
Abstract
Insulin secretion from pancreatic β cells is regulated by multiple stimuli, including nutrients, hormones, neuronal inputs, and local signalling. Amino acids modulate insulin secretion via amino acid transporters expressed on β cells. The granin protein VGF has dual roles in β cells: regulating secretory granule formation and functioning as a multiple peptide precursor. A VGF-derived peptide, neuroendocrine regulatory peptide-4 (NERP-4), increases Ca2+ influx in the pancreata of transgenic mice expressing apoaequorin, a Ca2+-induced bioluminescent protein complex. NERP-4 enhances glucose-stimulated insulin secretion from isolated human and mouse islets and β-cell–derived MIN6-K8 cells. NERP-4 administration reverses the impairment of β-cell maintenance and function in db/db mice by enhancing mitochondrial function and reducing metabolic stress. NERP-4 acts on sodium-coupled neutral amino acid transporter 2 (SNAT2), thereby increasing glutamine, alanine, and proline uptake into β cells and stimulating insulin secretion. SNAT2 deletion and inhibition abolish the protective effects of NERP-4 on β-cell maintenance. These findings demonstrate a novel autocrine mechanism of β-cell maintenance and function that is mediated by the peptide–amino acid transporter axis.
Similar content being viewed by others


Introduction
Amino acids play pivotal roles not just in protein synthesis, but also in vital cellular processes, including hormone secretion and cell maintenance1. Postprandial elevations in plasma amino acids enhance insulin release from pancreatic β cells2. Amino acid transporters (AATs) are membrane-bound transport proteins that import or export amino acids (i.e., symporters and antiporters, respectively) to regulate cellular and circulating amino acid levels3. Several AATs are expressed in β cells, and stimulate postprandial insulin secretion by mediating the influx of amino acids4,5. After being transported into β cells, glutamine and alanine maintain these cells by activating ATP generation and by suppressing oxidative and endoplasmic reticulum (ER) stress2,6,7. The synthesis of AATs is regulated by various metabolic signals, such as amino acid availability and amino acid starvation3,4; however, no endogenous substance that modulates the functional activity of AATs has been identified.
Altered intracellular Ca2+ concentration is a common physiological response downstream of cell-surface receptors8. Aequorin, a 21-kDa protein purified from luminous Aequorea, is a Ca2+-induced bioluminescent protein complex consisting of apoaequorin and coelenterazine as a luminous substrate9. Apoaequorin transgenic mice were generated to detect intracellular Ca2+ mobilisation under the control of the CAG promoter10. Using the pancreata of these transgenic mice, we identified a novel 19–amino acid peptide from human neuroendocrine medullary thyroid carcinoma TT cells: NERP-4, corresponding to VGF 489–50711,12. VGF, a granin protein also named secretogranin VII, comprises 615 (human) or 617 (mouse/rat) amino acids and functions in granule formation in neurons and endocrine cells, including β cells13,14,15,16. VGF also serves as a precursor of bioactive peptides that exhibit diverse biological activities, such as maintaining metabolic and glucose homeostasis14,17,18,19,20.
Here we demonstrate that NERP-4 enhances glucose-stimulated insulin secretion (GSIS) by elevating intracellular Ca2+ in β cells. NERP-4 administration reverses β-cell impairment by enhancing mitochondrial function and reducing oxidative and ER stress. Using LRC-TriCEPS technology, binding assays, and cellular functional experiments, we establish NERP-4 binds to the sodium-coupled neutral AAT SNAT2, stimulates amino acid uptake, and improves β-cell maintenance and function. The present study demonstrates a critical role of the NERP-4–SNAT2 axis in β-cell biology.
Results
NERP-4 is co-localised with insulin in β cells, thus stimulating Ca2+ influx
Aequorin-expressing tissues from apoaequorin transgenic mice were used to detect the elevation of intracellular Ca2+ concentrations by ligands of interest, such as novel compounds and peptides10. We used pancreatic pieces because the islets would lose their original function once they were incubated with coelenterazine for 3 h. NERP-4 evoked transient luminescence in a piece of pancreatic tissue from apoaequorin transgenic mice, suggesting an increase in intracellular Ca2+ level (Fig. 1b). NERP-4–induced Ca2+ influx into β cells was investigated in MIN6‐K8 cells, which are mouse insulinoma–derived β cells21 that have been used to study the mechanism of glutamine-amplified Ca2+ influx and insulin secretion22,23. NERP-4 enhanced Ca2+ influx into MIN6-K8 cells under high glucose (Fig. 1c), but not under low glucose (Supplementary Fig. 1a). To explore whether the rise in Ca2+ was due to extracellular Ca2+ influx via Ca2+ channels, we studied Ca2+ influx into MIN6-K8 cells treated with EGTA (a calcium chelator) or nifedipine (an L-type Ca2+ channel blocker). Both treatments suppressed NERP-4–induced Ca2+ influx (Fig. 1d, e), implying that NERP-4 mediated Ca2+ influx via Ca2+ channels.

a NERP-4 sequence in mouse VGF. NERP-4 is cleaved from VGF after a basic pair (lysine–lysine: KK) and before a single basic amino acid (arginine: R). b Representative profile of the relative luminescence evoked in the apoaequorin transgenic mouse pancreas. Arrows (A–D) represent the tested substances. Medium (A), 1 μM NERP-4 (B), 100 μM ATP (C), and 2.5% Triton X-100 (D) were administered before the time indicated by the corresponding arrow. ATP was used as a positive control. c–e Representative Fura-2-AM ratios in MIN6-K8 cells in response to NERP-4 with or without EGTA or nifedipine treatment (n = 8 cells), and average incremental AUC (iAUC) (9–20 min) of [Ca2+]i (n = 8 cells). Representative immunofluorescence images of NERP-4, insulin, and their merged images in C57BL/6 J mouse islet (f), rat islet (g), MIN6-K8 cell (h), and Vgf KO mouse islet (i). j A representative immunoelectron micrograph showing co-localisation of NERP-4 with insulin in C57BL/6 J mouse islets. Inset is a higher-magnification image (NERP-4: 5-nm gold particles; black arrowheads: insulin; 10-nm gold particles: white arrowheads). k RP-HPLC of immunoreactive NERP-4 extracted from human pancreas. The arrow indicates the elution position of synthetic human NERP-4. Results are representative of three (c–e) or two (f–j) independent experiments. Data are mean ± s.e.m (c–e). Unpaired two-tailed Student’s t-test (c–e). Scale bars, 50 μm (f, g, i), 5 μm (h), 50 nm (j). Source data are provided as a Source data file.
NERP-4 immunoreactivity was co-localised with insulin in C57BL/6 J mouse islets, rat islets, and MIN6-K8 cells (Fig. 1f–h). The specificity of an anti–NERP-4 antibody was confirmed in Vgf knockout (KO) mouse islets24 (Fig. 1i). Immunogold electron microscopy revealed co-localisation of NERP-4 with insulin in storage granules (Fig. 1j). NERP-4 was infrequent in cells producing glucagon or somatostatin in C57BL/6 J mouse islets (Supplementary Fig. 1b). Immunoreactive NERP-4 was identified in human pancreatic extracts by reverse-phase high-performance liquid chromatography (RP-HPLC). NERP-4 immunoreactivity measured by radioimmunoassay (RIA) was eluted at the same position as synthetic human NERP-4 (Fig. 1k). In C57BL/6 J mouse islets, Vgf mRNA levels were 2–3 orders of magnitude lower than those of Ins1 and Ins2 (encoding insulin 1 and insulin 2, respectively) (Supplementary Fig. 1c). These results indicate that NERP-4 is produced in β cells and co-localises with insulin. NERP-4 increases Ca2+ influx into β cells via Ca2+ channels.
NERP-4 stimulates insulin secretion
NERP-4 augmented insulin secretion in human and C57BL/6 J mouse islets and MIN6-K8 cells under high glucose, but not under low glucose (Fig. 2a–d). Co-administration of NERP-4 and NERP-2[VGF 310–347], another VGF-derived insulinotropic peptide, to Vgf knockdown MIN6-K8 cells additively increased GSIS (Supplementary Fig. 2a, b). In MIN6-K8 cells, NERP-4 enhanced the glucose-induced elevation of ATP concentration, but not that of intracellular cAMP concentration (Fig. 2e, f). Both EGTA and nifedipine treatments abolished NERP-4–induced GSIS in MIN6-K8 cells (Fig. 2g, h). Given that NERP-4 co-localised with insulin and directly enhanced GSIS by β cells, we investigated whether NERP-4 was secreted from β cells. High glucose stimulated both NERP-4 and insulin secretion (Fig. 2i, j). Anti–NERP-4 IgG administration to C57BL/6 J mouse islets suppressed GSIS below that observed with normal rabbit serum IgG (NRS IgG) (Fig. 2k). Thus, NERP-4 stimulates insulin secretion by enhancing Ca2+ influx into β cells in an autocrine fashion.

NERP-4 enhances GSIS in human islets (a, n = 5, 5, 6 biological replicates), islets from male (b, n = 13, 9, 13, 14 biological replicates) and female (c, n = 6, 6, 8, 8 biological replicates) C57BL/6 J mice, and MIN6-K8 cells (d, n = 4, 4, 5, 5 biological replicates). e ATP production in MIN6-K8 cells (n = 6 biological replicates). f Intracellular cAMP level in MIN6-K8 cells incubated with NERP-4 (n = 7 biological replicates). GSIS from MIN6-K8 cells treated with EGTA (g) or nifedipine (h) (n = 4 biological replicates). Glucose-induced secretion of NERP-4 (i) and insulin (j) from MIN6-K8 cells (n = 3 biological replicates). k Anti–NERP-4 IgG suppression of insulin secretion in C57BL/6 J mouse islets (n = 8 biological replicates). Results are pooled from three (b) or two (c, e, f, k) independent experiments, or are representative of three (a, d) or two (g–j) independent experiments. Data are mean ± s.e.m (a–k). One-way ANOVA and Tukey’s multiple comparisons test (a–h, k). Unpaired two-tailed Student’s t-test (i, j). Source data are provided as a Source data file.
