M) at 16 °C overnight. The ligated product was isolated using RNA Clean & Concentrator-5 (Zymo Research, R1016) to retain ≥200-nt RNAs and reverse transcribed in 25 µl with 50 pmol RT primer (GCA CCC GAG AAT TCC ANN NNN NNN) using SuperScript III (ThermoFisher, 18080093). After purification with 50 µl Ampure XP beads (Beckman Coulter, A63880), cDNA was PCR amplified using NEBNext High-Fidelity (NEB, M0541; 98 °C for 30 s; four cycles of: 98 °C for 10 s, 59 °C for 30 s, 72 °C for 12 s; six cycles of: 98 °C for 10 s, 68 °C for 10 s, 72 °C for 12 s; 72 °C for 3 min; with the following primers: CTA CAC GTT CAG AGT TCT ACA GTC CGA and GCC TTG GCA CCC GAG AAT TCC A). PCR products between 200 bp and 400 bp were isolated with a 1% agarose gel, purified with QIAquick Gel Extraction Kit (Qiagen, 28706), and amplified again with NEBNext High-Fidelity (NEB, M0541; 98 °C for 30 s; 3 cycles of: 98 °C for 10 s, 68 °C for 30 s, 72 °C for 14 s; six cycles of: 98 °C for 10 s, 72 °C for 14 s; 72 °C for 3 min; forward primer: AAT GAT ACG GCG ACC ACC GAG ATC TAC ACG TTC AGA GTT CTA CAG TCC GA; reverse primer: CAA GCA GAA GAC GGC ATA CGA GAT XXX XXX GTG ACT GGA GTT CCT TGG CAC CCG AGA ATT CCA, where XXXXXX represents the 6-nt sequencing barcode). The PCR product was purified in a 1% agarose gel and sequenced using a NextSeq 550 to obtain 79 + 79 nt, paired-end reads.
Sequencing data was aligned to the fly genome (dm6) with piPipes87. Briefly, before starting piPipes, sequences were reformatted to remove the degenerate portion of the 5′ adapter (nucleotides 1–15 of read 1). The reformatted reads were then aligned to fly rRNA using bowtie2 (v2.2.0). Unaligned reads were mapped to the fly genome (dm6) using STAR (v2.3.1), alignments with soft clipping of ends were removed with SAMtools (v1.0.0), and reads with the same 5′ end were merged to represent a single 5′-monophosphorylated RNA species.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data generated in this study have been deposited in the National Center for Biotechnology Information Short Read Archive database under accession code PRJNA879723. Fly genome sequence and annotation (build dm6/BDGP6.22 release 98) used in this study were downloaded from Ensembl at ftp://ftp.ensembl.org/pub/release-98/fasta/drosophila_melanogaster/ and ftp://ftp.ensembl.org/pub/release-98/gtf/drosophila_melanogaster/; fly rRNA sequences were downloaded from SILVA rRNA database at https://www.arb-silva.de/. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding authors upon request.
Code availability
Code used in this work is deposited at https://github.com/ildargv/Venkei_et_al_2023.
References
Ozata, D. M., Gainetdinov, I., Zoch, A., O’Carroll, D. & Zamore, P. D. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20, 89–108 (2019).
Girard, A., Sachidanandam, R., Hannon, G. J. & Carmell, M. A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202 (2006).
Aravin, A. A., Hannon, G. J. & Brennecke, J. The Piwi–piRNA pathway provides an adaptive defense in the transposon arms race. Science 318, 761–764 (2007).
Brennecke, J. et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103 (2007).
Das, P. P. et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol. Cell 31, 79–90 (2008).
Wu, P. H. et al. The evolutionarily conserved piRNA-producing locus pi6 is required for male mouse fertility. Nat. Genet. 52, 728–739 (2020).
Chen, P. et al. piRNA-mediated gene regulation and adaptation to sex-specific transposon expression in D. melanogaster male germline. Genes Dev. 35, 914–935 (2021).
