
Abstract
The histamine H4 receptor (H4R) plays key role in immune cell function and is a highly valued target for treating allergic and inflammatory diseases. However, structural information of H4R remains elusive. Here, we report four cryo-EM structures of H4R/Gi complexes, with either histamine or synthetic agonists clobenpropit, VUF6884 and clozapine bound. Combined with mutagenesis, ligand binding and functional assays, the structural data reveal a distinct ligand binding mode where D943.32 and a π-π network determine the orientation of the positively charged group of ligands, while E1825.46, located at the opposite end of the ligand binding pocket, plays a key role in regulating receptor activity. The structural insight into H4R ligand binding allows us to identify mutants at E1825.46 for which the agonist clobenpropit acts as an inverse agonist and to correctly predict inverse agonism of a closely related analog with nanomolar potency. Together with the findings regarding receptor activation and Gi engagement, we establish a framework for understanding H4R signaling and provide a rational basis for designing novel antihistamines targeting H4R.
Similar content being viewed by others


Introduction
Histamine, a biogenic amine chemical messenger, plays pivotal roles in various physiological and pathophysiological processes through binding and activating histamine receptors (H1R-H4R), members of the G-protein coupled receptors (GPCRs) superfamily1,2. H1R is the first identified histamine receptor and is closely linked to allergic reactions in humans. H1R is widely expressed throughout the body and primarily signals through the Gq pathway1. It has been successfully targeted for allergic disorders, leading to the development of a range of blockbuster drugs for alleviating allergic symptoms. Similarly, H2R is also widely expressed in the body and predominantly signals through the Gs pathway1. H2R has emerged as a successful target for blockbuster antihistamine drugs, revolutionizing the treatment of gastric acid-related conditions such as stomach ulcers.
On the other hand, H3R is mainly expressed in the central nervous system and is involved in e.g. cognitive functions, sleep-wake regulation, and energy homeostasis3. In 2014, pitolisant (Wakix®), an inverse agonist of H3R, received approval for the treatment of refractory narcolepsy4. Following the complete sequencing of the human genome, the H4R is the most recently identified histamine receptor. It is mainly expressed in immune cells including eosinophils, T cells, dendritic cells, basophils, and mast cells1,5. H4R plays a crucial role in mediating immune cell migration, cytokine release, and IL-17 production. Notably, H4R is specifically associated with prevailing inflammatory conditions such as psoriasis, atopic dermatitis, asthma, inflammatory bowel disease (IBD), and arthritis. Due to its significant impact on immune cells, H4R is seen as an attractive target for the treatment of these inflammatory diseases. Both H3R and H4R predominantly couple with Gi/o proteins1.
The successes of therapeutic targeting H1R, H2R, and H3R have inspired and fueled the field to develop numerous H4R antagonists and agonists for associated diseases. JNJ7777120 was first generated as a selective antagonist for H4R based on its ability to inhibit histamine-induced Gi activation6. It exhibits 1000-fold selectivity over H1R, H2R, and H3R. JNJ7777120 blocks histamine-induced chemotaxis in mouse mast cells and neutrophil infiltration, suggesting its potential for the treatment of inflammatory diseases. Later, JNJ7777120 was discovered to have the ability to recruit β-arrestin without activating G proteins7, i.e. being a biased agonist for H4R8, offering a promising approach to minimize undesired side effects. Several compounds targeting H4R have entered clinical trials for asthma, IBD, and arthritis, but so far not one has prevailed9.
In contrast to H1R, H2R, and H3R, structural information for H4R is currently absent. The first reported histamine receptor structure is the x-ray structure of antagonist doxepin-bound H1R solved in 201110. The structure reveals the overall framework of H1R and the mode of antagonist binding which serves as a template for designing and developing H1R antihistamines. It took a decade for the active cryo-EM structure of H1R to appear11. The active structure reveals a shrinkage of ligand binding pocket size via the ionic interaction of histamine with key residues in transmembrane helix 3, 6, and 7 (TM3,6 and 7), followed by an outward movement of TM6 to open the intracellular cavity for the Gq-protein to engage the GPCR, as the main mechanism for H1R activation. Later, a cryo-electron microscopy (cryo-EM) structure of antagonist famotidine-bound H2R via a fusion strategy12 and a crystal structure of antagonist PF03654746-bound H3R13 were subsequently reported, revealing the inactive conformations of H2R and H3R. Most recently, the cryo-EM structures of apo and antihistamine-bound H1R were reported14.
In this work, we resolve the cryo-EM structures of H4R/Gi complexes bound with the endogenous ligand histamine, and synthetic agonists clobenpropit, VUF6884, and clozapine. Through a combination of ligand binding experiments and functional assays, we reveal a distinctive ligand binding mode of H4R that significantly differs from that of H1R. We further uncover the mechanism of receptor activation and Gi coupling. The information unveiled by our study is not only essential for the molecular understanding of H4R signaling but also for the development of novel compounds that target H4R to treat inflammatory diseases.
Results
Overall architecture of H4R/Gi complex
We co-expressed human H4R with Gi1 protein, together with antibody fragment scFv16 that specifically recognizes the N-terminus of Gαi1, in Spodoptera frugiperda (Sf9) insect cells and purified the complex by a conventional membrane protein purification method of our lab11 (Supplementary Fig. 1, for details see methods). We solved the H4R/Gi complex bound with histamine, clobenpropit, VUF6884 and clozapine at resolutions of 3.07 Å, 3.06 Å, 3.01 Å and 3.21 Å, respectively, by the gold standard of FSC = 0.143 (Table 1 and Supplementary Fig. 2). The overall structures of the H4R/Gi complexes closely resemble the conventional GPCR/G-protein complex, where the receptor/G-protein interaction primarily occurs through the Gα subunit of Gi (Fig. 1). Local resolution analysis shows that the core of the Gβ subunit, the transmembrane domains of the GPCR and the Gi interface of scFv16 have the highest resolution. The alpha-helical domain (AHD) of Gαi is missing from the density map due to its high flexibility in the nucleotide-free state. The good density map of the receptor allows us to unequivocally assign residue 14–372 while lacking amino acids 204-292 of the intracellular loop 3 (ICL3) due to the high flexibility of this region. Of note, the clozapine-bound H4R/Gi complex has a slightly lower resolution than the histamine-, clobenpropit- and VUF6884-bound H4R/Gi complexes.

