
Abstract
African ancestry is a significant risk factor for prostate cancer and advanced disease. Yet, genetic studies have largely been conducted outside the context of Sub-Saharan Africa, identifying 278 common risk variants contributing to a multiethnic polygenic risk score, with rare variants focused on a panel of roughly 20 pathogenic genes. Based on this knowledge, we are unable to determine polygenic risk or differentiate prostate cancer status interrogating whole genome data for 113 Black South African men. To further assess for potentially functional common and rare variant associations, here we interrogate 247,780 exomic variants for 798 Black South African men using a case versus control or aggressive versus non-aggressive study design. Notable genes of interest include HCP5, RFX6 and H3C1 for risk, and MKI67 and KLF5 for aggressive disease. Our study highlights the need for further inclusion across the African diaspora to establish African-relevant risk models aimed at reducing prostate cancer health disparities.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is characterised by substantial ancestral disparity1, which together with significant heritability2, suggests an inherited genetic contribution. Specifically, men of African ancestry are at greatest risk, with African Americans 1.7 times more likely to receive a diagnosis and 2.1 times more likely to die from PCa than White American men3. Within Sub-Saharan Africa, mortality rates are up to 3.2 times greater than global estimates4. For southern Africa, PCa incidence rates largely reflect that reported for Northern America, however, mortality rates are 2.7-fold greater (age-standardised mortality rate of 22 per 100,000 men)4. Previously, we (and others) have demonstrated that southern Africa is not only home to genetically diverse populations5, but that Black South African men are at 2.1-fold increased risk for advanced PCa at presentation compared with African Americans (adjusted for age)6. Despite this, there is a notable lack of genetic data for populations across Africa.
As of 8 February 2023, the National Human Genome Research Institute-European Bioinformatics Institute (NHGRI-EBI) genome-wide association study (GWAS) Diversity Monitor reports that while Europeans contribute 95.85% to global GWAS, African Americans or Afro-Caribbeans contribute 0.49%, and Sub-Saharan Africans only 0.15%7. PCa GWAS research for Sub-Saharan Africa is no different, with further limitations including a focus on specific variants and study power8,9. As outlined in Fig. 1A, studies have been restricted to Ghana (474 cases, 458 controls; 2,837,019 variants)10, Uganda (560 cases, 480 controls; 118 known PCa risk loci and 17,125,421 imputed variants)11, South Africa (552 cases, 315 controls; 46 known PCa risk alleles)12, and a single study (MADCaP) that merged data from Ghana, Nigeria, and South Africa (399 cases, 403 controls; >1.5 million custom markers)13. Furthermore, studies associating rare variants with PCa pathogenesis within Sub-Saharan Africa are equally scarce. While a single study from Uganda associated pathogenic BRCA2, ATM, PALB2 and NBN variants with aggressive prostate disease14, pathogenic variants for BRCA2, ATM, CHEK2, and TP53, and rare early-onset/familial oncogenic variants of unknown pathogenicity for BRCA2, FANCA and RAD51C, were linked to advanced disease in Black South African men15. The latter study highlights a maximum 30% utility for current European-biased PCa germline testing gene panels for men from southern Africa.

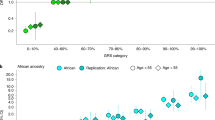
A Map showing populations across Africa where GWAS studies have been conducted for prostate cancer, and sample locations and sizes of African ancestry from the Human Genome Diversity Project (HGDP) and 1000 Genomes Project (1KGP) subset of gnomAD v3.1267 (red circles). Mortality rates of prostate cancer from GLOBOCAN 2020 are shown in the bottom left bar plot, with the population size of men in the region indicated in brackets below the region name4. Study references: aCook et al.10, bHarlemon et al.13, cTindall et al.12, dDu et al.11. B Admixture plot (K = 5, cross-validation error = 0.162) which was replicated in 10 out of 10 runs, including 1003 Africans, 20 Europeans, 20 Chinese individuals from the HGDP and 1KGP subset of gnomAD with our dataset of 781 South Africans.
Most recently, the largest meta-analysis of African ancestral PCa GWAS included samples from the Ghana and Uganda, with additional representation from the Democratic Republic of Congo (3,149 cases, 2,547 controls recruited from Africa out of a total of 19,378 cases and 61,620 controls)16. The study increased the number of known PCa risk alleles from 269 to 278, and combined to generate a multi-ancestry polygenic risk score (PRS)16,17. Demonstrating effective PCa risk stratification for men of African ancestry, including MADCaP validation, African men in the top PRS decile were further distinguished by aggressive disease. Notably and of relevance to this study, of the nine recently identified African-specific/predominant PCa risk variants identified, seven occurred in gene regions, including a protein truncating variant in the prostate-specific gene anoctamin 7 (ANO7 Ser914Ter), adding a third functionally relevant protein coding variant to the repertoire of known African-specific PCa risk alleles, including previously identified CHEK2 Ile448Ser18 and HOXB13 Ter285Lys19. Recognising that only 7.4% of the meta-analysis included men from Sub-Saharan Africa, which in turn represents only a fraction of the region with vast ethno-linguistic and genetic diversity20, the authors call for further studies across the African diaspora16.
Placing the Black South African population into global perspective, in this work we use data from 113 whole-genome sequenced men with predominantly high-risk PCa (HRPCa)21 to evaluate the use of polygenic scoring based on 278 known common risk PCa variants16.
To further assess for potentially functional variants associated with PCa and aggressive disease in 798 Black South African men, we conduct an exome-wide association study (EWAS; 247,870 variants), including both common variant (minor allele frequency (MAF) > 0.01) and gene-based rare variant (MAF ≤ 0.01) analyses.
