
Abstract
Histamine is a biogenic amine that participates in allergic and inflammatory processes by stimulating histamine receptors. The histamine H4 receptor (H4R) is a potential therapeutic target for chronic inflammatory diseases such as asthma and atopic dermatitis. Here, we show the cryo-electron microscopy structures of the H4R-Gq complex bound with an endogenous agonist histamine or the selective agonist imetit bound in the orthosteric binding pocket. The structures demonstrate binding mode of histamine agonists and that the subtype-selective agonist binding causes conformational changes in Phe3447.39, which, in turn, form the “aromatic slot”. The results provide insights into the molecular underpinnings of the agonism of H4R and subtype selectivity of histamine receptors, and show that the H4R structures may be valuable in rational drug design of drugs targeting the H4R.
Similar content being viewed by others

Introduction
Histamine, a biogenic neurotransmitter, exerts its pathophysiological functions in the central nervous system and peripheral tissues by stimulating histamine receptors belonging to the G protein-coupled receptor (GPCR) superfamily. Four histamine receptors (H1R–H4R) have been identified in humans, each with a different intracellular signaling mechanism1. In 1988 a Nobel prize was awarded for the discovery of H2R antagonists, following the pharmacological discovery of H1R and H2R2,3. The antagonists of these receptors have been developed for the treatment of allergic diseases and gastric acid secretion, respectively, and are still in use1,4. H3R was identified in the 80 s and has very low homology (<20%) with H1R and H2R5,6. It is a clinical target for central nervous system diseases, as it mainly regulates histamine function in the brain, and the H3R antagonist pitolisant has been used for the treatment of narcolepsy (Wakix®) and obstructive sleep apnea (Ozawade®)7.
H4R is the last histamine receptor to be identified8,9,10,11,12. Since H4R is mainly expressed in hematopoietic cells, such as monocytes, basophils, dendritic cells, and T-lymphocytes, it has been reported to participate in the release of cytokines, and consequently plays an important role in anti-inflammatory and immunomodulatory activities13,14. Therefore, H4R may be a target for chronic inflammatory diseases (CIDs), such as asthma, arthritis, and atopic dermatitis15,16. Preclinical and clinical studies involving H4R antagonists have demonstrated its therapeutic efficacy against CIDs, making H4R a promising therapeutic target4. H4R shares 34% of its amino acid sequence identity with H3R, and 50% in the transmembrane (TM) region. Because of their structural similarities, there are many ligands that have a high affinity for both receptors17,18. However, it is important to further explore selective H4R ligands to facilitate the development of more effective CID therapies.
In a previous study, we determined the crystal structure of the inactive form of H1R (H1Rdox), which is bound to doxepin, a first-generation antihistamine19. In the earlier study, the binding mode of doxepin and the reason for the low selectivity of first-generation antihistamines were investigated. Furthermore, the binding mode and specificity of second-generation antihistamines were determined using a computational approach. Recently, the cryo-electron microscopy (cryo-EM) structure of active H1R (H1Rhis) bound to an endogenous agonist histamine has been reported20. The structure revealed details of histamine binding in H1R. The activation mechanism of H1R was assessed further by examining the details of Gq protein engagement.
Here, we report the cryo-EM structures of H4R, which include H4R bound to an endogenous histamine ligand (H4Rhis) and H4R bound to the H3R/H4R selective agonist imetit (H4Rime). We elucidate the specific ligand recognition mechanism of H4R by comparing the binding mode of two agonists with different pharmacological characteristics. Furthermore, these results may provide insights that facilitate rational drug design targeting the H4R.
Results
Overall structures of the agonist-bound H4R-Gq complex
For the cryo-EM experiment, H4R and trimeric G proteins (Gαq, rat Gβ1, and bovine Gγ2) were co-expressed using Spodoptera frugiperda (Sf9) insect cells. H4R is known to primarily engage with Gαi and transduce Gi signals. We co-expressed and purified Gi-coupled H4R but failed to generate the complex (Supplementary Fig. 1b). However, the complex was successfully obtained by using the non-canonical miniGqiN as an accessory protein for structural determination. Furthermore, H4R has been reported to have a weak Gq signal (pEC50 = 6.8, Emax = 9% for Gq/pEC50 = 7.1, Emax = 26% for Gi)21; therefore, the H4R-Gq complex was used to determine the H4R structure.
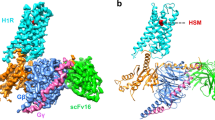
The H4R-Gq complex structures, H4Rhis and H4Rime, which were bound to histamine and imetit, were determined by a cryo-EM single-particle analysis at 3.0 Å and 3.1 Å resolutions, respectively (Fig. 1, Supplementary Fig. 2, and Table 1). In the H4Rhis and H4Rime structures, the side chains of most of the amino acids in the receptor and G protein regions could be assigned to the final EM map and refined with excellent geometry. Both the agonists used in the present study, histamine and imetit, were clearly identified, and a model was obtained to explain the recognition mechanisms of the agonists in H4R (Supplementary Fig. 3). In the overall structure of the H4R-Gq complex, similar to other GPCR-G protein complex structures, the Gq trimer bound to the intracellular region of H4R, and Nb3522 and scFv1623 bound to Gq and the Gq-Gβ interface, respectively, thereby stabilizing the complex (Fig. 1a, b, d, e). A local resolution analysis showed that the WD40 repeat core of the Gβ subunit, N-terminal region of the Gα subunit (GαHN), and scFv16 had the highest resolution, whereas the extracellular region of the receptor and a portion of the N-terminal region of Gβγ had a lower resolution (Supplementary Figs. 2e, f). In addition, the N/C-terminus, the intracellular loop 3 (ICL3), and some parts of the extracellular loops (ECL2, ECL3) of H4R were difficult to observe due to their high flexibility, and therefore, could not be modeled.

a, b Orthogonal views of the cryo-EM density map (a) and model (b) of the histamine-bound H4R-Gq complex. c Extracellular view of the histamine-bound H4R-Gq complex and chemical structure of histamine. d, e Orthogonal view of the cryo-EM density map (d) and model (e) of the imetit-bound H4R-Gq complex. f Extracellular view of the imetit-bound H4R-Gq complex and chemical structure of imetit. Cys873.25 and Cys164ECL2 form a disulfide bond and are shown as stick models (c, f). In (a)–(f), the complexes are colored by subunits. Histamine-bound H4R: pink, imetit-bound H4R: blue, Gq: purple, Gβ: navy, Gγ: green, Nb35: violet, scFv16: lime, histamine: yellow, and imetit: magenta.
The H4Rhis and H4Rime structures exhibited a canonical GPCR scaffold with seven transmembrane helices (TM1–7) and an intracellular amphipathic helix 8 (H8) (Fig. 1c, f, Supplementary Fig. 3c). The root mean square deviation of both structures was 0.32 Å for the overall structure and 0.60 Å for the receptor region, indicating similar conformations. Histamine and imetit occupied an orthosteric binding pocket in the middle of the TMs, which was covered by a C-terminal segment of ECL2 stabilized by the disulfide bonds of C873.25 and C164ECL2, which are common in other class A GPCRs (Fig. 1c, f).