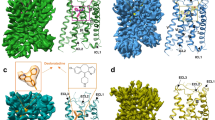
a–d Histamine-bound H4R/Gi, Clobenpropit-bound H4R/Gi, VUF6884-bound H4R/Gi, and Clozapine-bound H4R/Gi, respectively. Left panel, orthogonal views of the cryo-EM density map; right panel, model of the complex in the same view and color scheme as shown in the left panel. Ligands, histamine, Clobenpropit, VUF6884, and Clozapine were shown in a stick model with a density map (contour level of 0.4 in chimera) and in actual chemical structure in the middle of each sub-figure.
Histamine binding
The natural ligand, histamine, is well resolved in the ligand binding pocket of H4R, surrounded by D943.32, Y953.33, C983.36, Q3477.42, Y3196.51, F3447.39, and W3487.43. Histamine establishes direct polar interaction with D943.32, Y953.33, and F3447.39 (Fig. 2a, b). When bound to the receptor, histamine can act as a dication molecule where both the imidazole ring and the amine of the tails are charged15. The positively charged imidazole ring is positioned such that next to an interaction with D943.32, it also forms a cation-π interaction with F3447.39. We have additionally observed a small density adjacent to E1825.46, which is insufficient to accommodate a histamine molecule. However, based on its abundance in cells, we assigned this density as a phosphate ion, effectively linking the positively charged primary amine of histamine to E1825.46 through hydrogen-bond interaction. An alignment of the key residues of the pocket among the histamine receptor family shows that the pocket is more conserved in H3R and H4R than in H1R and H2R (Fig. 2c). Much to our surprise, histamine uses different strategies to engage H1R and H4R. In binding to H1R, the imidazole ring is orientated to TM5 and TM3, forming direct polar interactions with N1985.46 and T1123.37; in contrast, for H4R binding, the imidazole ring takes a completely opposite direction and is orientated to TM7 to interact with F3447.39 (Fig. 2d and Supplementary Fig. 3c). A key difference of the interaction network is the involvement of N1985.46 for histamine binding in H1R, while E1825.46 is distant from histamine and does not directly engage the ligand in H4R. A comparison of monoamine ligand binding modes shows that dopamine, adrenaline, and serotonin use identical modes as histamine in H1R to engage their respective GPCRs, while histamine in H4R uses a completely opposite binding orientation (Fig. 2d and Supplementary Fig. 3e, f). Despite the engaging difference in binding modes, all monoamines form key polar interactions with the conserved D3.32, highlighting the well-accepted importance of this residue in monoamine ligand binding.

a The ligand binding pocket of histamine. b A schematic map of histamine/receptor interaction. Green color, hydrophobic interaction; purple color, polar interaction. c Conservation of key residues of the ligand binding pocket among histamine receptor family. d A comparison of histamine binding between H4R and H1R (PDB:7dfl). e [3H] histamine and [3H] JNJ7777120 binding for H4R mutants. f pKi of histamine binding of H4R mutants. g pEC50 of Gi activation of H4R mutants by histamine. From e to g, data are presented as mean values ± SEM.; n = 4–9 independent experiments for e, n = 3–4 independent experiments for f, and n = 3–4 independent experiments for g. Each point in the figure represents an independent experiment. Source data are provided as a Source Data file.
We used molecular dynamic (MD) simulations to examine the stability of histamine binding in the pocket of H4R. Triplicate 200 ns runs show that histamine and the anion phosphate are very stable during the simulations (Supplementary Fig. 4c, d and Supplementary Table 1 and Supplementary Movie 1). A closer examination of a snapshot from the MD simulations reveals that the phosphate molecule sits at the gap between E182 and Y318, acting as a barrier that prevents histamine from escaping the cage formed by Y318, F344, W348, D94, and Y95 (Supplementary Fig. 4c). This arrangement leads to a highly stable histamine binding pose, as evidenced by minimal changes during the simulation (left upper panel of Supplementary Fig. 4d), further supported by the straight line from the RMSD analysis (left lower panel of Supplementary Fig. 4d). In contrast, simulations without the phosphate result in histamine flipping around in the binding pocket, as depicted in the snapshots of the simulation (Supplementary Fig. 4d, right upper panel). This dynamic behavior is reflected in the substantial fluctuation of the RMSD curves (the lower right panel of Supplementary Fig. 4d). Collectively, these findings suggest that the phosphate anion plays a crucial role in stabilizing histamine binding.
We employed radioligand binding experiments to investigate the contribution to ligand binding of each residue of the H4R ligand binding pocket. In [3H]histamine binding assays, none of the mutants bind to the radioligand with sufficiently high affinity for a precise evaluation of the contribution of each residue (Fig. 2e), while all mutants have a similar surface expression level compared to the wild-type H4R, as measured by an anti-HA ELISA (Supplementary Fig. 7b). We then custom-synthesized [3H]JNJ7777120, a highly selective and potent antagonist of H4R. The measured affinities (pKi) of all the examined ligands, determined through displacement of [3H]JNJ7777120, are 7.7, 8.0, 7.7, and 6.5 for histamine, clobenpropit, VUF6884, and clozapine, respectively (Table 2). These values align closely with the reported pKi values of 7.7, 7.9, 7.6, and 6.4 for histamine, clobenpropit, VUF6884, and clozapine, respectively16,17. Importantly, except for D943.32 and W3487.43, most H4R mutants maintain a substantial affinity for [3H]JNJ7777120 (Fig. 2e; Supplementary Figs. 3i and 5a), enabling a thorough assessment of the role played by each amino acid in ligand binding. The lack of [3H]JNJ7777120 binding by the D94A3.32 and D94N3.32 mutants implicates that the conserved D943.32 in TM3 also plays a crucial role in JNJ7777120 binding. This matches well with docking results for JNJ7777120 where D943.32 forms a key salt-bridge interaction with the amine of the methylpiperazine ring of JNJ7777120 (Supplementary Fig. 3g, h). In [3H]JNJ7777120-histamine competition binding experiments, F344A7.39 shows a dramatic decrease of histamine binding, consistent with the structural observation of a direct interaction between the imidazole ring with F3447.39. In addition, the Y95A3.33, E182A5.46, E182Q5.46, Y319A6.51, and Q347A7.42 mutants all show a substantial decrease of the pKi value of histamine (Fig. 2f and Supplementary Fig. 5b). We also used a BRET-based Gi-protein activation assay18 to evaluate the contribution of each key pocket residue on receptor activation. In agreement with the pKi binding data, G-protein activation by histamine is completely abrogated by the D94A3.32, D94N3.32, and W348A7.43 mutations, while F344A7.39 severely decreases receptor activation and all other mutants cause a substantial loss of H4R activation (Fig. 2g and Supplementary Fig. 8a).