NERP-4 prevents oxidative and ER stress and mitochondrial dysfunction induced by palmitate and cytokines
Lipotoxicity impairs β-cell function by inducing oxidative and ER stress in diabetes25. Because two other insulinotropic peptides, TLQP-21[VGF 556–576] and glucagon-like peptide-1 (GLP-1), protected β cells from lipotoxicity19,26, we investigated the roles of NERP-4 in β-cell maintenance in isolated C57BL/6 J mouse islets. Palmitate is a saturated fatty acid widely used to cause insulin secretory dysfunction, induction of ER stress, and apoptosis. Palmitate reduced GSIS and the mRNA levels of Ins1 and Ins2 (Fig. 3a–d). NERP-4 reversed these alterations (Fig. 3a–d). Palmitate increased the Vgf mRNA level and NERP-4 elevated it to an even greater extent (Fig. 3e). Palmitate increased the protein and mRNA levels of the ER stress marker CHOP, while NERP-4 reduced them (Fig. 3f, Supplementary Fig. 3). Palmitate reduced the protein and mRNA levels of the antioxidative response marker SOD2, while NERP-4 reversed these alterations (Fig. 3f, Supplementary Fig. 3). Nrf2 responds to oxidative stress by translocating from the cytosol to the nucleus and then inducing antioxidant genes27. Palmitate reduced Nrf2 translocation to the nucleus and Nrf2 mRNA level (Fig. 3g, Supplementary Fig. 3). NERP-4 reversed these alterations (Fig. 3g, Supplementary Fig. 3). Under non-stressed conditions, NERP-4 administration to isolated C57BL/6 J mouse islets increased the mRNA levels of Vgf, Sod2, and Nrf2 (Fig. 3e, Supplementary Fig. 3). NERP-4 also decreased the protein and mRNA levels of CHOP (Fig. 3f, Supplementary Fig. 3), increased the SOD2 protein level (Fig. 3f), and enhanced Nrf2 nuclear translocation (Fig. 3g).

a NERP-4 was administered at 0, 24, and 48 h to isolated C57BL/6 J mouse islets under palmitate. b GSIS from C57BL/6 J mouse islets (n = 5 biological replicates). c–e Ins1, Ins2, and Vgf mRNA amounts (n = 4 biological replicates). f Protein levels of CHOP and SOD2 (n = 3 biological replicates). g Nrf2 protein levels in nuclear and cytosolic fractions (n = 3 biological replicates). h–m C57BL/6 J mouse islets treated for 72 h with NRS IgG or NERP-4 IgG. h GSIS from C57BL/6 J mouse islets (n = 5 biological replicates). i–m Ins1, Ins2, Sod2, Nrf2, and Chop mRNA amounts (n = 3 biological replicates). Representative results of two independent experiments (b–m). Data are mean ± s.e.m (b–m). One-way ANOVA and Tukey’s multiple comparisons test (b–g). Unpaired two-tailed Student’s t-test (h–m). Source data are provided as a Source data file.
To investigate the role of endogenous NERP-4 in β-cell maintenance, we administered NERP-4 IgG to C57BL/6 J mouse islets for 3 days. NERP-4 neutralisation reduced GSIS and the mRNA levels of Ins1, Ins2, Sod2, and Nrf2, and increased the Chop mRNA level relative to those seen following 3-day NRS IgG administration (Fig. 3h–m). These results suggest that NERP-4 protects β cells from metabolic stress in an autocrine fashion.
Glucose oxidation and ATP production in mitochondria are necessary for insulin biosynthesis, transport, and secretion28,29. Mitochondrial dysfunction causes β-cell failure and the development of diabetes28,29. We next studied the role of NERP-4 in mitochondrial function in MIN6-K8 cells. NERP-4 increased the mitochondrial oxygen consumption rate (OCR) in palmitate-treated MIN6-K8 cells (Fig. 4a, b). Palmitate reduced ATP content, the mRNA levels of the ATP synthases Atp5e and Atp5j2, and GSIS in MIN6-K8 cells (Fig. 4c–e, Supplementary Fig. 4a), while NERP-4 reversed these reductions (Fig. 4c–e, Supplementary Fig. 4a). Palmitate increased the Vgf mRNA level while NERP-4 further elevated it (Fig. 4f). NERP-4 suppressed the palmitate-induced production of reactive oxygen species (ROS) (Fig. 4g). NERP-4 administration to naïve MIN6-K8 cells also increased ATP content, the mRNA levels of Atp5e and Atp5j2, and cell viability, and decreased ROS production (Fig. 4c–e, g, j).

a NERP-4 was administered at 0 and 24 h to MIN6-K8 cells under palmitate (b–g) or at 0 h under cytokines (h–k). b OCR analyses of MIN6-K8 cells (n = 3 biological replicates). The P values indicated the differences of basal respiration (1.6–14.8 min), ATP production (21.4–34.6 min), and maximal respiration (41.2–54.4 min) in palmitate-treated MIN6-K8 cells with or without NERP-4. c ATP production in MIN6-K8 cells treated with or without palmitate and NERP-4 (n = 6 biological replicates). Atp5e (d), Atp5j2 (e), and Vgf (f) mRNA amounts in MIN6-K8 cells (n = 4 biological replicates). g ROS production in MIN6-K8 cells (n = 3 biological replicates). h–k MIN6-K8 cells were treated with a cytokine cocktail and NERP-4 for 24 h. h Protein level of cleaved caspase-3 (n = 3 biological replicates). i GSIS from MIN6-K8 cells (n = 4 biological replicates). j Cell viability (n = 6, 6, 5, 4 biological replicates). k Representative TUNEL images and per cent ratio of TUNEL-positive cells (green) to DAPI-positive cells (blue) (n = 5 independent samples). Representative results of two independent experiments (b–k). Data are mean ± s.e.m. (b–k). Unpaired two-tailed Student’s t-test (b). One-way ANOVA and Tukey’s multiple comparisons test (c–k). Scale bar, 100 μm (k). Source data are provided as a Source data file.
We also investigated the roles of NERP-4 in a model of cytokine-induced β-cell impairment. Interleukin-1β was previously shown to induce oxidative and ER stress in β cells30. A cytokine cocktail (tumour necrosis factor-α, interleukin-1β, and interferon-γ) upregulated the apoptotic marker protein cleaved caspase-3 and its mRNA, reduced GSIS and cell viability, and induced cell death in MIN6-K8 cells (Fig. 4h–k, Supplementary Fig. 4b). NERP-4 reversed these alterations. A cytokine cocktail increased the Vgf mRNA level and NERP-4 further elevated it (Supplementary Fig. 4b). Collectively, these data indicate that NERP-4 suppresses oxidative and ER stress, thereby improving β-cell mitochondrial function.
NERP-4 reverses β-cell impairment in db/db mice
We next studied how NERP-4 impacted glucose metabolism and β-cell maintenance in obese, diabetic db/db mice. Insulin and NERP-4 immunoreactivities were reduced in db/db mice compared with db/+ mice (Fig. 5a, b). The mRNA levels of Vgf, Ins1, and Ins2 were lower in db/db mice compared with db/+ mice (Fig. 5c). Two-week NERP-4 administration to db/db mice decreased fasting blood glucose and plasma insulin levels compared with saline treatment, but did not change body weight or food intake (Fig. 6a, b, Supplementary Fig. 5a, b). Two weeks after NERP-4 administration, a glucose tolerance test (GTT) demonstrated lowered blood glucose and elevated plasma insulin (Fig. 6c, d), but an insulin tolerance test (ITT) showed no alteration in insulin sensitivity (Supplementary Fig. 5c). NERP-4 increased the number and mean diameter of insulin granules in β cells of db/db mice, as observed by transmission electron microscopy (TEM) (Fig. 6e, f). NERP-4 also increased the following in db/db islets: Ins1, Ins2, Iapp, Vgf, and Pdx-1 mRNA levels; insulin content; Ki67-positive cell number; and insulin staining intensity (Fig. 6g–o). NERP-4 did not change the Gcg mRNA level, plasma glucagon level, or the area of glucagon immunoreactivity (Supplementary Fig. 5d–f). Mitochondrial dynamics, such as fission and fusion, comprise a highly coordinated cycle that maintains the shape, distribution, and size of mitochondria and play a critical role in the regulation of pancreatic β-cell function28,29. Mitophagy acts to selectively degrade dysfunctional mitochondria31,32. NERP-4 increased the numbers of mitochondria and mitophagosomes and decreased mitochondrial size in β cells of db/db mice (Fig. 7a–d). NERP-4 upregulated the mRNA levels of mitochondrial markers of biogenesis (Pgc1α), fission (Drp1), fusion (Mfn1), and mitophagy (Park2 and Pink1) (Fig. 7e). NERP-4 reduced the protein and mRNA levels of CHOP (Fig. 7f, Supplementary Fig. 6), and increased the protein and mRNA levels of SOD2 and the mRNA level of Nrf2 (Fig. 7f, Supplementary Fig. 6). These results suggest that NERP-4 maintains β-cell function in db/db mice by improving mitochondrial dynamics and reducing oxidative and ER stress.

a Representative NERP-4 (red) and insulin (green) immunoreactivities in pancreatic islets of 15-week-old db/+ or db/db mice (n = 3 biological animals). b Relative fluorescence intensity of insulin or NERP-4 in pancreatic islets of 15-week-old db/+ mice (11 islets from three mice) or db/db mice (25 islets from three mice). c Vgf, Ins1, and Ins2 mRNA amounts in islets of 10-week-old db/+ mice and db/db mice (n = 4 biological animals). Data are mean ± s.e.m. Unpaired two-tailed Student’s t-test (b, c). Scale bar, 50 μm (a). Source data are provided as a Source data file.

a Daily administration of NERP-4 or saline for 14 days to db/db mice. Blood was collected on Days 0, 7, and 14. GTT and ITT were performed on Day 14. b Changes in blood glucose and plasma insulin concentrations (n = 28, 27 biological animals). c, d Blood glucose and plasma insulin concentrations and their areas under the curves (AUCs) in an intraperitoneal GTT (n = 7, 5 biological animals). e Representative TEM micrograph of β cells. f Number of insulin storage granules (n = 6, 8 biological replicates). Box plots show the diameters of insulin storage granules (saline, n = 448; NERP-4, n = 790). g–k Ins1, Ins2, Iapp, Vgf, and Pdx-1 mRNA amounts in pancreatic islets (n = 5 biological replicates). l Insulin content (n = 10 biological replicates). m Representative images of Ki67-positive cells (red) and insulin (green). n Number of Ki67-positive cells per islet (n = 3; saline, 145 islets from three mice and NERP-4, 147 islets from three mice). o Insulin intensity (saline, 78 islets from five mice and NERP-4, 69 islets from five mice). Results are pooled from four (b) or two (c, d, l) independent experiments or are representative of two independent experiments (e–k, m–o). Data are mean ± s.e.m (b–d, f–k, n). Centre line, median; box edges, first and third quartiles; whiskers, 1.5 times the interquartile range; outliers, individual points (f, l, o). Two-way ANOVA followed by Bonferroni’s post-test for multiple comparisons (b–d). Unpaired two-tailed Student’s t-test (f–l, n, o), Scale bars, 1 μm (e), 50 μm (m). Source data are provided as a Source data file.