Molecular basis for the recognition of histamine and imetit by H4R
Histamine and imetit share a common imidazole backbone, and imetit is characterized by a more extended structure with an additional isothiourea group, when compared with histamine (Fig. 1c, f). In the cryo-EM structure, histamine and imetit shared a common ligand-binding pocket surrounded by residues consisting of TM3, 4, 5, 6, and 7 (Fig. 2a, b, d, e). The binding mode of histamine and imetit was verified by a transforming growth factor (TGF)-α shedding assay using mutants of H4R, and was observed to be consistent with that in the model structure (Fig. 2c, f, Supplementary Fig. 4 and Supplementary Table 1)24.

a Detailed interactions between histamine and H4R. Histamine and contact residues are shown as yellow and pink sticks. b Schematic representation of histamine-binding interactions. Hydrogen bonds are shown as dashed lines, and hydrophobic interactions and amino acids are shown in green. c Mutant study to assess the histamine response by the TGFα shedding assay. Dashed lines in the mutant graphs represent the wild-type (WT) H4R response. Concentration-response curves are shown as the mean ± standard error of the mean (SEM) from three independent experiments performed in triplicates. In many data points, the error bars are smaller than the symbols and are therefore not visible. d Detailed interactions between imetit and H4R. Imetit and contact residues are shown as magenta and blue sticks. e Schematic representation of imetit-binding interactions. f Mutant study to assess the imetit response by the TGFα shedding assay. Details are the same as in (c).
In the structure of H4Rhis, the primary amino group of the ethylamine portion of histamine formed a salt bridge with Asp943.32 (Fig. 2a). This interaction is widely conserved in aminergic receptor structures, including the H1R structure. A loss of activity was observed in the D943.32A mutant of H4Rhis (Fig. 2c). In many aminergic receptors, Asp3.32 forms a hydrogen bond with the neighboring Trp/Tyr7.43, thereby stabilizing its side chain and facilitating interactions with the amine group of the ligand25. Hydrogen bonding between Asp943.32 and Trp3487.43 was also observed in H4Rhis, and the loss of signaling activity of the mutant W3487.43A indicated that the stability of Asp943.32 is important for ligand binding (Fig. 2a, c). The TM7 residues of Phe3447.39, Gln3477.42, and Trp3487.43 are located within the van der Waals radius of the primary amino group of histamine and form hydrophobic interactions with it. Furthermore, Gln3477.42 forms weak polar interactions with the imidazole group of histamine. These interactions are essential for histamine binding, and their importance was confirmed by the reduced activity of Phe3447.39 and Gln3477.42 in the Ala mutant (Fig. 2c).
On the opposite side of histamine from Asp943.32, Tyr3196.51 is conserved in aminergic receptors in a similar manner as Tyr/Phe6.51, suggesting that it is an essential residue for agonist and antagonist binding19,20,26,27. In H4Rhis, histamine forms hydrophobic contacts with this residue, and it was verified that the introduction of mutations to this residue is directly related to the decrease in receptor activity (Fig. 2a–c). Tyr/Phe6.51 of TM6 plays an important role in ligand binding by forming aromatic clusters with Trp6.48 and Phe6.52 in the aminergic receptor28. In addition to ionic binding to Asp3.32, many aminergic ligands form an aromatic stacking or hydrophobic interaction with this aromatic cluster. In H4Rhis, Trp3166.48 was further coordinated to the bottom of the ligand binding pocket and exhibited a stacking interaction with the imidazole ring of histamine. So far, H4R mutant experiments have already been conducted on some of the above-mentioned amino acid residues in previous studies29,30, and it was confirmed that there was no significant deviation from the experiments in the present study.
The imidazole ring of histamine was bound to a pocket consisting of Tyr953.33, Thr993.37, Asn1474.57, Glu1825.46, Trp3166.48, and Tyr3196.51. As mentioned above, the imidazole ring was stabilized by pi–pi stacking interactions from both the upper and lower sides, with Tyr953.33 as a lid, in addition to Trp3166.48 at the bottom of the pocket (Fig. 2a, b). In particular, the Y953.33A mutant dramatically reduced histamine activity, suggesting that it plays an essential role in binding (Fig. 2c). In addition, Glu1825.46 is an important residue in the binding of the imidazole ring. The nitrogen atom (Nτ) on the third position of the imidazole ring formed an ionic pair with Glu1825.46, suggesting the importance of Glu1825.46 along with that of Asp943.32 in the binding process. Previous studies have reported that mutants of this residue prevent histamine binding. This is in line with our signaling activity data (Fig. 2c)29,30. Although Thr993.37 and Asn1474.57, which form a pocket, do not interact directly with the ligand, the signaling activity of the T99A3.37 and N147A4.57 mutants was found to be 2.4- and 9.2-fold lower, respectively, than that of wild-type H4R. Thr993.37 is involved in pocket formation, and Asn1474.57 contributes to the stabilization of the interaction with histamine by forming a hydrogen bond with the neighboring Glu1825.46 and regulating the coordination of its side chain, as observed between Trp3487.43 and Asp943.32 (Fig. 2a–c, Supplementary Table 1). Position 4.57 is Asn4.57 in human H4R but His4.57 in pigs and dogs; therefore, altering the side chain conformation of Glu5.46 has been reported to be a possible reason for the species-specific differences in histamine affinity31.
In H4Rime, imetit was found to bind to a pocket in a manner similar to that of histamine (Fig. 2a, b, d, e). Imetit had two hydrogen bond donors, which were complementary to the two negative residues of the binding pocket, Asp943.32 and Glu1825.46, respectively. Mutagenesis experiments confirmed that these interactions are essential for imetit binding (Fig. 2f). Previous studies have suggested that the isothiourea group is a key determinant of the H4R activity enhancement in histaminergic compounds18. The cryo-EM structure of H4Rime revealed that the isothiourea group of imetit plays a significant role in ligand binding. This substructure of the agonist penetrated the subpocket that is formed by mostly aromatic and hydrophilic residues, such as Tyr3196.51, Phe3447.39, Gln3477.42, and Trp3487.43 (Fig. 2d, e). The two nitrogen atoms existing in the region formed a salt bridge with Asp943.32 for the primary amino group and a hydrogen bond with the oxygen atom of the main chain of Phe3447.39 for the methanimine group. The primary amino group not only interacted with Asp943.32, but also formed pi–cation interactions with Tyr3196.51, Phe3447.39, and Trp3487.43, which constitute the hydrophobic pocket, thereby increasing ligand binding stability. Mutant Y319A6.51 exhibited a 5.3-fold decrease in signaling, while mutants F344A7.39 and W348A7.43 had even greater effects when compared with wild-type H4R, indicating that the residues are crucial for imetit binding (Fig. 2f, Supplementary Table 1). The imidazole ring of imetit was tightly accommodated in a pocket consisting of TM3, 4, 5, and 6 (Fig. 2d, e). Furthermore, the hydrophobic interactions formed between imetit and Tyr953.33, Cys983.36, Thr993.37, and Asn1474.57 were shown to be crucial for its binding (Fig. 2f, Supplementary Table 1). Similar to histamine, the imidazole ring of imetit was stably coordinated in the pocket by forming stacking hydrophobic interactions with Tyr953.33 and Trp3166.48. However, unlike histamine, the imidazole group of imetit does not interact with Gln3477.42. This may be the difference between the two agonists in the Q347A7.42 mutant (Fig. 2c, f, Supplementary Fig. 4b).
