
Abstract
Histone H2B monoubiquitination (at Lys120 in humans) regulates transcription elongation and DNA repair. In humans, H2B monoubiquitination is catalyzed by the heterodimeric Bre1 complex composed of Bre1A/RNF20 and Bre1B/RNF40. The Bre1 proteins generally function as tumor suppressors, while in certain cancers, they facilitate cancer cell proliferation. To obtain structural insights of H2BK120 ubiquitination and its regulation, we report the cryo-electron microscopy structure of the human Bre1 complex bound to the nucleosome. The two RING domains of Bre1A and Bre1B recognize the acidic patch and the nucleosomal DNA phosphates around SHL 6.0–6.5, which are ideally located to recruit the E2 enzyme and ubiquitin for H2BK120-specific ubiquitination. Mutational experiments suggest that the two RING domains bind in two orientations and that ubiquitination occurs when Bre1A binds to the acidic patch. Our results provide insights into the H2BK120-specific ubiquitination by the Bre1 proteins and suggest that H2B monoubiquitination can be regulated by nuclesomal DNA flexibility.
Similar content being viewed by others

Introduction
Histones undergo various posttranslational modifications such as methylation, acetylation, and ubiquitination of lysine, which collectively regulate essentially all aspects of genome functions1. These modifications induce varied biological outputs depending on their chemical types and locations. Moreover, some histone modifications are known to affect the installation, removal, and biological outputs of other modifications, known as “crosstalk” between modifications2.
Ubiquitination of lysine residues is a major histone modification that regulates various genomic processes such as transcription, replication, and DNA repair3. In humans, histones are monoubiquitinated at multiple lysine residues, including H3K14, H3K18, H3K23, H2AK13, H2AK15, H2AK119, and H2BK120 (corresponding to H2BK123 in yeast). Among them, monoubiquitination of H2BK120 (H2BK120ub) and H2AK119 (H2AK119ub) are involved in transcriptional regulation but play opposite roles; H2BK120ub positively regulates transcription4, whereas H2AK119ub mediates transcriptional repression via the formation of facultative heterochromatin by polycomb group proteins5. H2BK120ub is also important for the repair and replication of genomic DNA4. Mechanistically, H2BK120ub stimulates the catalytic activity of histone H3K4 and H3K79 methyltransferases (Dot1L and MLL/Set1 family complexes, respectively)6,7,8,9,10,11,12,13, thus playing a central role in a modification crosstalk that establishes a transcriptionally active chromatin environment. Moreover, H2BK120ub itself induces an accessible, open chromatin conformation14 and recruits other chromatin regulators such as FACT (a histone chaperone)15 and the Swi/Snf complex (a chromatin remodeling ATPase)16 to regulate transcription elongation.
In yeast, H2BK123 is ubiquitinated by the Bre1 protein, which forms a heterodimer and acts as an E3 ubiquitin ligase enzyme17,18. In humans, two homologs of yeast Bre1 (Bre1A, also known as RNF20, and Bre1B, also known as RNF40) form a heterodimer responsible for H2BK120 ubiquitination, with Rad6A as the E2 enzyme10. The three Bre1 proteins share a C-terminal RING domain (Fig. 1a), which is required for the ubiquitination of H2BK120 in humans and H2BK123 in yeasts17,18,19,20. In yeast Bre1, the RING domain is minimally sufficient for nucleosomal H2BK123 ubiquitination21. Its basic residues are important for the activity22, presumably via interactions with the so-called “acidic patch”, a cluster of acidic residues from H2A and H2B and the hotspot of nucleosome recognition by various chromatin factors23. Structural studies have demonstrated that two histone H2A-specific ubiquitin ligases, the RING1B-Bmi1 complex (ubiquitinating H2AK119) and the BRCA1-BARD1 complex (ubiquitinating H2AK125, K127, and K129), recognize the acidic patch through their basic residues in the RING domains of catalytic subunits24,25,26. On the other hand, although the crystal structures of the RING domain of the yeast Bre1 homodimer27 and human Bre1A28 have been determined, structural insights into the H2BK120-specific ubiquitination by the Bre1 proteins have been elusive.

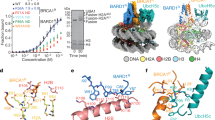
a Domain structures of yeast Bre1 and human Bre1A and Bre1B. An N-terminal helical domain (HD) and two coiled-coil regions (CC1 and CC2) are predicted based on the model structures calculated using AlphaFold. The residue numbers of the domain boundaries are shown. The arrows indicate the regions predicted to form intermolecular interactions. b Cryo-EM density map in two views, colored in accordance with the model in (c). c, d Atomic models of the two RING domains of Bre1A and Bre1B bound to the nucleosome in two views. In (c) (Model I) and (d) (Model II), RINGA and RINGB bind to the acidic patch, respectively. e Structure of the RING domain heterodimer (two views). Six basic residues that are important for nucleosome binding are shown. Zinc ions are shown as a sphere model. f Electrostatic surface potential of the RING domain heterodimer (two views). g Electrostatic surface potential of the nucleosome. The two Bre1-interaction interfaces (the acidic patch and the DNA backbone near SHL6.0) are indicated by green circles.
Accumulating evidence suggests important and complicated roles of the human Bre1 complex and H2BK120ub in cancers29. In many types of cancers, expression of Bre1A or Bre1B is downregulated, and a decrease in global H2BK120ub level is associated with poor prognosis30,31,32,33,34,35,36,37,38,39,40. Thus, Bre1A and Bre1B are generally considered tumor suppressors. On the other hand, in luminal breast cancer cells, Bre1A silencing reduces the proliferation and tumorigenic capacity of the cells, which suggests that it plays a tumor-promoting role31. Bre1A expression is also required for the development of MLL-rearranged leukemia, possibly because it facilitates the transcription elongation of leukemogenic genes driven by MLL-fusion proteins41. Interestingly, the knockdown of Bre1A, but not Bre1B, inhibited the proliferation of MLL-rearranged leukemia cells41. This result indicates the paralog-specific roles of human Bre1 proteins in cancer, but it has not been investigated whether the two Bre1 proteins have asymmetric functions.
To obtain structural insights into H2BK120-specific ubiquitination by Bre1 complexes and gain insights into its regulation and paralog-specific roles, we report the cryo-electron microscopy (cryo-EM) structure of the human Bre1 complex bound to the nucleosome. The structure reveals that the RING domains of Bre1A and Bre1B directly interact with the acidic patch and the DNA phosphates of the nucleosome through their basic residues. One RING domain exhibits flexible interaction with the nucleosomal DNA, suggesting that H2BK120 ubiquitination may be regulated by histone modifications that affect DNA flexibility. Mutational analyses reveal that the Bre1A subunit acts as the catalytic E3 enzyme by recruiting Rad6A and identify key residues for Rad6A binding. The orientation and location of the two RING domains are consistent with the H2BK120 specificity of the Bre1 complex but differ markedly from those of the H2A-specific ubiquitin ligases. Our study provides a structural framework for the regulation of H2BK120 ubiquitination by the Bre1 complex.