Results
Known risk alleles in Black South Africans with PCa
Published whole genome sequenced data generated for 113 Black South African PCa cases (average 45.4X coverage), filtered to represent no more than 8% non-African genetic ancestral contribution (Fig. 2A)21, were interrogated for known risk variants and previously associated variants from African GWAS. This included the recently reported set of 278 PCa risk variants16, and the top associated variants from the Ugandan11 and Ghanaian GWAS10. Biased towards HRPCa at presentation, defined as International Society of Urological Pathology (ISUP) grade group ≥3 (Gleason score ≥4 + 3, n = 87, 76.99%), the average age of the cohort was 66.9 years (standard deviation (SD) 8.43, range 45–99), with prostate specific antigen (PSA) levels highly elevated (33.63% PSA ≥ 100 ng/ml), as previously observed within the region6 (Supplementary Data 1). Of the 278 risk variants, 18 (6.47%) were absent, 21 (9.09%) fixed in our Southern African (SA) cohort, and eight were excluded from scoring due to uncertain risk variants (Supplementary Data 2). When compared to previously published risk allele frequencies (RAF) for African-ancestral (AA) controls16, 11 showed differences in RAF > 0.15, of which eight were more common in our SA population and three were more common in the published largely US-derived AA data (Fig. 3, Supplementary Data 2). The largest differences included rs111595856 (INHBB, RAFSA = 0.67, RAFAA = 0.398), rs35159226 (ZNF322, RAFSA = 0.562, RAFAA = 0.308), and rs8005621 (SALRNA1, RAFSA = 0.549, RAFAA = 0.348). Among the top 136 associated variants in the Ugandan GWAS, three variants showed differences in RAF > 0.15 compared to Ugandan cases and controls, rs6431219 (BIN1, RAFSA = 0.416, RAFUGPCS_Cases = 0.6, RAFUGPCS_Controls = 0.51), rs61005944 (ENSG00000237101, RAFSA = 0.527, RAFUGPCS_Cases = 0.31, RAFUGPCS_Controls = 0.22) and rs140698498 (RBFOX1, RAFSA = 1, RAFUGPCS_Cases = 0.87, RAFUGPCS_Controls = 0.8) (Supplementary Fig. 1). A total of 19 variants were more common in Ugandan controls than in our SA population (difference in RAF ranged from 0.001 to 0.094, Supplementary Data 3). Among the top 30 associated variants in the Ghanaian GWAS, only one had a difference in RAF > 0.15 (rs28747043, closest gene MTCO3P1 18.4 kb away, RAFSA = 0.181, RAFGhana_Controls = 0.371; Supplementary Fig. 2). A total of 19 of these variants were more common in Ghanaian controls than our SA cases (difference in RAF ranged from 0.0003 to 0.19; Supplementary Data 4).

A Summary of the data used for polygenic risk scoring (PRS), (B) sample filtering for the genotype data, (C) variant filtering for the exome wide association studies (EWAS), and (D) rare variant filtering for the gene-based analyses.

Comparison of the RAF for 267 out of 278 known risk variants between South African prostate cancer cases (N = 113) and African Ancestry controls (N = 61,620) as previously reported16. Gene labels in white boxes indicate variants that overlap a gene, while gene labels in grey indicate the closest genes to the variant. Note four risk variants had no risk allele frequency reported in the African Ancestry PCa cases16 and another seven variants in the South African PCa cases were excluded from the plot due to being indel repeats or having unclear risk variants.
Polygenic risk scores (PRS) in Black South Africans
The PRS was evaluated using two definitions of aggressive PCa, firstly Chen et al., 2023’s definition: ISUP 4-5 or PSA ≥ 20 ng/ml, which grouped our samples into N = 101 aggressive and N = 11 non-aggressive (one sample with missing PSA and ISUP excluded); and our definition: ISUP 3-5, grouping our samples into N = 87 aggressive and N = 18 non-aggressive (eight samples with missing ISUP excluded) (Fig. 2). A total of 231 non-fixed variants (Supplementary Data 2) were used to score the SA population with PLINK v1.922 using African and multiethnic weights, as previously published16. Using Chen et al., 2023’s definition of aggressiveness, for African weights, for the aggressive group the mean score was 0.034 (SD = 0.002, range 0.029–0.039) while that of the non-aggressive group was 0.034 (SD = 0.002, range 0.032–0.037). For multiethnic weights, the aggressive group’s mean score was 0.041 (SD = 0.002, range 0.035–0.046), and non-aggressive was 0.041 (SD = 0.002, range 0.039–0.045). Using our definition of aggressiveness, for African weights, the aggressive group had a mean score of 0.034 (SD = 0.002, range 0.029–0.039), while the non-aggressive group had a mean score of 0.034 (SD = 0.002, range 0.032–0.037). With multiethnic weights, we observed for the aggressive group a mean 0.041 (SD = 0.02, range 0.035–0.046) and for the non-aggressive group a mean 0.041 (SD = 0.02, range 0.039–0.045) (Supplementary Figs. 3, 4).
No significant associations were detected using either the multiethnic study’s definition of aggressiveness16: African score OR per SD = 1.38, 95% CI = 0.72–2.66; multiethnic score OR per SD = 1.24, 95% CI = 0.65–2.34; nor using our definition of HRPCa: African score OR per SD = 1.01, 95% CI = 0.6–1.71; multiethnic score OR per SD = 1.13, 95% CI = 0.67–1.9.
Common risk variants associated with PCa in Black South Africans
A total of 798 Black South Africans were genotyped on the Infinium HumanExome-12 v1.0 BeadChip array (Illumina, California, United States), screening 247,870 variants. A total of 781 remained after sample quality control (QC) filtering (Fig. 2B). The dataset was assessed for regional ancestral clustering using the Human Genome Diversity Project (HGDP) and 1000 Genomes Project (1KGP) subset of gnomAD v3.1.2, including 1,003 African, 20 European, and 20 Chinese samples. Using principal component analysis (PCA, Supplementary Fig. 5) and ADMIXTURE v1.3.0 for K = 1 to 10 with five-fold cross-validation (CV) and 10 replications each (Supplementary Fig. 6), with K = 5 generating the lowest CV error at 0.162 (Supplementary Fig. 7), we assessed for within-population substructure. Excluding for a single patient that clustered with Nigerian Yoruba and Esan (West African) populations, we confirm that our cohort represents a distinct southern African genetic ancestry (Fig. 1B). Recruited from Southern African Prostate Cancer Study (SAPCS) presentative urology clinics within South Africa, cases were defined as presenting with clinicopathologically confirmed PCa (N = 451) and controls with no histopathological evidence of cancer (i.e. no Gleason score, N = 292), with relatively even distribution across the age-representation (average 70.52, range 49–102 vs average 69.99, range 45-99, respectively). Further clinical characteristics are summarised in Supplementary Data 5. For exome-wide association analysis (EWAS) and gene-based analysis, a total of 37 men with unknown age at diagnosis were excluded, leaving 743 men.
After variant QC, 50,591 common variants (MAF > 0.01) remained for further case-control EWAS (Fig. 2C), with no variants reaching genome-wide significance (q < 0.05) (Fig. 4A). The QQ plot and genomic inflation factor (λ = 1.06) from the P-values of the EWAS indicated no population stratification in the data (Supplementary Fig. 8). Of the 17 SNPs with a P-value of <5E−04 (Table 1), six (35.3%) were in Chromosome 6 including two intronic variants in the HLA-complex P5 (HCP5) lncRNA gene (rs2244839, rs12660382), two within or close to MUC22 (rs1634718, rs1634725), and one each in LINC00243 (rs1264362) and RFX6 (rs339331). The RFX6 variant rs339331 is a known PCa variant (Fig. 4A), although not included in the set of 278 risk variants, was found to be in strong linkage (r2 = 0.95) with GPRC6A rs2274911 (Supplementary Fig. 9). The top-ranked SNPs included rs2244839 (P = 3.4E−05; OR 1.63, 95% CI:1.29–2.05) in HCP5, rs11009235 (P = 6.26E−05; OR 1.6, 95% CI:1.27–2.01) located 15.8 kb upstream and 44.9 kb downstream from IATPR and NRP1, respectively, and rs3865188 (P = 1.03E−04; OR 1.58, 95% CI: 1.25–1.99) 9.9 kb upstream of CDH13. Other noteworthy associated variants included rs7963300 (P = 1.43E−04; OR 2.15, 95% CI: 1.45−3.19), located within an unknown gene ENSG00000286069 approximately 74.7 kb upstream from a HOXC gene cluster (Supplementary Fig. 10), and the nonsynonymous rs114057260 (P = 4.18E−04; OR 0.34, 95% CI 0.19−0.62) in ZZEF1 which is the only predicted deleterious variant (PDV), defined as variants predicted to be deleterious by SIFT and damaging (or possibly damaging) by PolyPhen.