a Representative TEM images of mitochondria in β cells. Organelles surrounded by single or double membranes represent mitophagic vacuoles containing mitophagosomes (yellow arrows). Box plots displaying mitochondrial number (b) and size (c) (saline, 386 mitochondria from three mice and NERP-4, 417 mitochondria from three mice). d Quantification of the number of mitophagic vacuoles (12 cells from five mice). e Pgc1α, Drp1, Mfn1, Park2, and Pink1 mRNA amounts in pancreatic islets (n = 5 biological replicates). f Protein levels of CHOP and SOD2 (n = 4 biological replicates). Representative results of three (e) or two (f) independent experiments. Data are mean ± s.e.m (d–f). Centre line, median; box edges, first and third quartiles; whiskers, 1.5 times the interquartile range; outliers, individual points (b–c). Unpaired two-tailed Student’s t-test (b–f). Scale bar, 1 μm (a). Source data are provided as a Source data file.
SNAT2 is a target protein for NERP-4
To identify NERP-4 target candidates in MIN6-K8 cells, we used the ligand–receptor glycocapture technique LRC-TriCEPS33, a highly specific and sensitive pull-down assay involving binding to glycosylated membrane proteins. The volcano plot in Fig. 8a compares proteins bound by NERP-4 and NERP-2. Only SNAT2 and lysosome membrane protein 2 (SCRB2) satisfied the criteria for specific binding to TriCEPS (Fig. 8b). We detected NERP-4 binding in the cell membrane protein fraction of SNAT2-overexpressing HEK293 cells with a radioisotope-labelled C-terminally tyrosyl fragment of NERP-4[8–19], [125I]-Y-NERP-4[8–19], which was used in the RIA of NERP-4 (Fig. 8c). SCRB2 is a type III glycoprotein located primarily in limiting membranes of lysosomes and endosomes34. SCRB2 participates in membrane transportation and the reorganization of the endosomal/lysosomal compartment34. Our findings suggest that SCRB2 is not the target protein of NERP-4. AATs expressed on the cell membrane of β cells regulate β-cell behaviour by mediating amino acid uptake and release3,4. We therefore investigated SNAT2, a neutral AAT that enhances amino acid uptake into β cells to stimulate insulin secretion5. We studied the binding of NERP-4 to SNAT2. [125I]-Y-NERP-4[8–19] bound to SNAT2-overexpressing HEK293 cells in a concentration-dependent manner (Fig. 8d, Supplementary Fig. 7). [125I]-Y-NERP-4[8–19] binding was reduced by the addition of excessive NERP-4 (Fig. 8d), suggesting that the binding was specific for NERP-4. [125I]-Y-NERP-4[8–19] binding was detected in the membrane fraction of SNAT2-overexpressing HEK293 cells (Fig. 8d). SNAT1, SNAT2, and SNAT4 are system A AATs that facilitate the uptake of neutral amino acids34. We detected the mRNAs of Snat2, Snat3, Snat4, and Snat5, but not Snat1, in C57BL/6 J mouse islets and MIN6-K8 cells, consistent with a previous study22 (Fig. 8e). NERP-4 binding was detected only in SNAT2-overexpressing HEK293 cells, but not in HEK293 cells expressing other SNATs (Fig. 8f).

a Volcano plot depicting a comparison of proteins captured by NERP-4 or NERP-2. Data are shown at the protein level and are annotated using the UniProt mouse database. Y-axis = −Log10 (adjusted P value), X-axis = Log2 fold change compared to the other samples (n = 3 independent experiments). b Target candidates identified by the TriCEPS™-based ligand–receptor capture method. SCRB2: lysosome membrane protein 2. c Binding of [125I]-Y-NERP-4[8–19] to the membrane of SNAT2-OE HEK293 cells with or without unlabelled NERP-4 (n = 4 biological replicates). d Binding of [125I]-Y-NERP-4[8–19] with or without unlabelled NERP-4 in SNAT2-overexpressing (OE) HEK293 cells (n = 4 biological replicates). e mRNA levels of Snat1, Snat2, Snat3, Snat4, and Snat5 in C57BL/6 J mouse islets and MIN6-K8 cells (n = 5 biological replicates). f Binding of [125I]-Y-NERP-4[8–19] to mock, SNAT2-, SNAT3-, SNAT4-, or SNAT5-OE HEK293 cells (n = 3 biological replicates). Representative results of two independent experiments (c–f). Data are mean ± s.e.m (c–f). Differential protein abundance was tested using a statistical ANOVA model followed by multiple testing corrections (a, b). One-way ANOVA and Fisher’s LSD test, #P value, SNAT2 OE vs. SNAT2 OE plus unlabelled NERP-4; *P value, SNAT2 OE vs. mock (c). One-way ANOVA and Tukey’s multiple comparisons test (d, e). Two-way ANOVA and Tukey’s multiple comparisons test, *P = 0.0444, **P = 0.0051, ***P < 0.0001 vs. mock; not significant, SNAT3/SNAT4/SNAT5 vs. mock (f). Source data are provided as a Source data file.
NERP-4 increases amino acid uptake and GSIS via SNAT2
We next investigated the effect of NERP-4 on amino acid transport activity. Human and C57BL/6 J mouse islets and MIN6-K8 cells were administered NERP-4 with 1 µM [14C]-l-glutamine. NERP-4 increased the [14C]-l-glutamine uptake in both islets and MIN6-K8 cells (Fig. 9a–c). Next, MIN6-K8 cells were administered NERP-4 with 1 µM [14C]-l-alanine or 1 µM [3H]-l-proline. NERP-4 also increased their uptake (Fig. 9d, Supplementary Fig. 8a). NERP-4 increased the intracellular contents of glutamine, alanine, proline, glycine, and glutamic acid in MIN6-K8 cells (Supplementary Fig. 8b, Supplementary Table 1). Among system A transporters, proline is the preferential natural substrate of SNAT2, but not SNAT1 or SNAT435. To demonstrate that the effect of NERP-4 depends only on SNAT2, we performed SNAT2 deletion and inhibitor experiments. Snat2 knockdown in MIN6-K8 cells abrogated NERP-4–induced [14C]-l-glutamine and [14C]-l-alanine uptake (Fig. 9e, f, Supplementary Fig. 8c). α-Methylaminoisobutyric acid (MeAIB), a non-metabolisable l-alanine analogue, is an orthosteric inhibitor of system A transporter36. MeAIB also abrogated NERP-4–induced [14C]-l-glutamine and [14C]-l-alanine uptake (Fig. 9g, h). These results imply that NERP-4 acts only on SNAT2 to stimulate amino acid uptake. Glutamine and alanine are imported via several transporters in many types of cells5. In rat islets, SNAT2, SNAT4, ASC-1 (SLC1A10), ASCT2 (SLC1A5), and B0AT1 (SLC6A19) transport glutamine and alanine5. Snat2 knockdown reduced glutamine and alanine uptake by 13.6% and 30.3%, respectively, compared with siSCR treatment (Fig. 9e, f). MeAIB reduced glutamine and alanine uptake by 17.8% and 40.7%, respectively (Fig. 9g, h). These results suggest that transporters other than SNAT2 also mediate glutamine and alanine uptake in β cells.

NERP-4–induced [14C]-l-glutamine uptake into human islets (a, n = 4 biological replicates), C57BL/6 J mouse islets (b, n = 6, 5 biological replicates), and MIN6-K8 cells (c, n = 4 biological replicates). d [14C]-l-Alanine uptake into MIN6-K8 cells (n = 4 biological replicates). [14C]-l-Glutamine (e) and [14C]-l-alanine (f) uptake in siSnat2-MIN6-K8 cells (n = 4 biological replicates). [14C]-l-Glutamine (g) and [14C]-l-alanine (h) uptake by MIN6-K8 cells with or without NERP-4 and MeAIB (n = 4 biological replicates). i Representative Fura-2-AM ratios in siSnat2–MIN6-K8 cells in response to NERP-4 (n = 8 cells), and average iAUC (9–20 min) of [Ca2+]i (n = 8 cells). Effects of Snat2 knockdown (j, n = 6 biological replicates) and MeAIB (k, n = 8 biological replicates) on NERP-4–induced GSIS in MIN6-K8 cells. Effect of MeAIB on NERP-4–induced GSIS in human islets (l, n = 5 biological replicates) and C57BL/6 J mouse islets (m, n = 6 biological replicates). n Concentration dependence of MeAIB uptake into MIN6-K8 cells in the presence or absence of NERP-4 (n = 4 biological replicates). Inset shows 1 or 10 μM MeAIB uptake. All experiments were performed under 16.7 mM glucose. Representative results of two independent experiments (b–n). Data are mean ± s.e.m (a–n). One-way ANOVA and Tukey’s multiple comparisons test (a, c, e–h, j–n). Unpaired two-tailed Student’s t-test (b, d, i). Source data are provided as a Source data file.
Given that NERP-4 increased GSIS by stimulating amino acid uptake, we assumed that β-cell exposure to higher glutamine concentrations could mitigate the effects of NERP-4 on GSIS. We studied the insulinotropic activity of NERP-4 in MIN6-K8 cells at glutamine concentrations from 1 µM to 1 mM in amino acid–free HEPES Krebs–Ringer bicarbonate (HKRB) buffer. Glutamine concentrations equal to or higher than 1 µM increased GSIS in a concentration-dependent manner (Supplementary Fig. 8d). NERP-4 augmented GSIS at glutamine concentrations between 1 µM and 100 µM, but not at 1 mM glutamine (Supplementary Fig. 8d). Both Snat2 knockdown and MeAIB abrogated NERP-4–induced Ca2+ influx (Fig. 9i, Supplementary Fig. 8e, f) and GSIS (Fig. 9j, k) in MIN6-K8 cells. MeAIB also abolished NERP-4–induced GSIS in human and C57BL/6 J mouse islets (Fig. 9l, m). These findings reveal that NERP-4 acts specifically on SNAT2 to induce amino acid uptake, Ca2+ influx, and GSIS.
