
Abstract
Surface analysis by X-ray photoelectron spectroscopy and time-of-flight secondary ion mass spectrometry was applied to investigate the origin of the enhanced surface passivity and resistance to a chloride-induced breakdown provided by the protective ultrathin oxide films formed on Cr–Fe–Co–Ni–Mo single-phase fcc multi-principal element alloys. A bilayer structure of the oxide films is observed with the inner barrier layer mostly constituted of Cr(III) oxide and the outer layer enriched in Cr(III) hydroxides and Mo(IV,VI) oxides. The Mo(VI) and Mo(IV) species are mainly located in the outer and inner parts of the outer layer, respectively. Anodic passivation promotes mainly the growth of the inner layer on the alloy of higher Cr bulk content and the outer layer on the alloy of higher Mo bulk content. Passivation also promotes the enrichment of Cr(III) hydroxide and Mo(IV) and Mo(VI) oxides in the outer layer. Depth distribution analysis suggests that the ultra-thin protective inner barrier contains Cr(III)-depleted heterogeneities acting as weak sites for chloride attack, which are reinforced by the Mo(IV) oxide species concentrated close to the inner barrier layer. This elemental distribution provides an explanation for the reinforcement of the resistance to localized corrosion observed on these Cr–Fe–Co–Ni–Mo alloys.
Similar content being viewed by others

Introduction
Corrosion resistance is an important property needed for most industrial as well as everyday applications of functional alloys. One of the most effective means of protection against corrosion is surface passivity: the alloy becomes self-protected via the formation on its surface of an ultra-thin oxide film, the passive film, that isolates the material from its local environment1,2. The chemical structure, composition, and formation mechanisms of passive oxide films formed on conventional alloys have been widely investigated by surface analytical techniques such as X-ray photoelectron spectroscopy (XPS)3,4,5,6, time-of-flight secondary ion mass spectrometry (ToF-SIMS)4,5, and Auger electron spectroscopy (AES)3,6. For conventional corrosion-resistant alloys such as stainless steels and Ni-based stainless alloys, it has been shown that chromium and molybdenum, the two key elements added to enhance corrosion protection, are enriched in the protective surface oxide films4,5,6,7. The protecting effect of Cr is provided by the formation of a Cr oxide layer with extremely low dissolution kinetic8. The mechanism of the beneficial effect of Mo in combatting localized passivity breakdown is still under debate, being reported to act by impeding the penetration of aggressive ions9,10 or by promoting the self-repair of the oxide film11,12. The enrichment of chromium (and molybdenum for Mo-bearing alloys) is found to be further enhanced when the oxide film is immersed or electrochemically passivated by anodic polarization in the acidic and neutral aqueous environment due to the selective dissolution of iron species in the electrolyte3,4,6,7,13. Besides, anodic passivation also causes dehydroxylation of the oxide film4.
In many works, the protective oxide films formed on stainless alloys are reported to exhibit a stratified structure5,12,13,14, which can be described by a bilayer model5,12,14. The inner barrier layer, giving the alloy its high corrosion resistance, is enriched in Cr(III) oxide, while the outer layer can contain Cr(III) hydroxide, Fe(II,III) oxide/hydroxide, Mo(IV,VI) oxide and Ni(II) hydroxide, depending on the alloy composition4,5,12,13,14,15. Based on the fact that the formation of oxide is usually faster compared to cation diffusion, some researchers suggested that Ni2+, Fe2+, as well as Mo4+ cations, might be captured in the Cr oxide matrix and thus form a non-stoichiometric oxide, termed as nonequilibrium solute capture16,17,18.
The thickness of passive oxide films ranges between 1 and 3 nm, depending on the passivation conditions4,5,14,15,19,20. Thinning or thickening can also be observed after passivation, depending on the competition between the rates of oxide dissolution and oxide re-growth4,5. The alloy close to the metal/oxide interface exhibits a modified composition, resulting from the preferential oxidation of elements with higher affinity to oxygen such as Cr21 during oxide film formation and leading to enrichment in metallic nickel7,12,20.
Such systematic investigations of protective surface oxide films are much less advanced for multi-principal element alloys (MPEA)22,23,24,25,26. MPEAs have two prominent advantages over conventional alloys: (i) the alloying of several principal elements opens up a large composition space to explore, and ii) the high mixing entropy of MPEAs makes the formation of solid solution phases easier27,28, which can reduce the number of phases constituting the alloy and lead to specific properties29,30. For Mo-containing MPEAs, Dai et al. have studied FeCoCrNiMox as-cast alloys passivated in sulfuric acid and reported a bilayer structure of the passive film, similar to that found on traditional stainless alloys and composed of an inner layer enriched in chromium oxide and an outer oxide layer consisting of Cr/Fe/Mo oxides and hydroxides25. The existence of nonequilibrium solute capture is also suggested for Ni38Cr21Fe20Ru13Mo6W2 MPEA23. These studies contribute to our knowledge of the corrosion behavior and passive film composition of Mo-containing MPEAs and indicate that the elimination of second-phase particles and element segregation could further enhance the corrosion resistance, which has already been proved on traditional alloys31 but not on these Mo-containing MPEAs yet. Nevertheless, a more detailed surface analysis of the chemical structure and composition of the native and passive oxide films is necessary to clarify the origin of enhanced surface passivity obtained with these alloys and to better understand the influence of Mo24,25,26,32.
Herein, we report a detailed surface analysis of both native oxide-covered and electrochemically passivated surfaces of Cr–Fe–Co–Ni(–Mo) MPEAs having a stabilized single-phase fcc structure. The composition of the alloys was chosen based on previous thermodynamically obtained phase diagrams to ensure a maximum Cr and Mo content while retaining a pure fcc structure as reported separately32. The addition of Mo in these alloys markedly enhances the resistance to passivity breakdown and to initiation of localized corrosion32. XPS, including necessary analytical development, and ToF-SIMS were combined to investigate the composition, stratification, and thickness of the surface oxide films. A thorough analysis of the depth distribution of Mo in the oxide films was performed in order to improve our knowledge of the role of this element in reinforcing surface passivity and providing these alloys superior resistance to localized corrosion in a chloride-containing environment.