Binding of synthetic H4R agonists
We also evaluated the binding modes of three synthetic H4R agonists. The overall structures of histamine-, clobenpropit-, VUF6884- and clozapine-bound H4R are almost identical (Supplementary Fig. 3b) with a root mean square deviation (r.m.s.d.) of 0.299 Å over 215 pairs of Cα. While histamine only occupies half of the ligand binding pocket on the TM7 side, the synthetic agonists clobenpropit, VUF6884, and clozapine cover the whole orthosteric pocket (Supplementary Fig. 3a). Clobenpropit is a highly potent antagonist/inverse agonist of H3R and a partial agonist of H4R19,20. In the clobenpropit-bound H4R complex, its imidazole ring interacts with D943.32; on the other side, E1825.46 forms crucial salt-bridge interactions with both the N8 and N10 atom of the isothiourea group of clobenpropit (Fig. 3a, b). In addition, T993.37 also forms a polar interaction with the N10 atom of clobenpropit. Interestingly, like for histamine the positively charged imidazole ring of clobenpropit forms cation-π interactions with a π–π network formed by F3447.39, W3487.43, and Y3196.51 (Figs. 2a and 3a; and Supplementary Fig. 3d). In support of this, mutation of D943.32 completely abrogates receptor activity and mutation of F3447.39, Y3196.51, and W3487.43 strongly inhibit ligand binding (Fig. 3c; Supplementary Figs. 6a, b and 7a) and receptor activation (Fig. 3d; Supplementary Figs. 8b and 9a, b).

a The ligand binding pocket of Clobenpropit, VUF6884, and Clozapine. b A schematic map of Clobenpropit, VUF6884, and Clozapine/receptor interaction. Green color, hydrophobic interaction; purple color, polar interaction. c pKi of Clobenpropit, VUF6884 and Clozapine binding of H4R mutants. d pEC50 of Gi activation of H4R mutants by Clobenpropit, VUF6884 and Clozapine. The green bars indicate agonist and the red bars indicate inverse agonist. From c, d, data are presented as mean values ± SEM.; n = 3–4 independent experiments for c and n = 3–7 independent experiments for d. Each point in the figure represents an independent experiment. Source data are provided as a Source Data file.
Clozapine, an atypical antipsychotic medication approved by the FDA for the treatment of schizophrenia, functions as an antagonist of dopamine D4 receptor21,22. It is also a multi-target drug and binds with moderate to high affinity to a fair number of aminergic receptors, including serotonin 5-HT2A/2C receptor, H1R, and H4R. Interestingly, clozapine has been found to activate H4R, which might be related to the known side effect of agranulocytosis by clozapine20,23. VUF6884 is a more potent analog of clozapine at H4R24 and only differs from clozapine by the position of the chlorine atom and a substitution of the nitrogen atom with an oxygen atom at the dibenzodiazepine ring (Fig. 3b). In line with their structural similarity, both compounds exhibit a similar binding mode when interacting with H4R (Fig. 3a). The positively charged methyl-1-piperazinyl group forms a direct ionic interaction with D943.32 and is positioned to the π-π network formed by F3447.39, W3487.43, and Y3196.51, resembling the H4R interaction of the imidazole ring of histamine or clobenpropit. The dibenzodiazepine ring is positioned toward T1785.42 and E1825.46. Similar to the mutagenesis data observed with clobenpropit, mutant D943.32A/Q cannot interact anymore with the two ligands, while mutants in the π-π network significantly reduce receptor binding of VUF6884 and clozapine. Conversely, other mutations have minimal or negligible effects on receptor binding and activation by clozapine and its analog. (Fig. 3c, d). Interestingly, Q347A increases clozapine affinity and activity, while the effect is minimal with VUF6884. A closer examination of the binding poses of clozapine and VUF6884 shows that Q347 is closer to the dibenzodiazepine ring of clozapine than that of VUF6884 (Supplementary Fig. 4a). Mutation of Q347 to a small residue A (Q347A) may release the clash and accounts for the increase of clozapine activity.
Insight into H4R ligand recognition and receptor activity
A ligand interaction map of all ligands shows that the binding of histamine mainly involves residues from TM3 and TM7 (only the left half of the pocket), while the binding of the other agonists involves the whole pocket (TM3, TM4, ECL2, TM5, TM6, and TM7) (Fig. 4a). We conducted a comparison between the receptor binding data and the functional assay data regarding receptor activation. The comparison reveals a high degree of consistency between the binding data and the receptor activation data (Supplementary Fig. 10 and Table 2) and only minor differences were noticed. More importantly, in line with the cryo-EM observation, the site-directed mutagenesis studies revealed a distinct pattern for the interaction between various H4R agonists and H4R (Fig. 4b). As can be seen in the spider-web representation, the binding of histamine is most affected by the various mutations, especially the F3446.51A and the E182Q5.46 and E182A5.46 mutations (Fig. 4b). Clozapine seems least affected by the mutations, probably due to its relatively low affinity. The binding of clobenpropit or VUF6684 is also clearly affected by these mutations, but the drop in affinity is not as high as for histamine. Most interestingly, both E1825.46 mutants (E182Q5.46 and E182A5.46) convert the agonist clobenpropit into an inverse agonist (Fig. 4c and Supplementary Fig. 8b). Upon closer examination of clobenpropit receptor binding, it becomes evident that the negatively charged carboxyl group of E1825.46 forms a robust salt-bridge interaction with the positively charged N8 and N10 of the isothiourea group (Fig. 4d, left panel), effectively stabilizing the receptor in an active state. Conversely, mutations of the negatively charged E1825.46 to a neutral glutamine (Q) or alanine (A) disrupt the salt-bridge interaction, rendering the receptor incapable of maintaining an active state (Fig. 4d, middle and right panel). In fact, the site of E1825.46 has also been implicated in playing crucial roles in regulating H1R, H2R, and H3R activities. For instance, N192A5.46 totally abolished H1R activity11, while T190A5.46 and E206A5.46 have shown the importance for histamine interaction with H2R and H3R, respectively25,26. The structural insight into ligand recognition provides a clear explanation for the divergent receptor activities, facilitating the precise design of novel compounds that target H4R.