Comparison of agonist-binding characteristics between H4Rhis and H4Rime
Imetit was initially developed as an agonist for H3R32,33 but has also been reported to have a high binding affinity (Ki) for H4R (imetit Ki = 2.7 nM, histamine Ki = 8.1 nM)10. As described in the previous section, the residues constituting the orthosteric binding site in H4Rhis and H4Rime were almost identical. However, the binding mode of the two agonists was clearly different (Fig. 3, Supplementary Figs. 5a, b). Imetit has a larger surface area than histamine due to the substituted isothiourea moiety (histamine: 267.8 Å2 vs. imetit: 372.3 Å2) (Supplementary Fig. 5c). Although H4Rhis and H4Rime possess ligand-binding pockets of approximately the same size, several unique recognition mechanisms were established to make each agonist acceptable. First, remarkable differences in the binding features of both agonists were observed in the pockets around TM6 and 7. The H4Rime structure formed a distinct pocket (hereafter referred to as the “aromatic slot”) consisting of Tyr3196.51, Phe3447.39, Trp3487.43, and Gln3477.42, which was not observed in the H4Rhis structure. Compared to the histamine-bound structure, this pocket was generated by the Cβ–Cγ bond of Phe3447.39 that rotated approximately 90 degrees, like a turning doorknob, allowing it to accept a more extended imetit (Fig. 3b, c). The isothiourea moiety of imetit is likely protonated and charged. Both nitrogen atoms are equivalent and actively utilize this specific pocket to form multiple interactions with residues in the pocket. This may contribute to the increased binding strength of imetit. In contrast, the primary amine of histamine interacts relatively simply with Asp943.32 and Phe3447.39, while the isothiourea portion of imetit utilizes the entire aromatic slot, which may be one of the reasons why the binding affinity of imetit is approximately three times higher than that of histamine10.

a Structural alignment of the agonist-binding pocket of H4Rhis (pink) and H4Rime (blue). Histamine (yellow), imetit (magenta), and the surrounding residues of each agonist are shown as sticks. b, c Hydrophobic residue cluster of TM7 and 8 in H4Rhis (b) and H4Rime (c). The side chain rearrangement of Phe3447.39 forms the aromatic slot on H4Rime. Residues comprising the pocket are indicated by sticks and transparent spheres and are pink (H4Rhis) and blue (H4Rime).
The interaction between imetit and the aromatic slot and the overall molecular geometry of imetit caused its imidazole ring to coordinate 1.8 Å closer to TM3 than histamine. Imetit further exhibited a stronger interaction with peripheral residues such as Cys983.36 and Thr993.37, which may have also contributed to the binding affinity of imetit for H4R (Fig. 3a). The formation of a distinct ligand-specific binding pocket by the conformational change of the Phe side chain, as observed in the aromatic slot of H4R, has also been reported for the extended binding pocket in the crystal structure of the dopamine D2 receptor (in this case, Phe3.28), which is a similar aminergic receptor27. Since it is difficult to consider the structural plasticity of ligand-binding pockets in drug design, identifying the flexible ligand-binding pocket shown in our structure may make a significant contribution to rational drug design.
Subtype-selectivity of H4R and H1R
While the affinity of H1R and H2R for histamine is in the μM range, H3R and H4R are known to have a high affinity in nM range1,34. Histamine receptor subtypes therefore require different concentrations of histamine for activation; however, the molecular basis for the selectivity between these subtypes remains unknown given the lack of structural information on histamine receptors. Here, we assessed the selectivity between subtypes in histamine receptors by comparing the cryo-EM structure H1Rhis of histamine-bound H1R20 with the structures H4Rhis and H4Rime obtained in the present study.
The overall structure of the receptor region of H1Rhis and H4Rhis was relatively similar except for the extracellular ends of TM2 and 7 and the positions of the orthosteric binding pocket to which histamine bonds were almost identical (Fig. 4a). However, some of the residues that formed the ligand-binding pocket were different, which may have determined the affinity of histamine for both receptors. The most significant difference between the two receptors in histamine binding was the amino acid corresponding to position 5.46. Asn1925.46 of H1Rhis and Glu1825.46 of H4Rhis formed hydrogen bonds with Nτ atoms in the imidazole ring of histamine; however, in H4R, histamine exerted a stronger interaction with negatively charged Glu1825.46 by establishing an ionic association (Fig. 4b). In contrast, the corresponding amino acid Asn1925.46 in H1R is a neutral amino acid; therefore, the interaction was relatively weak, and the binding energy may have been lower than that in H4R. As described in the previous section, Glu1825.46 in H4R had its side chain coordination stabilized by hydrogen bonding with the neighboring Asn1474.57, whereas position 4.57 in H1R was Trp and could not form a hydrogen bond network with position 5.46. The side chain stabilization effect was therefore not achieved. These structural differences were thought to affect the stability of the interaction with the Nτ atom of histamine. In fact, the H1R mimic mutants E182N5.46 and N147W4.57 showed a loss of signaling activity for histamine, while the H3R type mutant N147Y4.57 at position 4.57 exhibited relatively similar EC50 compared to wild type H4R (WT 55.4 nM vs. mutant 82.9 nM), as verified by the TGFα shedding assay. This indicated that the configuration and interaction of the residues in positions 4.57 and 5.46 in histamine binding are some of key factors that influence the difference in affinity between H1R and H4R (Fig. 4e, f, Supplementary Fig. 4 and Supplementary Table 1).

a, b Structural superposition of H4Rhis (pink) and H1Rhis (orange, PDB 7DFL). (a) top view, (b) the agonist-binding pocket. The helices are shown as cylinders. The histamines of H4R and H1R are indicated by yellow and cyan sticks. c The aromatic slot on H4Rime (blue) and the corresponding region for H1Rhis (orange). Imetit and histamine are shown as magenta and cyan sticks. Residues comprising the pocket for H4Rime are indicated by sticks and for H1Rhis are shown as sticks and transparent spheres. d Sequence alignment of residues important for agonist binding in the histamine receptor family. Red triangles indicate residues corresponding to the aromatic slots in H4R. e, f TGFα shedding assay results of WT H4R and H4R mutants activated by histamine and imetit. The activities of the agonists are identified as pEC50 (e). Average Emax values were determined from the “log (agonist) vs. response -variable slope (three or four parameters)” function in GraphPad Prism 9 software (GraphPad Software Inc., San Diego, CA) (f). Data represent the mean ± SEM from n = 3 biologically independent experiments performed in triplicate. ND, not detected.