We used amino acid–free HKRB buffer in GSIS experiments. SNAT3, SNAT5, and ASCT2 function as exporters of glutamine under conditions in which extracellular glutamine is absent or very low37,38. To explore the source of amino acids that stimulate NERP-4–induced GSIS, we quantified amino acids released into the medium after GSIS experiments. MIN6-K8 cells released glutamine, alanine, proline, and other amino acids under both 2.8 mM and 16.7 mM glucose conditions (Supplementary Table 2). The concentrations of these amino acids were comparable between low and high glucose conditions. NERP-4 did not augment Ca2+ influx or GSIS under 2.8 mM glucose even when amino acids were present in the medium (Fig. 2b, c, Supplementary Fig. 1a). Another research group demonstrated that 2 mM glutamine administration to MIN6-K8 cells in medium containing 2.8 mM glucose did not affect GSIS or Ca2+ influx23. Together, these findings indicate that under high glucose conditions, NERP-4 functions as an insulinotropic peptide by enhancing uptake of amino acids released from β cells.
NERP-4 augmented [14C]-l-alanine uptake 1 min after its administration to MIN6-K8 cells (Supplementary Fig. 8g). To study the kinetic properties of SNAT2 activation by NERP-4, we determined the concentration-dependent uptake of [14C]-MeAIB in the range of 1 µM to 2 mM at 2 min in the presence or absence of NERP-4. [14C]-MeAIB uptake showed a good fit to a Michaelis–Menten curve, with a Km of 881.1 ± 220.7 µM and a Vmax of 2240 ± 242.3 pmol/mg protein/min (Fig. 9n). NERP-4 lowered the Km of [14C]-MeAIB (412.6 ± 113.5 µM) without changing the Vmax. Furthermore, MeAIB did not alter [125I]-Y-NERP-4[8–19] binding to MIN6-K8 cells (Supplementary Fig. 8h), supporting the finding that the NERP-4 binding site is different from the substrate binding site. Taken together, these results show that NERP-4 may modulate the binding affinity of SNAT2 to amino acids, implying that NERP-4 could act as a positive allosteric modulator (PAM).
NERP-4 prevents β-cell impairment via SNAT2
Insulin increases protein and mRNA levels of SNAT2, thereby enhancing the amino acid supply and upregulating insulin-dependent protein synthesis39. We found that insulin increased Snat2 mRNA in naïve MIN6-K8 cells and C57BL/6 J mouse islets (Supplementary Fig. 9a, b). NERP-4 also increased Snat2 mRNA in MIN6-K8 cells and C57BL/6 J mouse islets under non-stressed conditions (Supplementary Fig. 9c, d). SNAT2 is upregulated in β cells in diabetes as a response to the translational repression associated with ER stress4. We investigated Snat2 expression in palmitate-treated C57BL/6 J mouse islets and db/db mouse islets. The Snat2 mRNA level in C57BL/6 J mouse islets was increased by palmitate and was elevated to an even greater extent by NERP-4 (Fig. 10a). The Snat2 mRNA level was higher in db/db mice compared with db/+ mice (Fig. 10b). Two-week NERP-4 administration increased the Snat2 mRNA level in db/db islets (Fig. 10c). The Snat2 mRNA level was also increased in palmitate- or cytokine-treated MIN6-K8 cells after NERP-4 administration for 48 or 24 h, respectively (Supplementary Fig. 9e, f). SNAT2 upregulation induced by long-term NERP-4 administration under stress conditions could contribute to the effects of NERP-4 on β-cell viability and metabolism.

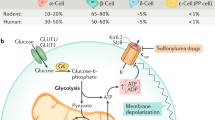
a, Snat2 mRNA amounts in mouse islets under palmitate treatment with or without NERP-4 (n = 4 biological replicates). b Snat2 mRNA amounts in 10-week-old db/+ mouse and db/db mouse islets (n = 4 biological animals). c Snat2 mRNA amounts in islets from db/db mice administered NERP-4 for two weeks (n = 5 biological replicates). d NERP-4 was administered at 24 and 48 h after the start of siSCR treatment or Snat2 knockdown to MIN6-K8 cells under palmitate (e–g) or at 48 h under cytokines (h). e OCR (n = 3 biological replicates). The P values indicated the differences of basal respiration (1.6–14.8 min), ATP production (21.4–34.6 min), and maximal respiration (41.2–54.4 min) in palmitate-treated siSnat2-MIN6-K8 cells with or without NERP-4. f ATP production (n = 4 biological replicates). g ROS production (n = 4 biological replicates). h Cell viability (n = 6 biological replicates). Representative results of two independent experiments (a–c, e–h). Data are mean ± s.e.m. (a–c, e–h). One-way ANOVA and Tukey’s multiple comparisons test (a, e–h). Unpaired two-tailed Student’s t-test (b, c). i Schematic of NERP-4 roles in pancreatic β cells. NERP-4 is processed from VGF, a granin protein that is critical for granule biogenesis in pancreatic β cells. NERP-4 is packed with insulin in secretory granules and secreted by glucose. NERP-4 binds to SNAT2 to stimulate amino acid uptake into β cells, thereby enhancing mitochondrial ATP production, glucose-induced Ca2+ mobilisation into β cells, and GSIS. NERP-4 expression is reduced in β cells in db/db mice. NERP-4 protects β cells from glucolipotoxicity by reducing ROS production and ER stress, thereby enhancing mitochondrial biogenesis and dynamics. Source data are provided as a Source data file.
To verify that SNAT2 was responsible for NERP-4–induced β-cell maintenance, we examined how NERP-4 impacted the effects of palmitate or cytokine treatment after Snat2 knockdown (Fig. 10d). Snat2 knockdown abolished the ability of NERP-4 to increase the OCR and the ATP production and to decrease ROS production in palmitate-treated MIN6-K8 cells (Fig. 10d–g). Snat2 knockdown also abolished the NERP-4–induced recovery of cell viability in cytokine-treated MIN6-K8 cells (Fig. 10h). Together, these data show that NERP-4 enhances GSIS and reduces β-cell impairment by enhancing both the expression and activity of SNAT2.
Discussion
This study demonstrated that the VGF-derived peptide NERP-4 potently enhanced glutamine, alanine, and proline uptake into β cells via SNAT2, thereby stimulating mitochondrial ATP production and GSIS (Fig. 10i). NERP-4 maintains β cells by reducing oxidative and ER stress in db/db mouse islets and palmitate-treated C57BL/6 J mouse islets. Our findings showed that these effects of NERP-4 on β-cell maintenance resulted from the interaction between NERP-4 and SNAT2.
The granin protein VGF is processed to multiple bioactive peptides by the prohormone convertases PC1/3 and PC2 in the dense core vesicles of endocrine and neuroendocrine cells17,18,20,40. To identify novel bioactive peptides, we analysed the peptides secreted from TT cells11, and isolated two C-terminally amidated VGF-derived peptides, NERP-1[VGF 281–306] and NERP-2. These two peptides were also expressed in human and mouse β cells41,42. NERP-2, but not NERP-1, enhanced GSIS by elevating intracellular Ca2+ in β cells41. NERP-4 shared no amino acid sequence homology with NERP-2. In the present study, NERP-4 additively enhanced GSIS when administered together with NERP-2, suggesting that the target protein of NERP-4 differs from that of NERP-2. VGF also produces three other insulinotropic peptides, namely TLQP-21, TLQP-62[VGF 556–617], and AQEE-30[VGF 588–617]19,43,44. Chronic administration of TLQP-21 to prediabetic Zucker diabetic fatty rats preserved islet β-cell mass and slowed diabetes onset19. C3aR1 knockout abolished the TLQP-21–induced anti-obesity effect seen in wild mice45. TLQP-62 induced a rapid increase in intracellular calcium mobilization, further increasing TLQP-62 secretion in a self-reinforcing manner43. AQEE-30 increased the phosphorylation of Akt and GSK3β in streptozotocin-treated β cells and suppressed β-cell death46. NERP-4 increased Vgf mRNA in db/db mouse islets, palmitate- or cytokine-treated β cells, and naïve β cells. VGF-derived peptides being induced by NERP-4 potentially mediate the long-term effects of NERP-4 on β-cell maintenance and function. Insulin itself has protective effects on β cells47. In the present study, NERP-4 recovered the suppression of insulin secretion caused by metabolic stress. NERP-4–induced insulin secretion could contribute to the effect of NERP-4 on β-cell maintenance.
To identify the NERP-4 target in β cells, we compared NERP-4 and NERP-2 in terms of the volcano plot results obtained by LRC-TriCEPS technology. NERP-4–SNAT2 interaction was verified by a binding assay, cellular functional experiments, and Snat2 knockdown. SNAT2 has 11 putative transmembrane domains with two potential N-glycosylated sites, and imports amino acids in a Na+-cotransport manner2,3,48. Considering the kinetics analysis of the NERP-4–SNAT2 interaction and the lack of MeAIB interference with the binding between NERP-4 and SNAT2, NERP-4 may act as a PAM of SNAT2. In the process of drug discovery, PAMs have been developed for ion channels, kinases, phospholipases, and G protein–coupled receptors49,50,51,52. The binding of allosteric modulators to these target proteins had little impact on either the orthosteric or allosteric binding pockets53. The only endogenous PAM that has been identified thus far is the glutamate transporter–associated protein 3–18 (GTRAP3-18), which acts directly on an AAT named excitatory amino acid carrier 1 (EAAC1)54. The interaction between NERP-4 and SNAT2 provides evidence that an endogenous peptide can modulate the activity of an AAT. Cryo-electron microscopy studies have determined the architectures and transport mechanisms of some AATs49,55,56. The topological structure and amino acid transport site of SNAT2 have yet to be determined. Future structural analyses of the molecular features of the SNAT2-binding site of NERP-4 could elucidate how NERP-4 modulates SNAT2 activity.
SNAT2 imports glutamine, alanine, proline, serine, and other amino acids57. Among the SNAT2 substrates, glutamine, alanine, and proline regulate insulin secretion2,58,59. SNAT3 and SNAT5 have bidirectional transport properties depending on the metabolic/nutritional status3: they import glutamine, asparagine, alanine, and histidine, and export glutamine and glycine57,60. In addition to its role in amino acid transfer, SNAT2 possesses a transceptor-like function61. It detects whether there are sufficient amounts of the amino acids that regulate its expression and stability61,62. SNAT2-mediated Na+ incorporation plays a potential role in membrane depolarisation and increases both intracellular Ca2+ levels and insulin secretion59. SNAT2 activation can also activate several transduction pathways that support cell growth and proliferation61,62,63. In the present study, NERP-4 increased SNAT2 expression, suggesting that NERP-4 not only contributes to the transport of Na+ and amino acids via SNAT2, but also increases SNAT2 expression that can enhance its downstream signalling.
