
Abstract
The design of human model systems is highly relevant to unveil the underlying mechanisms of aging and to provide insights on potential interventions to extend human health and life span. In this perspective, we explore the potential of 2D or 3D culture models comprising human induced pluripotent stem cells and transdifferentiated cells obtained from aged or age-related disorder-affected donors to enhance our understanding of human aging and to catalyze the discovery of anti-aging interventions.
Similar content being viewed by others
Introduction
The generation of human model systems is critical to reveal the mechanisms underlying the aging processes and to gain insights on interventions that extend the human organism’s health/life span. The use of non-vertebrate (yeast, worm, and fly) and vertebrate (zebrafish, mice, primates) models have already provided valuable insights on key proteins/pathways governing organismal aging (e.g., insulin/IGF-1, mTOR, AMPK, and sirtuins) and how their manipulation impacts health/life span1,2,3. Although these models can reproduce aspects of human aging and aging-associated diseases, they also have several limitations. First, non-vertebrate models have limited capacity to recapitulate the human complexity, in that they lack organs and systems that are important in human aging, such as a closed circulation system and an adaptive immune system. Second, the life span and rate of aging among most vertebrate species used in aging studies dramatically differs from humans4 and inbred and genetic models typically fail to account for the biological variability that exists amongst humans5. Although the basis for these differences remains largely unknown, it is likely that human-specific changes in epigenetics (e.g. DNA methylation), telomere length, protein composition, among others, contribute to those differences, and thus demand human models to complement our understanding. Third, although age-related diseases such as progeroid syndromes6, Parkinson´s disease (PD), Alzheimer´s disease (AD) may be an inspiration to elucidate the biology of aging, transgenic animal models do not fully recapitulate human age-related diseases with high fidelity, and can be expected to only partially predict the interface between aging and disease pathways.
Human cell models, such as primary cell samples isolated from young and old individuals, or from individuals affected by age-related diseases, can be a platform to study aging in a dish1. Yet, their limited availability (requires the participation of clinically/physically well-characterized human donors), inaccessibility of some tissues (e.g. brain, heart, blood vessels) and their finite in vitro expansion capacity (Hayflick limit) are major hurdles. Moreover, some primary cellular models fail to recapitulate some important aging aspects at tissue or organ levels because (i) the cells change phenotype once cultured in vitro, (ii) model cell lines are established by a small number of founding cells, and (iii) some crucial 3D spatial cell-cell interactions are absent7.
In this Perspective, we discuss the opportunities and limitations of using human induced pluripotent stem cells (iPSCs) and transdifferentiated cells to complement animal models, and in what aspects they might be better suited to understand the human aging process. Specifically, we discuss how reprogramming-based cell models from aging donors and patients with genetic and sporadic age-related diseases, including both iPSC-based and directly transdifferentiated cells, can advance our understanding of the human aging processes and help with the identification of anti-aging drugs. We emphasize recent progress in this area8,9,10,11,12,13,14,15,16 in which human cell models have proven to be very valuable for aging research as well as drug screening. These patient-specific cellular models often better reflect the dynamics and heterogeneity of aging processes across different human individuals whilst recapitulating the complex organization of tissue/organs at an unprecedented level. Finally, because cellular models are amenable to gene editing technologies, we explore how the introduction or correction of variants associated with aging-related disorders benefits current cell modeling research on aging.
From aging molecules to individuals, and aging-associated diseases
Aging is a naturally occurring complex process involved in every biological and physiological system. On the molecular level, progressive aging is associated with the accumulation of molecular damage and functional alterations. Examples include the accumulation of mutations in the DNA sequence and detrimental post-translational protein modifications (e.g. oxidation) that lead to protein misfolding, dysfunction of molecular pathways, and a gain of aberrant or toxic protein activity17. On a cellular level, persisting molecular damages can cause a decline in cellular functionality, impair the regenerative capacities of cells, and make individual cells more prone to death or to regressing towards cellular senescence18,19. In this Perspective, aged cells are referred to cells that present at least one of the nine hallmarks that represent common denominators of aging in different organisms such as genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, impaired stem cell fate and altered intercellular communication19. Loss of functional cells or the accumulation of malfunctioning and senescent cells can disrupt tissue and organ homeostasis and function. For example, senescent cells signal to their environment and alter the physiologic properties of surrounding cells, affecting their function, and altering the physiology of entire organs. Thus, aged tissues and organs usually not only perform less optimally but are also more vulnerable to develop chronic aging-related diseases (loss of resilience). On the level of an aging human individual, aging is typically perceived via phenotypic changes like for example graying of hair, wrinkling of skin or forgetfulness. However, for each individual person aging may manifest in different ways because different organs age at different biological rates20, dictating a higher incidence of developing aging-related diseases. Importantly, aging-associated diseases must be distinguished from impairments associated with normal aging, because lower cardiovascular fitness and a decline in cognitive abilities are normal, while myocardial infarction, AD, or cancer cannot be considered normal21.
In fact, aging is associated with a broad spectrum of different diseases, and one approach to establish a categorization of aging-associated diseases is by classifying them based on their interaction with normal biological aging, e.g. if (i) aging is primarily a major risk factor for the disease, (ii) if the disease causes pre-mature aging symptoms, or (iii) if the disease accelerates physiological categories of aging.