A Manhattan plot of -log10 p-values from an age-adjusted logistic regression, with 451 cases and 292 controls. B Manhattan plot of -log10 p-values from an age-adjusted logistic regression for high grade prostate cancer (ISUP 3-5) cases (N = 203) against low risk or no PCa (N = 540). Known risk variants (N = 9) from the recently described set of 278 variants16 are shown as red circles, and known cancer variants as summarised previously13 are shown as orange circles, while variants in both datasets are represented in red triangles. Variants labelled in a white box indicate the overlapping gene, while those labelled in grey are the closest genes.
Only 17 of the 278 known PCa risk variants16 were captured by the exome array data (Supplementary Data 2), with three SNPs found to be fixed for the risk allele (rs77482050, rs33984059, rs61752561), an additional two almost fixed (rs138708, rs17804499), two were fixed for the reference allele (rs77559646, rs74911261), and one rare (MAF < 0.01 rs76832527) in our SA study population. None of the nine remaining SNPs (MAF 0.015 to 0.49) showed risk association (all P > 0.25) (Fig. 4A). A total of 367 out of 2477 known cancer variants, summarised previously13, were genotyped in the exome array (Supplementary Data 6). Among these, the top associated variants included the RFX6 SNP rs339331 (P = 0.0002), intergenic variant rs9600079 (pseudogene RNU4-10P 37.5 kb downstream, closest protein-coding gene is KLF5 76 kb upstream, P = 0.0014), and CASC8/PCAT1 variant rs445114 (P = 0.0047).
Common risk variants associated with HRPCa in Black South Africans
Further classification of our study cohort as HRPCa (ISUP \(\ge\)3, N = 203) versus low-risk or no PCa (N = 461), again showed no genome-wide significance, while 25 SNPs had P < 5E−04 (Fig. 4B, Supplementary Fig. 11), of which three were among the top ranking PCa risk EWAS SNPs (rs11009235, rs2897495, rs7963300). Several of the top SNPs are in genes associated with PCa or PCa processes, including MKI67 (rs8473), PCSK6 (rs80278342), ABCB6 (rs60322991), and TCHP (rs11068997); or other cancer-associated processes, including GGA2 (rs1135045), H1-5 (rs11970638), and COL15A1 (rs2075662). The rs8473 SNP in MKI67 was strongly linked (r2 = 0.92) to rs1063535 in the same gene, and moderately linked (r2 = 0.4 to 0.64) to rs34750407, rs11016071, rs10082391, rs1050767, rs12777740, rs11016076, and rs7095325 (Supplementary Fig. 12). SNPs rs60322991 (ABCB6), rs877834 (NPVF), rs2075662 (COL15A1), and rs77944357 (ABCA10) have deleterious and potentially damaging effects based on SIFT and/or PolyPhen yet are benign or are lacking prediction by ClinVar (Table 2). The nine SNPs included in the 278 PCa risk allele panel showed non-significance (all P > 0.11), while the top associated known cancer variants were rs1859962 in CASC17 (P = 0.003; OR 1.49, 95% CI: 1.142–1.93) and rs3734805 in CCDC170 (P = 0.004; OR 2.0, 95% CI: 1.244–3.2) (Fig. 4B).
Genes associated with PCa and HRPCa in Black South African men
Derived from more than 12,000 European-biased sequenced genomes, the exomic array was designed with a focus on protein altering (nonsynonymous, slicing and nonsense) variants. These were selected based on a minimum of three observations across two or more datasets, and as such many rare variants have been included, allowing for further gene-based analyses (Fig. 2D). After improving rare variant genotype calls using zCall v3.423, gene-based analyses were performed using optimal unified sequence kernel association test (SKAT-O). SKAT-O adjusts for small sample size and retains power when variants in a region are causal and the effect is in a single direction (through burden tests) as well as when variants in a region have bidirectional effects and may contain noncausal variants (through SKAT)24.
Using this method, genes with rare variants associated with PCa risk (family-wise error rate (FWER) < 0.05; Supplementary Fig. 13, Supplementary Data 7) included H3C1 (rs199943654, P = 9.91e−05), MBP (rs61742941, P = 1.23e−04), and MTG1 (3 variants including predicted deleterious variant (PDV) rs138851534, P = 1.59e−04). Although no significance was observed for HRPCa (Supplementary Fig. 14), the top associated gene was EPS15 (3 rare variants, P = 1.47e−04). Through further analyses for common and rare variants (Supplementary Fig. 15, Supplementary Data 8), we show MBP to be significantly associated with PCa risk (1 rare and 5 common variants, P = 1.26e−04) and included the rare PDV rs61742941. For the HRPCa common and rare variants analysis (Supplementary Fig. 16, Supplementary Data 9), we found KLF5 to be significantly associated with aggressive disease (1 rare variant, 3 common variants, P = 1.52e−04), including the common PDV rs115503899.
PCa variance explained by exome SNPs
GREML was used to estimate the phenotypic variance explained by genetic variance (SNP heritability). The 49,534 common autosomal SNPs (post-QC) in GREML explained 48.19% (standard error (SE) 22.2%, P = 4.77E−03) of disease liability for PCa, and only 17.78% (SE 8.19%) when transformed for PCa prevalence of 0.001 (Table 3). Common and rare autosomal SNPs together (N = 80,421) explained 50.7% (SE 25.13%, P = 0.014) of disease liability for PCa and at a prevalence of 0.001, explained 18.7% (SE 9.27%).
Using the top 16 SNPs from the EWAS with all cases and controls, only 5.07% (SE 1.78%, P = 1.59E−25) of disease liability was explained. When stratifying the cohort by HRPCa versus low-risk/no-PCa at an estimated prevalence of 0.0004 for HRPCa, all 49,534 autosomal SNPs explained 16.15% (SE 11.17%, P = 0.065) of disease liability, 9.24% (SE 2.31%, P = 2.3E−44) of which could be explained using the top 25 SNPs from the HRPCa EWAS.