Results and discussion
Surface species identification
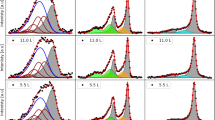
Figure 1 shows, as an example, the curve-fitted Cr 2p, Ni 2p, Fe 2p, Mo 3d, and Co 2p core level spectra of the passivated MPEA-15Cr10Mo surface (see method Section for the specific curve fitting procedure adopted in this work). The decomposed spectra indicate the presence of Cr(III)ox, Cr(III)hyd, Fe(II,III)ox,hyd, Ni(II)hyd, Mo(IV)ox, Mo(VI)ox, and an intermediate Mo species whose binding energy is between that of Mo(IV)ox and Mo(VI)ox, as well as metallic Cr, Fe, Co, Ni, Mo. The nature of the intermediate Mo species will be discussed in the Section “Distribution of Mo species and effect on passivity”. The same components were identified for the native and passive oxide films of all three alloys.

a Cr 2p, b Fe 2p, c Ni 2p, d Mo 3d, and e Co 2p.
Stratified structure of the oxide film
Figure 2 shows the ToF-SIMS negative ion depth profiles for the native oxide-covered and passivated MPEA-15Cr10Mo surfaces. CrO− (67.9354 amu), CrO3H2− (101.9409 amu), FeO2− (87.9248 amu), NiO2H− (88.9330 amu), MoO2− (129.8952 amu), MoO3− (145.8902 amu) and Ni2− (115.8707 amu) secondary ions were selected as representative ions for Cr(III)ox, Cr(III)hyd, Fe(II,III)ox,hyd, Ni(II)hyd, Mo(IV)ox, Mo(VI)ox and Ni(0)met species, respectively5,33. The Ni2− signal, characteristic of metallic Ni, was used to locate the metal/oxide interface at the position where 80% of the maximum intensity is reached5,33. The width of the oxide region corresponds to 35 and 41 s of sputtering time for the native and the passive oxides, respectively, suggesting an increase in thickness after passivation. When probing into the metallic substrate, the Ni2− signal exhibits a maximum (not very pronounced but significant) after the position of the metal/oxide interface. This has already been observed on Ni-based alloys5,20, Cantor alloy22, and stainless steel7,34, and is assigned to metallic Ni enriched in the alloy below the oxide film, as confirmed below.

a Native oxide surface and b passivated surface in 0.05 mol L−1 H2SO4 at 0 VMSE for 1 h.
The other signals in Fig. 2, characteristic of oxidized species, are used to determine the in-depth variation of the composition of the oxide films covering the alloy surface. The depth profiles of the different ions clearly evidence a stratification of the oxide films. A bilayer structure is observed with an outer part characterized by the presence of Ni and Cr hydroxides and Fe and Mo oxides and an inner part consisting mainly of Cr oxide. A closer examination of the outer oxide region shows that the NiO2H− profile always peaks before the other profiles. This suggests that Ni hydroxide concentrates at the surface of the oxide film. Signals characterizing Co oxides were not explored in this work because of the indistinguishable atomic mass difference between all CoOxHy− and NiOxHy+1− ions (0.01 amu) and their very low intensity compared to Ni signals. However, a work on Cantor alloy reported that the CoO2− signal peaks in the outer part of the native and passive oxide films22. The interface between the outer and inner parts of the oxide films can be located at the position where the CrO− signal, characteristic of the inner Cr oxide, reaches 80% of its maximum intensity by using the same convention as for locating the metal/oxide interface. This position corresponds to 17 and 19 s of sputtering time for the native and the passive oxides, respectively. Identical depth profiles were obtained in different zones of the sample surface, suggesting that, at the scale of the ToF-SIMS analyzed area (100 × 100 μm2), the composition and thickness measured in different zones of the oxide films do not present significant differences. Similar stratified structures and uniform distribution of the oxide film were also observed for MPEA-25Cr5Mo and MPEA-35Cr alloys, except for the absence of Mo oxides in the outer layer of Mo-free alloy MPEA-35Cr.
Based on these ToF-SIMS depth profiles, a bilayer model (shown in Fig. 3) of the attenuation of the emitted photoelectrons was applied to the three MPEAs in order to quantify the composition and thickness of the oxide film and the composition of the modified alloy at the metal/oxide interface22,35,36. In this bilayer model, the surface roughness and the gradients of concentration between layers are not taken into account, thus only sharp and flat interfaces are considered. As supported by the reproducibility of the ToF-SIMS depth profiles measured in different zones of the oxide films, both the inner and outer layers are hypothesized to be uniform in thickness with components homogeneously distributed within. The layer composition and thickness thus estimated for each uniform layer are therefore average compositions and equivalent thickness values. The inner layer is assumed to be only composed of Cr2O3 for simplification, which may seem arbitrary but in fact, is supported by the observation that the inner part is comprised mainly of Cr oxide as shown in Fig. 2. This is in agreement with recently published works on stainless steels4,12,37, Ni-Cr(-Mo)5,13 alloys and high entropy alloys22,25. The outer layer contains Ni and Cr hydroxides and Fe, Mo, and Co oxides/hydroxides, like those previously identified by XPS and ToF-SIMS for stainless steels4,12,34, the Cantor HEA alloy22 and Ni-based alloys5,14. Therefore, for calculations, the XPS intensities of the metallic Cr(0)met, Fe(0)met, Co(0)met, Ni(0)met, Mo(0)met components were assigned to the modified alloy under the oxide, the intensity of the Cr(III)ox component to the inner layer, and the intensities of the other oxide/hydroxide components to the outer layer, as indicated in Fig. 3. The model is applicable for all three MPEAs. For MPEA-35Cr, Mo oxides/hydroxides and Mo metal were not taken into account. The results obtained for the three MPEAs studied in this work are compiled in Table 1.

The interfacial roughness is neglected.