After being imported into β cells, glutamine and alanine are metabolised in the tricarboxylic acid (TCA) cycle to produce ATP, and eventually potentiate Ca2+ influx and insulin secretion2. Cytosolic glutamine is converted to glutamate, which yields the antioxidant glutathione, thereby reducing ROS production and improving mitochondrial dysfunction64,65. Glutamate also yields γ-aminobutyric acid, which contributes to β-cell survival3. Alanine is rapidly converted to pyruvate, and then to glutamate, aspartate, and lactate via the TCA cycle2. It is currently unknown which SNAT is responsible for alanine export from β cells. Proline plays key roles in protein structure and function, and also in the maintenance of cellular redox homeostasis66. Proline also stimulates insulin secretion through plasma membrane depolarisation as well as cell glycolytic and oxidative metabolism58,59. In this study, NERP-4 increased the intracellular contents of SNAT2 substrates and stimulated GSIS from MIN6-K8 cells prepared under amino acid–free conditions. Amino acids secreted from the cells potently mediated NERP-4–induced GSIS. Identifying a novel mechanism whereby the peptide–AAT axis regulates amino acid transport could increase our understanding of how amino acids regulate β-cell biology.
Glucotoxicity and lipotoxicity in type 2 diabetes stress β cells by increasing insulin biosynthesis6,25. β cells are susceptible to ROS-induced cellular stress and protein misfolding in the context of excessive caloric intake25,67. Mitochondrial quality is dynamically maintained by the regulation of biosynthesis, fission, fusion, and mitophagy68. Mitochondrial dysfunction and both oxidative and ER stress in β cells exacerbate diabetes6,31. In addition to insulin, β cells produce peptides that preserve β-cell longevity by protecting against metabolic stress69. In this study, NERP-4 administration to db/db mice upregulated genes related to mitochondrial biogenesis and dynamics. Under both stressed and non-stressed conditions, NERP-4 administration to MIN6-K8 cells improved β-cell viability and function. By contrast, NERP-4 neutralisation in isolated mouse islets aggravated oxidative and ER stress and impaired GSIS, suggesting that endogenous NERP-4 functions in β-cell behaviour.
The present study has expanded on the role of SNAT2 in β-cell maintenance. The biosynthesis of SNAT2 is regulated by the concentrations of insulin and amino acids4. Under ER stress in diabetes, ATF4 upregulates SNAT2 mRNA in β cells to stimulate amino acid influx and protein synthesis4,7. Palmitate induces oxidative stress by impairing cytosolic and mitochondrial Ca2+ homeostasis, thereby causing mitochondrial dysfunction and β-cell apoptosis70,71. In this study, NERP-4 upregulated SNAT2 mRNA in db/db islets, both palmitate- and cytokine-treated β cells, and naïve β cells. By contrast, Snat2 knockdown abolished the protective effects of NERP-4 on mitochondrial function, ROS production, and cell viability. In addition to the insulinotropic activity of NERP-4, which is mediated by SNAT2, NERP-4 protects β cells from oxidative and ER stress via SNAT2.
The present study revealed that NERP-4 acts on SNAT2 to regulate β-cell function and maintenance. NERP-4 is secreted together with insulin following glucose stimulation, and modulates SNAT2 activity in β cells. Understanding the NERP-4–SNAT2 axis may clarify how glucose and amino acids interact in β-cell biology, thus serving as a foundation for future research and therapeutic strategies in diabetes.
Methods
Animals
C57BL/6 J mice, db/+ and db/db mice, Wistar rats (Charles River Laboratories), Vgf KO mice (Dr. S.R. Salton, Mount Sinai School of Medicine)24, and apoaequorin transgenic mice10 were maintained under controlled temperature (21–23 °C) and light (light on: 08:00–20:00) conditions with free access to a standard diet (CLEA Japan, CE-2) and water. Male mice were used for the experiments. For GSIS studies, islets were isolated from both male and female C57BL/6 J mice. All animal experiments were performed in accordance with the Japanese Physiological Society guidelines for animal care and were approved by the Ethics Committee on Animal Experimentation of the University of Miyazaki. The mice and rats were humanely euthanized by intraperitoneal injection with a combination anesthetic, consisting of 0.3 mg/kg of medetomidine, 4.0 mg/kg of midazolam, and 5.0 mg/kg of butorphanol. Pancreas tissues and islets were collected after euthanasia.
Statement of ethics
Human tissues were studied after approval by the Ethics Committee of the University of Miyazaki (approval date: April 23, 2017; Approval No. O-136). Human pancreatic islets isolated from a heart-beating cadaveric donor (female, 53 years old) as described previously72 next-of-kin consent for research, were kindly provided by the University Hospital of Lille via the European Consortium for Islet Transplantation human islet distribution program supported by the Juvenile Diabetes Research Foundation. Islet purity was assessed as the percentage of endocrine clusters positive to dithizone staining (range: 80–90%). Participates provided written informed consent. Permissions for research use of human specimens and exportation of human islets were granted by the French Ministry of Higher Education and Research and the Agence de la biomédecine, respectively.
Cultured cells
MIN6-K8 cells were provided by Dr. Jun-ichi Miyazaki (Osaka University). MIN6-K8 cells are mouse insulinoma–derived β cells21 that have been used to study the mechanism of glutamine-amplified Ca2+ influx and insulin secretion22,23. HEK293 cells were from ATCC (CRL-1573). They were maintained in Dulbecco’s modified Eagle’s medium (DMEM, 044-29765, Wako) supplemented with 10% fetal bovine serum (FBS), 25 mM glucose, 4 mM glutamine, 100 U/mL benzylpenicillin, and 100 mg/mL streptomycin at 37 °C in a humidified atmosphere of 5% CO2.
Quantitative real-time PCR (RT-PCR)
mRNA was extracted from MIN6-K8 cells and mouse islets with a RibopureTM Kit (Thermo Fisher Scientific). First-strand cDNA was synthesised with high-capacity cDNA reverse transcription kits (Applied Biosystems). RT-PCR was performed with the TaqMan/Applied Biosciences primers shown in Supplementary Table 3. Quantitative RT-PCR was performed with TaqMan Fast Universal PCR Master Mix (Thermo Fisher Scientific) and a Thermal Cycler Dice Real Time System II (Takara Bio). PCR products were normalised to the expression of glyceraldehyde3-phosphate dehydrogenase (Gapdh) mRNA.
Digital PCR
Primers (Supplementary Table 3), QS3D Chips, and the ProFlex PCR System (Thermo Fisher Scientific) were used for amplification. QS3D Chips were analysed using QuantStudio 3D Analysis Suite Cloud Software (Thermo Fisher Scientific).
Short interfering RNA (siRNA) treatment
MIN6-K8 cells (1 × 105 cells/well in 24-well plates) were treated with mouse Snat2 siRNA (s85605, Thermo Fisher Scientific, 20 pmol/mL), mouse Vgf siRNA (s117809, Thermo Fisher Scientific, 20 pmol/mL), or scrambled control siRNA (siSCR) (4390843, Thermo Fisher Scientific) using Lipofectamine 2000 (Thermo Fisher Scientific) for 72 h. Knockdown efficacy was studied by RT-PCR.
Apoaequorin transgenic mice
We generated apoaequorin transgenic mice to explore transient luminescence caused by intracellular Ca2+ mobilisation as described elsewhere10. Mouse pancreatic tissues were minced in RPMI-1640 (Invitrogen) to prepare small pieces. In order to reconstitute aequorin, 1–2 mm3 pieces were incubated with coelenterazine (Molecular Probes) for 3 h. Pancreatic pieces were sequentially treated with 1 μM NERP-4, 100 μM ATP, and 2.5% Triton X-100. The light emission of apoaequorin was continuously monitored every second as relative luminescence units in an AutoLumat LB 953 luminometer (Berthold Technologies).
Measurement of intracellular Ca2+ influx
Naïve, siSCR-treated, or Snat2 knockdown MIN6-K8 cells were cultured in 35-mm culture plates (Becton Dickinson Labware). Cells were loaded with 1 μM fura-2 acetoxymethyl ester (Fura-2-AM, Dojindo) in HKRB containing 2.8 mM glucose for 30 min at 37 °C. Culture plates were placed on the stage of an integrated fluorescence microscope (BZ-X700, Keyence). Images were captured at 10-s intervals, and 340- and 380-nm excitation filters were used for Fura-2-AM dual-wavelength excitation-ratio imaging. For [Ca2+]i measurements, cells were exposed to HKRB containing 2.8 mM glucose for 30 min, then to HKRB containing 16.7 mM glucose with or without 10−10 M NERP-4. In some experiments, 1 mM EGTA (Nacalai Tesque), 10 μM nifedipine (Sigma-Aldrich), or 10 mM MeAIB (Abcam) was administered when 2.8 mM glucose started. [Ca2+]i was also measured in MIN6-K8 cells incubated under 2.8 mM glucose with 10−10 M NERP-4. The fluorescence ratio was recorded for 22 min. At the end of each experiment, cells were exposed to 35 mM KCl for 2 min. All data are expressed as per cent changes relative to the average fluorescence ratio in 2.8 mM glucose.
Antibody preparation
Acetyl AC-NERP-4[8–19] (Supplementary Table 6) was conjugated with maleimide-activated keyhole limpet hemocyanin (Pierce) and used to immunise rabbits, as described elsewhere20. Rabbits were immunized with each conjugate emulsified with an equal volume of Freund’s complete adjuvant. Anti–NERP-4 IgG and NRS IgG were purified from acetyl AC-NERP-4[8–19] antiserum and normal rabbit serum (FUJIFILM Wako Pure Chemical), respectively, over a Protein G Sepharose 4 Fast Flow column (Merck Millipore).
Immunohistochemistry and immunocytochemistry
Pancreata were infiltrated with 4% paraformaldehyde overnight at 4 °C and embedded in OCT compound (Sakura Finetek). MIN6-K8 cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT). Antibodies and staining conditions are described in Supplementary Table 4. For double immunofluorescence staining41, pancreata or MIN6-K8 cells were incubated overnight at 4 °C with the first primary antibodies and then overnight with the second primary antibodies (Supplementary Table 4). They were then incubated at RT with corresponding Alexa Fluor 488– or 594–conjugated secondary antibodies at RT for 1 h. Samples were observed using an AX-7 fluorescence microscope (Olympus) or C2 confocal microscope (Nikon). The fluorescence intensity and area were measured using ImageJ software (National Institutes of Health).