Diseases with aging as a major risk factor are characterized by increasing disease incidence with advancing age. The prevalence of cardiovascular diseases, including hypertension, coronary heart disease, heart failure, and stroke, increases from about 40% in men and women aged 40–59 years to 70–75% in those aged 60–79 years as they constitute the major cause of death of the elderly population22. Further, more than a third of new cancer cases occur in people aged 75 and over, with the highest age-specific incidence rates being at 85 to 89 years of age23. Cells that undergo cancer-like transformations bypass the inherent cellular programs that prevent such changes, and aging is the most important risk factor for cancer. Genetic mutations play a crucial role in cancer development, but the mere accumulation of DNA mutations over time is insufficient to explain this relationship, but rather failing safeguarding mechanisms such as impaired DNA damage repair mechanisms and failing immune pathways to prevent tumor development contribute to this relationship24. Nearly 60 years ago, Hayflick and Moorhead showed that cells cultured in vitro have a finite capacity for cell division25. They also noted that cultured cells enter a state of irreversible cell growth arrest, a process that they called replicative senescence. Cellular senescence is a major pathway suppressing the initiation of cancerous transformation, but already Hayflick suggested that replicative senescence contributed to organismal aging. Further, senescence can accelerate cancer progression once initiated through the senescence-associated secretory phenotype (SASP) that senescent cells utilize to mobilize the immune system26. Accumulation of cellular senescence is therefore proposed as a major link between aging, the immune system, and the progression of aging-driven diseases18,19. Several independent lines of research identified aging-related changes in the immune system to be associated with different diseases, constituting a major risk factor across the human body, and coined terms such as ‘inflammaging’ and ‘immunosenescence’24,27.
Diseases that cause premature aging phenotypes are markedly distinct from age-dependent diseases, and encompass several progeroid syndromes. The word “progeroid” comes from the Greek words “pro” (before, premature) and “gēras” (old age). Hutchinson-Gilford progeria syndrome (HGPS) is the most prominent and most prevalent form of progeroid syndromes. Patients with HGPS have accelerated markers of body-wide aging signatures (skin, cardiovascular, etc.) and they often die by heart attack or stroke28,29. HPGS is caused by a mutation in the LMNA gene, which interferes with canonical splicing of the gene and leads to a truncated form of the protein called progerin, while under normal conditions lamin A, the protein product of LMNA, is farnesylated and cleaved by a metallopeptidase30. Non-farnesylated progerin accumulates and thickens the nuclear lamina, leading to a loss of peripheral heterochromatin and dysfunctional nuclear pores, and loss of non-homologous and homologous DNA-repair cascades. The premature aging phenotypes may result from the destabilization of the nuclear lamina and impaired DNA-repair mechanisms, which are also critical in other premature aging diseases such as Werner syndrome31.
Diseases that accelerate measures of normal physiological aging have gained the attention of the aging research field more recently. This development was sparked by the novel ability to quantitatively measure the biological aging process through imaging, fluid biomarkers, and the discovery of epigenetic clocks32,33. With the advent of quantitative biological aging technologies, neuropsychiatric diseases such as post-traumatic stress disorder, major depressive disorder, schizophrenia, and bipolar disorder have been shown to accelerate the rate of physiological aging in affected individuals34,35. These psychiatric disorders cause a low level of chronic inflammation, the accumulation of oxidative stress markers, accelerated epigenetic aging and a decrease in the amount of mitochondrial genome copy numbers36. A more comprehensive understanding of the molecular-level relationships between neuropsychiatric conditions and ’classical’ aging-related neurodegenerative disorders is imperative, not only to enhance our comprehension of neuropsychiatric risk factors for aging-related neurodegenerative disorders, but also to benefit patients with neuropsychiatric conditions as they age more rapidly.
Modeling human aging with cell reprogramming models
Human iPSCs
Human iPSCs can provide a continuous supply of tissue-specific cell types, which can be issued to monitor aging processes using different strategies37. Interestingly, advanced human age does not impair the reprogramming of somatic cells into iPSCs38, and iPSCs generated from centenarians showed gene expression profiles and differentiation potentials similar to human embryonic stem cells39. While iPSCs are now largely considered to be globally rejuvenated, it is interesting to note that early-passage iPSCs retain the minimal but detectable epigenetic signature of aging that however diminishes quickly with passaging38. In the big picture however, the rejuvenation process that takes place during the reprogramming of old cells already during the early stages of iPSC reprogramming constitutes a limitation for using iPSC-based models to capture aging signatures of primary cells, e.g. from old donors40,41. However, many 2D and 3D protocols for a variety of cell type- and tissue-specific differentiation protocols are available for iPSCs, and they are very amenable to genetic manipulation. Thus, and despite their lack of endogenous donor-specific aging signatures, this outstanding versatility allows for mechanistic studies in iPSC relating to aging, such as how certain genes and pathways influence cellular aging starting from zero. During recent years several approaches have been explored to induce an aging phenotype in rejuvenated iPSC models, and we summarize some of these in the section below.
Inducing aging in non-diseased iPSCs-derived cells
Several strategies have been developed to induce an aging phenotype in wild-type iPSC-derived cells including: (i) long-term culture42,43,44, (ii) culture in aged extracellular matrix (ECM)11, (iii) telomere shortening41, and (iv) CRISPR/Cas9 technology45 (Fig. 1). These strategies have been used to age cardiomyocytes (CMs)11,42, neurons43 and mesenchymal stromal cells (MSCs)10. In vitro aging by long-term culture of cells was performed between 310 and 12 months43, and in some studies was done by culturing the iPSC-derived cells in aged ECM (e.g. decellularized ECM from 22–24 months old mice) for 3-4 months11. These strategies resulted in the appearance of age-associated markers, including a decrease in cell proliferation10, an increase in DNA damage and mitochondrial reactive oxygen species (ROS) generation41, high expression of senescence-associated genes10 and proteins (p2142 and SA-β-galactosidase10,42), DNA methylation changes at senescence-associated CG dinucleotides10, increase in lipofuscin accumulation11, among others. This indicates that long-term culture can successfully produce an at least partially aged phenotype in iPSCs-derived cells, and such iPSC-derived accelerated aging models were instrumental to: (i) investigate the metabolic changes occurring during replicative senescence10, (ii) study the role of ECM in inducing an aged phenotype11, (iii) dissect the role of senescence in post-mitotic cells42 and (iv) study the phenotype of aged midbrain dopamine neurons41. Because these aging models rely on the induction of aging by stressors that might not occur in physiologic conditions, their direct biological relevance to human aging, and as a risk factor for age-related disorders, remains limited (Fig. 1).