Discussion
In this study, motivated by limited studies having identified three African-specific protein-altering risk alleles19,25, we examined PCa risk and aggressive disease associations and have provided much needed evaluation of known PCa risk alleles within the under-represented region of southern Africa. While dwarfed in sample size compared to European-ancestral or African American GWAS studies, this study highlights significant resources and efforts needed to elucidate the genetic contribution to ancestrally-driven PCa health disparities across the African diaspora.
When examining the previously reported allele frequencies of the 278 known risk variants16 in our population, large differences in RAF (>0.15) were observed for 11 variants, including those in the genes INHBB (rs111595856), ZNF322 (rs35159226), SALRNA1 (rs8005621), FGF10 (rs1482675), HNF1B (rs11263763), PCAT19 (rs11673591), and TAB3 (rs5972255). These differences could mean that alternate oncogenic pathways or epigenetic regulation are at play in southern Africa. Importantly, although we were restricted in sample size and biased to aggressive PCa, we were unable to replicate previous findings of the multiethnic PRS association with aggressive disease16, nor with our definition of HRPCa. Notably, the variant rs72725854, which is the most strongly associated risk variant for PCa in men of African ancestry with an allele frequency of 6.1%26, was present at a frequency of 13.7% in our case-only South African population.
In the classic case-control PCa EWAS analysis, the top associations included variants in HCP5 and RFX6, and near IATPR and NRP1. The histocompatibility leucocyte antigen (HLA) complex P5 (HCP5) is a long non-coding RNA located in the HLA class I region and has shown aberrant expression in multiple cancers, including PCa27. A single study investigating HCP5 in PCa using tissues and cell lines, found high expression of HCP5 to be positively correlated to prostate tumour metastasis28. Expression of HCP5 acted as a sponge for miR-4656, preventing miR-4656 from suppressing the cell migration-inducing hyaluronidase 1 (CEMIP) gene, leading to upregulated expression of CEMIP, which plays a key role in tumour proliferation28,29. Functional experiments will be needed to explore whether the two associated variants from this study affect expression levels of HCP5. Conversely, the variant located between the lncRNA IATPR and NRP1 provides a potential proxy for a yet unknown gene-associated variant, Notably, IATPR has been found to promote cell migration and development in other cancers30,31, while NRP1 is an androgen-repressed gene that plays a role in cancer progression with its expression associated with prostate tumour grade and biochemical recurrence32.
Intriguingly, the member of the regulatory factor X family of transcription factors RFX6 gene variant rs339331 associated with a decreased PCa risk (C-allele OR 0.64, 95% CI:0.5–0.82, P = 4.6E−04) was in strong linkage with GPRC6A rs2274911 (r2 = 0.95). The rs339331 association replicates findings from previous studies of PCa in Ghanaian, Japanese, and Chinese men10,33,34. Conversely, the T-allele has been shown to increase HOXB13 binding to a transcriptional enhancer, which upregulates the expression of RFX6 associated with tumour progression, metastasis and biochemical relapse35, as well as upregulating GPRC6A expression36. Additionally, the A-allele at rs2274911 has been associated with increased PSA levels37. Representing the major allele in the current study (MAF = 0.7941) could potentially contribute (at least in part) to elevated PSA levels observed for Black South African men, irrespective of PCa status6. Notably, the intron variant rs339351 in the RFX6 gene is among the 278 known PCa risk variants16, differing in frequency by 0.039 (RAFSA = 0.783, RAFAA = 0.744). As noted for other PCa risk variants, such as European-specific HOXB13 p.Ile448Ser (rs138213197)38 and African-specific HOXB13 p.Ter285Lys (rs77179853)19,39, it is possible that different ancestral-specific causative variants may represent the same PCa gene.
In the HRPCa EWAS, the top associated variants were in several genes relevant to PCa, including MKI67, PCSK6, ABCB6, and TCHP. MKI67 encodes the Ki67 protein, a widely used diagnostic marker of proliferation in numerous human cancers, with increased expression associated with poor prognosis in localised PCa40. One study reported low PSA and high Ki67 expression in patients with HRPCa and TMPRSS2-ERG fusion gene, whereas high PSA and low Ki67 expression predominated in patients with low-risk disease and favourable outcomes41. Associating the T-allele of MKI67 rs8473 with reduced odds for HRPCa (P = 3.09E-04, OR = 0.64, 95% CI:0.5–0.82), warrants further investigation into the potential of this allele to reduce Ki67 expression. The PCSK6 variant rs80278342 is not regarded as a PDV in this study, but an isoform of PCSK6 has previously been identified as a plasma biomarker for PCa, and expression levels have been correlated with ERG tumour status and ISUP grade group42. ABCB6 codes for an ABC transporter, with expression levels linked to chemoresistance43. While increased ABCB6 expression has been reported in PCa, deregulated expression has been further associated with recurrent versus non-recurrent disease44. Although the correlation between ABCB6 expression levels and PCa grade has not yet been examined, increased ABCB6 has been associated with histological grade in gliomas45. The PDV rs11068997, associated with HRPCa in our study, is located within the tumour suppressor gene TCHP, shown to inhibit cell growth in PCa cells46. Other notable SNPs associated with HRPCa in our study and in genes representing cancer-associated processes include; GGA2 involved in cell growth47, H1-5 with transcriptional regulatory effects48, and COL15A1 shown to have tumour suppressive effects49. Lastly, the rs7963300 associated SNP upstream of a HOXC gene cluster, is of note as increased expression of HOX genes have been observed in prostate tumours50.
Since the distribution of several disease-associated alleles across a range of African populations has previously shown large variation51, it is plausible that, on top of potentially different oncogenic pathways or epigenetic regulation, germline PCa risks may also differ between regions within Sub-Saharan Africa. African American bias and within continental representation limited to a snapshot of largely west and east African ancestral diversity10,11,16,17 provides an explanation for the limited replication of previous GWAS findings in our SA-focused study. This is exemplified in the HOXB13 risk variant (rs77179853) found in men of West African ancestry being absent in Uganda and South Africa, and possibly arising after the Bantu migration from western to eastern and southern Africa around 1500–4600 years ago19. This variant was also absent in our cohort, along with another recently identified African-specific variant CHEK2 (rs17886163)18. Conversely, the ANO7 variant (rs60985508)18 was present (RAFSA = 0.363)52. Although further investigations are needed, this study highlights some avenues of interest for future germline studies of PCa across southern Africa.