Results
Overall structure
We reconstituted the trimeric Bre1A-Bre1B-Rad6A by mixing the coexpressed Bre1A-Bre1B heterodimer and individually expressed Rad6A and confirmed its nucleosome-binding and nucleosomal H2BK120 ubiquitination activities (Supplementary Fig. 1). Using this trimeric complex and in vitro assembled nucleosome with 147-bp DNA, we determined the cryo-EM structure of the Bre1 complex bound to the nucleosome (Supplementary Figs. 2–4, Table 1, and Supplementary Movie 1). When we calculated a reconstruction map from cryo-EM images at 2.8 Å using a conventional method, the density corresponding to the two Bre1 proteins were blurred (local resolution > 5 Å), suggesting a flexible binding mode of the Bre1 complex to the nucleosome. To better resolve the density of the Bre1 complex, we applied 3D Flexible Refinement (3DFlex) analysis42, which uses a motion-based deep neural network model of continuous heterogeneity. We obtained a map (the 3DFlex map) with an improved local resolution of the Bre1 complex (around 4.0 Å) with the latent space dimensionality K = 2, while the overall resolution did not improve. We also determined the structure of the free nucleosome at 2.51 Å resolution as a reference (Fig. 1, Supplementary Figs. 2, 3, and Table 1). Although we used the full-length Bre1A-Bre1B-Rad6A heterotrimer for grid preparation, only the C-terminal RING domains of Bre1A and Bre1B (hereafter designated as RINGA and RINGB, respectively) were visible in the reconstructed cryo-EM maps (Fig. 1a, b). This agrees with the model structures of the yeast and human Bre1 complexes calculated by AlphaFold43,44,45, in which the C-terminal RING domains are preceded by short flexible linkers and appear to move flexibly with respect to the other regions of the complex (Supplementary Fig. 5). Previous studies have reported that the yeast and human Bre1 complexes bind Rad6 proteins via their N-terminal regions13,21, which explains why the density of Rad6A was not observed in the current EM maps.
Consistent with the previous biochemical analysis of the yeast Bre1 complex22, one RING domain of the human Bre1 complex binds to the acidic patch (Fig. 1b–d). Unexpectedly, the other RING domain binds to the DNA phosphates of one DNA strand at SHL 6.0–6.5 (Fig. 1b–d). RINGA and RINGB have positively charged molecular surfaces (Fig. 1e, f); thus, they bind to the negatively charged acidic patch and DNA phosphate groups (Fig. 1g) through electrostatic interactions. Bre1 binding does not induce a large conformational change in the nucleosome, but a region of the nucleosomal DNA that contacts Bre1 undergoes a small shift (approximately 3 Å) (Supplementary Fig. 6). Since the local resolution of this DNA region is relatively low, as often observed in nucleosome structures, the functional significance of this small shift is unclear.
As the sequences of RINGA and RINGB are highly similar (with 86% identity; Supplementary Fig. 7), we were unable to assign the orientation of the two subunits to the density. Thus, we created two atomic models of the RINGA-RINGB complex bound to the nucleosome based on the obtained density. In Model I (Fig. 1c), RINGA and RINGB bind to the acidic patch and DNA phosphates, respectively, whereas in Model II (Fig. 1d), the two RING domains have opposite orientations. Based on our biochemical experiments, we propose that the Bre1 complex can bind to the nucleosome in both orientations and that Model I, in which RINGA binds to the acidic patch, represents the catalytically active form for H2BK120 ubiquitination (see below).
Bre1 activity may be regulated by the flexibility of the nucleosomal DNA
The 3DFlex analysis not only provides a density map with higher local resolution, but it also reveals flexible motions. Briefly, the flexible three-dimensional structure of a protein is represented as deformations of a single canonical density map, using a latent coordinate and a neural flow generator. This deformation results in a convected map that corresponds to a single conformation of the protein. The latent dimensions generated by the 3DFlex analysis represent possible structural flexibility, or motions, of the protein.
Figure 2a shows the distribution of the latent coordinates of the particles used for the 3DFlex analysis, and Fig. 2b–e show the atomic models constructed onto the convected densities on each side of the latent coordinate axes. Supplementary Fig. 8 shows the corresponding densities, and Supplementary Movies 2 and 3 show series of convected densities along the two axes. The two types of movements represented by the two dimensions both represent rotational movements with the region around the arginine anchor as the pivot point. The first dimension represents a larger movement, with 15° rotation of the two RING domains and about 11.6 Å displacement at the outer side of the DNA-binding subunit, while the second dimension represents a smaller movement with about 3° rotation and about 3 Å displacement (Fig. 2d, e). In contrast, the structural change of the nucleosomal DNA is minimal. This observation suggests that on binding to the nucleosome, the acidic-patch binding subunit stably associates with it, while the other subunit interacts with the nucleosomal DNA in a flexible manner.

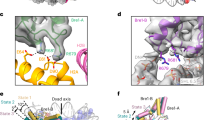
a Distribution of the latent coordinates. Coordinates that correspond to the structures shown in (b–e) are indicated. Green and orange circles represent the volume series shown in Supplementary Movies 2 and 3, respectively. b–e Conformational changes represented by the first (b, c) and second (d, e) dimensions. b, d An overall view. c, e A close-up view. f Residues whose modifications or substitutions (H2BS112GlcNAc, H3K36M, H3K37ac, H3Y41ph, H3R42A, H3K56ac, and H3R42me2a) upregulate Bre1 activity. H2BK120 itself is also shown.
A previous study using semi-synthesized nucleosomes identified multiple histone H3 residues that, when modified or mutated, stimulates H2BK120 ubiquitination46. These modifications and mutations, such as H3Y41 phosphorylation (H3Y41ph) and H3K56 acetylation (H3K56ac), are clustered near the DNA entry/exit site and tend to weaken histone-DNA contacts around the Bre1-bound site (Fig. 2f). Furthermore, addition of a linker histone, which stabilizes the conformation of the nucleosomal DNA, was shown to inhibit H2BK120 ubiquitination46. Based on these results, it was proposed that the local structure of the DNA entry/exit site regulates the ubiquitination activity46 Consistent with this, our 3DFlex analysis revealed the flexible binding of the distal Bre1 subunit to nucleosomal DNA. Taken together, these results suggest that the modifications which weaken histone-DNA contacts have an impact on the flexibility of DNA around SHL 6.0–6.5 region, potentially facilitating the productive binding of the Bre1 complex to the DNA. However, further studies are necessary to test this hypothesis and examine whether such a regulatory mechanism operates in a physiological context.