A Strategies to model human aging by (i) inducing aging in iPSC-derived cells, (ii) iPSCs from patients with aging-associated diseases and (iii) premature aging-syndrome iPSCs. B Choices and challenges in the use of iPSCs for human aging modeling.
iPSCs from aging-associated diseases
Human iPSCs carrying genetic mutations that occur in age-related diseases may provide invaluable information regarding the mechanisms governing aging. This has been explored in iPSC-derived CMs (reviewed in ref. 46) and iPSC-derived neurons (reviewed in ref. 47). For example, it has been shown that iPSC-derived CMs from individuals with end-stage hypertrophic cardiomyopathy or dilated cardiomyopathy aged faster than wild type iPSC-derived CMs as confirmed by significant shorter telomeres48. The accelerated aging profile in iPSC-derived CMs from individuals with dilated cardiomyopathies may be due to a dysfunctional activation of the platelet-derived growth factor signaling pathway49. Notably, most studies using iPSC-based neuronal models that carry disease-causing mutations applied additional stressors in tissue culture to elicit phenotypes, likely because rejuvenated iPSC-based models are young and fit, and thus do not develop pathologies spontaneously within reasonable culture periods of less than a few months to a year. Examples of such aging-related stressors include H2O2, 6-hydroxydopamine (6-OHDA), valinomycin, or carbonyl cyanide m-chlorophenyl hydrazone (CCCP) in iPSC models for PD50,51,52, proteasome inhibition in Huntington’s disease neurons53 or excitotoxicity in Ataxia54 (Fig. 1). Cocktail of stressors have been also used to induce senescence in neurons derived from iPSCs of amyotrophic lateral sclerosis (ALS) patient55. However, studies also report spontaneous disease phenotypes in iPSC models under standard culture conditions, which are likely downstream features of the mutated genes such as the constitutive active PD-causing LRRK2 kinase56,57. Taken together, while aging-related stressors help to elicit disease phenotypes in iPSC-derived models, a more complete induction of cellular age in iPSC-derived models seems favorable and represents a current focus in the field58,59.
iPSC models of premature aging syndromes
Human progeroid syndromes can provide additional insights into the mechanisms that control biological aging also during chronological aging (Fig. 1). iPSCs have been generated from patients suffering from Werner syndrome, Cockayne syndrome, xeroderma pigmentosum, ataxia telangiectasia, Fanconi anemia, and dyskeratosis congenita, all of which have defective DNA repair systems, and also from patients having alterations in the nuclear envelope such as HGPS (reviewed in47). In the context of this perspective, we will highlight the case of HGPS28,29. The extensive cardiovascular pathophysiology of HGPS ultimately contributes to most patients dying of myocardial infarction, heart failure, or stroke60, despite lacking major risk factors for cardiovascular disease (CVD)6. Interestingly, progerin accumulation is also observed in some tissues/cells during physiological aging (in living models without progeria) which makes this syndrome relevant for aging modeling61.
iPSCs derived from HGPS do not show a disease phenotype in their pluripotent state9,62 suggesting that the disease phenotype is only induced upon differentiation and LMNA expression. Thus, iPSCs derived from HGPS provide a unique approach to study how the cells get old besides being a source of aged cells. HGPS iPSCs have been used to derive smooth muscle cells (SMCs)9,62,63,64, endothelial cells (ECs)64,65,66, skin cells67, neural cells40,68, MSCs and their progenies69,70,71,72,73,74. In all these cellular models, several age-associated markers have been reported, including a decrease in cell proliferation64, increase in DNA damage40,64, abnormalities in nuclear morphology9,40, high expression of senescence-associated genes/proteins9, increased SA-β-galactosidase63,64, up-regulation of inflammatory cytokines63,64, shortened telomeres64, among others.
HGPS iPSCs were paramount to investigate molecular mechanisms of aging40,67,75. The first model of induction of cellular age in iPSC-derived neurons was largely inspired by HGPS, where overexpression of progerin in iPSC-derived neurons from PD patients resulted in cells that displayed age-dependent disease phenotypes40. When compared with wild type, HGPS-derived neurons exhibited a reduction in dendrite length, an increase in the expression of cleaved caspase-3 and a reduction in phosphorylated AKT, being the latter two associated with decreased neuronal survival40. HGPS-iPSCs may be also relevant for modeling skin aging, because HGPS individuals suffer from dermal hypopigmentation, a phenotype that is also common in the elderly67. Although melanocytes derived from HGPS-iPSCs did not exhibit changes in the expression of critical melanogenic markers, compared to their wild-type counterparts, they presented a reduction in melanin content and a decrease in melanosome maturity due to the accumulation of progerin67.
HGPS iPSC models have also been used as screening platforms to evaluate the mechanism and effects of pharmacological compounds that may be relevant both in the context of pathological as well as chronological aging. These models have been initially tested against drugs used in clinical trials71,76 and then against novel therapeutic drugs9. MSC derived from HGPS-iPSCs have been used to screen drugs, such as rapamycin76 and metformin71, able to reduce progerin accumulation70,71,76. Interestingly, the activation of retinoic acid (RA) signaling in MSCs by RA receptor agonists can decrease the expression of lamin A and progerin expression, while antagonists can increase their expression72. Importantly, the effect of RA agonists or antagonists is quite dependent on the microenvironment stiffness: stiffer substrates lead to a higher accumulation of RA receptor G isoform in the nucleus, culminating in an increased sensitivity of MSCs to RA signaling72. This observation suggests that both soluble and insoluble factors present in the microenvironments (which are highly dependent on the age of an individual and the type of tissue) can be determinants for the phenotype observed in progerin-expressing adult stem cells and on the efficacy of putative pharmacological treatments.