a A schematic summary of histamine, Clobenpropit, VUF6884, and Clozapine/receptor interactions. Green solid circle, hydrophobic interaction; purple solid circle, polar interaction; white emptied circle, no interaction. b Radar chart for affinities of ligands the four agonists to the wild-type and mutant H4R receptors, as measured by [3H]JNJ7777120 binding. c BRET-based Gi-protein activation assay of the wild-type and E182 H4R mutants. Data are presented as mean values ± SEM.; n = 6 independent experiments for WT, n = 7 independent experiments for E182A and n = 3 for E183Q. Source data are provided as a Source Data file. d A structural analysis of the interaction of clobenpropit with E182 and its mutants.
Antihistamine design of H4R
The disruption of the salt-bridge interaction between clobenpropit and E1825.46, observed in the E182Q5.46 mutant, results in the conversion of the agonist into an inverse agonist (Fig. 5a, left panel) within the context of the 2 mutant H4Rs. This intriguing finding raises the question of whether modifying the positively charged N8 and N10 groups of clobenpropit could potentially transform the modified compound into an inverse agonist for the wild-type H4R (Fig. 5a, right panel), which may have immediate therapeutic potential for associated informatory disease. Guided by this insight, we identified in our library VUF5202, which is differentiated from clobenpropit by a substitution of N10 and S11 with a carbon atom (Fig. 5b, middle panel), as a potential candidate for an inverse agonist for H4R. Previously, VUF5202 has been shown to act as an antagonist of H3R27, but has never been evaluated on H4R. Remarkably, while VUF5202 exhibits the same pKi as clobenpropit in the binding assay (Fig. 5b, left panel), VUF5202 acts as an inverse agonist for the wild-type (WT) H4R with a potency in the nanomolar range in the BRET-based G-protein activation assay (Fig. 5b, right panel). This successful identification of a novel inverse agonist for H4R underscores the precision of our structural analysis and provides a robust foundation for the design of new antihistamines to combat inflammatory diseases.

a A schematic diagram of the antihistamine design of H4R, utilizing key information of E182/ligand interaction. b VUF5202 exhibits inverse agonist activity. Data are presented as mean values ± SEM.; n = 3 independent experiments for the binding assay (both Clobenpropit and VUF5202), n = 6 for Clobenpropit BRET assay, and n = 3 for VUF5202 BRET assay. Source data are provided as a Source Data file.
Mechanism of H4R activation
A comparison of the antagonist PF03654746-bound inactive H3R13 with the histamine-bound active H4R shows that the most notable change is the outward movement of TM6 on the intracellular side in the active H4R, which allows the αH5 of Gαi to engage the intracellular cavity of the receptor (Fig. 6a), the most common feature of GPCR activation28. In addition, we also saw a small outward movement of TM7 on the extracellular side of the receptor. We also compared the histamine-bound active H4R with the AlphaFold29 prediction of apo H4R (inactive). The comparison shows a similar TM6 outward displacement when receptor activation (Supplementary Fig. 4b). In H1R activation, a “squash to activate and expand to deactivate” was proposed based on the shrinkage of the ligand binding pocket caused by histamine pulling key residues of TM3, TM5 and TM7 inside11. When comparing the extracellular side of histamine-bound H1R with the active state of H4R, no evident compression of the ligand binding pocket on H4R is observed (Supplementary Fig. 3c). Generally, receptor activation of class-A GPCR is mediated by a coordinated movement of conserved motifs such as C6.47W6.48xP6.50, P5.50I3.40F6.44, N7.49P7.50xxY7.53 and D3.49R3.50Y3.51. We observe TM7 to spin towards W3166.48 of the toggle switch (Fig. 6b) which in turn bends TM6 at the middle to allow the outward movement on the intracellular side. For the P5.50I3.40F6.44 motif, a signature movement of F3126.44 toward TM5 was observed (Fig. 6c). For the D3.49R3.50Y3.51 motif, we observed the movement of R1123.50 toward the center of intracellular cavity to allow the tip of the αH5 to engage the receptor (Fig. 6d). For the N7.49P7.50xxY7.53 motif, the most dominant movement is the shift of Y3587.53 toward TM3, a phenomenon seen in most class A GPCR activations (Fig. 6e).

a A comparison of the overall structure of the active H4R (histamine-bound) with the inactive H3R (PF03654746-bound, pdb:7f61). b–e A comparison of the CWxp, PIF, DRY, and NPxxY motif, respectively, between the active H4R (histamine-bound) with the inactive H3R.
G-protein engagement
H4R almost exclusively couples to Gi signaling which leads to a decrease of cAMP production and an increase of intracellular Ca2+. In the cryo-EM structures, the G-protein engagement is mainly mediated by the insertion of αH5 into the intracellular cavity of H4R. An analysis of the H4R-Gαi interaction shows that the hydrophobic interactions between a cluster of hydrophobic residues L353G.H5.25, L348G.H5.20, I344 G.H5.16, I343 G.H5.15 and a patch of the hydrophobic surface formed by L3086.40, L3056.37, L3016.33, L2015.65, I1975.61 of the H4R is the main driver (Fig. 7a and Supplementary Fig. 11a). This finding aligns with the analysis of multiple G-protein couplings on ADGRL330 and GPR11031, suggesting that hydrophobic interactions play a crucial role in determining Gi engagement. Interestingly, we also found the ICL2 of H4R to form extensive polar interaction with Gαi to stabilize the engagement, namely S1153.53, R123ICL2, and Q125 ICL2/H126 ICL2 of ICL2 form polar interactions with N347 G.H5.19, E33G.S1.01 and R32G.hns1.03 of Gαi, respectively (Fig. 7b). When comparing the engagements of H4R with Gi across different ligands, it is evident that while the overall engagements exhibit a high degree of similarity (r.m.s.d. = 0.603 Å over 1065 pairs of Cα atoms), there are slight differences observed in the clozapine-bound H4R/Gi complex compared to the histamine-, clobenpropit-, and VUF6884-bound H4R/Gi complexes (Fig. 7c). The most notable distinction is the 4° outward sway of the α N-terminal helix (αN) and a displacement of αH5 towards the receptor in the Clozapine-bound H4R/Gi complex (Supplementary Fig. 11b, c).

a The Gi engagement of H4R is featured by hydrophobic interaction between the αH5 of Gi and the intracellular side of the receptor. b The ICL2 of H4R forms extensive polar interactions with Gαi in the agonist-bound H4R-Gi complex. c A comparison of the overall Gαi conformations of histamine, Clobenpropit, VUF6884, and Clozapine-bound H4R/Gi complexes.