Surface composition quantification
As reported in Table 1, for the MPEA-25Cr5Mo and MPEA-15Cr10Mo quinary samples, the global equivalent thickness of the native oxide films is calculated to be 1.4–1.5 nm, in agreement with the thickness range reported in the literature for native oxide films formed on Fe- or Ni-based stainless alloys4,19,20,22. The outer layer is estimated to be 1.1–1.3 nm thick and the inner layer is 0.2–0.3 nm thick. Noteworthy is the very low equivalent thickness obtained for the inner layer, of the order of interlayer distances between oxide planes in crystalline compounds (the Cr–O bond lengths in Cr2O3 is 0.2 nm38). This very low equivalent thickness was obtained on the assumption of a uniform and dense (5.21 g.cm−3) Cr oxide layer. Published work on stainless steel has revealed that, in reality, the inner barrier layer can present a nanoscale heterogeneity in Cr enrichment due to a local lack of Cr supply at some sites during simultaneous oxide nucleation at multiple sites in the very first stages of oxide film formation39. By analogy, it is possible that the inner barrier layer of these MPEAs also presents heterogeneities, being locally thicker than the calculated equivalent thickness in some zones and locally depleted in Cr(III)ox in other zones, the latter corresponding to weak sites limiting the corrosion resistance. Consideration of a discontinuous or less dense oxide layer would result in a film thicker than calculated. In the outer oxide layer, chromium and, even more markedly molybdenum, are enriched, representing 38 and 15% of the metal cations versus 25 and 5% in bulk, respectively, for the MPEA-25Cr5Mo sample, and 17 and 21% versus 15 and 10%, respectively, for the MPEA-15Cr10Mo sample (Table 1). In the modified alloy region, nickel is enriched while chromium and iron are depleted as can be seen from the compositions for native surfaces in Table 1. This modified alloy composition confirms the preferential oxidation of chromium and iron, as observed by other authors for Fe- or Ni-based alloys and the Cantor alloy7,12,20,22.
Compared to the native oxide film, the passive films formed on these two quinary MPEAs are thicker, in the range 1.8–2.1 nm as listed in Table 1, in agreement with the trend observed by ToF-SIMS in Fig. 2. In the case of MPEA-25Cr5Mo alloy, containing more chromium, thickening is more pronounced in the inner layer5,12, reaching a value (0.6 nm) consistent with the formation of a continuous layer. In contrast, the passivation-induced thickening of the oxide film takes place mainly in the outer layer of the MPEA-15Cr10Mo sample that contains more molybdenum. Comparison of the chemical composition prior to and after anodic passivation, i.e., for the native and passive oxide films in Table 1, shows that passivation enhances the enrichment of both chromium and molybdenum in the outer oxide layer of these alloys (from 38 to 48 at.% Cr and from 15 to 21 at.% Mo for MPEA-25Cr5Mo, and from 17 to 45 at.% Cr and from 21 to 31 at.% Mo for MPEA-15Cr10Mo). Meanwhile, the iron and nickel contents in the outer layer decrease after passivation, in agreement with previous works on stainless steel and Ni-based alloy4,5,13,23. The comparison of the total amounts of Cr and Mo in the oxide layers shows a more pronounced passivation-induced enrichment in these elements for the MPEA-15Cr10Mo alloy (from 38 to 76 at%) than for the MPEA-25Cr5Mo alloy (from 53 to 68 at%) that may explain the larger thickening of the outer layer on the MPEA-15Cr10Mo alloy, despite the dissolution of Fe and Ni components. In the modified alloy region at the metal/oxide interface, nickel becomes slightly less enriched after anodic passivation while chromium and iron remain depleted as shown by the modified alloy composition for the passivated surface in Table 1. Interestingly, molybdenum is not further depleted in the modified alloy region after passivation despite its increased enrichment in the oxide film. These variations suggest the selective dissolution of nickel species in the electrolyte during the electrochemical passivation process4,5.
On the quaternary MPEA-35Cr alloy, also listed in Table 1, the native oxide film is thicker (2.1 nm) than on the Mo-containing quinary alloys (1.4–1.5 nm). However, and in contrast with the two quinary alloys, slight thinning is observed after passivation, down to a value (1.8 nm) similar to that obtained on the quinary alloy with higher Cr content (MPEA-25Cr5Mo) after passivation. Like on the quinary alloys, the iron and nickel contents in the outer layer decrease after passivation, indicating the selective dissolution of Fe and Ni oxides/hydroxides in the electrolyte when exposed to sulfuric acid solution4, and leading to significantly higher chromium content in the outer layer after passivation (74 at.% Cr). The modified alloy composition for the native MPEA-35Cr surface in Table 1 shows that the enrichment of nickel and depletion of chromium and iron are also found in the modified alloy underneath the native oxide film. Both are attenuated after anodic passivation, also suggesting the selective dissolution of Ni in the electrolyte4,5.
Distribution of Mo species and effect on passivity
Clarifying the location of Mo and its chemical states in the surface oxide film is a key point to understanding the role of this element in improving surface passivity. In some papers, the in-depth distribution of Mo species has been shown to be related to its valence state: the higher the valence, the more outward the position of the species12,34. In the present work, angle-resolved XPS (AR-XPS) measurements were carried out on the native and the passive oxide films formed on the MPEA-25Cr5Mo sample in order to get a deeper insight into the depth distribution and chemical state of the oxidized Mo species in the oxide layers. These angle-resolved measurements can also provide us with some clues regarding the nature of the intermediate Mo species revealed by the XPS curve fitting. Figure 4 shows the XPS spectra of the Mo 3d core level region measured on the native oxide-covered MPEA-25Cr5Mo surface at 90° and 40° take-off angles. The relative intensities of the Mo(IV)ox, Mo(VI)ox, and intermediate Mo species obtained at each take-off angle are reported in Table 2.

a 90° and b 40°.