Overall, despite the usefulness of premature aging syndrome iPSC models, the separation between pathological and physiological aging in cellular models remains a challenge, and further investigation is required to clarify this issue (Fig. 1).
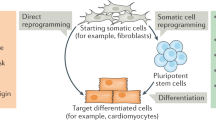
Direct transdifferentiation to preserve endogenous cellular aging features
A limitation in using iPSC-derived cells for aging research, and in particular for modeling age-dependent disorders is the fact that they are rejuvenated early embryonic-like cells that do not endogenously exhibit cellular aging features. For such studies, the direct cellular transdifferentiation from one somatic cell type to another, without undergoing an intermediary pluripotent or progenitor cell type, represents an alternative strategy. Directly transdifferentiated, or converted, cells broadly retain aging-associated features of the original somatic cells, and may be used to study aging77,78,79 (Fig. 2). Conceptually, transdifferentiation is comparable to iPSC reprogramming, in that lineage-determining pioneer transcription factors, often in combination with microRNAs, and signaling pathway modulators, facilitate cell type change80,81,82. In most cases, dermal fibroblasts are used as a starting cell type, and the timeline for direct transdifferentiation ranges between a few weeks to two months, and is thus comparable to differentiation protocols of iPSCs (once iPSC lines are established). Transdifferentiated cells are polyclonal cultures, originating from different fibroblasts in the culture, while iPSCs are clonal cells, each derived from a single founding fibroblast or blood cell. Therefore, transdifferentiated cells not only reflect interindividual variability but also capture the genetic mosaicism of the fibroblast culture of each patient, which may be advantageous in modeling conditions such as aging. Using such approaches, wild type83,84 or disease (PD, AD, Huntington´s disease, HGPS) fibroblasts12,13,79,85,86 have been transdifferentiated into induced neurons (iNs)12,13,15,85,86, astrocytes16,84, endothelial79, smooth muscle cells79 among others. For example, striatal medium spiny iNs directly reprogrammed from fibroblasts of patients with Huntington’s disease (HD) showed alterations in chromatin accessibility85, degeneration by impaired autophagic function85 and mitochondrial dysfunction86 in an age-dependent and repeat extension-dependent manner. HD patient iNs showed mutant huntingtin aggregates, DNA damage, spontaneous neuronal death, decline in mitochondrial function, aging-related changes in chromatin accessibility, and impaired autophagy85,86,87,88. Further, and in contrast to dopaminergic neurons derived from iPSCs generated from PD fibroblasts, dopaminergic iNs generated from the transdifferentiation of adult human dermal fibroblasts isolated from idiopathic PD patients endogenously showed altered autophagy, increased DNA damage and increased accumulation of phosphorylated α-synuclein13. PD-patient iNs exhibited lower basal chaperone-mediated autophagy compared with healthy donors and in stress-induced autophagy and they displayed an age-dependent accumulation of macroautophagic structures13. Sporadic AD patient-specific iNs showed markers of oxidative stress, DNA damage and destabilization of epigenetic neuronal identity programs12. These aging AD patient-based cellular models further showed that in AD, neurons have lost specialized neuronal functions and resilience to control cells12 and develop senescence and pro-inflammatory phenotypes that can compromise surrounding cells via bystander effects15.

A Direct transdifferentiation of skin cells (easy available) into somatic cells that are not easy to isolate (e.g. neurons, cardiomyocytes, etc.) can be achieved by reprogramming with cell-specific transcription factors, microRNAs, small molecules, epigenetic regulators, etc. The derived cells can be used for cell therapy, drug screening and disease modeling. B Choices and challenges in the use of transdifferentiated cells for human aging modeling.
Although cellular transdifferentiation offers an elegant strategy to generate aged cells, this strategy also has limitations. One of the primary limitations of direct conversion models is the finite number of starting material, as the method does not involve expandable stem cell stages. This results in lower cell numbers, which poses challenges for scaling up the system for material-demanding molecular assays or screening approaches that are very doable with iPSC-based models77,82 (Fig. 2). The reprogramming potential of fibroblast cell lines into other types of cells is largely contingent on the quality of the fibroblast culture. While the generation of high-quality fibroblast cell lines from skin punch biopsies is a routine practice in many labs and clinics today, some patient cell lines are exceedingly rare. In cases where existing fibroblast lines are of low quality or become depleted, the procurement of new biopsies may be necessary, which is not always feasible. In addition, the preservation of only skin-related signatures that are encoded in the starting fibroblasts (e.g., if they relate to sun exposure of the skin) are likely present also in transdifferentiated cells. This may cause artificial ‘false-positive’ aging signs in the transdifferentiated cells. In contrast, once an iPSC line is established, it is essentially immortal, providing a virtually perpetual cell source. The lower potential for genetic manipulation is a notable disadvantage of transdifferentiated cells compared to iPSCs, as iPSCs are very amenable to genetic engineering using CRISPR-based technologies to generate for example isogenic control cell models9. However, all CRISPR-related technologies including Cas9, Prime editing, and Base editing critically depend on the cell-intrinsic DNA damage repair mechanisms, and these tools are much less well-established in non-stem/-tumor cells such as fibroblasts and neurons, and their potentials are more limited due to the slow-growing and post-mitotic nature of these cells89,90.
2D versus 3D aging models
2D aging models
The majority of in vitro models for aging and age-related disorders make use of 2-dimensional (2D) cell cultures, which is mainly because they are: (i) relatively inexpensive, (ii) well established in cell biology research and (iii) easy to analyze using several molecular, biochemical and image-based assays. For example, such 2D culture systems were useful to investigate the mechanisms underlying the degeneration of aged HGPS-iPSC SMCs91, the aging program of iPSC-CMs cultured for extended periods in vitro42, the effect of ECM in the performance and induction of an aging program in human iPSC-derived CMs11, the maturation and drug response of aged hiPSC-derived neuronal networks43, the effect of substrate topography in inducing cell aging92 and the impaired function and high mechano-sensitivity of HGPS-iPSC ECs mediated by an increase in the activity of a transient receptor potential channel65,93. The scalability of 2D systems make them a useful system for large-scale drug testing. In addition, some morphological studies and imaging studies are easier in 2D than in 3D. Yet, 2D cell cultures have several limitations because the interaction with the underlying substrate prevails over the cell-cell and/or cell-ECM interactions, the apical-basal polarity is changed, cell migration is not constrained and the stiffness of the plastic dishes is not physiological ultimately impacting cell proliferation, migration and activity94,95.