The primary amino group in histamine also exhibited different binding for both receptors. The ionic interaction at position 3.32, which is common in aminergic receptors, was conserved in both H1Rhis and H4Rhis. In H1Rhis, however, the cationic amine moiety of histamine interacted relatively simply with Asp1073.32 and Tyr4316.51, whereas in H4Rhis, it interacted with the hydrophobic residue cluster on TM7 to gain greater binding affinity (Fig. 4b). Position 7.39 differed between H1R and H4R based on Ile and Phe, respectively, and comprised Phe3447.39 in H4R, which has a bulkier side chain that was present within a distance where it could interact with histamine. The H1R mimic mutant F344I7.39 did not exhibit histamine activity, which indicated that this residue plays a critical role in histamine recognition (Fig. 4e, f). We posit that the interaction between histamine and Gln3477.42 also affects histamine affinity. As previously mentioned, in the H4Rhis, Gln3477.42 forms a polar interaction with the imidazole ring of histamine. However, such binding is not observed in H1Rhis and the signaling activity of the H1R/H2R type mutant Q347G7.42 decreases about 46 times compared to that of the wild type (Fig. 4e, f, Supplementary Fig. 4 and Supplementary Table 1). Based on such observations, we consider the interaction of Gln3477.42 and histamine in H4R to be one of the reasons for the high affinity.
Imetit is an H3R/H4R selective agonist that does not bind to H1R/H2R and has a high binding affinity for H3R/H4R of 0.3 and 2.7 nM, respectively10,33. Since the publication of the structure of the first histamine receptor, doxepin-bound human H1R19, various computational chemistry approaches have been used to determine the molecular determinants of H3R/H4R ligand binding, and several binding modes have been predicted. However, in most reports, no determinant other than Glu1825.46 has been found to be important for subtype selectivity35,36. In the present study, we determined the structure of imetit-bound H4R, which can be compared with H1Rhis, to assess the mechanisms underlying subtype selectivity in histamine receptors. As mentioned above, a detailed investigation of histamine binding to H1R/H4R revealed that Glu1825.46 is undoubtedly one of the key residues involved in the selectivity of histamine receptors (Fig. 4b–f). Moreover, we identified the aromatic slot, a pocket that is crucial for the binding of the isothiourea region of imetit, as an important region for the selectivity of H1R and H4R (Fig. 4c). The aromatic slot consisting of Tyr3196.51, Phe3447.39, Gln3477.42, and Trp3487.43 was formed by the flexible orientation of the side chain of Phe in position 7.39, which is characterized by a mutually reinforced shape (Fig. 3b, c). The most important factor for the correct function of the aromatic slot is the flexibility of the side chain at position 7.39. In H3R/H4R, the residues corresponding to this position are both Phe, but in H1R/H2R, they are Ile and Leu, respectively. The formation and maintenance of the aromatic slot by conformational changes in these side chains are therefore unlikely (Fig. 4c, d). Furthermore, the orientations of Ile4547.39 and Tyr4587.43 in H1R are likely to cause steric hindrance with the two nitrogen atoms in the isothiourea group. The acceptance of the isothiourea group of imetit in H1R is further likely to be challenging given the restricted coordination of the Tyr4587.43 side chain in maintaining the Asp3.32–Trp/Tyr7.43 interaction that is common in aminergic receptors.
The importance of the aromatic slot in the selectivity of histamine receptors was also verified in mutant experiments. H1R mimic mutants F344I7.39 and W348Y7.43 exhibited a loss of imetit activity, thereby supporting the above structural findings (Fig. 4e, f). However, Q347G7.42 exhibited a slight increase in the imetit signal, which is consistent with the activity for Q347A7.42 mutant mentioned earlier. The conformation of the extended side chain of Gln3477.42 is thought to restrict the conformation of Trp3166.48, and the removal of this restriction by mutation of Gln3477.42 gives the Trp3166.48 rotamer more flexibility, suggesting that it may interact more closely with imetit. Since the interaction between Gln3477.42 and the imidazole ring of histamine was observed, we believe that its activity is reduced by the same mutations (Fig. 4e, f). We also found that this aromatic slot comprised a unique topology, not only for histamine receptors, but also for other aminergic receptors (Supplementary Fig. 6). The fact that residues in positions 7.39 and 7.42 of H4R were less conserved in other receptors supports that this sub-pocket in H4R is highly distinctive, and targeting this aromatic slot in H4R may be useful for the design of compounds specific to H4R (or H3R). Although the isothiourea group of imetit did not appear to be a substituent developed for the aromatic slot, our structure indicated that it was a substitution that could effectively exploit the slot.
As mentioned in the previous section, aromatic clusters on TM6 (Trp6.48, Phe/Tyr6.51, and Phe6.52) play an important role in the binding of agonists and antagonists at aminergic receptors25,28. In H1Rhis, the imidazole ring of histamine also interacted with this aromatic cluster and formed a hydrophobic interaction with Phe4356.55 within the Van Der Waals radius (Supplementary Fig. 7c). However, in H4R, some of the residues constituting this aromatic cluster were compact polar residues (Ser3206.52 and Thr3236.55) and created a wider polar subpocket between TM5 and 6 (Supplementary Fig. 7a, b). Since the residues in this position were distinguishable between H1R/H2R and H3R/H4R, the effective utilization of this pocket with aromatic substituents may have been important for generating selectivity in histamine receptor subtypes (Fig. 4d).
Activation mechanism of H4R
Since the inactive structure of H4R has not yet been elucidated, we assessed the activation mechanism of H4R by comparing the inactive structure of antagonist-bound H1R (H1Rdox)19 with our agonist-bound H4R structure H4Rime. Four motif structures (CWxP, PI(V)F, DR(E)Y, and NPxxY) that are important for GPCR activation are conserved in H4R; therefore, it is valid to compare H4Rime and inactive H1Rdox to assess these motif structures and the activation mechanism of H4R (Fig. 5a). Different pharmacological features exist for each ligand that binds to the GPCR. This is thought to be the result of the orientation of the side chains of this conserved local microswitch that affects the global movement of the helical backbone37,38. In agonist-bound H4Rime, a hydrophobic interaction between the agonist and Trp3166.48 (known as the “toggle switch”) of TM6 was observed and caused a downward swing in the side chain (Fig. 5b). This conformational change in Trp3166.48 promoted the rearrangement of the unique configuration of Pro1865.50-Val1023.40-Phe3126.44 (PIF motif in other GPCRs) and Asp1113.49-Arg1123.50-Tyr1133.51 (DR(E)Y motif), thus allowing movement outside the cytoplasmic end of TM6 (Fig. 5c, d, f). The NPxxY motif at the cytoplasmic end of TM7 also showed a conformational change in the side chain rotamer of Tyr3587.53. This conformational change moved TM7 toward the TM3 and 5 sides, a distance that allowed an interaction with the DR(E)Y motif (Fig. 5e). Thus, the conformational changes of the microswitch and global movement of the TM bundle, which are commonly observed in GPCRs, were conserved in our structure. This confirmed that our H4Rhis and H4Rime structures are the active conformations that interact with trimeric G proteins.