The gene-based analyses showed association between the genes H3C1, MBP, and MTG1 with PCa, and KLF5 with HRPCa. Modifications to the H3 histone plays a key role in epigenetic regulation, and their relevance to PCa and treatment options have been reviewed elsewhere53,54. In this study, significant association of H3C1 to PCa suggests that differences in transcription may exist, and since the variant was only present in controls it may suggest a protective effect against PCa. The MBP gene encodes the myelin basic protein, which is a constituent of the myelin sheath, and has no obvious role in PCa, however, mouse model studies have shown that neural progenitor cells can invade prostate tumours, triggering neurogenesis and promoting tumour growth and metastasis55. The MTG1 gene is involved in the regulation of mitochondrial translation56 and although it has no known direct role in PCa, mutations in mitochondria have been associated with PCa aggression57, including in Black South African men58. Conversely, KLF5 is a transcription factor that has been implicated in several cancers with opposing roles (tumour suppressor or oncogenic driver) depending on the context59. Expression levels and post-translational modifications of KLF5, specifically acetylation, are of key interest in PCa with therapeutic implications for chemoresistance60,61,62,63,64. While there are no PCa risk variants within KLF5, two risk SNPs in close proximity include snRNA rs7489409 (65 kb downstream) and lncRNA rs7336001 (344 kb downstream)16. A known cancer variant rs9600079 approximately 76 kb downstream from KLF5 showed slight association (P = 0.0017). Located at chromosome 13q22.1, this region is frequently deleted in PCa65. Although earlier PCa cell line and xenograft research found that mutations were rare, deletions and down-regulation of KLF5 was frequent66. In our recent study genome profiling SA derived prostate tumours, we describe a molecular taxonomy we call global mutational subtypes (GMSs), identifying a single KLF5 African-specific predicted cancer driver mutation21.
Acknowledging our limited sample size, in turn it cannot be ignored that our EWAS is not only comparable to the previous Ghanian and Uganda PCa GWAS10, but it provides an as yet unmet regionally focused SA alternative for both risk allele validation and discovery across Sub-Saharan Africa (Fig. 1A). While the HGDP and 1KGP subset from the gnomAD v3.1.2 dataset represents limited (20 genomes) representation across SA67 (Fig. 1A), we acknowledge and appreciate that through projects like H3Africa (www.h3africa.org)68 additional resources are becoming available, although the pace remains a fraction of the global effort. As with the exomic-array used in this study69, commercial genotyping arrays have been designed based on variant frequencies heavily skewed towards Europeans, with arrays from MADCaP and H3Africa tailored for African populations, with the MADCaP array specifically designed for PCa research13,68. Furthermore, GREML analyses showed that only 49.71% (±22.71%) of the variance in phenotype could be explained by the autosomal genomic variance, indicating that the genomic risk for PCa was not fully captured by the European-biased exome array. Finally, we appreciate that although we attempted to improve rare variant calls using the zCall23, the software can introduce false positives70 and as such, we call for caution when interpreting allele frequencies.
While we appreciate our limited sample size, we were unable to replicate previous findings of an association between multiethnic PRS to PCa aggression in our African population. Consequently, we call for further African-relevant whole-genome sequencing and genome-wide interrogation studies for establishing PRS of relevance for Sub-Saharan Africa. In our exome-wide association analyses, we identified several avenues of interest for further investigation, including HCP5, RFX6, and H3C1 for PCa, and MKI67 and KLF5 for HRPCa. Clearly, significant resources are necessary to elucidate the genomic variants contributing to ethnic disparity in PCa. The global inclusion of southern African data, a region with the most diverse human populations, will benefit not only the design of African-relevant cancer screening panels and further enhance ancestrally focused SNP arrays, but will also be important for accurate multiethnic PRS.
Methods
Ethics approvals and recruitment
Written informed consent was obtained from all participants, with study approval granted by the University of Pretoria Faculty of Health Sciences Research Ethics Committee (HREC #43/2010, with US Federal wide assurance FWA00002567 and IRB00002235 IORG0001762). Study participants were recruited at time of biopsy (diagnosis) from participating Southern African Prostate Cancer Study (SAPCS) urology clinics within the Gauteng and Limpopo Provinces of South Africa. The majority of men presented with a urological or associated complaint without a predetermined prostate specific antigen (PSA) test6. Men self-identifying ethno-linguistically, by two generations, as Black South Africans, where included in this study; firstly, irrespective of their PCa diagnosis to undergo whole exome genotyping (case-control study, N = 798), and secondly, selected for aggressive PCa at presentation and having undergone deep tumour/blood paired whole genome sequencing (N = 113), as recently published21. Genomic interrogation was performed under approval granted by the St. Vincent’s Sydney HREC (#SVH/15/227) and an executed material and data sharing agreement between the University of Pretoria in South Africa and University of Sydney in Australia. While data is shared, all data remains the property of the University of Pretoria, as chair of the SAPCS data sharing committee.
Interrogation of known PCa risk alleles in SAPCS
We interrogated population-matched whole genome sequenced data for the distribution of the 278 known risk variants16, and the top associated variants from the Uganda11 and Ghana10 GWAS within a cohort of men selected for bias towards aggressive PCa at presentation. Gene annotations were fetched through ANNOVAR from hg38 ensGene (GENCODE v43, last updated from UCSC 2023-02015)71. As recently described21, deep sequenced data (average 45.4X coverage) was generated for 113 Black South African PCa cases, which were filtered to include samples with no more than 8% non-African genetic ancestral contribution (Fig. 2A). A summary of clinical information is available in Supplementary Data 1.
We scored the South African cases via PLINK v1.922 using default settings based on their genotypes at 231 out of 278 available risk variants (Fig. 2A) using multiethnic and African ancestry weights16. Aggressiveness was defined as previously described (ISUP 4 or 5, or PSA ≥ 20 ng/ml)16, as well as using an alternative definition (ISUP 3-5), to use as the outcome variable in a logistic regression using the score (African or multiethnic) and age as covariates.
SAPCS EWAS data generation and genotype filtering
A total of 798 Black South Africans were genotyped on the Infinium HumanExome-12 v1.0 BeadChip array (Illumina, California, United States), assaying 247,870 variants (Fig. 2B–D). Genotypes were called using the Illumina GenomeStudio 2.0 software following previously published guides70,72. Briefly, non-pseudoautosomal variants with poor GenTrain scores were manually re-clustered based on visual inspection to improve the accuracy of genotype calls. No samples were removed as all sample call rates were >0.97.
Several quality control steps were conducted to prepare the dataset for exome-wide association analyses (EWAS) (Fig. 2C). Following manual re-clustering, variants that had a poor GenTrain score (<0.7) and poor call frequency (<0.95) were excluded (N = 3743). The remaining 244,127 variants were exported to PLINK format. The strand was converted using scripts by William Rayner from the Wellcome Centre for Human Genetics, Oxford website (https://www.well.ox.ac.uk/~wrayner/strand/), in the process removing two single nucleotide polymorphisms (SNPs) that did not reach the required 90% threshold for mapping to the genome. The variants coordinates were converted from hg18 to hg38 using UCSC’s webtool liftOver (http://genome.ucsc.edu/cgi-bin/hgLiftOver), and 43 variants not in hg38 were removed. Triallelic or duplicated variants were removed (N = 826). Among duplicated variants pairs, the variant with the higher call rate was kept. Heterozygous chromosome X SNPs (N = 38) were removed. Variants with MAF < 0.01 (N = 192,606; Supplementary Data 10) and 21 SNPs that failed the Hardy-Weinberg exact test (threshold 1E−6) were removed, leaving 50,591 variants.