Discussion
Histamine receptors have long been recognized as successful targets for treating immune-related disorders and allergies, with antihistamines against H1R being widely prescribed for allergy relief1. Clinical successes have been also achieved with H2R and H3R for respectively the treatment of gastric ulcers and narcolepsy1. Despite the lack of clinical success thus far, H4R holds promise for developing new therapeutic solutions for allergic and inflammatory diseases1,5. The medicinal chemistry field has benefited from the first structure of antagonist-bound H1R many years ago10. Subsequently, the active structure of H1R provides further insight into the distinct mechanisms by which agonists and antagonists modulate receptor activity11, thereby bolstering the design and advancement of novel antihistamines targeting H1R.
However, until now, the structural information of H4R has been absent, and most H4R antihistamines have been discovered following high throughput screening, fragment-based screening, or successful, classical scaffold hopping strategies1,5. Our structural study of H4R offers new important information for new structure-based approaches. Our data on H4R-Gαi complexes, first of all, reveal a completely different ligand binding mode for histamine compared to its interaction with H1R11 (Fig. 2d). In H1R, the imidazole of histamine is pointing towards TM3/TM5 side and forms crucial ionic interaction with N1985.46, while in H4R, histamine only occupies the pocket on the TM7 side where a π-π network formed by F3447.39, W3487.43 and Y3196.51, leaving the pocket on the TM3/TM5 side empty. Other, larger H4R agonists, like clobenpropit and clozapine, occupy also the TM3/TM5 side of the binding pocket. We assigned a phosphate molecule to an unidentified density within the histamine binding pocket. MD simulations revealed that the phosphate molecule played a crucial role in stabilizing the binding of histamine to the receptor Concurrently, our investigation does not rule out the possibility that other anions may also play a role in stabilizing histamine binding.
One of the most interesting discoveries of our study is the observation that mutation on the single residue E1825.46 converts clobenpropit from a partial agonist into an inverse agonist of H4R. E1825.46 is not directly involved in histamine binding, but plays a crucial role in the binding of the other 3 tested agonists (Figs. 2a and 3a, b). Particularly, it forms a salt-bridge interaction with the positively charged N8/N10 of clobenpropit, which positions the ligand to activate the receptor. A slight change of the net charge at this position (E1825.46 to Q1825.46) without altering the overall conformation causes the loss of the key salt-bridge interaction, resulting in a complete inactivation of the receptor while retaining the affinity of the clobenpropit (Fig. 4b–d). This demonstrates the tight connection between receptor activation of H4R and subtle changes in ligand binding. The identification of E1825.46 as a regulator of receptor activation carries substantial implications for the design of novel antihistamines targeting H4R. The development of new selective H4R inverse agonists is generally considered of interest for inflammatory and allergic conditions1. The discovery of VUF5202 as a novel inverse agonist for H4R is a proof of concept for this principle for novel antihistamine design for H4R. Moreover, VUF5202 has a completely different chemical scaffold compared to the known H4R antihistamines JNJ7777120 and Toreforant32, both of which didn’t succeed in clinical trials9, highlighting the potential of developing innovative antihistamines for H4R via this new structural insight.
While our manuscript was under review, a similar study reported the histamine- and imetit-bound H4R in complex with Gq33, an unusual coupling partner of H4R. Compared to this study, we reveal the coupling information with the Gi protein, which is considered the primary transducer of H4R. Moreover, clobenpropit, VUF6884, and Clozapine are large H4R ligands, having different backbones compared to the small agonist’s histamine and imetit. Therefore, our structures could provide additional information for designing novel ligands/modulators targeting H4R.
Collectively, we successfully determined the cryo-electron microscopy (cryo-EM) structures of the H4R/Gi complex in association with 4 different (partial) agonists, unveiling a distinctive mode of histamine binding specific to H4R. Furthermore, we have discovered that E1825.46 plays a crucial role in determining ligand efficacy and H4R activation and discovered VUF5202 as a novel inverse agonist for H4R. Together with the mechanistic insights into GPCR activation and Gi engagement, our study provides a structural basis for the understanding of H4R signaling and offers a logical foundation for the development of novel antihistamines targeting H4R.
Methods
Constructs
The human H4R gene, optimized for codon usage, was incorporated into the pFastBac1 baculovirus expression vector. The gene sequence included an HA-signal peptide sequence at the N-terminus and a LgBiT fusion at the C-terminus, followed by a Tobacco etch virus (TEV) cleavage site and two maltose-binding protein (MBP) domains. To enhance protein expression and folding, the ICL3 loop of H4R (residues 215-286) was removed. Additionally, the C-terminal fusion of human Gβ1 with HiBiT34 was cloned into a separate pFastBac plasmid, as described in the VIP1R paper. The pFastBac plasmid also contained clones of the human dominant-negative Gαi1 (bearing the G203A/A326S mutant for the histamine- and Clozapine-bound H4R, and the S47N/G203A/A326S/E245A mutant for the Clobenpropit- and VUF6884-bound H4R), wild-type human Gβ1, wild-type human Gγ2, and the scFv16 encoding the single-chain variable fragment of mAb16, as previously described. Site-directed mutagenesis was performed by polymerase chain reaction (PCR) using the N-terminal HA-epitope tagged human H4R (GenBank: NM_021624) as a template. PCR products were subcloned into the mammalian expression plasmid pcDEF3 using flanking BamHI and XbaI restriction sites and verified by DNA sequencing.
Protein expression and purification
To express the proteins, Spodoptera frugiperda (Sf9) cells were co-infected with baculoviruses carrying H4R, Gαi1, Gβ1, Gγ2, and scFv16 at a ratio of 1:100 (virus volume to cell volume). The cells were harvested 48 h post-infection. The cell pellets were resuspended in a buffer containing 20 mM Hepes, 150 mM NaCl, 10 mM MgCl2, 20 mM KCl, 5 mM CaCl2 at pH 7.5, supplemented with 0.5 mU/mL apyrase, and homogenized by douncing approximately 30 times. Throughout the purification process, ligands including HSM, Clobenpropit, VUF6884, and Clozapine were added at concentrations of 100 μM, 10 μM, 5 μM, and 5 μM, respectively. After incubating the lysate at room temperature for 1 h, 0.5% (w/v) lauryl maltose neopentylglycol (LMNG) and 0.1% (w/v) cholesteryl hemisuccinate TRIS salt (CHS) were added to solubilize the membranes, followed by incubation at 4 °C for 2 h. The lysate was then subjected to ultracentrifugation at 65,000 × g and 4 °C for 40 min. The supernatant was incubated with an amylose column for 2 h, washed with a buffer containing 25 mM Hepes, pH 7.5, 150 mM NaCl, 0.01% LMNG, and 0.002% CHS, and eluted with the same buffer supplemented with 10 mM maltose. The eluate was concentrated and treated with homemade TEV protease overnight at 4 °C. Subsequently, the sample was separated on a Superdex 200 Increase 10/300 GL gel filtration column using a buffer composed of 25 mM Hepes, pH 7.5, 150 mM NaCl, 0.00075% (w/v) LMNG, 0.00025% glyco-diosgenin (GDN), and 0.0002% (w/v) CHS. The peak corresponding to the H4R/Gi complex was concentrated to approximately 10 mg/mL and snap-frozen for subsequent cryo-EM grid preparation.