3D aging models
Tissue-engineered models
To model age-related changes in cell-cell interactions, 3D cell cultures are believed to better recapitulate the physiology of an aged organ/tissue. 3D tissue models containing different cell types allow for the investigation of how cellular interactions might be affected by the presence of aged cells. Further, hetero-chronic cell models, i.e. mixing both aged cells with non-aged cells, has the potential to better understand how cell-cell communication might be involved in the aging process of a tissue and to evaluate the importance of the ECM components in regulating (enhancing or reverting) cellular aging. For example, it is known that senescent cells communicate with proliferative cells via the senescence-associated secretory phenotype (SASP) that includes a temporally-regulated moiety of soluble factors such as interleukin-1a, IL-6, IL-8, transforming growth factor β, extracellular vesicles, lipid mediators96. These factors can induce a senescence phenotype in proliferative cells97 as well as induce ECM remodeling96. By recapitulating some of the complex microenvironment found in living tissues, 3D culture systems comprising defined cell types and ECM components might be a better model for studying intercellular communication, ECM remodeling by aged cells and the impact of biomechanical forces in cellular activity.
Recently, studies have investigated the impact of 3D culture systems to study cell aging biology. Pseudo-3D systems, i.e. systems that are in the transition between a 2D and a 3D culture system but do not have the complexity of a 3D culture system, have been used to investigate in vitro single or multiple physiological parameters in aged cells. For example, aged cells (HGPS-iPSC SMCs) were cultured in microfluidic systems, under flow conditions, to investigate their response to biomechanical forces9,63. Under biomechanical stress, HGPS-iPSC SMCs displayed an exacerbated inflammatory profile which was reversed by pharmacological treatment with inhibitors of the enzyme hydroxymethylglutaryl-coenzyme A (lovastatin) or farnesyl transferase (lonafarnib)63. Another example of a pseudo-3D system was based in the co-culture of ECs with HGPS-iPSC SMCs in a microfluidic system, under flow conditions. It was demonstrated that HGPS-iPSC SMCs were vulnerable to flow shear stress and detached from the substrate by the action of matrix metalloproteinase 13 (MMP13)9. In other cases, a 3D culture system has been adopted to create tissue engineered blood vessels (TEBVs)8,98. TEBVs comprised a perfused 3D tubular structure with an outer layer composed of HGPS-iPSC SMCs and an inner layer composed of HGPS-iPSC ECs or wild-type iPSC ECs. These 3D systems have shown reduced vasoactivity and medial wall thickening (through extracellular deposition), with HGPS-iPSC SMCs having increased calcification and increased detachment relative to control cells. In a separate study, the direct neuronal reprogramming of AD patient fibroblasts, cultured in a 3D hydrogel, by miRNAs was able to capture age-related neuropathology and the interplay between Aβ accumulation, tau dysregulation and neuronal death99.
Overall, these tissue-engineered models are very promising to recapitulate in vitro aging profiles that are dependent on intercellular communication, ECM and biomechanical forces. Significant advances in this area are expected in the coming years due to the need for better understanding aging in a more complex network and because of the progresses in tissue/organ printing technology that allows the preparation of 3D tissues with high resolution.
Organoids
Organoids are three-dimensional multicellular cultures that mimic various features of an organ such as the structure and cellular composition94,100,101. Compared to conventional 2D cultures, organoids are complex 3D cellular systems can be originated from progenitor cells differentiated from pluripotent stem cells (or other sources101) and cultured in suspension or embedded in ECM. They rely in morphogenesis processes initiated by the organoid initiating cell (e.g. primary cell, tissue stem cell, or pluripotent stem cell) and, in some cases, a permissive ECM that allows cellular remodeling, a feature that sometimes is absent in tissue engineered organs (Fig. 3). These 3D cellular systems have the potential to provide near-physiological models to study human aging101. Organoids allow more degrees of freedom in long-term cultures, giving rise to cellular diversity, complex cell–cell interactions and unique physical structures. More recent advances in this area described the fusion of different organoids to form an assembloid94. For example, different organoids reflecting different brain regions or tissue are cultured together and allowed to form connections.

A Creation of 3D aged models by organoids. Initially, iPSCs are differentiated into organoid-initiating cells followed by the culture of these cells in a 3D permissive ECM. B Choices and challenges in the use of 3D models for human aging modeling.
Recent studies have used organoids to study cellular aging14,102,103 and to identify treatments to ameliorate age-related diseases104,105. For example, brain organoids generated from iPSCs isolated from patients suffering from ataxia-telangiectasia (carrying a mutation in the ataxia-telangiectasia-mutated (ATM) gene that encodes the ATM kinase) have been used to investigate the impact of ATM kinase absence in the nervous system14. This is particularly relevant because the process of neurodegeneration in the disease is poorly understood and the animal models available fail to display clear symptoms of neurodegeneration. Brain organoids showed an increase of senescent cells located mostly within astrocyte populations, increased expression of pro-inflammatory genes as well as premature neuronal degeneration and dysfunction. Importantly, inhibition of cGAS and STING-dependent inflammation effectively suppressed self-DNA-induced SASP activation and rescued the neuropathological features in brain organoids. In a separate study, brain organoids generated from AD iPSC-derived cells or γ-irradiated brain organoids generated from wild-type cells, have been also used to screen drugs104,105. Moreover, aged colon organoids were obtained from human iPSCs to investigate whether DNA damage response was inversely correlated with an increase in the production of growth hormone102. Cellular aging was confirmed by an increase of p16, DNA damage, decrease of telomere length, increase in senescence-associated β-galactosidase activity and expression. Interestingly, aged colon organoids showed increased expression of growth hormone production at gene and protein levels and this production correlated with attenuated DNA damage response.