SAPCS EWAS cohort characterisation
After excluding for a single patient presenting with prostate metastasis with squamous cell primary carcinoma (Fig. 2B), the remaining 797 SAPCS EWAS samples were checked for genetic duplicates. Identity-by-descent (IBD) was calculated using the –genome function in PLINK v1.922. Eight pairs of genetic duplicates were identified (PI_HAT ≥ 0.99), and all 16 individuals were removed from further analysis. The maximum PI_HAT in remaining pairs of individuals was 0.29, so no further samples were removed. To check for genetic admixture or non-African genetic fractions, the exome array variants were extracted from published African ancestral genomes (N = 1003) as well as 20 randomly selected individuals from European (CEU) and Han Chinese (CHB) populations each from the human genome diversity project (HGDP) and 1000 genome project (1KGP) subset of gnomAD v3.1.267. The extracted data was merged with the data in the current study and pruned for SNPs based on linkage disequilibrium (LD), using a 50 SNP window moving 5 SNPs at a time, at a variance inflation factor of 1.5 (--indep 50 5 1.5) in PLINK v1.922. The remaining 77,372 SNPs were then used for a principal component analysis (PCA) using PLINK v1.922 and plotted with ‘ggplot2’ in RStudio v4.1.173. While no individual showed substantial non-African genetic contribution, a single study participant clustering near the Nigerian Yoruba and Esan west African populations was excluded (Supplementary Fig. 5), leaving a total of 780 samples. The distribution of samples from this study and previous PCa GWAS studies in Africa10,11,12,13 were plotted on a map using the R package ‘rnaturalearth’74.
To further assess for African-specific ancestral fractions, an unsupervised ADMIXTURE v1.3.0 analysis75 was performed using the same dataset. The ADMIXTURE analysis was conducted for K = 1 to 10 with five-fold cross-validation (CV) and 10 replications each. The tool pong v1.576 was used to plot ancestry proportions with a greedy approach set to 0.95 (Supplementary Fig. 6). The K = 5 ADMIXTURE run produced the lowest cross-validation error at 0.16182 (Supplementary Fig. 7).
As age is a significant PCa risk factor, 37 samples were further excluded from the EWAS for lack of reported age at diagnosis, leaving a total of 451 clinicopathologically confirmed cases (70.52 years SD ± 9.21, range 49-102) and 292 controls either with or without benign prostate hyperplasia (69.99 years SD ± 9.07, range 45–99) (Supplementary Data 5). Cases were relatively evenly distributed across the ISUP grade grouping, representing high (ISUP 3 to 5, 54.58%) and low-risk PCa (ISUP 1 to 2, 45.43%). As previously observed for this study population6, PSA levels were significantly elevated, with 44.95% of cases presenting with PSA > 100 ng/ml, and only 20.85% of controls presenting with a PSA level less than the global standard for PCa diagnosis (4 ng/ml)77.
EWAS analysis and statistics
As the controls in this study were recruited based on a referral to a urologist clinic and were negative at histopathological examination of resected biopsy cores (on average 12 per patient), one cannot ignore the possibility of missed PCa diagnoses in these individuals. Therefore, in parallel to a classic case-control EWAS analysis (451 cases, 292 controls), we designed an additional EWAS focused on distinguishing HRPCa (ISUP 3-5, N = 203) versus low-risk or no PCa (LRPCa/NoPCa, N = 461). Cases that were missing an ISUP grading were excluded from this analysis.
RStudio v4.1.1 and PLINK v1.9 were used for analyses and visualisation22,78. A logistic regression using an additive genetic model accounting for age was used. A q-value false-discovery rate (FDR) cut-off of 0.05, calculated in R using the package ‘qvalue’79, was used to determine genome-wide significance. Manhattan, quantile-quantile (QQ), and regional association plots were generated using ‘ggplot2’ in RStudio73, with gene annotations for the canonical transcripts fetched through the R package ‘biomaRt’80 from Ensembl Human GRCh38.p13, version 108 and GENCODE v43 via UCSC81. African ancestry minor allele frequencies were fetched from gnomAD v3.1.267.
For the 17 out of 278 known PCa risk variants16, the three variants from the Ghana study10, and the previously summarised known cancer variants13 that were available in our exome array data, the risk allele frequency in cases and controls, age-adjusted odds ratio (OR), 95% confidence intervals (CI), and P-values were calculated in PLINK v1.922. For SNPs where the risk allele was the major allele in our population, the inverse odds ratio (and 95% CI) was calculated to reflect the odds of the risk allele in cases compared to controls. Three of the 17 known PCa risk SNPs were rare variants that were processed with zCall v3.423 (see below) to improve genotype calls. There were no changes to the allele frequencies of the three SNPs post-processing.
Gene-based analyses with rare variants
To improve the genotype calls of rare variants (minor allele frequency (MAF) < 0.01), zCall v3.423 was used with a default z value of 7 (global concordance=99.19%). Quality control followed that of EWAS filtering, including strand flipping, converting coordinates from hg18 to hg38, removing triallelic or duplicated variants, and removing heterozygous chromosome X variants (Fig. 2D). A total of 31,445 rare variants (0 < MAF ≤ 0.01) remained. Processing the data with zCall reduced the total number of fixed variants from 163,838 to 162,678 SNPs (Supplementary Data 10). Variant annotations for the canonical transcripts were fetched via the R package ‘biomaRt’80 from Ensembl Human GRCh38.p13 version 108. The R package ‘SKAT’ version 2.2.4 was used to conduct optimal unified sequence kernel association tests (SKAT-O)24. SNPs were grouped by genes, and two analyses were conducted per phenotype: (1) SKAT_CommonRare to test for the combined effect of common and rare variants, using 71,908 common and rare SNPs across 15,593 genes; and (2) SKATBinary to test for rare variant associations, using 31,345 rare SNPs across 11,740 genes. The phenotypes tested were as per the EWAS analyses: PCa risk (cases vs controls) and HRPCa risk (HRPCa vs LRPCa/No PCa). The analyses were conducted chromosome by chromosome, using age as a covariate with N = 1000 resampling used in the null model. Significant genes were identified using the family-wise error rate (FWER) multiple testing correction (cut-off 0.05) built into the package.