As can be seen in Table 2, for the native oxide film, the relative intensity of Mo(VI)ox is higher at 40° than at 90° take-off angle, supporting its enrichment near the outer surface of the oxide film. The relative intensity of Mo(IV)ox is also higher at 40° take-off angle (although the increase is lower than for Mo(VI)ox), indicating a deeper Mo(IV)ox location in the oxide film. This is supported by the ToF-SIMS data (Fig. 2) that show a less steep decrease of the MoO2− intensity in the profile than of the MoO3− profile when probing the oxide region inwards. In contrast, the relative intensity of intermediate Mo species in Table 2 is lower at 40° than at 90° take-off angle, clearly pointing to their location underneath the Mo(IV)ox and Mo(VI)ox species. For the passive oxide film, Table 2 shows that the sequence is the same although the difference in variation observed for the Mo(IV)ox species suggests that these species may be distributed closer to the intermediate Mo species than to the Mo(VI)ox species. These results clearly support that the oxidized Mo species are distributed from the outer surface of the oxide films to the metal/oxide interface according to the following sequence: Mo(VI)ox then Mo(IV)ox then intermediate Mo.
The existence of an intermediate doublet between the Mo(IV)ox and Mo(VI)ox doublets in XPS Mo 3d spectra have been reported in many published works, with BEs varying between 230.2 and 231.8 eV13,15,40,41. Three assignments were commonly made and described in the literature: hydroxylated Mo(IV)4,15, Mo(V) oxide13,41, or an intrinsic satellite of Mo(IV)ox due to final state screening effect40,42. Although there has been no experimental confirmation, the presence of this peak could also be explained by Mo(IV) captured in the Cr oxide matrix as recently proposed by the nonequilibrium solute capture theory16,18. Above all, the formation of mixed oxides of the Cr2−xMoxO3 type is ruled out since not consistent with the expected chemical state.
In order to discriminate the role of each component, bilayer, and trilayer models were adopted in order to determine the equivalent thicknesses corresponding to each component as well as the distribution of molybdenum in the different layers. Indeed, it is worth discussing whether a continuous Mo oxide layer can be formed at the outer layer/inner layer interface in the oxide film since Mo is known to be beneficial for increasing the resistance to passivity breakdown and localized corrosion19.
The ToF-SIMS depth profiles in Fig. 2 reveal that the location of the maximum of the MoO2− signal is above that of CrO− but beneath that of MoO3− (assigned to Mo(VI)). Assuming that the Mo(IV) species at 229.2 eV is localized at the outer/inner oxide interface as suggested by the AR-XPS and ToF-SIMS observations, it is possible to determine the equivalent thickness of this layer using a three-layer model of intensity attenuation. The calculated thickness ranges between 0.04 and 0.12 nm, depending on the alloy sample and native oxide-covered or passivated surface state. This value is well below the expected thickness for a complete monolayer and suggests that the Mo(IV) species forms a discontinuous layer. From this observation, it could be considered that Mo(IV) is located at the termination of the Cr oxide constituting the inner layer.
Concerning intermediate Mo, the AR-XPS observations in Table 2 shows that this species is located deeper in the films than the Mo(IV) species, which is located at the outer/inner oxide interface as shown by the location of maximum MoO2− intensity in Fig. 2. Besides, as supported also by the shoulder observed after the maximum of the MoO2− ions ToF-SIMS depth profile (Fig. 2), it is suggested that this intermediate Mo species is mainly located in the outermost part of the inner layer, close to the outer/inner oxide interface.
Thus, two hypotheses can be advanced to explain the presence of the intermediate Mo species in the Cr inner oxide layer: (a) Mo is captured in the Cr oxide as proposed by the nonequilibrium solute capture theory16,17,18,23, (b) the equivalent thickness calculated for the inner Cr(III)ox-enriched layer of the two Mo-bearing alloys being low, e.g., 0.3 nm for the passive oxide film on MPEA-15Cr10Mo, this inner layer of the passive film, commonly regarded as a protective barrier layer on conventional alloys4,9,12, may present weak sites locally depleted in Cr(III)ox and richer in Fe as observed on stainless steel39. Mo would reinforce the barrier layer of the passive film by substituting Fe in these Cr(III)ox-depleted weak sites.
Using the XPS intensity of the intermediate Mo components, which was attributed to the outer oxide layer in Table 1, the percentage of Mo(IV) in the Cr oxide matrix of the inner layer is calculated to be 8 at.% and 6 at.% for the native and passive oxide films on the MPEA-25Cr5Mo sample, respectively, and 18 at.% and 19 at.% for the native and passive oxide films on the MPEA-15Cr10Mo sample. These values are high, which tend to eliminate a “doping effect” of Mo(IV) cations captured in the Cr(III) oxide matrix. Besides, the ToF-SIMS profiles (Fig. 2) show clearly that the largest part of Mo is located above the inner layer. The depth distribution of Mo in the inner layer is not homogeneous and never reaches the metallic substrate, which seems to be in contradiction with the nonequilibrium solute capture theory. On the contrary, the intensity shoulder observed after the maximum of the MoO2− ion depth profile when probing into the inner layer (Fig. 2) suggests that the Mo(IV) oxide in the inner layer may be located mainly in part near the inner/outer oxide interface. Based on this ToF-SIMS observation, the low equivalent thickness of the Cr oxide layer, and the observed presence of nanoscale weak sites enriched in Fe oxide soluble species in the oxide film on stainless steel39, we infer that the inner layer of these MPEAs may also present Cr(III)ox-depleted weak sites, possibly thinner compared to the calculated average thickness. However, instead of being enriched only in Fe species, Mo(IV) oxide would concentrate in these weak sites thus stabilizing the Fe species.
Besides, works on conventional Fe- and Ni-based alloys43,44 have shown that Mo(IV) species located in the inner part of the surface oxide films promote outward cationic oxide growth by forming a barrier against the anionic transport of oxygen towards the oxide/metal interface. Thus, our explanation of the high corrosion resistance obtained with these Mo-bearing quinary alloys is that, although the surface may not be completely covered by a protective Cr(III)-enriched inner layer and thus may expose local Cr(III)ox-depleted sites due to local lack of Cr supply at the early stage of oxide film formation39, these weak sites can be enriched with the Mo oxide species concentrated at the interface between outer and inner layers as depicted in Fig. 5. The Mo(IV) species that reinforce the Cr(III)ox-depleted weak points of the barrier inner layer could block the inward ingress of Cl− anions, thus markedly improving the resistance to passivity breakdown of the studied MPEAs in chloride-containing corrosive environments.