Subcellular fractionation
NERP-4–induced Nrf2 translocation into the nucleus was studied in isolated mouse islets. NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fischer Scientific) was used to prepare cytosol and nuclear fractions. The cytosol extracts and nuclear extracts were transferred to a new prechilled tube and stored at –80 °C until used. The protein contents of the extracts were determined by a Bradford assay.
Western blotting
Proteins (20 µg) extracted from HEK293 cells, MIN6-K8 cells, and mouse islets were analysed by western blot with the indicated antibodies (Supplementary Table 4), as described elsewhere73. The cells and islets were ruptured with RIPA buffer (Nacalai Tesque). The protein samples were resolved by SDS‒PAGE and transferred onto PVDF membranes, blocked for 1 h with 5% skim milk, and incubated overnight at 4 °C with primary antibodies. After three washes with TBST, the membranes were incubated for 1 h with the appropriate secondary antibodies and developed using chemiluminescent substrates. Fusion Edge software (Vilber Lourmat) was used for quantification.
Double immunoelectron microscopy
Double immunoelectron microscopy was performed as described elsewhere74. Ultrathin sections of the pancreata of C57BL/6 J mice were incubated with guinea pig anti-insulin antibody for 12 h at 4 °C, followed by incubation with 10 nm of gold-labelled goat anti–guinea pig IgG (BB International) for 3 h at RT. They were next incubated with anti–NERP-4 antibody for 12 h at 4 °C and further with 5 nm of gold-labelled goat anti–rabbit IgG (BB International) for 3 h at RT. They were stained with uranium acetate–lead citrate and examined using an H-7600 transmission electron microscope (Hitachi).
NERP-4 secretion from MIN6-K8 cells
MIN6-K8 cells at 80% confluence were incubated in HKRB containing 2.8 mM or 16.7 mM glucose for 30 min. The supernatant was applied to a Sep-Pak C-18 cartridge (Waters), and peptides bound to the resin were eluted with a 60% acetonitrile solution containing 0.1% trifluoroacetic acid (TFA), as described elsewhere11. A portion of the eluate was subjected to radioimmunoassay for NERP-4.
Quantification and chromatographic characterisation of NERP-4 in human pancreas
Noncancerous human pancreatic tissues (total wet weight 7.95 g) were obtained from four patients undergoing surgery for pancreatic cancer. The study was carried out in accordance with the principles of the Declaration of Helsinki, and written informed consent was obtained. The tissues were subjected to the peptide isolation procedure, as described previously11. The pancreatic extract was applied to a Sep-Pak C-18 cartridge. The eluted peptides were subjected to RP-HPLC analysis using a TSKgel ODS-120T column (Tosoh) and a linear gradient of 10–60% acetonitrile containing 0.1% TFA for 80 min at a flow rate of 1.0 mL/min.
Radioimmunoassay (RIA)
RIA was carried out using antiserum for NERP-4 and [125I]-Y-NERP-4[8–19]. The half-maximum-inhibition amount of ligand binding in the RIA was 30 fmol/tube of NERP-4. The specificity of NERP-4 RIA was examined for 20 peptides, including the VGF-derived peptides listed in Supplementary Table 5.
Islet isolation
Mouse pancreatic islets were isolated as described previously75. Each minced pancreas was digested in 10 mL collagenase P (1 mg/mL in HBSS, Roche Diagnostics) for 15 min at 37 °C. The pellet was resuspended in RPMI-1640 supplemented with 10% FBS, 1% penicillin–streptomycin, 2 mM glutamine, and 11 mM glucose. Pancreatic islets were selected under a stereomicroscope.
Insulin secretion and content in MIN6-K8 cells and islets
Naïve, siSCR-treated, and Snat2 or Vgf knockdown MIN6-K8 cells (2 × 105 cells/well) were pre-incubated for 30 min in HKRB containing 2.8 mM glucose, and then incubated for 30 min in HKRB containing 16.7 mM glucose with or without 10−10 M NERP-4. EGTA (1 mM), nifedipine (10 μM), glutamine (1 μM to 1 mM, Nacalai Tesque), MeAIB (10 mM), or NERP-2 (1 μM)42 was administered in experiments described in the text. Ten size-matched islets from C57BL/6 J mice were pre-incubated in 2.8 mM glucose for 1 h, then incubated for 1 h with 2.8 mM or 16.7 mM glucose, or with 16.7 mM glucose containing 10−8 M NERP-4. To examine the effect of palmitate plus low glutamine, islets were cultured in RPMI-1640 medium supplemented with 0.5 mM palmitate (Sigma-Aldrich), 1 mM glutamine, 10% FBS, 100 U/mL benzylpenicillin, and 100 mg/mL streptomycin, and were treated with 10−8 M NERP-4 daily for 3 d. MIN6-K8 cells were cultured in DMEM supplemented with 0.5 mM palmitate, 2 mM glutamine, 10% FBS, 100 U/mL benzylpenicillin, and 100 mg/mL streptomycin, and were treated with 10−10 M NERP-4 daily for 2 d. To examine the effects of cytokines, MIN6-K8 cells were treated with a cytokine cocktail (50 ng/mL mouse tumour necrosis factor-α, 10 ng/mL mouse interleukin-1β, and 50 ng/mL mouse interferon-γ) (Mitltenyi Biotec) and 10−10 M NERP-4 in DMEM (045-30285, Wako) supplemented with 2 mM glutamine, 10% FBS, 100 U/mL benzylpenicillin, and 100 mg/mL streptomycin for 24 h. To measure insulin contents, MIN6-K8 cells and islets were treated with 1% hydrochloric acid–ethanol for 24 h at 4 °C after ultrasonic homogenisation. Mouse insulin and human insulin were measured with an ultra-sensitive Mouse Insulin ELISA Kit (Morinaga Institute of Biological Science) and a Mercodia Insulin ELISA Kit (Mercodia), respectively. Protein content was determined with a Protein Assay Kit I (Bio-Rad). All GSIS experiments were conducted in amino acid–free buffer.
Neutralisation of NERP-4 in experiments with mouse islets
To study the short-term effect of NERP-4 IgG, C57BL/6 J mouse islets were pre-incubated for 3 h with 3 μg/mL anti–NERP-4 IgG or NRS IgG in HKRB buffer containing 2.8 mM glucose. Islets were incubated for 1 h with 3 μg/mL anti–NERP-4 IgG, 3 μg/mL NRS IgG, or 3 μg/mL NRS IgG plus 10−8 M NERP-4 in HKRB buffer containing 16.7 mM glucose. To study the long-term effect of NERP-4 IgG, C57BL/6 J mouse islets cultured in RPMI-1640 medium containing 1 mM glutamine were treated with 3 μg/mL anti–NERP-4 IgG or NRS IgG daily for 3 d. The islets were stimulated by 16.7 mM glucose for 1 h after pre-incubation for 1 h in 2.8 mM glucose.
ATP assay
Naïve, siSCR-treated or Snat2 knockdown MIN6-K8 cells were incubated with 2.8 mM or 16.7 mM glucose with or without 10−10 M NERP-4 for 30 min. The cells were treated with 0.5 mM palmitate in DMEM containing 2 mM glutamine for 48 h, and treated with 10−10 M NERP-4 once daily during palmitate treatment. They were lysed and the ATP content was determined with a CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega) or a Colorimetric/Fluorometric Assay Kit (BioVision).
Measurement of intracellular cAMP
MIN6-K8 cells were pre-incubated with HKRB buffer containing 1 mM glucose and 250 µM 3-isobutyl-1-methylxanthine (Sigma-Aldrich) for 1 h at 37 °C. They were treated with 2.8 mM glucose or 16.7 mM glucose with or without 10−10 M NERP-4 for 30 min. cAMP was measured with a cAMP Biotark enzyme immunoassay system (GE Healthcare).
Cell viability assay and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay
Cell viability was studied with a Cell Counting Kit-8 (Dojindo). Cells were treated with a cytokine cocktail (50 ng/mL IFN-γ, 50 ng/mLTNFα, 10 ng/mL IL-1β; Supplementary Table 6) and 10−10 M NERP-4 in DMEM containing 2 mM glutamine for 24 h. A TUNEL assay was carried out using an In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics). TUNEL-positive cells were counted in five random areas.
TriCEPS™-based ligand–receptor glycocapture
The putative target protein for NERP-4 was identified with TriCEPS™-based ligand–receptor capture (LRC-TriCEPS, P05201; Dualsystems Biotech)33. In brief, 300 μg NERP-4 or NERP-2 used as a control was reacted with TriCEPS™ reagent in 25 mM HEPES buffer for 2 h at 22 °C. Cell surface proteins in MIN6-K8 cells (1.2 × 108) were oxidised with 1.5 mM NaIO4. TriCEPS™-coupled NERP-4 or NERP-2 was incubated with cell surface proteins for 90 min; the proteins were then purified by solid-phase chromatography. Proteins were reduced, alkylated, and digested with trypsin. Tryptic peptides were analysed on a Thermo LTQ Orbitrap XL spectrometer fitted with an electrospray ion source. The experiment was performed two times. Progenesis software was used for raw file alignment and feature detection, the Comet search engine was used for spectra identification, and the Trans proteomic pipeline was used for statistical validation of putative identifications and protein inference33. Upon protein inference, relative quantification of controls and ligand samples was performed on the basis of ion intensity. Differential protein abundance was tested using a statistical ANOVA model followed by multiple testing corrections. The results are presented as a volcano plot, with the X-axis representing the mean ratio fold change (on a log2 scale) and the Y-axis representing the statistical significance P value of the ratio fold change for each protein (on a −log10 scale). The criteria for considering a protein as a candidate for interacting with the ligand of interest were an adjusted P value < 0.01 and a fold change >3.5 for NERP-4 compared to NERP-2, according to the manufacturer’s recommendation33.
Stable expression of SNAT family in HEK293 cells
Human SNAT2/SLC38A2 (Myc-DDK–tagged)-CMV 6-entry vector was purchased from Origene (RC201892). Human SNAT3, SNAT4 and SNAT5 were amplified by PCR and inserted into the BglII and XhoI sites of pCMV6-Entry with an In-Fusion HD Cloning Kit (Z9640N, Takara Bio). These constructs were transfected into HEK293 cells with ViaFect (Promega). The transfected cells were cultured in a medium containing 0.5 mg/mL G418 (Promega) for 2 weeks, and single colonies were subsequently isolated.