Choi, H., Wang, Z. & Dean, J. Sperm acrosome overgrowth and infertility in mice lacking chromosome 18 pachytene piRNA. PLoS Genet. 17, e1009485 (2021).
Aravin, A. et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207 (2006).
Grivna, S. T., Beyret, E., Wang, Z. & Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 20, 1709–1714 (2006).
Lau, N. C. et al. Characterization of the piRNA complex from rat testes. Science 313, 363–367 (2006).
Vagin, V. V. et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324 (2006).
Pal-Bhadra, M. et al. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303, 669–672 (2004).
Saito, K. et al. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev. 20, 2214–2222 (2006).
Sienski, G., Donertas, D. & Brennecke, J. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151, 964–980 (2012).
Huang, H. et al. AGO3 slicer activity regulates mitochondria-nuage localization of Armitage and piRNA amplification. J. Cell Biol. 206, 217–230 (2014).
Le Thomas, A. et al. Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev. 28, 1667–1680 (2014).
Post, C., Clark, J. P., Sytnikova, Y. A., Chirn, G. W. & Lau, N. C. The capacity of target silencing by Drosophila PIWI and piRNAs. RNA 20, 1977–1986 (2014).
Han, B. W., Wang, W., Li, C., Weng, Z. & Zamore, P. D. Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348, 817–821 (2015).
Mohn, F., Handler, D. & Brennecke, J. Noncoding RNA. piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348, 812–817 (2015).
Senti, K. A., Jurczak, D., Sachidanandam, R. & Brennecke, J. piRNA-guided slicing of transposon transcripts enforces their transcriptional silencing via specifying the nuclear piRNA repertoire. Genes Dev. 29, 1747–1762 (2015).
Wang, W. et al. Slicing and binding by Ago3 or Aub trigger Piwi-bound piRNA production by distinct mechanisms. Mol. Cell 59, 819–830 (2015).
Gainetdinov, I., Colpan, C., Arif, A., Cecchini, K. & Zamore, P. D. A single mechanism of biogenesis, initiated and directed by PIWI proteins, explains piRNA production in most animals. Mol. Cell 71, 775–790 e775 (2018).
Gunawardane, L. S. et al. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315, 1587–1590 (2007).
Homolka, D. et al. PIWI slicing and RNA elements in precursors instruct directional primary piRNA biogenesis. Cell Rep. 12, 418–428 (2015).
Yang, Z. et al. PIWI slicing and EXD1 drive biogenesis of nuclear piRNAs from cytosolic targets of the mouse piRNA pathway. Mol. Cell 61, 138–152 (2016).
Pane, A., Wehr, K. & Schupbach, T. Zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Dev. Cell 12, 851–862 (2007).
Ge, D. T. et al. The RNA-binding ATPase, Armitage, couples piRNA amplification in Nuage to phased piRNA production on mitochondria. Mol. Cell 74, 982–995 e986 (2019).
Munafo, M. et al. Daedalus and Gasz recruit Armitage to mitochondria, bringing piRNA precursors to the biogenesis machinery. Genes Dev. 33, 844–856 (2019).
Yamashiro, H. et al. Armitage determines Piwi-piRISC processing from precursor formation and quality control to inter-organelle translocation. EMBO Rep. 21, e48769 (2020).
Blumenstiel, J. P. & Hartl, D. L. Evidence for maternally transmitted small interfering RNA in the repression of transposition in Drosophila virilis. Proc. Natl Acad. Sci. USA 102, 15965–15970 (2005).
Brennecke, J. et al. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 322, 1387–1392 (2008).
de Vanssay, A. et al. Paramutation in Drosophila linked to emergence of a piRNA-producing locus. Nature 490, 112–115 (2012).
de Albuquerque, B. F., Placentino, M. & Ketting, R. F. Maternal piRNAs are essential for germline development following de novo establishment of endo-siRNAs in Caenorhabditis elegans. Dev. Cell 34, 448–456 (2015).