a Activation motifs in the H4R structure. Each motif residue is shown as a blue stick. b–g Structural comparison between the active H4Rime (blue) and antagonist-bound inactive H1Rhis (gray, PDB 3RZE). (b) CwxP motif, (c) PI(V)F motif, (d) DR(E)Y motif, (e) NPxxY motif. Imetit and doxepin are shown as magenta and khaki sticks. The residues involved in receptor activation are indicated by sticks, and conformational changes are shown as red arrows. Microswitch rearrangement allowed movement outside the cytoplasmic ends of TM5 and 6 (f). g Gln3477.42 coordination and its effect on surrounding residues.
Recently, the inactive structures of the antagonist-bound histamine H2 receptor (H2R) and the histamine H3 receptor (H3R) were published39,40, and comparison of our agonist-bound H4R structure with the PF-03654746-bound inactive H3R structure suggests that the activation mechanism in H3R/H4R is different from that in H1R. As mentioned above, in the case of many aminergic receptors, the agonist interacts with the toggle switch, causing a downward swing of its side chain and rearrangement of the downstream structure to the active form. In the case of H3R/H4R, however, the present structure shows that the side chain of the toggle switch is not moved perpendicularly but horizontally (Fig. 5g). In H4R, the agonist formed the hydrophobic interaction with Trp3166.48 and changes its side chain about 90° to the TM3 side, which may serve as a trigger in receptor activation. Here, the residue at position 7.42 may play an important role in controlling the conformational change of the toggle switch horizontally. The side chain of Gln3477.42 in the cryo-EM structure of H4R is extended to the TM6 side, which is consistent with Leu4017.42 conformation in the H3R structure. In most aminergic receptors, position 7.42 is conserved in Gly (only muscarinic acetylcholine receptors have Cys), but only H3R/H4R have Leu/Gln, which have larger side chains than Gly (Supplementary Fig. 8). In other words, the toggle switch within the H3R/H4R appears to have a unique movement, characterized by a lateral movement that maintains the “knock over” state to prevent steric hindrance with Leu/Gln7.42.
Discussion
In this study, we present the cryo-EM structures of the histamine H4 receptor bound by two agonists histamine and imetit, respectively, which exhibit distinct pharmacological properties. Although Gq signal activity could not be measured directly in the in-house measurement, weak Gq coupling was reported in the previous study21. The tethering system used in the present study effectively brings GPCRs and trimeric G proteins into close proximity, which is thought to make the binding with Gq stronger, enabling structural analysis.
In histamine receptor subtypes, H4R has low sequence similarities with H1R and H2R but has a more similar sequence identity with H3R. Given the overlap of the sequences, it would be difficult to develop a ligand that is H4R-selective and H3R-nonselective. Since the active structure of H3R is currently unknown, we generated a homology model of active state H3R based on the H4R structure revealed in the present study and compared their ligand recognition sites (Supplementary Fig. 9). Although the orthosteric binding sites in both receptors were expected to be similar, there were differences in some of the residues involved in ligand recognition (Fig. 4d, Supplementary Figs. 9a–c). Histamine had almost the same affinity for H3R/H4R, but OUP-16, which is an imidazole derivative agonist, exhibited 16-fold selectivity for H4R (H3R Ki = 2 μM / H4R Ki = 125 nM) (Supplementary Fig. 10)15. To investigate the difference in selectivity, we also performed a docking simulation of OUP-16 for the cryo-EM structure of H4R obtained in the present study (Supplementary Fig. 9d). Comparing the docking result of OUP-16 and homology model of H3R may explain the differences in their affinity. The docking simulation indicated that the guanidine moiety of OUP-16 engages in hydrogen bonding with Asp94. The imidazole ring on the opposite side of the agonist exhibits an alternative binding mode compared to the imidazole ring of histamine and is oriented toward the extracellular ends of TM5 and 6. Furthermore, the hydrogen bond with Glu182 is maintained and the interaction with Ser320 is established. The entire imidazole ring is occupied within the sub-pocket formed between TM4, 5, and 6. Within this pocket, however, there were different residues (Y/N4.57, T/S6.52, M/T6.55) for H3R/H4R, which may have resulted in the differences in pocket shape and properties in the two receptors (Supplementary Fig. 9a–c, d). The coordination of the hydrophobic and massive residues Tyr1674.57 and Met3786.55 next to Glu2065.46, which is essential for hydrogen bonding with histamine in H3R, implies that the introduction of an additional sub-pocket is not tolerated. Therefore, the design of ligands that take into account the shape of the orthosteric binding pocket specific to H3R and H4R would allow for their respective selectivity to be generated. JNJ-7777120 was the first H4R-selective antagonist to be discovered and has contributed significantly to our understanding of the pathophysiological functions of H4R (Supplementary Fig. 10)41. In addition, the compound has interesting pharmacological characteristics, such as biased activity toward β-arrestin42,43. Several antagonists derived from this compound have been demonstrated to be effective against pruritus, dermatitis, asthma, and arthritis in preclinical studies in animal models. Clinical trials have been conducted in some cases44,45. Since ligands that achieve such pharmacological effects require a high selectivity for H4R, the structures of H4R revealed in the present study may provide information that could facilitate the development of H4R-selective ligands. We also performed a docking study on JNJ-7777120 (Supplementary Fig. 9e). The results show that the H4R-specific sub-pocket provides space to accept the chloroindole ring of JNJ-7777120, which is not present in other histamine receptors, including H3R, and is expected to be an important region that allows subtype selectivity to be acquired. In addition, mutant experiments in previous reports have demonstrated that Asn1474.57, Phe169ECL2, Leu1755.39, and Ser1795.43, which correspond to this sub-pocket, decrease the activity of JNJ-777120, which could support our structural considerations31,46.
Acquiring the inactive structure of H4R is a future challenge that may yield a better understanding of the antagonist binding and the activation mechanism of H4R. The structural, biological, and pharmacological results of the H4R obtained in this study provide details on the molecular mechanisms of agonism in H4R and key insights into the mechanism of selectivity in histamine receptor subtypes. A rational structure-based design for effective drugs against CIDs may be developed further using the structural information on H4R presented herein.
Methods
Constructs
The full-length human H4R (residues 1–390) was subcloned into a pFastBac1 plasmid with an N-terminal hemagglutinin (HA) signal peptide, a FLAG tag, and 8× His tag, followed by a thermostabilized apocytochrome b562RIL (bRIL) epitope47 to facilitate protein expression and stability (Supplementary Fig. 1a). An HRV 3 C protease cleavage site and linker residues were inserted between the H4R and bRIL. The artificial Gq used in the present study was based on the mini-Gs skeleton, which has two dominant-negative mutations and the alpha-helical domain truncation48. The 35 amino acids of the N-terminal were replaced with the corresponding sequence of Gαi1 to facilitate scFv16 binding49,50. Such an approach was used to obtain the structures of Gq-bound 5-HT2A receptor/ghrelin receptor and G11-bound M1 receptors51,52,53. Furthermore, Gi (DNGi1) corresponding to Supplementary Fig. 1b was generated by introducing four dominant-negative mutations, S47N, G203A, A326S, and E245A50. To improve the stability and homogeneity of the receptor-G protein complex, we employed the NanoBiT tethering system with a large fragment of NanoLuc (LgBiT) and high affinity 11 amino acid peptide (HiBiT) tag attached to the C-terminus of the receptor and Gβ1, respectively54. Gq, Nb35, scFv16, and Ric8A were cloned into the pFastBac1 plasmid. His10-rat Gβ1 with C-terminal HiBiT and bovine Gγ2 were subcloned into the pFastBac Dual vector.