Developed bilayer model with Mo(IV)-sealed weak sites in the inner layer for the Mo-bearing alloys MPEA-25Cr5Mo and MPEA-15Cr10Mo.
Methods
Sample preparation
Samples of composition Cr35Fe20Co5Ni40, Cr15Fe10Co5Ni60Mo10, and Cr25Fe25Co5Ni40Mo5 (at.%), all having single-phase fcc structure and similar grain size (69 ± 5, 61 ± 5, and 66 ± 3 µm, respectively) after homogenization, were used. They are hereafter denoted MPEA-35Cr, MPEA-15Cr10Mo, and MPEA-25Cr5Mo, respectively. Design, production, structural properties as well as corrosion resistance properties were reported separately32. The surfaces were prepared by mechanical polishing with SiC paper of 1200, 2400, and 4000 grades followed by 3, 1, and 0.25 µm diamond paste finish, subsequently cleaned in ultrasonic baths of acetone, ethanol, and deionized water (Merk Millipore, resistivity of 18.2 MΩ cm) for 5 min each, and dried in a flow of compressed air. The air-formed oxide films covering the samples after this procedure are the native oxide films discussed hereafter.
The passive oxide films were prepared in a 3-electrode cell composed of a saturated mercury sulfate reference electrode (MSE, ESHE = EMSE + 0.64 V), a Pt wire used as a counter electrode, and the alloy sample constituting the working electrode of 1.13 cm2 area delimited by a Viton O-ring. A VersaSTAT4 potentiostat (AMETEK) served as controller. Electrochemical passivation was carried out at room temperature in 0.05 mol L−1 H2SO4 electrolyte prepared from ultrapure chemicals (VWR) and deionized water. The electrolyte was deaerated by bubbling Ar gas for 30 min before the passivation treatment in order to minimize dissolved gas. Anodic passivation was performed by stepping the potential to 0 VMSE for 1 h after stabilization at open circuit potential (OCP) for 15 min. The selected passivation potential corresponds to the minimum passive current density measured in the passive range32.
Surface analysis
The analytical approach adopted in this work consists of an elemental depth profiling by ToF-SIMS associated with identification and quantification by XPS of the different species present in the stratified surface oxide layers and underlying modified alloy.
In-depth elemental analysis was performed with an IONTOF ToF-SIMS 5 spectrometer (base pressure of about 10−8 mbar). The depth profiles were recorded in dual-beam mode. A pulsed 25 keV Bi+ primary ion source delivering 1.2 pA of target current over a 100 × 100 µm2 area was used for analysis. The Bi+ primary ion flux was sufficiently low to ensure analysis in static SIMS conditions. Analysis was interlaced with sputtering using a 0.5 keV Cs+ sputter beam delivering 17 nA of target current over a 300 × 300 μm2 area. Both ion beams were at the incidence of 45° with respect to the sample surface and well-aligned to ensure analysis from the center of the sputtered crater. Negative ions were recorded because of their higher sensitivity to fragments coming from oxide matrices. For each surface, depth profiles were recorded at least in two different zones in order to verify the uniformity of the oxide films. Data acquisition and processing were done with the IONSPEC 6.5 software.
XPS analysis was performed with a Thermo Electron ESCALAB 250 spectrometer (base pressure of 10−9 mbar) using a monochromatic Al Kα X-ray source (hν = 1486.6 eV). High-resolution spectra of the Cr 2p, Fe 2p, Co 2p, Ni 2p, Mo 3d, O 1s, S 2p, and C 1s core levels were recorded at pass energy of 20 eV with a step size of 0.1 eV. Photoelectrons were collected at 90° and 40° emission angles.
Data processing (curve fitting) was performed with CasaXPS, subtracting a Shirley-type background for the metallic element spectra and a linear background for the S 2p spectra. Due to the overlaps between the 2p3/2 and 2p1/2 spin-orbit doublets of Cr and Fe and between the 3d5/2 and 3d3/2 doublets of Mo, the whole Cr 2p, Fe 2p, and Mo 3d core level regions were decomposed. For Ni, there was no overlap between the 2p3/2 and 2p1/2 doublets, thus only the Ni 2p3/2 peak was decomposed. The Co 2p region is overlapped by the much broader Ni L3M23M23 and Fe L3M45M45 Auger lines, thus the whole Co 2p core level spectrum was decomposed to take Auger peaks into account. However, owing to low content (5 at.%) in the studied alloys, the oxidized Co signal in the Co 2p core level region had much lower intensity than the overlapping Auger peaks, impeding a decomposition reliable enough to identify the oxidized cobalt species and discuss the passivation-induced Co content variation in the oxide films.
Spectral decomposition by curve fitting was performed by applying constraints on binding energy (BE), full width at half maximum (FWHM), the intensity ratio of spin–orbit doublets, and lineshape. Metallic component peaks were obtained from in-house references prepared for oxide-free pure metals. They were fitted with LF (α, β, w, m) lineshapes representing an asymmetric Lorentzian curve convoluted by a Gaussian curve or saved as lineshapes without detailed decompositions. The α and β parameters define the asymmetry, w the damping of the tail, and m (integer value between 0 and 499) the width of the Gaussian. Non-metallic component peaks were fitted with symmetric Gaussian/Lorentzian lineshape products GL(x) where x is the percentage of Lorentzian character.