Kidwell, M. G., Kidwell, J. F. & Nei, M. A case of high rate of spontaneous mutation affecting viability in Drosophila melanogaster. Genetics 75, 133–153 (1973).
Kidwell, M. G. & Kidwell, J. F. Selection for male recombination in Drosophila melanogaster. Genetics 84, 333–351 (1976).
Kidwell, M. G., Kidwell, J. F. & Sved, J. A. Hybrid dysgenesis in Drosophila melanogaster: a syndrome of aberrant traits including mutation, sterility and male recombination. Genetics 86, 813–833 (1977).
Ronsseray, S., Anxolabehere, D. & Periquet, G. Hybrid dysgenesis in Drosophila melanogaster: influence of temperature on cytotype determination in the P–M system. Mol. Gen. Genet. 196, 17–23 (1984).
Khurana, J. S. et al. Adaptation to P element transposon invasion in Drosophila melanogaster. Cell 147, 1551–1563 (2011).
Teixeira, F. K. et al. piRNA-mediated regulation of transposon alternative splicing in the soma and germ line. Nature https://doi.org/10.1038/nature25018 (2017).
Wakisaka, K. T., Ichiyanagi, K., Ohno, S. & Itoh, M. Diversity of P-element piRNA production among M′ and Q strains and its association with P–M hybrid dysgenesis in Drosophila melanogaster. Mobile DNA 8, 13 (2017).
Moon, S. et al. A robust transposon-endogenizing response from germline stem cells. Dev. Cell 47, 660–671.e663 (2018).
Srivastav, S. P. et al. Har-P, a short P-element variant, weaponizes P-transposase to severely impair Drosophila development. eLife 8, e49948 (2019).
Hardy, R. W. et al. Cytogenetic analysis of a segment of the Y chromosome of Drosophila melanogaster. Genetics 107, 591–610 (1984).
Livak, K. J. Organization and mapping of a sequence on the Drosophila melanogaster X and Y chromosomes that is transcribed during spermatogenesis. Genetics 107, 611–634 (1984).
McKee, B. D. & Satter, M. T. Structure of the Y chromosomal Su(Ste) locus in Drosophila melanogaster and evidence for localized recombination among repeats. Genetics 142, 149–161 (1996).
Kalmykova, A. I., Dobritsa, A. A. & Gvozdev, V. A. Su(Ste) diverged tandem repeats in a Y chromosome of Drosophila melanogaster are transcribed and variously processed. Genetics 148, 243–249 (1998).
Belloni, M., Tritto, P., Bozzetti, M. P., Palumbo, G. & Robbins, L. G. Does Stellate cause meiotic drive in Drosophila melanogaster? Genetics 161, 1551–1559 (2002).
Bozzetti, M. P. et al. The Ste locus, a component of the parasitic cry-Ste system of Drosophila melanogaster, encodes a protein that forms crystals in primary spermatocytes and mimics properties of the beta subunit of casein kinase 2. Proc. Natl Acad. Sci. USA 92, 6067–6071 (1995).
Aravin, A. A. et al. Dissection of a natural RNA silencing process in the Drosophila melanogaster germ line. Mol. Cell. Biol. 24, 6742–6750 (2004).
Aravin, A. A. et al. The small RNA profile during Drosophila melanogaster development. Dev. Cell 5, 337–350 (2003).
Aravin, A. A. et al. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 11, 1017–1027 (2001).
Nishida, K. M. et al. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA 13, 1911–1922 (2007).
Nagao, A. et al. Biogenesis pathways of piRNAs loaded onto AGO3 in the Drosophila testis. RNA 16, 2503–2515 (2010).
Quenerch’du, E., Anand, A. & Kai, T. The piRNA pathway is developmentally regulated during spermatogenesis in Drosophila. RNA 22, 1044–1054 (2016).
Chen, P., Luo, Y. & Aravin, A. A. RDC complex executes a dynamic piRNA program during Drosophila spermatogenesis to safeguard male fertility. PLoS Genet. 17, e1009591 (2021).