Although organoids offer the possibility to study human aging at the organ level, they have some limitations such as: (i) fail to completely recapitulate the aging profile at transcriptional and protein level, (ii) limited vascularization, (iii) low reproducibility and (iv) difficulty to stimulate the cells biomechanically100,101 (Fig. 3).
Exploiting the power of human aging modeling: future directions
Although the field of human aging modeling in a dish by reprogrammed cells started more than 10 years ago with the use of iPSCs, it evolved for the use of direct conversion technologies and, more recently, for the use of tissue-engineered models and organoids. The number of studies describing the use of cellular transdifferentiation remains limited, mainly in that they focused mostly in the direct conversion of patient fibroblasts to neuronal cells12,13,15,85,86. It is expected that in the near future similar strategies will be used for the derivation of post-mitotic cells such as CMs or glial cells, which will help to advance our understanding of the human aging process. Other progresses are also expected in the use of iPSC-derived cells or transdifferentiated cells for the generation of 3D models of aging. Given the development of direct transdifferentiation protocols to generate different cell types of the same tissue, such as neurons and astrocytes of the brain, also multicellular 3D models based on age-preserved tissue models become possible. It will be interesting to build multicellular age-equivalent models for aging and age-related disorders of the brain, but also use them to build heterochronic modeling paradigms, which can contain combinations of young and old cells from patient and control donors.
The use of these cells for the generation of tissue-engineered models or organoids is yet in early stages with the first studies reported in the last 5 years14,63,98,102,103,105. These 3D models may be important to unravel the role of insoluble cues, such as ECM composition and/or substrate topography in aging processes. Recent studies have shown that the stiffness106, as well as the composition of the ECM11,107, are important regulators of the aging processes. In addition, it will be important to evaluate the crosstalk between aged cells with proliferative cells in a 3D environment to dissect how aging can be propagated. Next, in vivo xenotransplantation approaches have been shown to overcome limitations related to necrotic cores, and incomplete vascularization and maturation of iPSC-derived organoids, which will become relevant for long-term organoid models of aging108,109,110. Further, 3D live cell bioprinting approaches may be relevant to produce multiple types of artificial organs and tissues. In this case, a 3D printer add, layer by layer, cells and biologics to achieve tissues with a well-defined and organized 3D structure111. More advanced 3D tissues/organs will allow for the development of screening platforms for the identification of anti-aging drugs and can provide models of human age-related diseases that cannot be recapitulated in animals.
References
Brunet, A. Old and new models for the study of human ageing. Nat. Rev. Mol. Cell Biol. 21, 491–493 (2020).
Kimura, K. D., Tissenbaum, H. A., Liu, Y. & Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946 (1997).
Howitz, K. T. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003).
Jones, O. R. et al. Diversity of ageing across the tree of life. Nature 505, 169–173 (2014).
Belsky, D. W. et al. Quantification of biological aging in young adults. Proc. Natl Acad. Sci. USA 112, E4104–E4110 (2015).
Hamczyk, M. R., del Campo, L. & Andres, V. Aging in the cardiovascular system: lessons from hutchinson-gilford progeria syndrome. Annu Rev. Physiol. 80, 27–48 (2018).
Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014).
Atchison, L. et al. iPSC-derived endothelial cells affect vascular function in a tissue-engineered blood vessel model of Hutchinson-Gilford progeria syndrome. Stem Cell Rep. 14, 325–337 (2020).
Pitrez, P. R. et al. Vulnerability of progeroid smooth muscle cells to biomechanical forces is mediated by MMP13. Nat. Commun. 11, 4110 (2020).
Fernandez-Rebollo, E. et al. Senescence-associated metabolomic phenotype in primary and iPSC-derived mesenchymal stromal cells. Stem Cell Rep. 14, 201–209 (2020).
Ozcebe, S. G., Bahcecioglu, G., Yue, X. S. & Zorlutuna, P. Effect of cellular and ECM aging on human iPSC-derived cardiomyocyte performance, maturity and senescence. Biomaterials 268, 120554 (2021).
Mertens, J. et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 28, 1533–1548.e1536 (2021).
Drouin-Ouellet, J. et al. Age-related pathological impairments in directly reprogrammed dopaminergic neurons derived from patients with idiopathic Parkinson’s disease. Stem Cell Rep. 17, 2203–2219 (2022).
Aguado, J. et al. Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell 20, e13468 (2021).
Herdy, J. R. et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell 29, 1637–1652.e1636 (2022).
Gatto, N. et al. Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell 20, e13281 (2021).
Gladyshev, V. N. et al. Molecular damage in aging. Nat. Aging 1, 1096–1106 (2021).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Oh, H. S. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Guo, J. et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct. Target Ther. 7, 391 (2022).
Yazdanyar, A. & Newman, A. B. The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin. Geriatr. Med 25, 563–577 (2009).
Ugai, T. et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat. Rev. Clin. Oncol. 19, 656–673 (2022).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Liberale, L. et al. Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J. Am. Coll. Cardiol. 79, 837–847 (2022).
Eriksson, M. et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003).
De Sandre-Giovannoli, A. et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055 (2003).
Pitrez, P. R., Rosa, S. C., Praca, C. & Ferreira, L. Vascular disease modeling using induced pluripotent stem cells: Focus in Hutchinson-Gilford Progeria Syndrome. Biochem. Biophys. Res. Commun. 473, 710–718 (2016).
Oshima, J., Sidorova, J. M. & Monnat, R. J. Jr Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 33, 105–114 (2017).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Tian, Y. E. et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 29, 1221–1231 (2023).