Binding of the acidic patch and DNA phosphates
The detailed interactions between the acidic patch and RINGA or RINGB are shown in Fig. 3, Supplementary Fig. 9, and Supplementary Movie 4, and the density around this region is shown in Supplementary Fig. 4. Basic residues that are homologous between RINGA and RINGB (Figs. 1e and 3b and Supplementary Figs. 7 and 9) mediate the interactions with the acidic patch as follows. R953A and R978B (Bre1A and Bre1B residues are indicated with superscripted “A” and “B,”, respectively) in Models I and II, respectively, form salt bridges with H2AE61, H2AD90, and H2AE92. These arginine residues serve as the canonical “arginine anchor” in a manner similar to that of many other chromatin factors that bind the nucleosome23. R955A in Model I and R981B in Model II also form salt bridges with H2AE61 and H2AE64 at the position of the so-called variant arginine type 123. In addition, the side chains of K936A in Model I and K962B in Model II are located at the interface with H2B, possibly contributing to nucleosome binding by forming intermolecular interactions.

a Structure of Model I. The magnified regions in (b, c) are indicated. b Interactions between RINGA and the acidic patch in Model I. Hydrophilic interactions (salt bridges and hydrogen bonds) are indicated by gray dotted lines. c Basic residues of RINGB near the DNA phosphates in Model I. d A schematic representation of the protein constructs used in the experiment shown in (e). TS, Twin-Strep-tag. e H2BK120 ubiquitination assay of the wild type (WT) and mutants possessing substitutions at basic residues. The signals were normalized usig the normalization control. The mean and standard deviation of six (for WT) or three (for mutants) independent results are shown. Source data are provided as a Source Data file.
The other RING domain of the Bre1 complex binds the DNA phosphates around SHL 6.0–6.5 (Fig. 3a, c and Supplementary Fig. 9). K962B, R979B, R981B, and R982B in Model I and K936A, R953A, R955A, and K956A in Model II face DNA phosphates. Although the lower local resolution around this region hinders the precise modeling of their side chain atoms (Supplementary Fig. 4), these residues possibly mediate the interaction with DNA by forming salt bridges. We note that among these residues, R953A, R955A, R979B, and R979B mediate interactions with the acidic patch when the Bre1 complex binds to the nucleosome in the opposite direction.
Basic residues of the RING domains important for H2BK120 monoubiquitination
To examine the roles of the basic residues in RINGA and RINGB, we prepared mutant Bre1 complexes carrying alanine mutations at the positions of K936A, R953A, R955A, K962B, R979B, and R981B. To focus on the functional role of the RING domains and facilitate enzymatic analysis, we created constructs expressing RINGA or RINGB fused to Twin-Strep-tag and Rad6A and coexpressed each of them with the GST-fused RING domain of the other Bre1 protein (Fig. 3d and Supplementary Fig. 10a). A similar fusion strategy was used to analyze the activity of the yeast Bre1 complex21,22. To purify the heterodimeric complexes, the coexpressed proteins were purified by serial affinity chromatography steps using Strep-Tactin and glutathione resins and then used for enzymatic analysis.
The results of the enzymatic analysis using GST-RINGA and Twin-Strep-Rad6A-RINGB are shown in Fig. 3e, whereas those of the enzymatic analysis using GST-RINGB and Twin-Strep-Rad6A-RINGA are shown in Supplementary Fig. 10b. All six mutations showed reduced activity, albeit at varying degrees. The K936AA, R953AA, and R955AA mutations almost completely abolished the activity in both combinations. The K962AB and R981AB mutations showed milder effects but still showed reduced activity in both combinations. The R979AB mutation resulted in a mild reduction when GST-RINGA and Twin-Strep-Rad6A-RINGB were used, while it almost completely abolished the activity when GST-RINGB and Twin-Strep-Rad6A-RINGA were used. These results demonstrate the importance of the interaction between the basic residues of the two RING domains and the acidic patch and nucleosomal DNA observed in the current structure for H2BK120 ubiquitination.
Bre1A as a catalytic subunit
The mutational analysis results showed that mutations on Bre1B had milder effects, which suggests that Bre1A and Bre1B play asymmetrical functions in catalysis, even though their sequences are similar. Consistent with this, a previous paper noted that the ectopic expression and knockdown of Bre1A, not Bre1B, affected the H2BK120ub level in HEK293T cells19, although the data on Bre1B was not shown in this paper. Thus, we addressed whether the RING domains of Bre1A and Bre1B may perform asymmetrical functions. Many RING-type E3 enzymes, including the H2A-specific enzymes RING1B and BRCA1, are known to bind non-catalytic RING proteins to form RING domain heterodimers, in which the catalytic RING subunit recruits the E2 enzyme and ubiquitin for site-specific ubiquitination47. To examine if Bre1 proteins can asymmetrically interact with its cognate E2 protein, Rad6A, we first created model structures of Rad6A bound to the Bre1 RING domains by superposing with the structures of a known RING E3-E2-ubiquitin complex, the RNF4-UbcH5A ubiquitin complex48 (PDB ID 4AP4), followed by the superposition of human Rad6A49 (PDB ID 6CYO) on UbcH5A (Fig. 4a, Supplementary Movie 5). Previous reports have created model structures of yeast and human Bre1 complexes with bound Rad6 in a similar manner, which were supported by cross-linking and mass spectrometry analyses or a mutational assay22,28. Our human Bre1-Rad6A model suggests that if RINGA binds Rad6, the interaction may involve Bre1A residues C924A, N926A, M927A, T948A, R949A, T952A, Q954A, K956A, P958A, and K959A. Bre1A and Bre1B have different amino acids only at three (M927A/T953B, T948A/G974B, and T952A/A978B) of these positions (Fig. 4b, Supplementary Fig. 7). Furthermore, in the model structure, the side chains of T948A and T952A are located within the hydrogen-bonding distance of the N65 and K66 side chains of Rad6A (Fig. 4c). These observations suggest that the two positions, T948A/G974B and T952A/A978B, are likely located at the interaction interface with Rad6A and that the amino acids at these positions may be involved in the asymmetrical functions of Bre1A and Bre1B.

a Structural modeling of the RINGA-RINGB-Rad6A complex. The current structure of the RINGA-RINGB complex is superposed on that of the RNF4 dimer bound to UbcH5A and ubiquitin (PDB ID 4AP4, ubiquitin not shown), and then the structure of Rad6A (PDB ID 6CYO) is further superposed on that of UbcH5A. b RINGA residues that are not conserved in RINGB. The RINGA structure is shown as a surface representation, and 10 non-conserved residues are colored red. Three residues near a possible Rad6A binding region are labeled. c Close-up view of the RINGA-RINGB-Rad6A model in (b). d H2BK120 ubiquitination assay of the wild type (WT) and mutants possessing substitutions at the possible Rad6A binding region. The constructs shown in Fig. 2d and their mutants were used. GA: T948GA-T952AA. TT: G974TB-A978TB. The mean and standard deviation of six (for WT) or three (for mutants) independent results are shown. Source data are provided as a Source Data file. e The binding affinities of the WT and mutants for the nucleosome. The constants between the Bre1 complexes and the nucleosome are shown with their 1-sigma confidence interval. The values were calculated with four (for GA + TT) or three (for the others) independent results.