Protein expression and complex purification
Recombinant baculoviruses were prepared using the Bac-to-Bac baculovirus expression system (Invitrogen, Carlsbad, CA). Spodoptera frugiperda (Sf9) insect cells were grown to a density of 2–3 × 106 cells/mL and co-infected with H4R, Gq, Gβ1Gγ2, Nb35, scFv16, and Ric8A viral stocks at a multiplicity of infection ratio of 8:2:2:1:1:1. They were harvested 48 h later. Cell pellets were resuspended in a buffer containing 20 mM of HEPES (pH 7.5), 100 mM of NaCl, 10% glycerol, 5 mM of MgCl2, 5 mM of CaCl2, 0.25 mM of TCEP, protease inhibitor cocktail (Nacalai Tesque), 25 mU/mL of apyrase, and 50 μM of H4R agonists (histamine or imetit) followed by incubation for 1 h at room temperature. Cell membranes were collected by centrifugation and solubilized in Solubilization Buffer containing 20 mM of HEPES (pH 7.5), 100 mM of NaCl, 10% glycerol, 5 mM of MgCl2, 5 mM of CaCl2, 0.25 mM of TCEP, protease inhibitor cocktail, 25 mU/mL of apyrase, 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) with 0.05% (w/v) cholesteryl hemisuccinate (CHS, Sigma-Aldrich), and 50 μM of agonists, for 2 h at 4 °C.
Insoluble materials were removed by centrifugation, and the supernatants were incubated with TALON metal affinity resin (Clontech) overnight at 4 °C. The resin was washed with 10 column volumes (CV) of Wash Buffer containing 20 mM of HEPES (pH 7.5), 100 mM of NaCl, 10% glycerol, 2 mM of MgCl2, 2 mM of CaCl2, 20 mM of imidazole, 0.25 mM of TCEP, 0.01% (w/v) LMNG, 0.01% (w/v) GDN (Anatrace), 0.001% (w/v) CHS, and 20 μM of agonists. The complex was then eluted with 3 CV of Elution Buffer containing 20 mM of HEPES (pH 7.5), 100 mM of NaCl, 10% glycerol, 2 mM of MgCl2, 2 mM of CaCl2, 250 mM of imidazole, 0.25 mM of TCEP, 0.01% (w/v) LMNG, 0.001% (w/v) GDN (Anatrace), 0.001% (w/v) CHS, and 20 μM of agonists. The N-terminal FLAG tag and bRIL epitope were cleaved by an HRV 3 C protease for 3 h at 4 °C. Finally, the complex was concentrated to 0.5 mL using the Amicon Ultra-15 concentrator (Millipore) and subjected to size-exclusion chromatography on a Superdex 200 Increases 10/300 column (GE Healthcare) pre-equilibrated with a buffer containing 20 mM of HEPES (pH 7.5), 100 mM of NaCl, 2 mM of MgCl2, 2 mM of CaCl2, 0.25 mM of TCEP, 0.0015% (w/v) LMNG, 0.0005 (w/v) GDN, 0.00015% (w/v) CHS, and 20 μM of agonists. The fractions containing the monomeric complex were collected and concentrated to approximately 10 mg/mL for electron microscopy experiments. Expression and purification of the H4R-Gi complex (corresponding to Supplementary Fig. 1b) was performed similar to that for Gq above. However, since Nb35 does not bind to Gi, Nb35 co-expression was not performed.
Cryo-EM grid preparation and data collection
The Quantifoil R1.2/1.3 holy carbon copper grid (Quantifoil) was glow-discharged at 7 Pa with 10 mA for 10 s using a JEC-3000FC sputter coater (JEOL) prior to use. A 3-μL aliquot was applied to the grid, blotted for 3.5 s with a blot force of 10 in 100% humidity at 8 °C, and plunged into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM data collection for screening sample quality and grid conditions was performed using a Glacios Cryo-transmission electron microscope operated at 200 kV accelerating voltage with a Falcon4 camera (Thermo Fisher Scientific) at the Institute for Life and Medical Sciences, Kyoto University. After several screening sessions, data were collected using a Titan Krios (Thermo Fisher Scientific) at 300 kV accelerating voltage equipped with a direct K3 electron detector, Gatan BioQuantum energy filter (slit width of 20 eV) (Gatan), and Cs corrector (CEOS, GmbH), which were installed at the Institute for Protein Research, Osaka University. Data collection was carried out using SerialEM software55 at a norminal magnification of ×81,000 (calibrated pixel size of 0.88 Å pixel−1) with a total exposure time of 5.0 s (50 frames) with a defocus range of –0.8 to –2.0 μm. The detailed imaging conditions are described in Table 1.
Cryo-EM data processing
All image processing was performed using cryoSPARC56. We manually inspected and curated the micrographs after patch-based motion correction and contrast transfer function (CTF) estimation. The particles for the histamine-bound-H4R-Gq complex were selected using the Blob particle picker and initial two-dimensional (2D) classification yielded templates for subsequent template picking. A subset of the selected particles was used as a training set for Topaz, which was used to repick the particles from the micrographs57. The particles for the imetit-bound H4R-Gq complex were picked using the automated procedure in crYOLO58. The picked particles were subjected to a 2D classification to discard particles in poorly defined classes. Ab initio reconstruction was performed in cryoSPARC asking for four classes, which resulted in one good class and three trash classes. Multiple rounds of heterogeneous refinement were then performed against the four ab initio models to remove bad particles. The selected particles were extracted at full pixel size and subjected to non-uniform (NU) refinement59. Further classification without alignment was performed using a soft protein mask to create a better-looking ligand binding region for the histamine-bound-H4R-Gq complex.
The densities were further Improved by per-particle defocus refinement and another round of non-uniform refinement to generate the final map. Resolutions were estimated using the ‘gold standard’ criterion (FSC = 0.143). The local resolution was calculated in cryoSPARC. Map sharpening was reevaluated with the Phenix autosharpen tool60,61. These maps were used for modeling. The validation on the coordinate refinement (FSCwork/FSCtest) was calculated using servalcat62. The processing strategy is described in Supplementary Fig. 2.
Model building and refinement
Model building was facilitated by both the previous cryo-EM structures of the GHSR-Gq complex (PDB 7F9Z) and H1R bound to histamine (PDB 7DFL)20,53. The receptor, Gq trimer, Nb35, and scFv16 models were manually built in Coot63, followed by several rounds of real-space refinement using Phenix64. All molecular graphics were prepared using CueMol (http://www.cuemol.org) and UCSF ChimeraX65. The 3D reconstruction and model refinement statistics are summarized in Table 1.