Grid preparation and cryo-EM data collection
A protein complex sample (~10 mg/mL) of approximately 3–5 µL was loaded onto Cu holey carbon grids (Quantifoil R1.2/1.3) that were pre-treated with glow charging (Quantifoil GmbH). The loaded grids were then vitrified by rapidly plunging them into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). The Vitrobot settings used were as follows: blot force 10, blot time 5 s, humidity 100%, and temperature 4 °C. The prepared grids, containing evenly distributed particles in thin ice, were placed into a FEI 300 kV Titan Krios transmission electron microscope (TEM) equipped with a Gatan Quantum energy filter. Imaging was performed using a Gatan K2 Summit direct electron detector employing a super-resolution counting model, with a pixel size of 0.55 Å at a magnification of 64,000×. The energy filter slit was adjusted to 20 eV. Each image consisted of 40 frames, with a total exposure time of 7.3 s and a dose rate of 1.5 e/Å2/s (resulting in a total dose of 60 e/Å2). The nominal defocus value ranged from –1.2 to –2.2 µm.
Data processing
The cryo-electron microscopy (cryo-EM) data were processed using a standard pipeline established in our laboratory35. Initially, the raw movies were binned once (1.1 Å) and corrected for motion using MotionCor236. Subsequently, the contrast transfer function (CTF) parameters were estimated using CTFFIND 4.137. Particle picking was performed using crYOLO38, followed by reference-free 2D classification in RELION39. The well-defined 2D features obtained from this classification were used to generate an initial model using cryoSPARC’s ab initio method40. The generated initial model served as a reference for further refinement steps in RELION. A 3D classification was conducted, resulting in 3–4 classes. The best class, displaying clear secondary structure features, was selected for Non-uniform Refinement in cryoSPARC.Subsequently, a no-alignment 3D classification was performed in RELION, employing 6–10 classes and applying a mask on the complex. Bayesian polishing41 and additional rounds of Non-uniform Refinement were carried out to enhance the map quality. The resolution of the final map was estimated using the gold standard Fourier Shell Correlation (FSC) criterion at FSC = 0.143. Local resolution estimations were performed using an implemented program in cryoSPARC.
Model building
We employed AlphaFold prediction29 of human H4R (AF-Q9H3N8-v1) as initial models to guide the process of model rebuilding against the electron microscopy map. The docking of these models into the density map was performed using UCSF Chimera42. Iterative manual adjustments were carried out in Coot to refine the models, followed by Rosetta cryo-EM refinement43 and Phenix real space refinement44 to further improve the structural accuracy. For the visualization and preparation of structural figures, UCSF ChimeraX45 and PyMOL (https://pymol.org/2/) were utilized.
Molecular docking
The docking methodology employed in this study follows a similar approach to previous research46. Initially, the histamine-bound H4R structure was utilized as the starting model and prepared/minimized using established protocols. 3D model files (in SDF format) for the candidate ligands were obtained from PubChem. The candidate ligands were then positioned within the ligand binding pocket using the triangle matcher, with the London docking score used for assessment. Refinement steps were performed utilizing a rigid receptor and GBVI/WSA docking scoring.
Synthesis of [3H]JNJ7777120
As described early47, a precursor for radiolabeling, (5-Chloro-1H-indole-2-yl)-(piperazine-1-yl)-methanonex (0.15 mg, 39.6 µmol) was dissolved in 117 µL of [3H]methyl nosylate (Perkin Elmer, 854 MBq/mL in acetonitrile). The reaction mixture was heated for 15 min at 70 °C. Next, the reaction was allowed to cool to ambient temperature, diluted with 2 mL of HPLC eluent and injected onto preparative HPLC (Jasco PU-2080 Pump, Jasco UV-2075 UV detector (Jasco, Utrecht, The Netherlands) mounted with a Luna C18 10*250 mm, 100 Å, 10 µm column en eluted with 25/75 acetonitrile/water, 0.2% DIPEA at 5 mL/min, UV was measured at 225 nm. The product eluted at 52 to 54 min and was collected in a solution of 60 mL of water. The total mixture was purged over a Sep-Pak tC18 (Waters, Milford, USA) which was pre-washed with 10 mL of ethanol and 20 mL of water, successively. After trapping of [3H]JNJ7777120, the Sep-Pak was washed with 20 mL of water and [3H]JNJ7777120 was obtained with elution of the Sep-Pak with 2 mL of ethanol.
The concentration of [3H]JNJ7777120 in ethanol was determined using beta counting (Hidex 300 SL, Turku, Finland) and found to be 19.7 MBq/mL. The product was analyzed with HPLC (Jasco PU-2080 Pump, Jasco UV-2075 UV detector mounted with a Lablogic (Sheffield, UK) β-RAM Scintilation detector) using a Luna C18, 4.6*250 mm, 100 Å, 5 µm column which was eluted with 35/65 acetonitrile/water, 0.1% DIPEA at 1 mL/min. UV was measured at 225 nm. The radiochemical purity was 97.9% and no chemical impurities were observed. The molar activity was 2.57 MBq/nmol, based on the used [3H]methyl nosylate.