Klattenhoff, C. et al. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 138, 1137–1149 (2009).
Ryazansky, S. S. et al. RNA helicase Spn-E is required to maintain Aub and AGO3 protein levels for piRNA silencing in the germline of Drosophila. Eur. J. Cell Biol. 95, 311–322 (2016).
Kibanov, M. V. et al. A novel organelle, the piNG-body, in the nuage of Drosophila male germ cells is associated with piRNA-mediated gene silencing. Mol. Biol. Cell 22, 3410–3419 (2011).
Raj, A. & Tyagi, S. Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol. 472, 365–386 (2010).
Fingerhut, J. M., Moran, J. V. & Yamashita, Y. M. Satellite DNA-containing gigantic introns in a unique gene expression program during Drosophila spermatogenesis. PLoS Genet. 15, e1008028 (2019).
Pena, J. T. et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat. Methods 6, 139–141 (2009).
Yamaguchi, S. et al. Structure of the Dicer-2-R2D2 heterodimer bound to a small RNA duplex. Nature 607, 393–398 (2022).
Van Doren, M., Williamson, A. L. & Lehmann, R. Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr. Biol. 8, 243–246 (1998).
Xiol, J. et al. RNA clamping by vasa assembles a piRNA amplifier complex on transposon transcripts. Cell 157, 1698–1711 (2014).
Wang, W. et al. The initial uridine of primary piRNAs does not create the tenth adenine that is the hallmark of secondary piRNAs. Mol. Cell 56, 708–716 (2014).
Schulz, C. et al. A misexpression screen reveals effects of bag-of-marbles and TGFβ class signaling on the Drosophila male germ-line stem cell lineage. Genetics 167, 707–723 (2004).
Kawase, E., Wong, M. D., Ding, B. C. & Xie, T. Gbb/Bmp signaling is essential for maintaining germline stem cells and for repressing bam transcription in the Drosophila testis. Development 131, 1365–1375 (2004).
Shivdasani, A. A. & Ingham, P. W. Regulation of stem cell maintenance and transit amplifying cell proliferation by tgfβ signaling in Drosophila spermatogenesis. Curr. Biol. 13, 2065–2072 (2003).
Reichholf, B. et al. Time-resolved small RNA sequencing unravels the molecular principles of microRNA homeostasis. Mol. Cell 75, 756–768 e757 (2019).
Kingston, E. R. & Bartel, D. P. Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 29, 1777–1790 (2019).
Mohn, F., Sienski, G., Handler, D. & Brennecke, J. The rhino-deadlock-cutoff complex licenses non-canonical transcription of dual-strand piRNA clusters in Drosophila. Cell 157, 1364–1379 (2014).
Pane, A., Jiang, P., Zhao, D. Y., Singh, M. & Schupbach, T. The Cutoff protein regulates piRNA cluster expression and piRNA production in the Drosophila germline. EMBO J. 30, 4601–4615 (2011).
Baumgartner, L., Handler, D., Platzer, S., Duchek, P. & Brennecke, J. The Drosophila ZAD zinc finger protein Kipferl guides Rhino to piRNA clusters. eLife 11, e80067 (2022).
Chen, Y. A. et al. Cutoff suppresses RNA polymerase II termination to ensure expression of piRNA precursors. Mol. Cell 63, 97–109 (2016).
Andersen, P. R., Tirian, L., Vunjak, M. & Brennecke, J. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549, 54–59 (2017).
Zhang, Z. et al. The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157, 1353–1363 (2014).
Hur, J. K. et al. Splicing-independent loading of TREX on nascent RNA is required for efficient expression of dual-strand piRNA clusters in Drosophila. Genes Dev. 30, 840–855 (2016).
Desset, S., Buchon, N., Meignin, C., Coiffet, M. & Vaury, C. In Drosophila melanogaster the COM locus directs the somatic silencing of two retrotransposons through both Piwi-dependent and -independent pathways. PLoS ONE 3, e1526 (2008).