The compositional design of the MPEAs studied in this work introduces difficulties in the XPS data processing that were overlooked in previous studies of the same system25,26. The proportions of the alloying elements being considerable, the overlapping of Auger peaks in the core level region of interest is not negligible. In the present case, the large proportions of Ni and Fe make the decomposition methods of the Fe 2p and Co 2p core level spectra adopted on Fe-4,9 or Co-based alloys45 inapplicable. Besides, in view of the weak intensities measured for Co, it is not possible to apply the methodology previously proposed for surface analysis of CrCoFeMnNi Cantor alloy based on the decomposition of the 3p core level spectra22. Thus, a specific XPS spectral decomposition methodology was developed involving the subtraction of overlapping Auger peaks. In order to apply this methodology, measurements on pure metal substrates were performed in order to extract the fitting parameters for the Auger lineshapes to be subtracted from the core level spectra of interest.
Figure 6 shows the reference Cr 2p, Ni 2p, Fe 2p, Mo 3d, and Co 2p core level XPS spectra obtained for oxide-free surfaces of pure Cr, Ni, Fe, Mo, and Co samples, respectively, as well as the Co L2M23M45 and Ni L3M23M45 Auger peaks present in the Fe 2p BE region. The Cr 2p (Fig. 6a), Ni 2p (Fig. 6b), and Mo 3d (Fig. 6d) spectra were well-fitted with LF lineshapes. BE values and FWHM values are listed in Table 3 and are in good agreement with literature4,5,12,46. The experimental Fe 2p (Fig. 6c) and Co 2p (Fig. 6e) spectra were saved as lineshapes without detailed decomposition and used as such in subsequent curve fitting of the spectra obtained with the alloys in order to avoid complicating the spectral decomposition with too many peaks when taking into account multiplet splitting or plasmon loss feature46,47. Similarly, the experimental spectra of the Co L2M23M45 (Fig. 6f) and Ni L3M23M45 (Fig. 6g) Auger peaks were saved as lineshapes and used as such in subsequent spectral decomposition for the alloys.

a Cr 2p, b Fe 2p, c Co 2p, d Ni 2p, e Mo 3d core levels and f Co L2M23M45 and g Ni L3M23M45 Auger transitions.
The BE values and FWHM values of the component peaks used in curve fitting shown in Fig. 1 are also compiled in Table 3. These values were selected based on literature data through a self-consistent procedure and were fixed in this work for all oxide/hydroxide components except Cr oxide, and on our own reference measurements of corresponding pure substances for all metal components as presented above and for Cr oxide. The same parameters were used for all spectral decompositions in this work.
The Cr 2p spectrum (Fig. 1a) was decomposed into three spin-orbit doublets assigned to Cr(0)metal, Cr(III)ox, and Cr(III)hyd respectively. The 2 p3/2–2 p1/2 doublets at 574.0–583.3 eV for Cr(0)met and at 577.3–587.0 eV for Cr(III)hyd are in good agreement with literature4,5,46. The reference lineshape and parameters obtained for pure Cr oxidized in gaseous oxygen under the controlled UHV environment of the XPS spectrometer were used to take into account the multiplet structure of the Cr(III)ox component48.
The Ni 2p spectrum (Fig. 1b) was decomposed into four peaks. The main peak at 852.8 eV and satellite peak at 859.0 eV were assigned to metallic Ni(0)met according to our reference measurement. The lower intensity peak at 856.1 eV and satellite peak at 861.5 eV were assigned to Ni(II)hyd49. No NiO was observed, in agreement with results reported for stainless steels7,12,50, Cantor alloy22, and Ni–Cr(-Mo) alloy5,14,20, explained by the reduction of NiO to metallic Ni in literatures33,50.
The Fe 2p spectrum (Fig. 1c) was fitted with five spin–orbit doublets and two Auger peaks. The lineshape of metallic Fe was fixed with the BE and FWHM reference values obtained in the present work. The 2 p3/2–2 p1/2 doublets at 709.4–722.4 eV and 712.9–725.9 eV were assigned to Fe(II) and its associated satellite, respectively46,51. Fe(III) is represented by principal 2 p3/2–2 p 1/2 peaks at 711.1–724.4 eV, and satellites at 714.2–727.5 eV46,47,51. In addition, the overlapping Auger lineshapes of cobalt and nickel, shown in Fig. 6f, g, respectively, were added in the curve fitting with BEs fixed at the reference values reported in Table 3. In agreement with Fig. 1b, e, only the Auger lineshapes of metallic nickel and cobalt were used since metallic nickel and cobalt largely dominate the contribution to the Ni 2p3/2 and Co 2p spectra compared to oxidized nickel and cobalt. The BE of the Auger peaks was constrained by the BE difference between the 2p3/2 peak and the Auger peak obtained from the reference measurements. The value (BEAuger–BE2p3/2) is 140.4 eV for Ni and −65.4 eV for Co. The area of the two Auger peaks was constrained by the ratio between the area (in cps) of 2p3/2 peaks and the Auger peak, which is equal to 0.615 for pure nickel and 0.350 for pure cobalt according to our reference measurements (Table 4). Thus, for each curve fitting, the area of the Auger peaks was determined according to the area of the corresponding 2p3/2 peak in the same measurement, i.e., Ni 2p3/2 peak in Fig. 1b and Co 2p3/2 peak in Fig. 1e for the passivated MPEA-15Cr10Mo surface.
The Mo 3d spectrum (Fig. 1d) was fitted with four 3 d5/2–3 d3/2 doublets, assigned to Mo(0)met, Mo(IV)ox, Mo(VI)ox6,40,41 at 227.8–231.0 eV, 229.3–232.3 eV, and 232.6–235.7 eV, respectively, and to an intermediate component located at 231.4–234.4 eV. The nature of the latter will be discussed further on. For surfaces passivated in sulfuric acid, an S 2s peak located at 233.0 eV and assigned to sulfate adsorbed from the solution was added in the curve fitting of the Mo 3d region. The peak area of S 2s was determined from the S 2p3/2 peak area and the reference area ratio between S 2p3/2 and S 2 s obtained on the passivated MPEA-35Cr sample (Table 4).