Heritability estimates
SNP-based heritability was calculated using genome-based restricted maximum likelihood (GREML) in GCTA v1.92.082, using autosomal variants at a prevalence of 0.001 based on the 5-year prevalence of PCa in South Africa (39,863 cases out of 29,216,012 men)4, as well as an approximate prevalence of 0.0004 for high risk PCa based on the fraction of cases with ISUP 3-5 (43%) in this study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data analysed in this study were obtained from the European Genome-Phenome Archive (EGA; https://ega-archive.org/) under overarching accession EGAS00001006425, with access to the Southern African Prostate Cancer Study (SAPCS) Dataset (EGAD00001009067) granted by the SAPCS Data Access Committee (DAC). Exomic genotyping summary statistics have been deposited in the GWAS Catalog database (www.ebi.ac.uk/gwas) under accession code GCST90296485 for cases versus control data and GCST90296486 for high-risk PCa versus low-risk PCa and control. Polygenic risk scores are available in the PGS Catalog database (https://www.pgscatalog.org/) under accession code PGP000516. Source data are provided with this paper. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Mahal, B. A. et al. Prostate cancer racial disparities: a systematic review by the prostate cancer foundation panel. Eur. Urol. Oncol. 5, 18–29 (2022).
Hjelmborg, J. B. et al. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol. Biomarkers Prev. 23, 2303–2310 (2014).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249 (2021).
Petersen, D. C. et al. Complex patterns of genomic admixture within southern Africa. PLoS Genet. 9, e1003309 (2013).
Tindall, E. A. et al. Clinical presentation of prostate cancer in black South Africans. Prostate 74, 880–891 (2014).
Mills, M. C. & Rahal, C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 52, 242–243 (2020).
Acheampong, E. et al. Association of genetic variants with prostate cancer in Africa: a concise review. Egyptian J. Med. Hum. Genet. 22, 1–9 (2021).
Rotimi, S. O., Rotimi, O. A. & Salhia, B. A review of cancer genetics and genomics studies in Africa. Front. Oncol. 10, 606400 (2020).
Cook, M. B. et al. A genome-wide association study of prostate cancer in West African men. Hum. Genet. 133, 509–521 (2014).
Du, Z. et al. Genetic risk of prostate cancer in Ugandan men. Prostate 78, 370–376 (2018).
Tindall, E. A. et al. Addressing the contribution of previously described genetic and epidemiological risk factors associated with increased prostate cancer risk and aggressive disease within men from South Africa. BMC Urol. 13, 74 (2013).
Harlemon, M. et al. A custom genotyping array reveals population-level heterogeneity for the genetic risks of prostate cancer and other cancers in Africa. Cancer Res 80, 2956–2966 (2020).
Matejcic, M. et al. Pathogenic variants in cancer predisposition genes and prostate cancer risk in men of African ancestry. JCO Precis. Oncol. 4, 32–43 (2020).
Gheybi, K. et al. Evaluating germline testing panels in southern african males with advanced prostate cancer. J. Natl. Compr. Canc. Netw. 21, 289–296 e283 (2023).
Chen, F. et al. Evidence of novel susceptibility variants for prostate cancer and a multiancestry polygenic risk score associated with aggressive disease in men of African ancestry. Eur. Urol. 84, 13–21 (2023).
Conti, D. V. et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 53, 65–75 (2021).
Conti, D. V. et al. Two novel susceptibility loci for prostate cancer in men of African ancestry. J. Natl Cancer Inst. 109 (2017).
Darst, B. F. et al. A rare germline HOXB13 variant contributes to risk of prostate cancer in men of African ancestry. Eur. Urol. 81, 458–462 (2022).
Soh, P. X. Y. & Hayes, V. M. Common genetic variants associated with prostate cancer risk: the need for African inclusion. Eur. Urol. In press.
Jaratlerdsiri, W. et al. African-specific molecular taxonomy of prostate cancer. Nature 609, 552–559 (2022).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Goldstein, J. I. et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics (Oxford, England) 28, 2543–2545 (2012).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Na, R. et al. The HOXB13 variant X285K is associated with clinical significance and early age at diagnosis in African American prostate cancer patients. Br. J. Cancer 126, 791–796 (2022).
Walavalkar, K. et al. A rare variant of African ancestry activates 8q24 lncRNA hub by modulating cancer associated enhancer. Nat. Commun. 11, 3598 (2020).
Zou, Y. & Chen, B. Long non-coding RNA HCP5 in cancer. Clin. Chim. Acta 512, 33–39 (2021).
Hu, R. & Lu, Z. Long non‑coding RNA HCP5 promotes prostate cancer cell proliferation by acting as the sponge of miR‑4656 to modulate CEMIP expression. Oncol. Rep. 43, 328–336 (2020).
Li, L., Yan, L. H., Manoj, S., Li, Y. & Lu, L. Central role of CEMIP in tumorigenesis and its potential as therapeutic target. J. Cancer 8, 2238–2246 (2017).
Yan, M. et al. Long noncoding RNA linc-ITGB1 promotes cell migration and invasion in human breast cancer. Biotechnol. Appl. Biochem. 64, 5–13 (2017).
Dai, L. et al. LncRNA ITGB1 promotes the development of bladder cancer through regulating microRNA-10a expression. Eur. Rev. Med. Pharmacol. Sci. 23, 6858–6867 (2019).
Tse, B. W. C. et al. Neuropilin-1 is upregulated in the adaptive response of prostate tumors to androgen-targeted therapies and is prognostic of metastatic progression and patient mortality. Oncogene 36, 3417–3427 (2017).
Takata, R. et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat. Genet. 42, 751–754 (2010).
Wang, N. N. et al. Susceptibility loci associations with prostate cancer risk in northern Chinese men. Asian Pac. J. Cancer Prev. 14, 3075–3078 (2013).
Huang, Q. et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 46, 126–135 (2014).
Wang, M. et al. Replication and cumulative effects of GWAS-identified genetic variations for prostate cancer in Asians: a case-control study in the ChinaPCa consortium. Carcinogenesis 33, 356–360 (2012).
Qi, N. et al. rs2274911 polymorphism in GPRC6A associated with serum E2 and PSA in a Southern Chinese male population. Gene 763, 145067 (2020).
Ewing, C. M. et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 366, 141–149 (2012).
Marlin, R. et al. Mutation HOXB13 c.853delT in Martinican prostate cancer patients. Prostate 80, 463–470 (2020).
Berlin, A. et al. Prognostic role of Ki-67 score in localized prostate cancer: A systematic review and meta-analysis. Urol. Oncol. 35, 499–506 (2017).
Hammarsten, P. et al. Immunoreactivity for prostate specific antigen and Ki67 differentiates subgroups of prostate cancer related to outcome. Mod. Pathol. 32, 1310–1319 (2019).
Couture, F. et al. PACE4-altCT isoform of proprotein convertase PACE4 as tissue and plasmatic biomarker for prostate cancer. Sci. Rep. 12, 6066 (2022).
Minami, K. et al. Expression of ABCB6 is related to resistance to 5-FU, SN-38 and vincristine. Anticancer Res. 34, 4767–4773 (2014).
Karatas, O. F., Guzel, E., Duz, M. B., Ittmann, M. & Ozen, M. The role of ATP-binding cassette transporter genes in the progression of prostate cancer. Prostate 76, 434–444 (2016).