Li, C. et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137, 509–521 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Gainetdinov, I. et al. Terminal modification, sequence, length, and PIWI-protein identity determine piRNA stability. Mol. Cell 81, 4826–4842 e4828 (2021).
Fu, Y., Wu, P. H., Beane, T., Zamore, P. D. & Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics 19, 531 (2018).
Glockner, F. O. et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176 (2017).
Chou, M. T. et al. Tailor: a computational framework for detecting non-templated tailing of small silencing RNAs. Nucleic Acids Res. 43, e109 (2015).
Zhang, Z., Theurkauf, W. E., Weng, Z. & Zamore, P. D. Strand-specific libraries for high-throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9 (2012).
Han, B. W., Wang, W., Zamore, P. D. & Weng, Z. piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome- and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 31, 593–595 (2015).
Acknowledgements
We thank the Bloomington Drosophila Stock Center and the Developmental Studies Hybridoma Bank for reagents. We thank Z. Zhang and N. Lau for their helpful discussions and the Yamashita lab members for comments on the paper. The research was supported by the Howard Hughes Medical Institute (Y.M.Y., P.D.Z. and S.E.J.), National Institute of Health (NIH R01 HD109667 to J.K.K., R35 GM136275 to P.D.Z., R01GM097363 to A.A.A. and R35 GM130272 to S.E.J.) and the Whitehead Institute for Biomedical Research (Y.M.Y.).
Author information
Authors and Affiliations
Contributions
Z.G.V. and Y.M.Y. conceived the project. Z.G.V., I.G., S.E.J., J.K.K., P.D.Z. and Y.M.Y. designed experiments and interpreted the results. Z.G.V., I.G., A.B., C.B., J.M.F. and Y.M.Y. conducted experiments. Z.G.V., I.G., M.R.S., C.P.C., T.W.W., G.W.B. and S.F. analysed data. P.C. and A.A.A. contributed critical information in the course of the investigation. Z.G.V., I.G., Y.M.Y. and P.D.Z. wrote and edited the paper with the input from other authors. Y.M.Y. and P.D.Z. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Gregory Hannon, P. Jeremy Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Expression of sense and anti-sense Su(Ste) precursor RNA in testis.
a, b, smRNA-FISH for sense (red) and antisense (green) Su(Ste) precursor transcripts in the apical tip of the testis (a, lower magnification; b, higher magnification, not the same tissue). Hub (*), DAPI (blue). Bar: 20 µm in a, 5 µm in b. c, Metaplot of stranded RNA-seq coverage in Su(Ste) loci (Supplementary Table 1). The data are shown for three independent biological samples. Source numerical data are available in source data.
Extended Data Fig. 2 Su(Ste) piRNA precursor accumulates in zuc mutant testis.
a, b, Representative images of smRNA-FISH for antisense Su(Ste) precursor RNAs (green) in adult testes of the indicated genotypes. Arrows point to cytoplasmic precursor RNAs; arrowheads point to nuclear transcripts. DAPI (blue), bars 20 µm (a and b) and 5 µm (a′ and b′). Experiments were repeated three times with similar results.
Extended Data Fig. 3 Su(Ste) piRNA precursor is not upregulated upon knockdown of aub, vasa, ago3, or piwi.
a–d, Su(Ste) piRNA precursor transcript testes of the indicated genotypes. Magnified regions of the niche are shown in a′, b′, c′, d′. The region of GSCs/SGs is indicated by a yellow dotted line, SC region by cyan lines. Arrowheads point to nuclear transcripts. Hub (*), DAPI (blue), bars 20 µm (a–d) and 5 µm (a′–d′). e, Bias in nucleotide composition (sequence logo) of transposon- and Su(Ste)-derived (Supplementary Table 1) piRNAs in control testis from 0–5-day-old y1w1118/Y; nos-gal4:VP16 males. The data are the mean of two independent biological samples. Source numerical data are available in source data.