The Co 2p region (Fig. 1e) is also overlapping with a broad Ni Auger peak and a Fe Auger peak. The very low intensity of the oxidized cobalt species and the relatively high intensity and energy range of the overlapping Auger peaks lead to considerable uncertainty in the identification and quantification of the oxidized cobalt species. The BE and FWHM values of the reference lineshapes obtained on pure metals were applied for the 2 p3/2–2 p1/2 doublet of Co(0)met, and for the Ni L3M23M23 Auger peak and Fe L3M45M45 Auger peaks. Peaks located at 781.0–796.9 eV and 786.8–802.7 eV represent the main 2 p3/2–2 p1/2 doublets and the associated satellite peaks of Co(II)ox/hyd52,53.
Data availability
The relevant data are available from the corresponding authors upon reasonable request.
References
Kruger, J. Passivity of metals—a materials science perspective. Int. Mater. Rev. 33, 113–130 (1988).
Maurice, V. & Marcus, P. Progress in corrosion science at atomic and nanometric scales. Prog. Mater. Sci. 95, 132–171 (2018).
Olsson, C.-O. A. & Hörnström, S. E. An AES and XPS study of the high alloy austenitic stainless steel 254 SMO® tested in a ferric chloride solution. Corros. Sci. 36, 141–151 (1994).
Lynch, B. et al. Passivation-Induced Cr and Mo enrichments of 316L Stainless Steel Surfaces and EFfects Of Controlled pre-oxidation. J. Electrochem. Soc. 167, 141509 (2020).
Wang, Z. et al. XPS and ToF-SIMS investigation of native oxides and passive films formed on nickel alloys containing chromium and molybdenum. J. Electrochem. Soc. 168, 041503 (2021).
Marcus, P. & Olefjord, I. A round robin on combined electrochemical and AES/ESCA characterization of the passive films on Fe-Cr and Fe-Cr-Mo alloys. Corros. Sci. 28, 589–602 (1988).
Olsson, C.-O. A., Malmgren, S., Gorgoi, M. & Edström, K. Quantifying the metal nickel enrichment on stainless steel. Electrochem. Solid-State Lett. 14, C1 (2011).
Strehblow, H.-H. Passivity of Metals. in Advances in Electrochemical Science and Engineering 271–374 (John Wiley & Sons, Ltd., 2002). https://doi.org/10.1002/3527600787.ch4.
Wang, Z., Seyeux, A., Zanna, S., Maurice, V. & Marcus, P. Chloride-induced alterations of the passive film on 316L stainless steel and blocking effect of pre-passivation. Electrochim. Acta 329, 135159 (2020).
Vignal, V., Olive, J. M. & Desjardins, D. Effect of molybdenum on passivity of stainlesssteelsin chloride media using ex situ near field microscopy observations. Corros. Sci. 41, 869–884 (1999).
Newman, R. C. The dissolution and passivation kinetics of stainless alloys containing molybdenum—1. Coulometric studies of Fe-Cr and Fe-Cr-Mo alloys. Corros. Sci. 25, 331–339 (1985).
Maurice, V. et al. Effects of molybdenum on the composition and nanoscale morphology of passivated austenitic stainless steel surfaces. Faraday Discuss. 180, 151–170 (2015).
Zhang, X., Zagidulin, D. & Shoesmith, D. W. Characterization of film properties on the Ni-Cr-Mo Alloy C-2000. Electrochim. Acta 89, 814–822 (2013).
Marcus, P. & Grimal, J. M. The anodic dissolution and passivation of NiCrFe alloys studied by ESCA. Corros. Sci. 33, 805–814 (1992).
Olefjord, I., Brox, B. & Jelvestam, U. Surface Composition of Stainless Steels during Anodic Dissolution and Passivation Studied by ESCA. J. Electrochem. Soc. 132, 2854–2861 (1985).
Yu, X. et al. Nonequilibrium solute capture in passivating oxide films. Phys. Rev. Lett. 121, 145701 (2018).
Cwalina, K. L. et al. In operando analysis of passive film growth on Ni-Cr and Ni-Cr-Mo alloys in chloride solutions. J. Electrochem. Soc. 166, C3241–C3253 (2019).
Sherman, Q. C., Voorhees, P. W. & Marks, L. D. Thermodynamics of solute capture during the oxidation of multicomponent metals. Acta Mater. 181, 584–594 (2019).
Sugimoto, K. & Sawada, Y. The role of molybdenum additions to austenitic stainless steels in the inhibition of pitting in acid chloride solutions. Corros. Sci. 17, 425–445 (1977).
Lim, A. S. & Atrens, A. ESCA studies of Ni-Cr alloys. Appl. Phys. A 54, 343–349 (1992).
Steffen, J. & Hofmann, S. Oxidation of NiCr and NiCrFe alloys at room temperature. Surf. Interface Anal. 11, 617–626 (1988).
Wang, L. et al. Study of the surface oxides and corrosion behaviour of an equiatomic CoCrFeMnNi high entropy alloy by XPS and ToF-SIMS. Corros. Sci. 167, 108507 (2020).
Quiambao, K. F. et al. Passivation of a corrosion resistant high entropy alloy in non-oxidizing sulfate solutions. Acta Mater. 164, 362–376 (2019).
Rodriguez, A. A. et al. Effect of molybdenum on the corrosion behavior of high-entropy alloys CoCrFeNi2 and CoCrFeNi2Mo0.25 under sodium chloride aqueous conditions. Adv. Mater. Sci. Eng. 2018, 1–11 (2018).
Dai, C. et al. X-ray photoelectron spectroscopy and electrochemical investigation of the passive behavior of high-entropy FeCoCrNiMox alloys in sulfuric acid. Appl. Surf. Sci. 499, 143903 (2020).
Wang, W. et al. Effect of Mo and aging temperature on corrosion behavior of (CoCrFeNi)100-Mo high-entropy alloys. J. Alloy. Compd. 812, 152139 (2020).
Yeh, J.-W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Senkov, O. N., Miller, J. D., Miracle, D. B. & Woodward, C. Accelerated exploration of multi-principal element alloys with solid solution phases. Nat. Commun. 6, 6529 (2015).