To directly test whether T948A/G974B and T952A/A978B play important roles, we purified Bre1-Rad6 fusion complexes with the Bre1A subunit carrying a single mutation or double Bre1B-like mutations as follows: T948GA, T952AA, or T948GA-T952AA (hereafter referred to as GA). We then analyzed their nucleosomal H2BK120 ubiquitination activity. As shown in Fig. 4d, the T948GA, T952AA, and GA mutants all exhibited significantly reduced activity. To further validate the importance of threonine at these positions, we created two Bre1 complexes, one containing the Bre1B subunit carrying Bre1A-like dual mutations (G974TB-A978TB, hereafter referred to as TT) and the wild-type Bre1A subunit, and the other carrying Bre1B-like Bre1A and Bre1A-like Bre1 subunits (GA + TT). As shown in Fig. 4d, the both mutant complexes exhibited slightly higher activity than the wild-type complex. We speculate that the effect of the inactivating GA mutation in the GA + TA mutant was masked by the activating TT mutation, which for some unknown reason endowed Bre1B with a higher activity than the wild-type Bre1A. We further measured the nucleosome affinities of these mutations by microscale thermophoresis and found that the wild-type complex and the GA and TT mutants had comparable affinities, while the affinity of the GA + TT mutant was reduced (Fig. 4e and Supplementary Fig. 11). These results show that the higher activities of the TT and GA + TT mutants are not the result of the higher nucleosome affinities. Collectively, our mutational analyses strongly suggest that Bre1A functions as a catalytic subunit to recruit the E2-ubiqution complex for H2BK120 mono ubiquitination and that both T948A and T952A are important for Rad6A binding.
A model for H2BK120-specific ubiquitination
Figure 5a and Supplementary Movie 5 show the model structure of the RINGA-RINGB-Rad6A ubiquitin bound to the nucleosome based on Model I, where RINGA binds the acidic patch. In this model, the lysine residue closest to ubiquitin G76 is H2BK120 (Fig. 5b, Supplementary Movie 5), and the distance between the Cα atom of H2BK120 and the carbonyl carbon of ubiquitin G76 is 8.6 Å. The second closest lysine is H2BK116, whose Cα atom is located 10.8 Å away from the carbonyl carbon of ubiquitin G76, and might be too farther away or not ideally oriented for ubiquitin transfer (Fig. 5b). Unlike the C-terminal ubiquitination sites of H2A, which are structurally flexible, H2BK117 and H2BK120 are located on the C-terminal α-helix of H2B. The helical structure should exhibit limited backbone flexibility, which may hinder the access of H2BK117 to the ubiquitination catalytic center. Taken together, our model structure of the RINGA-RINGB-Rad6A-ubiquitin complex bound to the nucleosome is consistent with the H2BK120 specificity of the human Bre1 complex.

a Model structure of the RINGA-RINGB-Rad6A-ubiquitin complex bound to the nucleosome in two views. b Close-up view of ubiquitin and H2B. Two lysine residues (H2BK120 and H2BK116) near G76 of ubiquitin are shown. H2BS112, whose GlcNAcylation stimulates H2BK120 ubiquitination, is also shown. c Proposed mechanistic model. The wild-type Bre1 complex can bind to the nucleosome in two orientations, but H2BK120 ubiquitination occurs only when Bre1A binds to the acidic patch, as RINGA, but not RINGB, can recruit Rad6A and ubiquitin. Bre1B with G974TB-A978TB double substitution can recruit Rad6A and ubiquitin; thus, H2BK120 ubiquitination occurs in both binding modes.
GlcNAcylation at H2BS112 was shown to facilitate H2BK120 ubiquitination by the Bre1 complex46,50. In our model H2BS112 is located close to Rad6A (Fig. 5b). Thus, H2BS112GlcNAc may facilitates H2BK120 ubiquitination by interacting with Rad6, thereby increasing the binding affinity or correctly positioning Rad6A and ubiquitin suitable for ubiquitination.
Our structural model also suggests that the catalytic RING domain should bind to the acidic patch to position the E2 enzyme and ubiquitin near H2BK120 for ubiquitination, as the other RING domain contacting the DNA phosphates is farther away from H2BK120 and faces the opposite direction. Thus, when the wild-type Bre1 complex ubiquitinates H2BK120, the catalytically active RINGA should bind to the acidic patch. Meanwhile, the results of our mutational experiments also showed that when Bre1A-type mutations (G974TB and A978TB) were introduced, RINGB could function as the catalytic E3 enzyme. This suggests that RINGB also binds to the acidic patch, which agrees with the high sequence similarity between RINGA and RINGB and the perfect conservation of the two arginine residues that binds to the acidic patch (R953A/R979B and R955A/R981B).
Based on these observations, we propose a mechanistic model of how the human Bre1 complex ubiquitinates H2BK120 (Fig. 5c). The Bre1 complex interacts with the nucleosome in at least two regions: the acidic patch and the DNA phosphates around SHL 6.0–6.5. The two regions are recognized via the basic surfaces of the two RING domains. The Bre1 complex interacts with the nucleosome in two possible orientations of their RING domains, and H2BK120 ubiquitination occurs only when RINGA binds to the acidic patch. R953A and R955A are the canonical arginine anchor and variant arginine type 1, respectively, for recognizing the acidic patch and are thus essential for H2BK120 ubiquitination. On the other hand, basic residues in RINGB, such as K962B, R979B, and R981B, have a rather supportive role in orienting the catalytic RINGA domain and providing additional affinity for the nucleosome via interaction with the nucleosomal DNA phosphates. When Bre1A-type mutations (G974TB and/or A978TB) are introduced, the mutant Bre1 complex can catalyze H2BK120 ubiquitination in both orientations, as both RING domains can recruit Rad6A and ubiquitin.