Zhao, S. G. et al. Increased expression of ABCB6 enhances protoporphyrin IX accumulation and photodynamic effect in human glioma. Ann. Surg. Oncol. 20, 4379–4388 (2013).
Vecchione, A. et al. MITOSTATIN, a putative tumor suppressor on chromosome 12q24.1, is downregulated in human bladder and breast cancer. Oncogene 28, 257–269 (2009).
Uemura, T., Kametaka, S. & Waguri, S. GGA2 interacts with EGFR cytoplasmic domain to stabilize the receptor expression and promote cell growth. Sci. Rep 8, 1368 (2018).
Li, H. et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood 123, 1487–1498 (2014).
Mutolo, M. J. et al. Tumor suppression by collagen XV is independent of the restin domain. Matrix Biol. 31, 285–289 (2012).
Morgan, R. et al. Targeting HOX transcription factors in prostate cancer. BMC Urol. 14 17 (2014).
Choudhury, A. et al. High-depth African genomes inform human migration and health. Nature 586, 741–748 (2020).
Jiang, J. et al. ANO7 African-ancestral genomic diversity and advanced prostate cancer. Prostate Cancer Prostatic Dis (2023).
Jones, K. et al. Epigenetics in prostate cancer treatment. J. Transl. Genet. Genom. 5, 341–356 (2021).
Sugiura, M. et al. Epigenetic modifications in prostate cancer. Int. J. Urol. 28, 140–149 (2021).
Mauffrey, P. et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 569, 672–678 (2019).
Barrientos, A. et al. MTG1 codes for a conserved protein required for mitochondrial translation. Mol. Biol. Cell 14, 2292–2302 (2003).
Hopkins, J. F. et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 8, 656 (2017).
McCrow, J. P. et al. Spectrum of mitochondrial genomic variation and associated clinical presentation of prostate cancer in South African men. Prostate 76, 349–358 (2016).
Diakiw, S. M., D’Andrea, R. J. & Brown, A. L. The double life of KLF5: Opposing roles in regulation of gene-expression, cellular function, and transformation. IUBMB Life 65, 999–1011 (2013).
Xing, C. et al. Different expression patterns and functions of acetylated and unacetylated Klf5 in the proliferation and differentiation of prostatic epithelial cells. PLoS ONE 8, e65538 (2013).
Jia, J. et al. KLF5 downregulation desensitizes castration-resistant prostate cancer cells to docetaxel by increasing BECN1 expression and inducing cell autophagy. Theranostics 9, 5464–5477 (2019).
Li, Y. et al. TGF-beta causes docetaxel resistance in prostate cancer via the induction of Bcl-2 by acetylated KLF5 and protein stabilization. Theranostics 10, 7656–7670 (2020).
Che, M. et al. Opposing transcriptional programs of KLF5 and AR emerge during therapy for advanced prostate cancer. Nat. Commun. 12, 6377 (2021).
Zhang, B. et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat. Commun. 12, 1714 (2021).
Kluth, M. et al. 13q deletion is linked to an adverse phenotype and poor prognosis in prostate cancer. Genes Chromosomes Cancer 57, 504–512 (2018).
Chen, C., Bhalala, H. V., Vessella, R. L. & Dong, J. T. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate 55, 81–88 (2003).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Mulder, N. et al. H3Africa: current perspectives. Pharmgenomics Pers. Med. 11, 59–66 (2018).
Grove, M. L. et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS ONE 8, e68095 (2013).
Guo, Y. et al. Illumina human exome genotyping array clustering and quality control. Nat. Protoc. 9, 2643–2662 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Zhao, S. et al. Strategies for processing and quality control of Illumina genotyping arrays. Brief Bioinform. 19, 765–775 (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Massicotte, P., South A. rnaturalearth: World Map Data from Natural Earth. R package version 0.3.4 edn (2023).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Behr, A. A., Liu, K. Z., Liu-Fang, G., Nakka, P. & Ramachandran, S. pong: fast analysis and visualization of latent clusters in population genetic data. Bioinformatics (Oxford, England) 32, 2817–2823 (2016).
David MK & Leslie SW. Prostate Specific Antigen. [Updated 2022 Nov 10]. In: StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK557495/ (StatPearls Publishing, Treasure Island, FL, 2023)
Team R. RStudio: Integrated Development for R (RStudio, PBC, 2020).
Storey J. D., Bass A. J., Dabney A., Robinson D. qvalue: Q-value estimation for false discovery rate control.). R package version 2.30.0 edn (2022).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Karolchik, D. et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–496 (2004).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Acknowledgements
The authors are grateful to the patients and their families who have contributed to this study, to co-founding SAPCS members the late Professor Philip A. Venter and retired urologist Dr. Richard Monare from the University of Limpopo in South Africa, as well as the Medical Research Council (MRC) of South Africa for SAPCS seed-funding; without their contribution, this research would not be possible. This study was supported by a Cancer Association of South Africa (CANSA) grant to M.S.R.B. and V.M.H., the National Health and Medical Research Council (NHMRC) of Australia via a Project grant (APP1165762) to V.M.H. and Ideas grants (APP2001098) to V.M.H. and M.S.R.B. and (APP2010551) to V.M.H., as well as partially via the U.S.A. Congressionally Directed Medical Research Programs (CDMRP) Prostate Cancer Research Program (PCRP) through an Idea Development Award (PC200390, TARGET Africa) to V.M.H. and a HEROIC Consortium Award (PC210168, HEROIC PCaPH Africa1K) to V.M.H. and M.S.R.B., acknowledging our co-leads Professors Gail S. Prins (University of Illinois at Chicago) and Mungai Peter Ngugi (University of Nairobi, Kenya). V.M.H. is further supported by the Petre Foundation via the University of Sydney Foundation, Australia.
Author information
Authors and Affiliations
Contributions
V.M.H. and M.S.R.B. co-conceived the study. V.H.H. and D.C.P. designed the experimental approach. P.X.Y.S. led the statistical analyses, with statistical and computational assistance provided by K.G., J.J. and W.J. S.v.Z., R.C. and S.B.A.M. recruited patients and provided critical clinical review. N.M., D.C.P., S.M.P. and M.S.R.B. collated the specimens and provided both quality control and administrative data support. D.C.P. prepared the DNA for analysis and quality control. P.X.Y.S. generated the figures and provided the data analytics, with further interpretation provided by V.M.H. M.S.R.B. as the SAPCS Clinical Director, S.B.A.M. as the SAPCS Urological Director and V.M.H. as the SAPCS Scientific Director provided expertise-specific supervision. P.X.Y.S. drafted the manuscript with supervision from V.M.H., with all authors reviewing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Soh, P.X.Y., Mmekwa, N., Petersen, D.C. et al. Prostate cancer genetic risk and associated aggressive disease in men of African ancestry. Nat Commun 14, 8037 (2023). https://doi.org/10.1038/s41467-023-43726-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-43726-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