Molecular docking
The docking study was performed using H4Rime with the Glide program66 in the Schrödinger Suite 2022-4 (Schrödinger LLC). The coordinates of OUP-16 and JNJ-7777120 were initially built using LigPrep in the Schrödinger Suite at pH 7.0 and the OPLS3 force field. Each ligand was docked into the binding pocket of H4R, resulting in ten conformations per ligand. The most reliable binding poses were selected based on the interaction energy and visual inspection.
TGFα shedding assay
The mutants were prepared using the primers listed in Supplementary Table 2. The activity of the endogenous agonist (histamine) and its derivative (imetit) for H4R mutants on G protein signaling was determined using a TGFα shedding assay. In brief, a pCAGGS plasmid encoding the human wild-type or a mutant H4R (human, full-length, and untagged), together with pCAGGS plasmids that encoded the chimeric Gαq/i1 subunit and alkaline phosphatase-tagged TGFα (AP-TGFα; human codon-optimized), were transfected into HEK293A cells by a Lipofectamine® 3000 (LFA) transfection reagent (Thermo Fisher Scientific) (125 ng of the H4R plasmid, 125 ng of the Gαq/i1 plasmid, 625 ng of the AP-TGFα plasmid, and 18.2 μL of LFA per well in a six-well culture plate). The chimeric Gαq/i1 subunit comprises the Gαq backbone and Gαi1-derived six-amino acid C-terminus and couples with Gi-coupled H4R but induces a Gq-dependent TGFα shedding response24. After culturing for 1 day and incubating at 37 °C in a 5% CO2 incubator, the transfected cells were harvested by trypsinization, neutralized with Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (FCS) and penicillin–streptomycin, washed once with Hank’s balanced salt solution (HBSS) containing 5 mM of HEPES (pH 7.4), and resuspended in 6 mL of the HEPES-containing HBSS. The cell suspension was seeded into a 96-well plate at a volume of 90 mL per well (typically, 48 wells per transfected cells) and incubated for 30 min in the CO2 incubator. Test compounds (10×, diluted in 0.01% BSA and 5 mM of HEPES-containing HBSS, 10 mL volume) were added to duplicate wells and incubated for 1 h. After centrifugation, the conditioned medium (80 mL) was transferred to an empty 96-well plate. AP reaction solution [10 mM of p-nitrophenylphosphate (p-NPP), 120 mM of Tris–HCl (pH 9.5), 40 mM of NaCl, and 10 mM of MgCl2] was dispensed into the cell culture plates and plates containing conditioned media (80 mL). Absorbance was measured at 405 nm before and after a 1-h incubation period at 25 °C using a microplate reader (iMark™ Microplate Absorbance Reader; BioRad). Ligand-induced AP-TGFα release was calculated as previously described24. The vehicle-treated AP-TGFα release signal was set as a baseline unless otherwise specified. AP-TGFα release signals were fitted with a four-parameter sigmoidal concentration-response curve, from which EC50 and Emax values were obtained, using GraphPad Prism 9 software (GraphPad Prism). Negative values of logarithmically transformed EC50 values (pEC50) were used to calculate the mean and standard error of independent experiments.
To estimate the expression level of each H4R mutant, a FLAG tag was added to the N-terminus of all mutants. Transfection of the genes into the cells was performed as described above, and the FLAG-tagged mutant genes were transfected at the same time as all the genes required for the TGFα shedding assay. One day after gene transfer, transfected cells were harvested and stained with anti-DYKDDDDK tag monoclonal antibody (FUJIFILM Wako Pure Chemical Corporation) as primary antibody and Goat anti-mouse IgG antibody conjugated with Alexa Fluor™ 488 (Thermo Fisher Scientific) as a secondary antibody for 30 min on ice in the dark. Stained cells were replaced with FACS buffer (PBS(-) with 2% FCS and 0.1% NaN3), and cells were assayed using a flow cytometer (Guava EasyCyte Plus, Millipore). The expression levels of the mutant were estimated in terms of the mean fluorescence intensity (MFI) ratio (MFI of the mutant compared to that of the WT (Supplementary Fig. 4a).
Statistical analysis
All functional study data were analyzed using Prism 9 (GraphPad) and are presented as mean ± standard error of the mean (SEM) of three independent experiments. Statistical analyses were performed using Prism 9 (GraphPad) with one-way analysis of variance followed by Dunnett’s post-hoc test. Values with p < 0.05 are considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps and atomic coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and wwPDB under accession numbers EMD-33785 and 7YFC [https://doi.org/10.2210/pdb7YFC/pdb] for the histamine-bound H4R-Gq complex, EMD-33786 and 7YFD [https://doi.org/10.2210/pdb7YFD/pdb] for the imetit-bound H4R-Gq complex. Raw movies were deposited to EMPIAR data base (https://www.ebi.ac.uk/empiar/) with accession numbers EMPIAR-11708. Source data are provided with this paper.
References
Panula, P. et al. International union of basic and clinical pharmacology. XCVIII. histamine receptors. Pharmacol. Rev. 67, 601–655 (2015).
Ash, A. S. F. & Schild, H. O. Receptors mediating some actions of histamine. Br. J. Pharmacol. Chemother. 27, 427–439 (1966).
Black, J. W., Duncan, W. A. M., Durant, C. J., Ganellin, C. R. & Parsons, E. M. Definition and antagonism of histamine H2-Receptors. Nature 236, 385–390 (1972).
Thurmond, R. L. The histamine H4 receptor: From orphan to the clinic. Front. Pharmacol. 6, 1–11 (2015).
Arrang, J. M., Garbarg, M. & Schwartz, J. C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302, 832–837 (1983).
Lovenberg, T. W. et al. Cloning and functional expression of the human histamine H3 receptor. Mol. Pharmacol. 55, 1101–1107 (1999).
Schwartz, J. C. The histamine H3 receptor: From discovery to clinical trials with pitolisant. Br. J. Pharmacol. 163, 713–721 (2011).
Oda, T., Morikawa, N., Saito, Y., Masuho, Y. & Matsumoto, S. I. Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J. Biol. Chem. 275, 36781–36786 (2000).
Nakamura, T., Itadani, H., Hidaka, Y., Ohta, M. & Tanaka, K. Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem. Biophys. Res. Commun. 279, 615–620 (2000).
Liu, C. et al. Cloning and pharmacological characterization of a fourth histamine receptor (H4) expressed in bone marrow. Mol. Pharmacol. 59, 420–426 (2001).
Morse, K. L. et al. Cloning and characterization of a novel human histamine receptor. J. Pharmacol. Exp. Ther. 296, 1058–1066 (2001).
Nguyen, T. et al. Discovery of a novel member of the histamine receptor family. Mol. Pharmacol. 59, 427–433 (2001).
Gutzmer, R. Pathogenetic and therapeutic implications of the histamine H4 receptor in inflammatory skin diseases and pruritus. Front. Biosci. S3, 985 (2011).