Constantinides, C. et al. Brain ageing in schizophrenia: evidence from 26 international cohorts via the ENIGMA Schizophrenia consortium. Mol. Psychiatry 28, 1201–1209 (2023).
Yang, R. et al. A DNA methylation clock associated with age-related illnesses and mortality is accelerated in men with combat PTSD. Mol. Psychiatry 26, 4999–5009 (2021).
Fries, G. R. et al. Accelerated aging in bipolar disorder: a comprehensive review of molecular findings and their clinical implications. Neurosci. Biobehav Rev. 112, 107–116 (2020).
Singh, V. K., Kalsan, M., Kumar, N., Saini, A. & Chandra, R. Induced pluripotent stem cells: applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 3, 2 (2015).
Lo Sardo, V. et al. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 35, 69–74 (2017).
Lapasset, L. et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253 (2011).
Miller, J. D. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705 (2013).
Vera, E., Bosco, N. & Studer, L. Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep. 17, 1184–1192 (2016).
Acun, A., Nguyen, T. D. & Zorlutuna, P. In vitro aged, hiPSC-origin engineered heart tissue models with age-dependent functional deterioration to study myocardial infarction. Acta Biomater. 94, 372–391 (2019).
Odawara, A., Katoh, H., Matsuda, N. & Suzuki, I. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Sci. Rep. 6, 26181 (2016).
Ebert, A. et al. Proteasome-dependent regulation of distinct metabolic states during long-term culture of human iPSC-derived cardiomyocytes. Circ. Res 125, 90–103 (2019).
Acun, A. & Zorlutuna, P. CRISPR/Cas9 edited induced pluripotent stem cell-based vascular tissues to model aging and disease-dependent impairment. Tissue Eng. Part A 25, 759–772 (2019).
Sacchetto, C., Vitiello, L., de Windt, L. J., Rampazzo, A. & Calore, M. Modeling cardiovascular diseases with hiPSC-derived cardiomyocytes in 2D and 3D cultures. Int J. Mol. Sci. 21, 3404 (2020).
Soria-Valles, C. & Lopez-Otin, C. iPSCs: on the road to reprogramming aging. Trends Mol. Med. 22, 713–724 (2016).
Chang, A. C. Y. et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl Acad. Sci. USA 115, 9276–9281 (2018).
Lee, J. et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340 (2019).
Reinhardt, P. et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367 (2013).
Seibler, P. et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976 (2011).
Rakovic, A. et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 288, 2223–2237 (2013).
Nekrasov, E. D. et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 11, 27 (2016).
Koch, P. et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480, 543–546 (2011).
Fathi, A. et al. Chemically induced senescence in human stem cell-derived neurons promotes phenotypic presentation of neurodegeneration. Aging Cell 21, e13541 (2022).
Schwab, A. J. et al. Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Rep. 9, 1839–1852 (2017).
Sanchez-Danes, A. et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395 (2012).
Cornacchia, D. & Studer, L. Back and forth in time: Directing age in iPSC-derived lineages. Brain Res. 1656, 14–26 (2017).
Mertens, J., Reid, D., Lau, S., Kim, Y. & Gage, F. H. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293 (2018).
Gordon, L. B. et al. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA 319, 1687–1695 (2018).
Revechon, G. et al. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 7, 4405 (2017).
Liu, G. H. et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 472, 221–225 (2011).
Ribas, J. et al. Biomechanical strain exacerbates inflammation on a progeria-on-a-chip model. Small 13 (2017).
Xu, Q. et al. Vascular senescence in progeria: role of endothelial dysfunction. Eur. Heart J. Open 2, oeac047 (2022).
Estronca, L. et al. Induced pluripotent stem cell-derived vascular networks to screen nano-bio interactions. Nanoscale Horiz. 6, 245–259 (2021).
Matrone, G. et al. Dysfunction of iPSC-derived endothelial cells in human Hutchinson-Gilford progeria syndrome. Cell Cycle 18, 2495–2508 (2019).
Lo Cicero, A. et al. Pathological modelling of pigmentation disorders associated with Hutchinson-Gilford Progeria Syndrome (HGPS) revealed an impaired melanogenesis pathway in iPS-derived melanocytes. Sci. Rep. 8, 9112 (2018).
Nissan, X. et al. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2, 1–9 (2012).
Xiong, Z. M., LaDana, C., Wu, D. & Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging (Albany NY) 5, 288–303 (2013).
Blondel, S. et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 7, e2105 (2016).
Egesipe, A. L. et al. Metformin decreases progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech. Dis. 2, 16026 (2016).
Ivanovska, I. L. et al. Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol. Biol. Cell 28, 2010–2022 (2017).
Pacheco, L. M. et al. Progerin expression disrupts critical adult stem cell functions involved in tissue repair. Aging (Albany NY) 6, 1049–1063 (2014).
Cho, S. et al. Progerin phosphorylation in interphase is lower and less mechanosensitive than lamin-A,C in iPS-derived mesenchymal stem cells. Nucleus 9, 230–245 (2018).
Studer, L., Vera, E. & Cornacchia, D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 16, 591–600 (2015).
Blondel, S. et al. Induced pluripotent stem cells reveal functional differences between drugs currently investigated in patients with hutchinson-gilford progeria syndrome. Stem Cells Transl. Med. 3, 510–519 (2014).
Mertens, J. et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718 (2015).
Huh, C. J. et al. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 5, e18648 (2016).
Bersini, S., Schulte, R., Huang, L., Tsai, H. & Hetzer, M. W. Direct reprogramming of human smooth muscle and vascular endothelial cells reveals defects associated with aging and Hutchinson-Gilford progeria syndrome. Elife 9, e54383 (2020).
Wang, H., Yang, Y., Liu, J. & Qian, L. Direct cell reprogramming: approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 22, 410–424 (2021).
Weintraub, H. et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl Acad. Sci. USA 86, 5434–5438 (1989).