Discussion
The structural comparison between the current Bre1 complex (Supplementary Fig. 12a) and two other histone ubiquitin ligases recognizing the nucleosome (the RING1B-Bmi1 complex ubiquitinating H2AK119, shown in Supplementary Fig. 12b, and the BRCA1-BARD1 complex ubiquitinating H2AK125, K127, and K129, shown in Supplementary Fig. 12c) revealed a clear similarity and difference between their nucleosome recognition mechanisms. In all structures, the catalytic RING domain recognizes the acidic patch using the conserved arginine residues as the arginine anchor, clarifying a common mechanism adopted by all structurally characterized histone ubiquitin ligases that recognize the nucleosome. In contrast, the orientation of the catalytic RING domain in the Bre1 complex differs substantially from that of the two H2A ubiquitin ligases, consistent with their different residue specificity. Moreover, the non-catalytic RING domains interact with the nucleosome differently. Unlike the Bre1 complex, whose non-catalytic RING domain (RINGB) binds to DNA phosphates, the non-catalytic RING domains of the two H2A ubiquitin ligases (Bmi1 and BARD1) do not possess the basic surface and thus do not bind to DNA. Instead, Bmi1 contacts residues 76–79 of histone H3, whereas BARD1 interacts mainly with the H2B/H4 cleft, resulting in distinct residue specificities on the H2A C-terminal region. In summary, the two H2A ubiquitin ligases bind to nucleosomes in similar but distinct ways to specifically ubiquitinate different H2A lysine residues, whereas the Bre1 complex binds in a nearly opposite direction for H2BK120-specific ubiquitination.
To date, our understanding of the functional differences between two Bre1 proteins in human has been limited. However, a previous study on MLL-rearranged leukemia suggested they have asymmetric functions41. Consistently, by mutational experiment here we have identified Bre1A, but not Bre1B, as the catalytic subunit responsible for recruiting the cognate E2 enzyme (Rad6A) and ubiquitin. Supporting our findings, data from the DepMap database51 reveals that the knockout of Bre1A has a larger impact on the survival of cancer cell lines compared to the knockout of Bre1B (average gene effect scores of −0.82 and −0.44, respectively) (Supplementary Data 1). Further studies are required to fully comprehend the distinct activities and physiological roles of the two Bre1 proteins in human under both normal and pathogenic conditions.
While this manuscript was under review, a structural study on yeast and human Bre1 complexes bound to the nucleosome was reported by Deng et al.52. Another study on the yeast Bre1-nucleosome complex was also reported as a preprint by Zhao et al.53. Deng et al. used a complex chemical trapping strategy to covalently attach the Bre1 complex and Rad6 to the nucleosome, while Zhao et al. used a fusion protein containing two tandem RING domains of Bre1 followed by Rad6. Nevertheless, the structures reported by these studies are very similar to ours using the full-length human Bre1 complex without any protein engineering; one RING subunit binds stably to the acidic patch via arginine anchors, and the other binds flexibly to the nucleosomal DNA. The three studies complement with each other to show that the effects of a chemical trapping strategy and fusion formation on the structures are minimal, and together reveal the mechanism of specific H2B ubiquitination conserved between human and yeast. It should also be noted that Deng et al. modeled the heterodimeric Bre1 complex in one orientation with the Bre1A subunit bound to the acidic patch. Since our structural and biochemical analyses have now identified Bre1A as the likely catalytic subunit, the structure reported by Deng et al. is assumed to represent a catalytically active state, providing an insight into the Bre1A-Rad6A interactions. In this structure, T948A and T952A are located proximal to Rad6A, consistent with our proposal that these two threonine residues are responsible for Rad6A binding.
Methods
AlphaFold calculation
The model structures of the full-length yeast Bre1 homodimer and human Bre1 heterodimer were calculated using the program LocalColabFold45, with AlphaFold243 as the calculation engine.
Protein and DNA sequences
The protein and nucleosomal DNA sequences used in this study are shown in Supplementary Data 2.
Purification of the Bre1A-Bre1B-Rad6A complex
The full-length human Rad6A sequence was cloned into a modified pET32 plasmid (Roche) and expressed as a fusion protein with the N-terminal thioredoxin-His6-SUMOstar tag. The plasmid was introduced in Rosetta2(DE3) pLysS cells, and the protein expression was induced by adding 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) and culturing overnight at 20 °C. The harvested cells were suspended in 20 mM Na phosphate at pH 7.3, 500 mM NaCl, 1 mM DTT, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 1% Triton X-100 and disrupted by sonication. Clarified lysate was loaded onto a cOmplete His-Tag column (Roche). The protein was eluted with 20 mM Na phosphate at pH 7.3, 500 mM NaCl, 1 mM DTT, and 500 mM imidazole. For the reconstitution of the Bre1A-Bre1B-Rad6A complex, the tag-fused Rad6A was flash-frozen in liquid nitrogen and stored at −80 °C. For the histone H2B ubiquitination assay, the tag-fused Rad6A was mixed with SUMOstar protease and dialyzed against 20 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 1 mM DTT overnight. The digested sample was then mixed with an equal amount of water and loaded onto tandemly connected HisTrap HP and HiTrap Q columns (Cytiva) equilibrated with 20 mM Tris-HCl at pH 7.5, 100 mM NaCl, 20 mM imidazole, and 1 mM DTT. After disconnecting the HisTrap column, the bound Rad6A protein was eluted from the HiTrapQ column with a linear gradient from 100 mM to 1 M of NaCl in 20 mM Tris-HCl at pH 7.5, 20 mM imidazole, and 1 mM DTT. The fractions containing Rad6A were pooled, concentrated, buffer exchanged to 10 mM HEPES-Na at pH 7.5, 150 mM NaCl, 1 mM DTT, flash-frozen with liquid nitrogen, and stored at −80 °C.
For the structural study, the full-length sequences of human Bre1A and Bre1B were first individually cloned into pACEBac1 (Geneva Biotech) with the N-terminal His6-SUMOstar tag and the N-terminal Twin-Strep-His6-SUMOstar tag, respectively. The two expression cassettes were then integrated into a single pACEBac1 plasmid to construct a bicistronic vector expressing both proteins. Baculovirus production in Sf9 cells was conducted using the Bac-to-Bac system (ThermoFisher), in accordance with the manufacturer’s instructions. The full-length Bre1A-Bre1B complex was expressed using the bicistronic baculovirus in High Five cells, which were collected 48 h after viral infection. The cell pellets were suspended in 20 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM DTT, 0.1 m PMSF, 0.5% Triton X-100, and cOmplete ULTRA protease inhibitor cocktail (Roche) and disrupted by sonication. Clarified lysate was loaded onto a Strep-Trap HP column (Cytiva), and the complex was eluted in a buffer containing 20 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM DTT, and 2.5 mM desthiobiotin. To reconstitute the trimeric Bre1A-Bre1B-Rad6A complex, the eluted fractions containing the tagged Bre1A-Bre1B complex were mixed with the tag-fused Rad6A and SUMOstar protease and incubated at 4 °C overnight. The tag-removed, reconstituted ternary complex was then loaded onto a HiTrap Heparin column (Cytiva) equilibrated with 20 mM Tris-HCl at pH 7.5, 100 mM NaCl, and 1 mM DTT, and eluted with a linear gradient from 100 mM to 1 M NaCl in 20 mM Tris-HCl at pH 7.5 and 1 mM DTT. The complex was further purified with a Superose 6 column (Cytiva) equilibrated with 20 mM HEPES-Na at pH 7.5, 200 mM NaCl, and 1 mM DTT, concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C.