Cramp, S. et al. Identification and hit-to-lead exploration of a novel series of histamine H4 receptor inverse agonists. Bioorganic Med. Chem. Lett. 20, 2516–2519 (2010).
Marson, C. M. Targeting the histamine H4 receptor. Chem. Rev. 111, 7121–7156 (2011).
Zampeli, E. & Tiligada, E. The role of histamine H 4 receptor in immune and inflammatory disorders. British J. Pharmacol. 157, 24–33 (2009).
Sadek, B. & Stark, H. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology 106, 56–73 (2016).
Corrêa, M. F. & Fernandes, J. P. D. S. Histamine H4 receptor ligands: Future applications and state of art. Chem. Biol. Drug Des. 85, 461–480 (2015).
Shimamura, T. et al. Structure of the human histamine H 1 receptor complex with doxepin. Nature 475, 65–72 (2011).
Xia, R. et al. Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat. Commun. 12, 1–9 (2021).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e25 (2019).
Rasmussen, S. G. F. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nat. 2011 4777366 477, 549–555 (2011).
Maeda, S. et al. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, (2018).
Inoue, A. et al. TGFα shedding assay: An accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021–1029 (2012).
Michino, M. et al. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 67, 198–213 (2015).
Zhuang, Y. et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 184, 931–942.e18 (2021).
Im, D. et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 11, 1–11 (2020).
Kooistra, A. J., Kuhne, S., De Esch, I. J. P., Leurs, R. & De Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 170, 101–126 (2013).
Jongejan, A. et al. Delineation of agonist binding to the human histamine H4 receptor using mutational analysis, homology modeling, and ab initio calculations. J. Chem. Inf. Model. 48, 1455–1463 (2008).
Shin, N. et al. Molecular modeling and site-specific mutagenesis of the histamine-binding site of the histamine H4 receptor. Mol. Pharmacol. 62, 38–47 (2002).
Lim, H. D. et al. Molecular determinants of ligand binding to H4R species variants. Mol. Pharmacol. 77, 734–743 (2010).
van der Goot, H., Schepers, M., Sterk, G. & Timmerman, H. Isothiourea analogues of histamine as potent agonists or antagonists of the histamine H3-receptor. Eur. J. Med. Chem. 27, 511–517 (1992).
Garbarg, M. et al. S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist.J. Pharmacol. Exp. Ther. 263, 304–310 (1992).
Mehta, P. et al. Enigmatic histamine receptor h4 for potential treatment of multiple inflammatory, autoimmune, and related diseases. Life 10, (2020).
Feng, Z., Hou, T. & Li, Y. Docking and MD study of histamine H4R based on the crystal structure of H1R. J. Mol. Graph. Model. 39, 1–12 (2013).
Schultes, S. et al. Mapping histamine H4 receptor-ligand binding modes. Medchemcomm 4, 193–204 (2013).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 55, 114–120 (2019).
Robertson, M. J. et al. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 29, 1188–1195 (2022).
Peng, X. et al. Structural basis for recognition of antihistamine drug by human histamine receptor. Nat. Commun. 13, (2022).
Jablonowski, J. A. et al. The first potent and selective non-imidazole human histamine H4 receptor antagonists. J. Med. Chem. 46, 3957–3960 (2003).
Rosethorne, E. M. & Charlton, S. J. Agonist-biased signalling at the histamine H4 receptor: JNJ7777120 recruits beta-arrestin without activating G proteins. Mol. Pharmacol. 79, 749–757 (2010).
Nijmeijer, S. et al. Detailed analysis of biased histamine H4 receptor signalling by JNJ 7777120 analogues. Br. J. Pharmacol. 170, 78–88 (2013).
Thurmond, R. L. et al. Clinical and preclinical characterization of the histamine H(4) receptor antagonist JNJ-39758979. J. Pharmacol. Exp. Ther. 349, 176–184 (2014).
Murata, Y. et al. Phase 2a, randomized, double-blind, placebo-controlled, multicenter, parallel-group study of a H4 R-antagonist (JNJ-39758979) in Japanese adults with moderate atopic dermatitis. J. Dermatol. 42, 129–139 (2015).
Wifling, D. et al. Molecular determinants for the high constitutive activity of the human histamine H4 receptor: Functional studies on orthologues and mutants. Br. J. Pharmacol. 172, 785–798 (2015).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Nehmea, R. et al. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One 12, 1–26 (2017).
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. W. & Tate, C. G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536, 104–107 (2016).
Liang, Y. L. et al. Dominant negative g proteins enhance formation and purification of agonist-gpcr-g protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 12–20 (2018).
Kim, K. et al. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 182, 1574–1588.e19 (2020).
Maeda, S., Qu, Q., Robertson, M. J., Skiniotis, G. & Kobilka, B. K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science (80-.). 364, 552–557 (2019).
Wang, Y. et al. Molecular recognition of an acyl-peptide hormone and activation of ghrelin receptor. Nat. Commun. 12, 5064 (2021).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 1–10 (2020).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T., Kelley, K., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. Nat. Commun. 11, 1–12 (2020).
Wagner, T. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 1–13 (2019).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Terwilliger, T. C., Sobolev, O. V., Afonine, P. V. & Adams, P. D. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. Sect. D Struct. Biol. 74, 545–559 (2018).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Yamashita, K., Palmer, C. M., Burnley, T. & Murshudov, G. N. Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallogr. Sect. D Struct. Biol. 77, 1282–1291 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D Struct. Biol. 74, 531–544 (2018).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Friesner, R. A. et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Acknowledgements
This work was supported by a Grant-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology (19H00923 (S.I.) and 23K06357 (D.I.)) and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from Japan Agency for Medical Research and Development (AMED) under the grant number JP21am0101079 and JP23ama121007 (S.I.). This study was also supported by the Takeda Science Foundation and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (D.I.). This work was performed in part under the Collaborative Research Program as the Visiting Fellow of Institute for Protein Research, Osaka University, VFCR–23-02 (S.I.) and in part using the cryo-electron microscope under the Collaborative Research Program of Institute for Protein Research, Osaka University, CEMCR-23-02 (H.A.).
Author information
Authors and Affiliations
Contributions
D.I., H.A., and S.I. designed the experiments. D.I. prepared the cryo-EM samples, and Y. Shiimura helped with the construction of Gq-heterotrimer expression. D.I., J.K., Y.F.F., Y. Sugita, T.N., T.K., and H.A. performed the cryo-EM analysis. H.H., A.I., and H.A. performed the TGFα shedding assay. D.I., H.A., and S.I. wrote the paper with assistance from all of the authors. T.K., H.A., and S.I. supervised the project. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Javier Garcia-Nafria and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Im, D., Kishikawa, Ji., Shiimura, Y. et al. Structural insights into the agonists binding and receptor selectivity of human histamine H4 receptor. Nat Commun 14, 6538 (2023). https://doi.org/10.1038/s41467-023-42260-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42260-z
This article is cited by
-
Structural basis of ligand recognition and design of antihistamines targeting histamine H4 receptor
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