Mertens, J., Marchetto, M. C., Bardy, C. & Gage, F. H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17, 424–437 (2016).
Kim, Y. et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 23, 2550–2558 (2018).
Quist, E. et al. Transcription factor-based direct conversion of human fibroblasts to functional astrocytes. Stem Cell Rep. 17, 1620–1635 (2022).
Oh, Y. M. et al. Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nat. Neurosci. 25, 1420–1433 (2022).
Victor, M. B. et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 21, 341–352 (2018).
Lee, S. W. et al. Longitudinal modeling of human neuronal aging reveals the contribution of the RCAN1-TFEB pathway to Huntington’s disease neurodegeneration. Nat. Aging 4, 95–109 (2023).
Pircs, K. et al. Distinct subcellular autophagy impairments in induced neurons from patients with Huntington’s disease. Brain 145, 3035–3057 (2022).
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
Davis, J. R. et al. Efficient prime editing in mouse brain, liver and heart with dual AAVs. Nat. Biotechnol. 42, 253–264 (2024).
Zhang, H., Xiong, Z. M. & Cao, K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl Acad. Sci. USA 111, E2261–E2270 (2014).
Pitrez, P. R. et al. Substrate topography modulates cell aging on a progeria cell model. ACS Biomater. Sci. Eng. 4, 1498–1504 (2018).
Lo, C. Y. et al. An upregulation in the expression of vanilloid transient potential channels 2 enhances hypotonicity-induced cytosolic Ca(2)(+) rise in human induced pluripotent stem cell model of Hutchinson-Gillford Progeria. PLoS ONE 9, e87273 (2014).
Pasca, S. P. The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018).
Kraehenbuehl, T. P., Langer, R. & Ferreira, L. S. Three-dimensional biomaterials for the study of human pluripotent stem cells. Nat. Methods 8, 731–736 (2011).
Fafian-Labora, J. A. & O’Loghlen, A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 30, 628–639 (2020).
Borghesan, M. et al. Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 27, 3956–3971.e3956 (2019).
Atchison, L., Zhang, H., Cao, K. & Truskey, G. A. A tissue engineered blood vessel model of hutchinson-gilford progeria syndrome using human iPSC-derived smooth muscle cells. Sci. Rep. 7, 8168 (2017).
Sun, Z. et al. Endogenous recapitulation of Alzheimer’s disease neuropathology through human 3D direct neuronal reprogramming. bioRxiv https://doi.org/10.1101/2023.05.24.542155 (2023).
Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater. 6, 402–420 (2021).
Hu, J. L., Todhunter, M. E., LaBarge, M. A. & Gartner, Z. J. Opportunities for organoids as new models of aging. J. Cell Biol. 217, 39–50 (2018).
Chesnokova, V. et al. Local non-pituitary growth hormone is induced with aging and facilitates epithelial damage. Cell Rep. 37, 110068 (2021).
Rai, M. et al. Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging. Cell Metab. 33, 1137–1154.e1139 (2021).
Shakhbazau, A., Danilkovich, N., Seviaryn, I., Ermilova, T. & Kosmacheva, S. Effects of minocycline and rapamycin in gamma-irradiated human embryonic stem cells-derived cerebral organoids. Mol. Biol. Rep. 46, 1343–1348 (2019).
Park, J. C. et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 12, 280 (2021).
Segel, M. et al. Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature 573, 130–134 (2019).
Schuler, S. C. et al. Extensive remodeling of the extracellular matrix during aging contributes to age-dependent impairments of muscle stem cell functionality. Cell Rep. 35, 109223 (2021).
Mansour, A. A. et al. Erratum: an in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 772 (2018).
Revah, O. et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–326 (2022).
Schafer, S. T. et al. An in vivo neuroimmune organoid model to study human microglia phenotypes. Cell 186, 2111–2126.e2120 (2023).
Murphy, S. V., De Coppi, P. & Atala, A. Opportunities and challenges of translational 3D bioprinting. Nat. Biomed. Eng. 4, 370–380 (2020).
Acknowledgements
This work was funded by FEDER through the Program COMPETE and by Portuguese fund through FCT in the context of the projects 2022.07615.PTDC and PTDC/BTM-SAL/5174/2020, as well as the European project RESETaging (ref. 952266), the National Institute on Aging grants AG056306 and AG062429, the Epstein Family Research Collaboration Fund and the Alzheimer’s Disease Cooperative Study (ADCS) at UCSD, and the Alzheimer’s Association Research Grant AARG-22-972303. The authors thank the project “CHAngeing – Connected Hubs in Ageing: Healthy Living to Protect Cerebrovascular Function” funded by the European Union’s Horizon Europe program (Excellence Hubs - HORIZON-WIDERA-2022-ACCESS-04-01) under grant agreement No. 101087071. This work was also supported by “REGENERAR - Improving the Effectiveness and Safety of Epigenetic” funded by the European Union’s Horizon Europe program (HORIZON.3.1 - The European Innovation Council (EIC)) under grant agreement No. 101129812. And this work was also funded by “DREAMs - Drug REpurposing with Artificial intelligence for Muscular disorderS” funded by the European Union’s Horizon Europe program (HORIZON-HLTH-2022-DISEASE-06-04-two-stage) under grant agreement No. 101080229. The dissemination of results herein reflects only the author’s view, and the European Commission is not responsible for any use that may be made of the information it contains.
Author information
Authors and Affiliations
Contributions
P.P., L.M., O.B., X.N., J.M., and L.F. co-wrote this perspective. L.F. conceived the figures. All authors edited and proofread the manuscript and authorized the submission of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Antonella Consiglio, Janelle Drouin-Ouellet and the other anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pitrez, P.R., Monteiro, L.M., Borgogno, O. et al. Cellular reprogramming as a tool to model human aging in a dish. Nat Commun 15, 1816 (2024). https://doi.org/10.1038/s41467-024-46004-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46004-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