Bracq, G. et al. Combining experiments and modeling to explore the solid solution strengthening of high and medium entropy alloys. Acta Mater. 177, 266–279 (2019).
Granberg, F. et al. Mechanism of radiation damage reduction in equiatomic multicomponent single phase alloys. Phys. Rev. Lett. 116, 135504 (2016).
Fargas, G., Anglada, M. & Mateo, A. Effect of the annealing temperature on the mechanical properties, formability and corrosion resistance of hot-rolled duplex stainless steel. J. Mater. Process. Technol. 209, 1770–1782 (2009).
Wang, X. et al. Enhanced passivity of Cr-Fe-Co-Ni-Mo multi-component single-phase face-centred cubic alloys: design, production and corrosion behaviour. Corros. Sci. 200, 110233 (2022).
Wang, Z. et al. Thermal stability of surface oxides on nickel alloys (NiCr and NiCrMo) investigated by XPS and ToF-SIMS. Appl. Surf. Sci. 576, 151836 (2022).
Dou, Y., Han, S., Wang, L., Wang, X. & Cui, Z. Characterization of the passive properties of 254SMO stainless steel in simulated desulfurized flue gas condensates by electrochemical analysis, XPS and ToF-SIMS. Corros. Sci. 165, 108405 (2020).
Jabs, T., Borthen, P. & Strehblow, H.-H. X‐ray photoelectron spectroscopic examinations of electrochemically formed passive layers on Ni‐Cr alloys. J. Electrochem. Soc. 144, 1231 (1997).
Gardin, E. et al. Early stage of marine biofilm formation on duplex stainless steel. Biointerphases 15, 041014 (2020).
Olefjord, I. & Wegrelius, L. Surface analysis of passive state. Corros. Sci. 31, 89–98 (1990).
The Materials Project. Materials Data on Cr2O3 by Materials Project. (Data retrieved from the Materials projects databset mp-19399, https://doi.org/10.17188/1194409, United States, 2021), (2020).
Ma, L., Wiame, F., Maurice, V. & Marcus, P. Origin of nanoscale heterogeneity in the surface oxide film protecting stainless steel against corrosion. npj Mater. Degrad. 3, 1–9 (2019).
Scanlon, D. O. et al. Theoretical and experimental study of the electronic structures of MoO3 and MoO2. J. Phys. Chem. C 114, 4636–4645 (2010).
Clayton, C. R. & Lu, Y. C. Electrochemical and XPS evidence of the aqueous formation of Mo2O5. Surf. Interface Anal. 14, 66–70 (1989).
Gulino, A., Parker, S., Jones, F. H. & Egdell, R. G. Influence of metal–metal bonds on electron spectra of MoO2 and WO2. J. Chem. Soc. Faraday Trans. 92, 2137–2141 (1996).
Henderson, J. D. et al. Investigating the transport mechanisms governing the oxidation of Hastelloy BC-1 by in situ ToF-SIMS. Corros. Sci. 159, 108138 (2019).
Wang, L., Seyeux, A. & Marcus, P. Ion transport mechanisms in the oxide film formed on 316L stainless steel surfaces studied by ToF-SIMS with 18O2 isotopic tracer. J. Electrochem. Soc. 167, 101511 (2020).
Hanawa, T., Hiromoto, S. & Asami, K. Characterization of the surface oxide film of a Co–Cr–Mo alloy after being located in quasi-biological environments using XPS. Appl. Surf. Sci. 183, 68–75 (2001).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis. Appl. Surf. Sci. 257, 2717–2730 (2011).
Grosvenor, A. P., Wik, S. D., Cavell, R. G. & Mar, A. Examination of the bonding in binary transition-metal monophosphides MP (M = Cr, Mn, Fe, Co) by X-ray photoelectron spectroscopy. Inorg. Chem. 44, 8988–8998 (2005).
Zanna, S., Mercier, D., Gardin, E., Allion-Maurer, A. & Marcus, P. EPS for bacterial anti-adhesive properties investigated on a model metal surface. Colloids Surf. B 213, 112413 (2022).
Mansour, A. N. Characterization of β‐Ni(OH)2 by XPS. Surf. Sci. Spectra 3, 239–246 (1994).
Cui, Z. et al. Influence of temperature on the electrochemical and passivation behavior of 2507 super duplex stainless steel in simulated desulfurized flue gas condensates. Corros. Sci. 118, 31–48 (2017).
McIntyre, N. S. & Zetaruk, D. G. X-ray photoelectron spectroscopic studies of iron oxides. Anal. Chem. 49, 1521–1529 (1977).
Foelske, A. & Strehblow, H.-H. Passivity of cobalt in borate buffer at pH 9.3 studied by x-ray photoelectron spectroscopy. Surf. Interface Anal. 29, 548–555 (2000).
Wang, J. et al. Three-dimensional interconnected Co(OH)2 nanosheets on Ti mesh as a highly sensitive electrochemical sensor for hydrazine detection. N. J. Chem. 43, 3218–3225 (2019).
Acknowledgements
This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (ERC Advanced Grant agreement No. 741123).
Author information
Authors and Affiliations
Contributions
The study was conceived by D.M., V.M., and P.M. The materials were previously produced by L.P., M.L.-B., and I.G. X.W. performed surface preparation, experiments, and data acquisition, and analysis under the supervision of D.M., S.Z., and A.S. All authors discussed the data interpretation. X.W. wrote the paper’s first draft, revised by all. All authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Mercier, D., Zanna, S. et al. Origin of enhanced passivity of Cr–Fe–Co–Ni–Mo multi-principal element alloy surfaces. npj Mater Degrad 7, 13 (2023). https://doi.org/10.1038/s41529-023-00330-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-023-00330-z
This article is cited by
-
Molybdenum effects on the stability of passive films unraveled at the nanometer and atomic scales
npj Materials Degradation (2024)
-
Recent research progress on the passivation and selective oxidation for the 3d-transition-metal and refractory multi-principal element alloys
npj Materials Degradation (2023)