The DNA sequences encoding the RING domain of Bre1A or Bre1B were cloned into pGEX-6P-1 (Cytiva) to express GST-RINGA or GST-RINGB and cloned into a modified pET plasmid (Merck) to express Twin-Strep-Rad6A-RINGA or Twin-Strep-Rad6A-RINGB. To prepare the RING domain heterodimers for ubiquitination assay, two plasmids (pGEX-6P-RINGA and pET-Twin-Strep-Rad6A-RINGB or pGEX-6P-RINGB and pET-Twin-Strep-Rad6A-RINGA) were simultaneously introduced into Rosetta2(DE3)pLysS cells. Protein expression was induced with 0.2 mM IPTG and overnight culture at 16 °C. The harvested cells were suspended in 40 mM K2HPO4, 10 mM KH2PO4, 500 mM NaCl, 1 mM DTT, 0.1 mM PMSF, and 0.5% Triton X-100 and lysed by sonication. The clarified lysate was applied to a StrepTrap column (Cytiva), and the bound heterodimers were eluted with 50 mM Tris-HCl at pH 7.5, 500 mM NaCl, 1 mM DTT, 2.5 mM desthiobiotin. The eluents were further purified with a GSTrap column (cytiva) and eluted with 50 mM Tris-HCl at pH 8.0, 500 mM NaCl, and 10 mM reduced glutathione. The purified heterodimers were flash-frozen in liquid nitrogen and stored at −80 °C. To express the His6-Twin-Strep-Rad6A-RINGB for microscale themophoresis experiments, a DNA sequence encoding six histidine residues was inserted into pET-Twin-Strep-Rad6A-RINGB The RING domain heterodimers with the His6 tag were expressed and purified in the same way as those without the His6 tag.
The plasmids encoding GST- and His-tagged human E1 (pGEX-6P-HsE1-His8) and ubiquitin (pET26b-Ub) were kind gifts from Dr. Yusuke Sato (Tottori University). pGEX-6P-HsE1-His8 was introduced into Rosetta2(DE3)pLysS cells, and protein expression was induced by adding 0.3 mM IPTG and culturing overnight at 20 °C. The harvested cells were suspended in 40 mM K2HPO4, 10 mM KH2PO4, 20 mM imidazole, 500 mM NaCl, 1 mM DTT, 0.1 mM PMSF, and 0.5% Triton X-100 and lysed by sonication. The clarified lysate was applied to a HisTrap column, and the protein was eluted with a linear gradient from 20 mM to 500 mM of imidazole in 40 mM K2HPO4, 10 mM KH2PO4, 500 mM NaCl, and 1 mM DTT. Fractions containing E1 were dialyzed against 20 mM Tris-HCl at pH 7.5 and 50 mM NaCl overnight. The dialyzed sample was applied to a HiTrap Q column (Cytiva) and eluted with a linear gradient from 50 mM to 500 mM NaCl in 20 mM Tris-HCl at pH 7.5. The eluted sample was applied to a GSTrap column; eluted with 50 mM Tris-HCl at pH 8.0, 50 mM NaCl, and 10 mM glutathione; flash-frozen in liquid nitrogen; and stored at −80 °C.
Nucleosome reconstitution
The nucleosome was reconstituted with human core histones and 147- or 185-bp DNA fragments containing the Widom 601 spacing sequence, as described previously54. The 185-bp (final 6.1 μM) or 147-bp (final 5.6 μM) DNA fragment was mixed with a histone octamer at a molar ratio of ~1:1.1 and then dialyzed against 125 mL of 10 mM HEPES-Na pH 7.5, 2 M KCl, and 1 mM DTT for 1 h. Thereafter, 875 mL of 10 mM HEPES-Na pH 7.5 and 1 mM DTT was gradually added to facilitate nucleosome reconstitution. The nucleosome samples were further dialyzed against 10 mM HEPES-Na pH 7.5, 50 mM KCl, and 1 mM DTT. Finally, the centrifuged supernatant was concentrated and stored at 4 °C.
Electrophoretic analysis of the Bre1-nucleosome complex
To monitor the formation of the Bre1-nucleosome complex, the nucleosome and trimeric Bre1 complex were mixed at different molar ratios and NaCl concentrations in 20 mM HEPES-Na at pH 7.5, 1 mM DTT, and 0.5 mM EDTA. The mixtures were incubated for an hour at 4 °C and then analyzed using electrophoresis on non-denaturing 4% acrylamide gel in 0.5% TBE buffer at 150 V for 40 minutes. The gels were stained with SYBR Gold (Thermo Fisher).
Ubiquitination assay
For the ubiquitination assay using the trimeric Bre1A-Bre1B-Rad6A complex, 1.2 μM 147- or 185-bp nucleosome, 100 nM E1, 0.5–1.5 μM Bre1 complex, and 36 μM ubiquitin were mixed in 50 mM Tris-HCl at pH 8.0, 50 mM NaCl, 50 mM KCl, 3 mM ATP, 10 mM MgCl2, and 1 mM DTT and incubated at 30 °C for 90 min. For the ubiquitination assay of the truncated RINGA-RINGB heterodimers, 1 μM 147-bp nucleosome, 100 nM E1, 10 μM RINGA-RINGB heterodimer or its mutants, and 36 μM ubiquitin were mixed in 50 mM Tris-HCl at pH 8.0, 125 mM NaCl, 3 mM ATP, 10 mM MgCl2, 1 mM DTT, and incubated at 30 °C for 1, 2, or 4 hours. Reactions were stopped by the addition of 3× SDS loading buffer and analyzed by Western blotting or using Wes (ProteinSimple). For Western blotting, Ubiquityl-Histone H2B (Lys120) (D11) Rabbit mAb (No. 5546; Cell Signaling) was used as the primary antibody (1:1000 dilution), goat anti-rabbit IgG-HRP (sc-2004; Santa Cruz Biotechnology) as the secondary antibody (1:2000 dilution), and ECL Prime (Cytiva) as the chemiluminescent reagent. The images were recorded using LAS-3000 mini (Fujifilm). For Wes analysis, Ubiquityl-Histone H2B (Lys120) (D11) Rabbit mAb was used as the primary antibody (1:250 dilution), and Anti-Rabbit Detection Module (DM-001; ProteinSimple) was used without dilution following the manufacturer’s protocol. As the normalization control, a single reaction mix using the wide type RINGA-RINGB heterodimer was created in a large volume and loaded each time. The data were quantified using Compass for SW v 6.2.0 (ProteinSimple).