Extended Data Fig. 4 Long 5′ monophosphorylated RNAs from piRNA producing loci 42AB and petrel and from nos, bam, bgcn genic loci.
a, Frequency of 0–20-nt overlaps between 5′ monophosphorylated long RNAs and piRNAs on opposite genomic strands in control testis from 0–5-day-old y1w1118/Y; nos-gal4:VP16 males. The standard score (number of standard deviations from the mean) and the corresponding p value (two-sided Z-test) of the 10-nt overlap (Z10) is shown. Data are for all possible permutations of two small RNA data sets and two 5′ monophosphorylated long RNA data sets (n = 2 × 2 = 4). b, Change in steady-state abundance of 5′ monophosphorylated long RNA data sets in nos>armiRNAi males (n = 2 for control; n = 2 for nos>armiRNAi) and in zucEY11457/− mutants (n = 3 for control; n = 3 for zucEY11457/−); p values are shown for the two-sided Mann-Whitney test. Source numerical data are available in source data.
Extended Data Fig. 5 Su(Ste) precursor transcripts and piRNAs in XXY ovaries.
a, b, Germaria and early egg chambers of XX (a) and XXY (b) females with magnified inserts of germaria shown in a′ and b′. Antisense Su(Ste) piRNA precursor transcript (green), DAPI (blue), bars 20 µm. c, Relative abundance of act5C mRNA and antisense Su(Ste) piRNA precursor transcript in XX and XXY ovaries, and in XY testis, determined by qRT-PCR, normalized to Gapdh (n = 3). Boxplots show the median and interquartile range (IQR). Source numerical data are available in source data.
Extended Data Fig. 6 gfp-Ste reporter is silenced in the ovary of XXY females.
Representative images of GFP (green) in testis from XY males (a–c) or germaria from XX (d–f) or XXY (g–h) females. DAPI (blue), bars 20 µm. Asterisk indicates the hub in a–c. Experiments were repeated three times with similar results.
Extended Data Fig. 7 Repression of Ste protein in armiRNAi males from XXY mothers.
Representative images of Anti-Ste and anti-Tubulin Western blotting of testes from the indicated genotypes. Source numerical data and unprocessed blots are available in source data. Experiments were repeated twice with indistinguishable results.
Extended Data Fig. 8 Su(Ste) piRNAs make Ste piRNAs in XXY ovaries.
a, Nascent transcripts (GRO-seq) at a Ste locus in w1 XX ovaries. Data are from ref. 79 for all (uniquely and multiply mapping) reads without apportioning to other Ste loci. b, Length profile of Ste-derived small RNAs in XXY ovaries. The data are the mean of three independent biological samples. c, Ping-pong signature—that is, frequent 10-nt overlap on opposite genomic strands—between Su(Ste) and Ste-derived piRNAs in XXY ovaries. The data are the mean of three independent biological samples. The standard score (number of standard deviations from the mean) and the corresponding p value (two-sided Z-test) of the 10-nt overlap (Z10) is shown. Source numerical data are available in source data.
Supplementary information
Source data
Source Data Figs. 1–3, 5 and 6 and Extended Data Figs. 1, 3–5 and 8 (download XLS )
Source numerical data for Figs. 1–3, 5 and 6 and Extended Data Figs. 1, 3–5 and 8.
Uncropped scans of western blotting data for Fig. 3 and Extended Data Fig. 7 (download PDF )
Uncropped scans of western blotting data for Fig. 3i and Extended Data Fig. 7.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Venkei, Z.G., Gainetdinov, I., Bagci, A. et al. A maternally programmed intergenerational mechanism enables male offspring to make piRNAs from Y-linked precursor RNAs in Drosophila. Nat Cell Biol 25, 1495–1505 (2023). https://doi.org/10.1038/s41556-023-01227-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-023-01227-4
This article is cited by
-
Spatially revealed roles for lncRNAs in Drosophila spermatogenesis, Y chromosome function and evolution
Nature Communications (2024)