Radioligand binding experiments
Two million HEK293T cells were seeded in 100 mm tissue-culture dishes in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% FBS, penicillin (100 IU/mL), and streptomycin (100 μg/mL) at 37 °C with 5% CO2. The next day, cells were transiently transfected with 2.5 μg DNA encoding for human wild-type HA-H4R or mutant HA-H4R and 2.5 μg empty pcDEF3 using 30 μg 25 kDa linear polyethylenimine. After 48 h, cells were washed and collected with ice-cold phosphate-buffered saline (PBS) and centrifuged at 1900 × g for 10 min at 4 °C. Cell pellets were stored at –20 °C. Next, cell pellets were resuspended in binding assay buffer (50 mM Tris-HCI, pH 7.4) and sonified for 15 s before each experiment. Radioligand competition binding was measured on 50 μL cell homogenates expressing wild-type or mutant H4R using 25 μL [3H] histamine or [3H] JNJ7777120, and 25 μL buffer or unlabeled ligands. Nonspecific radioligand binding was determined in the presence of 10 μM JNJ7777120. After 2 h at 25 °C, the incubations were terminated by rapid filtration over a 0.5% PEI-coated 96-well GF/C filter plate through three rapid wash steps with ice‐cold wash buffer (50 mM Tris‐HCl, pH 7.4) using a Perkin Elmer 96‐well Filtermate-harvester (Perkin Elmer, Groningen, the Netherlands). The GF/C filter plates were dried at 52 °C for 1 h and 25 μL Microscint‐O scintillation liquid was added per well. Filter‐bound radioactivity was measured using a Microbeta2 plate counter (Perkin Elmer) after a 120 min delay.
Data for competition binding were analyzed by nonlinear regression analysis using GraphPad Prism 9.5.1. IC50 values were obtained by fitting the data from the competition studies to a one-site competition model. The Ki of unlabeled ligands was calculated using the Cheng-Prusoff equation with radioligand binding affinity values determined by the homologous displacement equation. Competition binding graphs represented the pooled data from at least three independent experiments performed in duplicate.
BRET-based Gαi activation assay and anti-HA ELISA
For the BRET-based Gαi activation assay, two million HEK293T cells were seeded in 100 mm tissue-culture dishes. The next day, 1 μg plasmid encoding for wild-type or mutant HA-H4R was transiently cotransfected with 1.5 μg bicistronic plasmid encoding for a BRET-based Gαi sensor18 using 20 μg 25 kDa linear polyethylenimine. An empty pcDEF3 vector was added to normalize the total amount of DNA to 5 μg per 100 mm dish. At 24 h after transfection, 50,000 cells per well were transferred into 0.01% Poly-L-Lysine (PLL) precoated white and transparent 96-well plates (Greiner, #655083) and further maintained for 24 h at 37 °C with 5% CO2. Cells in the white plates were washed with HBSS and incubated with agonists for H4R and furimazine (Nano-Glo®, Promega). After 40 min at room temperature (RT), luminescence was measured using the CLARIOstar Plus Microplate reader at 535-20 and 470-80 nm. The BRET ratio was determined as the acceptor emission divided by the donor emission. At least three independent experiments were performed in duplicate, and data were normalized to the vehicle using GraphPad Prism 9.5.1. Significant analysis was performed using a one-way AVONA test under the multiple comparisons of Dunnett (****p < 0.0001, ***p = 0.0002, **p = 0.02, *p = 0.03).
To measure (mutant) H4R protein expression transfected cells in the transparent plates were washed with TBS buffer (50 mM Tris and 150 mM NaCI) and fixed with 4% PFA for 30 min at RT. Cells were incubated overnight at 4 °C with anti-HA (Sigma, Cat# 11867423001, diluted 1000-fold from stock), followed by incubation with anti-rat-HRP (diluted 1000-fold from stock) for 2 h at RT. Cells were washed twice between all antibody incubations finally and the absorption at 450 nm was measured using the CLARIOstar Plus Microplate reader after the addition of substrate solution (Mix TMB and H2O2).
Molecular dynamics simulation
The cryo-EM structure of histamine-bound H4R (receptor only) was used as the initial model in the MD simulation. The ICL3 break (204-292) was filled with residues AAGAAA. The model was prepared and parameterized in CHARMM-GUI48,49. Protonation states of all titratable residues were assigned at pH 7.0. Histamine was bi-protonated according to a previous report15. PO4 was protonated as HPO4-2 according to Protonate3D analysis50. The H4R model was inserted into a lipid bilayer containing POPC (palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and cholesterol at a 4:1 ratio. The membrane had dimensions of 65 × 65 Å, with 22.5 Å of water on the top and bottom (resulting in final system dimensions of approximately 65 x 65 x 120 Å). The ion concentration was set to 0.15 M KCl (see Supplementary Table 1 for the details of the system setting). The Amber force fields were configured as follows: protein FF19SB, lipid LIPID17, water TIP3P, and ligand GAFF2. Simulations were conducted using the Amber20 package51. The system underwent initial energy minimization for solvent and all atoms, followed by heating to 300 K over 300 ps and equilibration for 700 ps. Subsequently, three independent production runs of 200 ns each were performed with a time step of 2 fs. During simulations, the Particle Mesh Ewald algorithm calculated long-range electrostatic interactions, while a cutoff of 10 Å was applied for short-range electrostatic and van der Waals interactions. SHAKE algorithm constraints were applied to all bonds involving hydrogens. Temperature (300 K) and pressure (1 atm) were controlled by the Langevin thermostat and Berendsen barostat, respectively. Trajectory analysis and visualization were carried out using VMD52, and video recording was facilitated by VMD.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data produced or analyzed in this study are included in the main text or the Supplementary Figs./tables. Source data are provided in this paper. The cryo-EM density maps and atomic coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) under accession numbers EMD-36712 and 8JXT for H4R/Histamine/Gi complex; EMD-36716 and 8JXX for H4R/Clobenpropit/Gi complex; EMD-36715 and 8JXW for H4R/VUF6884/Gi complex and EMD-36714 and 8JXV for H4R/Clozapine/Gi complex. The MD simulation data were deposited to Zenodo (ID: 10802634) Source data are provided in this paper.
References
Panula, P. et al. International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 67, 601–655 (2015).
Parsons, M. E. & Ganellin, C. R. Histamine and its receptors. Br. J. Pharmacol. 147, S127–S135 (2006).
Leurs, R., Bakker, R. A., Timmerman, H. & de Esch, I. J. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat. Rev. Drug Discov. 4, 107–120 (2005).
Lamb, Y. N. Pitolisant: a review in narcolepsy with or without cataplexy. CNS Drugs 34, 207–218 (2020).
Leurs, R., Chazot, P. L., Shenton, F. C., Lim, H. D. & de Esch, I. J. Molecular and biochemical pharmacology of the histamine H4 receptor. Br. J. Pharmacol. 157, 14–23 (2009).
Thurmond, R. L. et al. A potent and selective histamine H4 receptor antagonist with anti-inflammatory properties. J. Pharmacol. Exp. Ther. 309, 404–413 (2004).