Microscale thermophoresis
The microscale thermophoresis assay was performed using a Monolith NT.115 instrument (NanoTemper Technologies). For the MST assay, the His6-tagged RING domain heterodimers (200 nM) labeled with RED-Tris-NTA and a series of twofold-diluted nucleosome or DNA (from 0.82 nM to 26.5 μM) were incubated in the binding buffer (10 mM HEPES-Na pH 7.5, 25 mM KCl, 10% glycerol, 0.05% Tween 20, and 1 mM DTT) for 30 min at room temperature and centrifuged at 15,000 × g for 10 min. Premium capillaries were filled with the samples to obtain measurements with an extinction power of 80-100% and medium MST power at 25 °C. Thermophoresis data were analyzed using MO. Affinity Analysis software ver. 2.3 (NanoTemper Technologies).
Cryo-EM sample preparation and data acquisition
Th nucleosome (147 bp DNA) and Bre1 complex (Bre1A-Bre1B-Rad6A) were mixed in a 1:4 molar ratio. Final concentrations of 4.5 µM NCP(400 µg/mL dsDNA) and 18 µM BRE1 complex were incubated in buffer (20 mM HEPES at pH 7.5, 30 mM NaCl, 1 mM DTT, and 1 mM EDTA) at 4 °C for an hour. The sample was then diluted three times, and 3 µl of the diluted sample was applied onto Cu-Rh grids (quantifoil) and plunge-frozen in liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific). The images were obtained with a 300 kV Titan Krios G3i microscope (Thermo Fisher Scientific) equipped with a K3 direct electron detector (Gatan), installed at the University of Tokyo, Japan. The data sets were acquired with the SerialEM software55, with a defocus range of −0.8 to −1.8 µm. The data acquisition statistics are shown in Table 1.
Cryo-EM data processing
The cryo-EM data were processed using cryoSPARC v4.2.156 (Supplementary Fig. 2). Particles were automatically picked with a blob picker module using 500 motion-corrected, CTF-estimated micrographs, and then subjected to multiple rounds of two-dimensional (2D) classification. The resultant particles were used for the training by Topaz57, which was used to pick particles from all micrographs. After several rounds of 2D classifications on the Topaz-picked particles, multiple rounds of ab initio reconstruction and heterogenous refinement with cryoSPARC were conducted to remove junk particles. The resultant particles were used for Non-uniform refinement followed by 3D classification without focus mask to be classified into 10 classes, of which two showed a clear density for the two RING domains of the Bre1 complex. The particle belonging to the two classes (83,434 particles) were combined and subjected to Non-uniform refinement, yielding a 2.82 Å map of the Bre1-nucleosome complex. The particles belonging to the other eight classes (341,993 particles) were combined, subjected to a 2D classification followed by Non-uniform refinement, yielding 2.51 Å map of the free nucleosome. The resolution was estimated based on the gold standard Fourier shell correlation curve (FSC) at a 0.143 criterion.
3DFlex analysis
The 3DFlex analysis was performed using CryoSPARC. For the 3DFlex training, the particles were downsampled to a box size of 128 pixels (from 320 pixels), with the Nyquist limit of 5.25 Å. A tetrahedral mesh was created using default parameters. The training was performed with two latent dimensions and the overall rigidity of 0.1. The 3DFlex map used for the model building was calculated with the trained model and the original, non-downsampled particles.
Model building and refinement
A model structure of the RINGA-RINGB heterodimer (calculated using AlphaFold2) and the nucleosome structure (PDB 1KX5) were fit into the density using UCSF Chimera58 and then manually modified with Coot59. Real-space refinement was performed using the phenix.real_space_refine module in Phenix against the 3DFlex map for the Bre1-nucleosome complex and against the Non-uniform map (without sharpening) for the free nucleosome, with the nonbonded_weight parameter set to 200 to reduce clashes.
Figure preparation
Structural figures were prepared using PyMOL v2.5.0 (https://pymol.org/2/) and ChimeraX v1.460.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps were deposited to the Electron Microscopy Data Bank under accession code EMD-34274 and EMD-34275. The atomic coordinates were deposited to the Protein Data Bank under accession code 8GUI (Model I), 8GUJ (Model II), and 8GUK (Free nucleosome). Source data are provided with this paper.
References
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Zhang, T., Cooper, S. & Brockdorff, N. The interplay of histone modifications – writers that read. EMBO Rep. 16, 1467–1481 (2015).
Vaughan, R. M., Kupai, A. & Rothbart, S. B. Chromatin Regulation through Ubiquitin and Ubiquitin-like Histone Modifications. Trends Biochem. Sci. 46, 258–269 (2021).
Fuchs, G. & Oren, M. Writing and reading H2B monoubiquitylation. Biochimica et Biophysica Acta (BBA) - Gene Reg Mechan. 1839, 694–701 (2014).
Schuettengruber, B., Bourbon, H.-M., Di Croce, L. & Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57 (2017).
Sun, Z.-W. & Allis, C. D. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108 (2002).
Dover, J. et al. Methylation of Histone H3 by COMPASS Requires Ubiquitination of Histone H2B by Rad6. J. Biol. Chem. 277, 28368–28371 (2002).
Briggs, S. D. et al. Trans-histone regulatory pathway in chromatin. Nature 418, 498–498 (2002).
Ng, H. H., Xu, R.-M., Zhang, Y. & Struhl, K. Ubiquitination of Histone H2B by Rad6 Is Required for Efficient Dot1-mediated Methylation of Histone H3 Lysine 79. J. Biol. Chem. 277, 34655–34657 (2002).
Zhu, B. et al. Monoubiquitination of Human Histone H2B: The Factors Involved and Their Roles in HOX Gene Regulation. Mol. Cell 20, 601–611 (2005).
Lee, J.-S. et al. Histone Crosstalk between H2B Monoubiquitination and H3 Methylation Mediated by COMPASS. Cell 131, 1084–1096 (2007).
McGinty, R. K., Kim, J., Chatterjee, C., Roeder, R. G. & Muir, T. W. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812–816 (2008).
Kim, J. et al. RAD6-Mediated Transcription-Coupled H2B Ubiquitylation Directly Stimulates H3K4 Methylation in Human Cells. Cell 137, 459–471 (2009).
Fierz, B. et al. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 7, 113–119 (2011).
Pavri, R. et al. Histone H2B Monoubiquitination Functions Cooperatively with FACT to Regulate Elongation by RNA Polymerase II. Cell 125, 703–717 (2006).
Shema-Yaacoby, E. et al. Systematic Identification of Proteins Binding to Chromatin-Embedded Ubiquitylated H2B Reveals Recruitment of SWI/SNF to Regulate Transcription. Cell Rep. 4, 601–608 (2013).