Binding assay
SNAT2-expressing HEK293 cells were incubated in HEPES buffer (DMEM containing 0.1% bovine serum albumin, 0.1% NaN3 and 50 mM HEPES (pH 7.4)) containing 0.5–16 nM [125I]-Y-NERP-4[8–19] with or without unlabelled NERP-4 for 1 h at 37 °C. The radioactivity of cell lysates was measured with an automatic γ-counter (AccuFLEXγ ARC-7001, Hitachi). Nonspecific binding was determined in the presence of a 500-fold excess of unlabelled NERP-4. Cell membrane proteins (4 µg) were extracted from SNAT2-expressing HEK293 cells, as described previously76. Cells were scraped into HEPES buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2, and 0.25 M sucrose) and homogenized on ice. The homogenate was centrifuged at 800 × g at 4 °C for 20 min and the supernatant was centrifuged twice at 100,000 × g at 4 °C for 1 h. The membrane proteins were incubated with 2 nM [125I]-Y-NERP-4[8–19] for 3 h at 25 °C. Bound [125I]-Y-NERP-4[8–19] was isolated by vacuum filtration, and the radioactivity was measured. SNAT3-, SNAT4-, or SNAT5-expressing HEK293 cells were also studied in the above binding assay.
Transport assay and inhibition experiments
MIN6-K8 cells and islets were preincubated with HKRB or HBSS buffer supplemented with 2.8 mM glucose for 1 h at 37 °C. They were incubated for 2, 5, or 10 min with HKRB or HBSS buffer containing 2.8 mM or 16.7 mM glucose and 1 μM [14C]-l-glutamine (NEC451, PerkinElmer), 1 μM [14C]-l-alanine (MC-466, Moravek), or 1 μM [3H]-l-proline (NET483001MC, PerkinElmer) with or without NERP-4 (MIN6-K8 cells, 10−10 M and islets, 10−8 M). In an inhibition experiment of SNAT2, 10 mM MeAIB was administered to MIN6-K8 cells for 30 min. Uptake of glutamine, alanine, and MeAIB into siSnat2 MIN6-K8 cells was also studied. The kinetic parameters of SNAT2 uptake activation in MIN6-K8 cells were determined by the uptake of [14C]-MeAIB (NEC671050UC, PerkinElmer) for 2 min at concentrations of 1, 10, 100, 500, 1000, and 2000 μM in HKRB buffer containing 16.7 mM glucose, with or without NERP-4 (10−10 M). The uptake values were plotted against [14C]-MeAIB concentration and fitted to a Michaelis–Menten curve. The Michaelis constant (Km) and the maximum velocity values (Vmax) were determined by nonlinear regression using the enzyme kinetics module of GraphPad Prism 7 statistical software (GraphPad). Radioactivity was measured in Tri-Carb 2810TR (PerkinElmer).
Amino acid determination
MIN6-K8 cells were preincubated for 1 h in HKRB containing 2.8 mM glucose. To measure amino acids released into the medium, MIN6-K8 cells (80–90% confluency in a 100 mm cell culture dish) were incubated for 30 min in HKRB containing 2.8 mM or 16.7 mM glucose. Supernatants were applied to 30 kDa Amicon Ultra (Merck Millipore) to remove albumin, and the eluates were subsequently lyophilised. To measure the intracellular contents of amino acids in MIN6-K8 cells (4 × 106 cells/well in a 6-well plate), they were incubated for 5 min in HKRB containing 16.7 mM glucose with or without NERP-4. After washing the cells 3 times with cold PBS, the cell lysates were collected with RIPA buffer (Nacalai Tesque) and stored at –80 °C until used. Amino acids were determined by liquid chromatography/mass spectrometry (SRL Inc.).
Administration of NERP-4 to db/db mice
db/db mice (8-week-old male, n = 35 for each group) were subcutaneously injected with NERP-4 (600 nmol/kg) or saline once daily (16:00) for 2 weeks. Blood samples were collected from their tail veins after a 16-h fast once per week. Two weeks after the start of NERP-4 or saline administration, db/db mice were intraperitoneally administered glucose (0.5 g/kg BW) or insulin (1.5 U/kg BW, Humulin, Eli Lilly) after a 16-h fast. Blood was collected at time points from 0 to 120 min after administration.
TEM
Pancreata of db/db mice were fixed with a mixture of 2% paraformaldehyde and 2.5% glutaraldehyde overnight at 4 °C. They were then embedded in epoxy resin. Ultrathin sections mounted on gold meshes were stained with uranyl acetate–lead citrate and examined using an HT7700 transmission electron microscope (Hitachi)6. Sizes of insulin granules (saline, 594; and NERP-4, 305) and mitochondria (saline, 386; and NERP-4, 417) of each of three db/db mice administered NERP-4 or saline were measured.
Mitochondrial respiration
The OCR was determined using an Extracellular Flux Analyzer XFe (Agilent Technologies). MIN6-K8 cells (3 × 104 cells/well) were placed in minimal XF assay medium (Agilent Technologies) containing 16.7 mM glucose with palmitate (0.5 mM) or palmitate plus NERP-4 (10−10 M) for 48 h. Basal respiration, ATP-coupled respiration, proton leakage and maximal respiratory capacity were measured by adding their respective inhibitors as described elsewhere77: the electron transport chain inhibitor oligomycin (4 μM), the proton ionophore FCCP (2 μM), rotenone (5 μM) and antimycin A (5 μM) (Agilent Technologies). All OCR measurements were corrected for non-mitochondrial OCR.
Measurement of ROS production
Naïve, siSCR-treated, or Snat2 knockdown MIN6-K8 cells were seeded on a 96-well black plate (Costar, Corning). The cells were treated with 0.5 mM palmitate in DMEM containing 2 mM glutamine for 48 h, and were administered 10−10 M NERP-4 once daily during palmitate treatment. The cells were loaded with 50 μM 2,7-dichlorodihydrofluorescein diacetate (Dojindo) for 30 min at 37 °C. Fluorescence intensity was measured using a VICTOR Nivo (PerkinElmer) with excitation at 488 nm and emission at 522 nm.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 7 statistical software (GraphPad) using two-way ANOVA followed by Bonferroni’s post-test for multiple comparisons, One-way ANOVA and Tukey’s multiple comparisons test, one-way ANOVA and Fisher’s LSD test, and the unpaired two-tailed Student’s t-test. Outliers were identified by ROUT method with Prism in all experiments. The statistical results were the same in all the experiments when outliers were included or excluded. We showed the figures that include outliers. All data are expressed as means ± s.e.m. P < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data needed to reproduce the findings described in this manuscript can be found in the manuscript figures and supplementary materials. Source data are provided with this paper.
References
Gutiérrez-Preciado, A., Romero, H. & Peimbert, M. An evolutionary perspective on amino acids. Nat. Educ. 3, 29 (2010).
Newsholme, P., Bender, K., Kiely, A. & Brennan, L. Amino acid metabolism, insulin secretion and diabetes. Biochem. Soc. Trans. 35, 1180–1186 (2007).
Kandasamy, P., Gyimesi, G., Kanai, Y. & Hediger, M. A. Amino acid transporters revisited: new views in health and disease. Trends Biochem. Sci. 43, 752–789 (2018).
Javed, K. & Fairweather, S. J. Amino acid transporters in the regulation of insulin secretion and signalling. Biochem. Soc. Trans. 47, 571–590 (2019).
Bröer, S. Amino acid transporters as modulators of glucose homeostasis. Trends Endocrinol. Metab. 33, 120–135 (2022).
Hasnain, S. Z., Prins, J. B. & McGuckin, M. A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 56, R33–R54 (2016).
Krokowski, D. et al. A self-defeating anabolic program leads to β-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J. Biol. Chem. 288, 17202–17213 (2013).
Pinto, M. C. et al. Calcium signaling and cell proliferation. Cell Signal. 27, 2139–2149 (2015).
Shimomura, O., Johnson, F. H. & Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell Comp. Physiol. 59, 223–239 (1962).
Yamano, K. et al. Identification of the functional expression of adenosine A3 receptor in pancreas using transgenic mice expressing jellyfish apoaequorin. Transgenic Res. 16, 429–435 (2007).
Sasaki, K., Takahashi, N., Satoh, M., Yamasaki, M. & Minamino, N. A peptidomics strategy for discovering endogenous bioactive peptides. J. Proteome Res. 9, 5047–5052 (2010).
Eipper, B. A., Stoffers, D. A. & Mains, R. E. The biosynthesis of neuropeptides: peptide alpha-amidation. Annu. Rev. Neurosci. 15, 57–85 (1992).
Lewis, J. E., Brameld, J. M. & Jethwa, P. H. Neuroendocrine role for VGF. Front. Endocrinol. 6, 3 (2015).
Stephens, S. B. et al. The prohormone VGF regulates β cell function via insulin secretory granule biogenesis. Cell Rep. 20, 2480–2489 (2017).
Ferri, G. L., Levi, A. & Possenti, R. A novel neuroendocrine gene product: selective VGF8a gene expression and immuno-localisation of the VGF protein in endocrine and neuronal populations. Brain Res. Mol. Brain Res. 13, 139–143 (1992).
Canu, N., Possenti, R., Ricco, A. S., Rocchi, M. & Levi, A. Cloning, structural organization analysis, and chromosomal assignment of the human gene for the neurosecretory protein VGF. Genomics 45, 443–446 (1997).
Dalbøge, L. S. et al. Evaluation of VGF peptides as potential anti-obesity candidates in pre-clinical animal models. Peptides 136, 170444 (2021).
Namkoong, C. et al. NERP-2 regulates gastric acid secretion and gastric emptying via the orexin pathway. Biochem. Biophys. Res. Commun. 485, 409–413 (2017).
Stephens, S. B. et al. A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta-cell survival and function. Cell Metab. 16, 33–43 (2012).
Yamaguchi, H. et al. Peptidomic identification and biological validation of neuroendocrine regulatory peptide-1 and −2. J. Biol. Chem. 282, 26354–26360 (2007).
Iwasaki, M. et al. Establishment of new clonal pancreatic β-cell lines (MIN6-K) useful for study of incretin/cyclic adenosine monophosphate signaling. J. Diabetes Investig. 1, 137–142 (2010).