Rosethorne, E. M. & Charlton, S. J. Agonist-biased signaling at the histamine H4 receptor: JNJ7777120 recruits β-arrestin without activating G proteins. Mol. Pharmacol. 79, 749–757 (2011).
Nijmeijer, S. et al. Detailed analysis of biased histamine H4 receptor signalling by JNJ 7777120 analogues. Br. J. Pharmacol. 170, 78–88 (2013).
Thurmond, R. L. et al. in Histamine and Histamine Receptors in Health and Disease (eds Y. Hattori & R. Seifert) 301–320 (Springer International Publishing, 2017).
Shimamura, T. et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65–70 (2011).
Xia, R. et al. Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat. Commun. 12, 2086 (2021).
Robertson, M. J. et al. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 29, 1188–1195 (2022).
Peng, X. et al. Structural basis for recognition of antihistamine drug by human histamine receptor. Nat. Commun. 13, 6105 (2022).
Wang, D. et al. Molecular mechanism of antihistamines recognition and regulation of the histamine H(1) receptor. Nat. Commun. 15, 84 (2024).
Ratnala, V. R. et al. Solid-state NMR evidence for a protonation switch in the binding pocket of the H1 receptor upon binding of the agonist histamine. J. Am. Chem. Soc. 129, 867–872 (2007).
Smits, R. A., Leurs, R. & de Esch, I. J. Major advances in the development of histamine H4 receptor ligands. Drug Discov. Today 14, 745–753 (2009).
Lim, H. D. et al. Molecular determinants of ligand binding to H4R species variants. Mol. Pharmacol. 77, 734–743 (2010).
Schihada, H., Shekhani, R. & Schulte, G. Quantitative assessment of constitutive G protein-coupled receptor activity with BRET-based G protein biosensors. Sci. Signal. 14, eabf1653 (2021).
Yokoyama, H. et al. Clobenpropit (VUF-9153), a new histamine H3 receptor antagonist, inhibits electrically induced convulsions in mice. Eur. J. Pharmacol. 260, 23–28 (1994).
Liu, C. et al. Cloning and pharmacological characterization of a fourth histamine receptor (H(4)) expressed in bone marrow. Mol. Pharmacol. 59, 420–426 (2001).
Nucifora, F. C. Jr., Mihaljevic, M., Lee, B. J. & Sawa, A. Clozapine as a Model for Antipsychotic Development. Neurotherapeutics 14, 750–761 (2017).
Meltzer, H. Y. An overview of the mechanism of action of clozapine. J. Clin. Psychiatr. 55, 47–52 (1994).
Oloyede, E. et al. Clozapine haematological monitoring for neutropenia: a global perspective. Epidemiol. Psychiatr. Sci. 31, e83 (2022).
Smits, R. A. et al. Characterization of the histamine H4 receptor binding site. Part 1. Synthesis and pharmacological evaluation of dibenzodiazepine derivatives. J. Med. Chem. 49, 4512–4516 (2006).
Gantz, I. et al. Molecular basis for the interaction of histamine with the histamine H2 receptor. J. Biol. Chem. 267, 20840–20843 (1992).
Uveges, A. J. et al. The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J. Pharmacol. Exp. Ther. 301, 451–458 (2002).
Govoni, M. et al. A chemical switch for the modulation of the functional activity of higher homologues of histamine on the human histamine H3 receptor: effect of various substitutions at the primary amino function. J. Med. Chem. 49, 2549–2557 (2006).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Qian, Y. et al. Structural insights into adhesion GPCR ADGRL3 activation and G(q), G(s), G(i), and G(12) coupling. Mol. Cell 82, 4340–4352 e4346 (2022).
Zhu, X. et al. Structural basis of adhesion GPCR GPR110 activation by stalk peptide and G-proteins coupling. Nat. Commun. 13, 5513 (2022).
Boyle, D. L. et al. Toreforant, an orally active histamine H(4)-receptor antagonist, in patients with active rheumatoid arthritis despite methotrexate: mechanism of action results from a phase 2, multicenter, randomized, double-blind, placebo-controlled synovial biopsy study. Inflamm. Res. 68, 261–274 (2019).
Im, D. et al. Structural insights into the agonists binding and receptor selectivity of human histamine H(4) receptor. Nat. Commun. 14, 6538 (2023).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Xu, Z. et al. Structural basis of sphingosine-1-phosphate receptor 1 activation and biased agonism. Nat. Chem. Biol. https://doi.org/10.1038/s41589-021-00930-3 (2021).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Method. 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Wagner, T. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218 (2019).
Fernandez-Leiro, R. & Scheres, S. H. W. A pipeline approach to single-particle processing in RELION. Acta Crystallogr. D Struct. Biol. 73, 496–502 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Method. 14, 290–296 (2017).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, https://doi.org/10.7554/eLife.17219 (2016).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Wang, N. et al. Structural basis of leukotriene B4 receptor 1 activation. Nat. Commun. 13, 1156 (2022).
Engelhardt, H. et al. Detailed structure-activity relationship of indolecarboxamides as H4 receptor ligands. Eur. J. Med. Chem. 54, 660–668 (2012).
Wu, E. L. et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Labute, P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 75, 187–205 (2009).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
This work was supported by the Startup Funds of HIT Center for Life Sciences; and the National Natural Science Foundation of China (32070048 to Y.H.). Shuang Shi was supported by a grant from China (CSC grant number 202006310016). R.X. was supported by “the Fundamental Research Funds for the Central Universities”.
Author information
Authors and Affiliations
Contributions
R.X. made the constructs, expressed and purified the proteins and assembled the H4R/G-protein complex, prepared and screened the grids, analyzed the data, and prepared the figures. S.S. conducted ligand binding, BRET-based Gi activation, and surface expression experiments, analyzed the data, and prepared the figures. H.F.V. supervised ligand binding experiment, BRET assay, and analyzed data. Z.X., Y.Q., Y.D., J.L. and K.C. cultured the cells and prepared the plasmids. A.W. synthesized and provided [3H]JNJ7777120. A.Z. and C.G. collected cryo-EM data. R.L. designed the experiments, supervised the project, analyzed the data, and wrote the manuscript with Y.H. Y.H. designed experiments, solved the structures, analyzed data, supervised the project, and wrote the manuscript with R.L. All authors contributed to the data interpretation and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Arun Shukla and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, R., Shi, S., Xu, Z. et al. Structural basis of ligand recognition and design of antihistamines targeting histamine H4 receptor. Nat Commun 15, 2493 (2024). https://doi.org/10.1038/s41467-024-46840-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46840-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