Wood, A. et al. Bre1, an E3 Ubiquitin Ligase Required for Recruitment and Substrate Selection of Rad6 at a Promoter. Mol. Cell 11, 267–274 (2003).
Hwang, W. W. et al. A Conserved RING Finger Protein Required for Histone H2B Monoubiquitination and Cell Size Control. Mol. Cell 11, 261–266 (2003).
Kim, J., Hake, S. B. & Roeder, R. G. The Human Homolog of Yeast BRE1 Functions as a Transcriptional Coactivator through Direct Activator Interactions. Mol. Cell 20, 759–770 (2005).
Kim, J. & Roeder, R. G. Direct Bre1-Paf1 Complex Interactions and RING Finger-independent Bre1-Rad6 Interactions Mediate Histone H2B Ubiquitylation in Yeast. J. Biol. Chem. 284, 20582–20592 (2009).
Turco, E., Gallego, L. D., Schneider, M. & Köhler, A. Monoubiquitination of Histone H2B Is Intrinsic to the Bre1 RING Domain-Rad6 Interaction and Augmented by a Second Rad6-binding Site on Bre1. J. Biol. Chem. 290, 5298–5310 (2015).
Gallego, L. D. et al. Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6–Bre1. Proc. Natl. Acad. Sci. USA. 113, 10553–10558 (2016).
McGinty, R. K. & Tan, S. Principles of nucleosome recognition by chromatin factors and enzymes. Curr. Opin. Struc. Biol. 71, 16–26 (2021).
McGinty, R. K., Henrici, R. C. & Tan, S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 (2014).
Witus, S. R. et al. BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-020-00556-4 (2021).
Hu, Q. et al. Mechanisms of BRCA1–BARD1 nucleosome recognition and ubiquitylation. Nature 596, 438–443 (2021).
Kumar, P. & Wolberger, C. Structure of the yeast Bre1 RING domain. Proteins 83, 1185–1190 (2015).
Foglizzo, M., Middleton, A. J. & Day, C. L. Structure and Function of the RING Domains of RNF20 and RNF40, Dimeric E3 Ligases that Monoubiquitylate Histone H2B. J. Mol. Biol. 428, 4073–4086 (2016).
Marsh, D. J., Ma, Y. & Dickson, K.-A. Histone Monoubiquitination in Chromatin Remodelling: Focus on the Histone H2B Interactome and Cancer. Cancers 12, 3462 (2020).
Prenzel, T. et al. Estrogen-Dependent Gene Transcription in Human Breast Cancer Cells Relies upon Proteasome-Dependent Monoubiquitination of Histone H2B. Cancer Res. 71, 5739–5753 (2011).
Tarcic, O. et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 24, 694–704 (2017).
Shema, E. et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 22, 2664–2676 (2008).
Urasaki, Y., Heath, L. & Xu, C. W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 7, e36775 (2012).
Dickson, K.-A. et al. The RING finger domain E3 ubiquitin ligases BRCA1 and the RNF20/RNF40 complex in global loss of the chromatin mark histone H2B monoubiquitination (H2Bub1) in cell line models and primary high-grade serous ovarian cancer. Hum. Mol. Genet. 25, 5460–5471 (2016).
Hooda, J. et al. Early Loss of Histone H2B Monoubiquitylation Alters Chromatin Accessibility and Activates Key Immune Pathways That Facilitate Progression of Ovarian Cancer. Cancer Res. 79, 760–772 (2019).
Tarcic, O. et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 14, 1462–1476 (2016).
Zhang, K. et al. Loss of H2B monoubiquitination is associated with poor-differentiation and enhanced malignancy of lung adenocarcinoma: Loss of H2Bub1 enhances lung adenocarcinoma malignancy. Int. J. Cancer 141, 766–777 (2017).
Wang, Z.-J. Decreased histone H2B monoubiquitination in malignant gastric carcinoma. WJG 19, 8099 (2013).
Lee, J. H. et al. RNF20 Suppresses Tumorigenesis by Inhibiting the SREBP1c-PTTG1 Axis in Kidney Cancer. Mol. Cell Biol. 37, e00265–17 (2017).
Hahn, M. A. et al. The tumor suppressor CDC73 interacts with the ring finger proteins RNF20 and RNF40 and is required for the maintenance of histone 2B monoubiquitination. Hum. Mol. Genet. 21, 559–568 (2012).
Wang, E. et al. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc. Natl. Acad. Sci. 110, 3901–3906 (2013).
Punjani, A. & Fleet, D. J. 3DFlex: determining structure and motion of flexible proteins from cryo-EM. Nat. Methods, https://doi.org/10.1038/s41592-023-01853-8 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, R. et al. “Protein complex prediction with AlphaFold-Multimer” (preprint, Bioinformatics, 2021); https://doi.org/10.1101/2021.10.04.463034.
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Wojcik, F. et al. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun. 9, 1394 (2018).
Buetow, L. & Huang, D. T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642 (2016).
Plechanovová, A., Jaffray, E. G., Tatham, M. H., Naismith, J. H. & Hay, R. T. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (2012).
de Oliveira, J. F. et al. Mechanistic insights revealed by a UBE2A mutation linked to intellectual disability. Nat. Chem. Biol. 15, 62–70 (2019).
Fujiki, R. et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480, 557–560 (2011).
Tsherniak, A. et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017).
Deng, Z. et al. Mechanistic insights into nucleosomal H2B monoubiquitylation mediated by yeast Bre1-Rad6 and its human homolog RNF20/RNF40-hRAD6A. Mol. Cell 83, 3080–3094.e14 (2023).
Zhao, F., Hicks, C. W. & Wolberger, C. Mechanism of histone H2B monoubiquitination by Bre1. bioRxiv, 2023.03.27.534461 (2023).
Sato, K. et al. Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat Commun. 12, 6605 (2021).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struc. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Pettersen, E. F. et al. UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
We thank Dr. Yusuke Sato (Tottori University), who kindly provided the plasmids encoding human E1 and ubiquitin. This study was supported by a grant from the Japan Society for the Promotion of Science KAKENHI (grant No. 21H05161) to T.S. The study was partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research), funded by the Japan Agency for Medical Research and Development (grant No. JP20am0101115, support No. 1061).
Author information
Authors and Affiliations
Contributions
T.S. conceived the project. S.O., K.U., C.O., S.K., and T.S. prepared the samples. S.O., K.S., T.N., O.N., and T.S. collected cryo-EM images. S.O., K.S., K. H., and T.S. solved the cryo-EM structures, with T.N.‘s advice. S.O., K.U., C.O., and T.S. performed the biochemical analyses. S.O., K.U., K.O., and T.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Onishi, S., Uchiyama, K., Sato, K. et al. Structure of the human Bre1 complex bound to the nucleosome. Nat Commun 15, 2580 (2024). https://doi.org/10.1038/s41467-024-46910-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46910-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