Hashim, M. et al. Inhibition of SNAT5 induces incretin-responsive state from incretin-unresponsive state in pancreatic β-cells: study of β-cell spheroid clusters as a model. Diabetes 67, 1795–1806 (2018).
Han, G. et al. Glutamate is an essential mediator in glutamine-amplified insulin secretion. J. Diabetes Investig. 12, 920–930 (2021).
Hahm, S. et al. Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron 23, 537–548 (1999).
Ye, R., Onodera, T. & Scherer, P. E. Lipotoxicity and β cell maintenance in obesity and type 2 diabetes. J. Endocr. Soc. 3, 617–631 (2019).
Müller, T. D. et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130 (2019).
Ulasov, A. V., Rosenkranz, A. A., Georgiev, G. P. & Sobolev, A. S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 291, 120111 (2022).
Wiederkehr, A. & Wollheim, C. B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell. Endocrinol. 353, 128–137 (2012).
Kaufman, B. A., Li, C. & Soleimanpour, S. A. Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol. Asp. Med. 42, 91–104 (2015).
Hasnain, S. Z. et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 20, 1417–1426 (2014).
Twig, G. et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008).
Anello, M. et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48, 282–289 (2005).
Frei, A. P. et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nat. Biotechnol. 30, 997–1001 (2012).
Canton, J., Neculai, D. & Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13, 621–634 (2013).
Mackenzie, B. & Erickson, J. D. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflug. Arch. 447, 784–795 (2004).
Prentki, M. & Renold, A. E. Neutral amino acid transport in isolated rat pancreatic islets. J. Biol. Chem. 258, 14239–14244 (1983).
Zielińska, M., Dąbrowska, K., Hadera, M. G., Sonnewald, U. & Albrecht, J. System N transporters are critical for glutamine release and modulate metabolic fluxes of glucose and acetate in cultured cortical astrocytes: changes induced by ammonia. J. Neurochem. 136, 329–338 (2016).
Bröer, A. et al. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 73, 2184–2194 (1999).
Kashiwagi, H., Yamazaki, K., Takekuma, Y., Ganapathy, V. & Sugawara, M. Regulatory mechanisms of SNAT2, an amino acid transporter, in L6 rat skeletal muscle cells by insulin, osmotic shock and amino acid deprivation. Amino Acids 36, 219–230 (2009).
Helle, K. B. The granin family of uniquely acidic proteins of the diffuse neuroendocrine system: comparative and functional aspects. Biol. Rev. 79, 769–794 (2004).
Moin, A. S. et al. Neuroendocrine regulatory peptide-2 stimulates glucose-induced insulin secretion in vivo and in vitro. Biochem. Biophys. Res. Commun. 428, 512–517 (2012).
Matsuo, T. et al. Localization of neuroendocrine regulatory peptide-1 and-2 in human tissues. Regul. Pept. 163, 43–48 (2010).
Petrocchi-Passeri, P. et al. The VGF-derived peptide TLQP-62 modulates insulin secretion and glucose homeostasis. J. Mol. Endocrinol. 54, 227–239 (2015).
Hannedouche, S. et al. Identification of the C3a receptor (C3AR1) as the target of the VGF-derived peptide TLQP-21 in rodent cells. J. Biol. Chem. 288, 27434–27443 (2013).
Cero, C. et al. The neuropeptide TLQP-21 opposes obesity via C3aR1-mediated enhancement of adrenergic-induced lipolysis. Mol. Metab. 6, 148–158 (2017).
Hirakida, H. et al. VGF nerve growth factor inducible has the potential to protect pancreatic β-cells. J. Endocrinol. 257, e220267 (2023).
White, M. F. & Kahn, C. R. Insulin action at a molecular level − 100 years of progress. Mol. Metab. 52, 10130 (2021).
Ge, Y., Gu, Y., Wang, J. & Zhang, Z. Membrane topology of rat sodium-coupled neutral amino acid transporter 2 (SNAT2). Biochim. Biophys. Acta Biomembr. 1860, 1460–1469 (2018).
Kato, T. et al. Structural insights into inhibitory mechanism of human excitatory amino acid transporter EAAT2. Nat. Commun. 13, 4714 (2022).
Melancon, B. J. et al. Allosteric modulation of 7 transmembrane spanning receptors: theory, practice and opportunities for CNS drug discovery. J. Med. Chem. 55, 1445–1464 (2012).
Han, B., Salituro, F. G. & Blanco, M. J. Impact of allosteric modulation in drug discovery: innovation in emerging chemical modalities. ACS Med. Chem. Lett. 11, 1810–1819 (2020).
Conn, P. J., Christopolous, A. & Lindsley, C. W. Allosteric modulators of GPCRs as a novel approach to treatment of CNS disorders. Nat. Rev. Drug Discov. 8, 41–54 (2009).
Chen, C. J. et al. How do modulators affect the orthosteric and allosteric binding Pockets? ACS Chem. Neurosci. 13, 959–977 (2022).
Lin, C. I. et al. Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3-18. Nature 410, 84–88 (2001).
Garaeva, A. A. et al. Cryo-EM structure of the human neutral amino acid transporter ASCT2. Nat. Struct. Mol. Biol. 25, 515–521 (2018).
Yan, R., Zhao, X., Lei, J. & Zhou, Q. Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 568, 127–130 (2019).
Bröer, S. The SLC38 family of sodium-amino acid co-transporters. Pflug. Arch. 466, 155–172 (2014).
McClenaghan, N. H., Barnett, C. R. & Flatt, P. R. Na+ cotransport by metabolizable and nonmetabolizable amino acids stimulates a glucose-regulated insulin-secretory response. Biochem. Biophys. Res. Commun. 249, 299–303 (1998).
Turbitt, J. et al. NKCC transport mediates the insulinotropic effects of taurine and other small neutral amino acids. Life Sci. 316, 121402 (2023).
Chaudhry, F. A. et al. Molecular analysis of system N suggests novel physiological roles in nitrogen metabolism and synaptic transmission. Cell 99, 769–780 (1999).
Hyde, R., Cwiklinski, E. L., MacAulay, K., Taylor, P. M. & Hundal, H. S. Distinct sensor pathways in the hierarchical control of SNAT2, a putative amino acid transceptor, by amino acid availability. J. Biol. Chem. 282, 19788–19798 (2007).
Hoffmann, T. M. et al. Effects of Sodium and Amino Acid substrate availability upon the expression and stability of the SNAT2 (SLC38A2) amino acid transporter. Front. Pharmacol. 9, 63 (2018).
Pinilla, J. et al. SNAT2 transceptor signalling via mTOR: a role in cell growth and proliferation? Front. Biosci. 3, 1289–1299 (2011).
Carlessi, R. et al. Glutamine deprivation induces metabolic adaptations associated with beta cell dysfunction and exacerbate lipotoxicity. Mol. Cell Endocrinol. 491, 110433 (2019).
Curi, R. et al. Molecular mechanisms of glutamine action. J. Cell Physiol. 204, 392–401 (2005).
Vettore, L. A., Westbrook, R. L. & Tennant, D. A. Proline metabolism and redox; maintaining a balance in health and disease. Amino Acids 53, 1779–1788 (2021).
Porte, D. Jr. & Kahn, S. E. β-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 50, S160–S163 (2001).
Supale, S., Li, N., Brun, T. & Maechler, P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol. Metab. 23, 477–487 (2012).
Khan, D., Moffett, R. C., Flatt, P. R. & Tarasov, A. I. Classical and non-classical islet peptides in the control of β-cell function. Peptides 150, 170715 (2022).
Ly, L. D. et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 49, e291 (2017).
Ciregia, F. et al. Palmitate-induced lipotoxicity alters acetylation of multiple proteins in clonal β cells and human pancreatic islets. Sci. Rep. 7, 13445 (2017).
Vantyghem, M. C. et al. Ten-year outcome of islet alone or islet after kidney transplantation in type 1 diabetes: a prospective parallel-arm cohort study. Diabetes Care 42, 2042–2049 (2019).
Zhang, W. et al. Neuromedin U suppresses insulin secretion by triggering mitochondrial dysfunction and endoplasmic reticulum stress in pancreatic β-cells. FASEB J. 34, 133–147 (2020).
Wang, Q. P., Guan, J. L. & Shioda, S. Immunoelectron microscopic study of beta-endorphinergic synaptic innervation of GABAergic neurons in the dorsal raphe nucleus. Synapse 42, 234–241 (2001).
Zhang, W. et al. Neuromedin U uses Gαi2 and Gαo to suppress glucose-stimulated Ca2+ signaling and insulin secretion in pancreatic β cells. PLoS One 16, e0250232 (2021).
Misu, H. et al. Deficiency of the hepatokine selenoprotein P increases responsiveness to exercise in mice through upregulation of reactive oxygen species and AMP-activated protein kinase in muscle. Nat. Med. 23, 508–516 (2017).
Kristinsson, H. et al. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 7, 4657 (2017).
Acknowledgements
The authors thank Mikiya Miyazato and Kenji Mori (National Cerebral and Cardiovascular Center Research Institute) for suggesting the useful methods employed in this study. The authors thank Stephen R.J. Salton (Mount Sinai School of Medicine, NY) for providing Vgf−/− mice, and Junichi Miyazaki (Osaka University) for providing MIN6-K8 cells. The authors thank Yoshiteru Goto, Itsuki Morinaga, and Eiko Kurata (University of Miyazaki) for their technical support. Part of this work was performed at the Frontier Science Research Centre, University of Miyazaki. This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI (25293216, and 15K09439) and the Agency for Medical Research and Development–Core Research for Evolutional Science and Technology (AMED-CREST, 19gm0610016h0006) (to M.N.).
Author information
Authors and Affiliations
Contributions
W.Z., A.Miura., A.Moin., H.S., N.Minamino. and M.Nakazato. designed the study; W.Z., A.Miura., A.Moin., K.Shimizu., Y.M., R.T., S.Hirako., S.S., C.J., Y.K., K.Sasaki., N.Minamino., V.G., J.K.C. and F.P. conducted the experiments and analysed the data; W.Z., A.Miura., H.S., and M.Nakazato. wrote the manuscript. All authors commented on and approved the final version of the manuscript. M.Nakazato. is the guarantor of this work, has full access to all the data in the study, and takes responsibility for the integrity and accuracy of the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Alessandro Bartolomucci and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Miura, A., Abu Saleh, M.M. et al. The NERP-4–SNAT2 axis regulates pancreatic β-cell maintenance and function. Nat Commun 14, 8158 (2023). https://doi.org/10.1038/s41467-023-43976-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-43976-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
