
Abstract
The valence electronic structure of magnetic centers is one of the factors that determines the characteristics of a magnet. This may refer to orbital degeneracy, as for jeff = 1/2 Kitaev magnets, or near-degeneracy, e.g., involving the third and fourth shells in cuprate superconductors. Here we explore the inner structure of magnetic moments in group-5 lacunar spinels, fascinating materials featuring multisite magnetic units in the form of tetrahedral tetramers. Our quantum chemical analysis reveals a very colorful landscape, much richer than the single-electron, single-configuration description applied so far to all group-5 GaM4X8 chalcogenides, and clarifies the basic multiorbital correlations on M4 tetrahedral clusters: while for V strong correlations yield a wave-function that can be well described in terms of four V4+V3+V3+V3+ resonant valence structures, for Nb and Ta a picture of dressed molecular-orbital jeff = 3/2 entities is more appropriate. These internal degrees of freedom likely shape vibronic couplings, phase transitions, and the magneto-electric properties in each of these systems.
Similar content being viewed by others
Introduction
A magnet is a collection of magnetic moments; its characteristic properties are determined by the nature of those moments and by how they mutually interact. To shape the properties of magnetic materials according to specific requirements we therefore need to (i) understand and (ii) have some degree of control over magnetic moments—inner structure and the way they interact with each other. It turns out that both—inner morphology and mutual interaction—depend on the subtle interplay of electronic correlations, spin-orbit couplings (SOCs), and crystal-field effects (CFEs). The combined action of these three factors received enormous attention in recent years. New insights and new ideas in this research area have led to new physical models, new concepts, and new research paths, as for example Kitaev’s spin model1 and extensive associated work2.
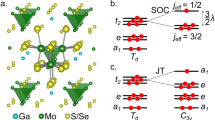
Here we reveal what lies behind effective moments in each of the group-5 GaM4 X8 lacunar spinels (M = V, Nb, Ta and X = S, Se), fascinating materials displaying remarkable magnetic3, magneto-electric4,5, and transport6,7 properties. The characteristic structural feature of this family of compounds is that the transition-metal ions are clustered as M4 tetrahedra (see Fig. 1). The latter can be then viewed as effective (magnetic) sites of a fcc lattice and their electronic structure can be described in terms of Td point-group symmetry-adapted cluster orbitals—a1/2, e, and t1/2. From electronic-structure calculations based on density functional theory (DFT), an \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) valence electron configuration was inferred8,9,10. While indications of genuine many-body physics are available from both ab initio quantum chemical investigations11,12,13 and dynamical mean field theory (DMFT)14, an in-depth profile of correlation effects across the 3d-4d-5d lacunar-spinel series is missing, which is the scope of our present quantum chemical study.

From the extended solid, a [M4X28Ga6]25− cluster is cut for quantum chemical analysis (M = V, Nb, Ta, and X = S, Se). Each transition-metal ion M is surrounded by six ligand ions X in a distorted octahedron. Xi and Xo labels indicate X atoms inside and outside the M4 tetrahedron, respectively, referring to the basis set assignment in Supplementary Table 2. The small spheres indicate embedding capped effective core potentials (cECPs). Inset: valence level representations analyzed in this study.
Besides clarifying essential electronic-structure features, the multiconfigurational wave-function analysis that we provide—in terms of either localized, site-centered or multisite orbitals—makes these materials a distinct correlated-electron model system, as illustrative but much more captivating than other platforms typically employed to illustrate electronic correlations, as e.g., the H2 molecule for variable interatomic separation15,16. Using as indicator for the strength of correlations the weight of ionic configurations in the ground-state wave-function, we picture (i) what strong correlations mean in the 3d vanadates (GaV4S8, GaV4Se8, AlV4S8) and (ii) the notion of moderate correlations and ‘dressed’ jeff = 3/2 objects in the 4d (GaNb4S8, GaNb4Se8) and 5d (GaTa4Se8) variants. Even for the heavier cations, when expressing the multiconfigurational wave-functions in terms of delocalized multisite orbitals, the weight of the \((...){t}_{2}^{1}\) configuration presently assumed to correctly describe the ground-state amounts to only 60%. Impressively, that shrinks to as little as 20% for 3d electrons. Yet, SOCs are still effective—even in the vanadates, those give rise to a spin-orbit-induced splitting of ≈10 meV for the ground-state term. Also peculiar here is the near-degeneracy of the ground and a higher-spin state, which should be possible to evidence by either spectroscopy or pressure experiments. All these electronic-structure features for the 3d case—massive correlations, scaled down but still detectable spin-orbit fine structure, and close proximity of high-spin states—outline a few important differences between 3d and 4d/5d group-5 lacunar spinels, i.e., the starting point in understanding the differences in their magnetic properties.
Results
High-temperature, tetrahedral-symmetry multiplet structure
The M4-tetrahedron multiplet structure, as computed for the high-temperature \(F\bar{4}3m\) cubic lattice arrangement of group-5 lacunar spinels, is displayed in Fig. 2. The multiconfigurational complete active space self-consistent field (CASSCF) method17 with an active space of seven electrons in twelve orbitals [(7e,12o)-CAS] was applied. Those orbitals are depicted in Supplementary Figure 1.

The excitation energies were calculated for (a) [V4S28Ga6]25−, (b) [Nb4Se28Ga6]25−, and (c) [Ta4Se28Ga6]25− embedded clusters (CAS(7e,12o)). Different spin states S (S = 1/2, 3/2, 5/2) are shown in green, red, and blue, respectively. SOC was accounted for on top of the CASSCF/NEVPT2 wave functions. The corrections brought by NEVPT2 are minor for the Nb- and Ta-based compounds and therefore not depicted (see Supplementary Tables 4 and 5).
For GaV4S8 (Fig. 2a), this (7e,12o)-CASSCF yields a high-spin (S = 3/2) ground state. Accounting for dynamical correlation effects in the scheme of post-CASSCF N-electron valence perturbation theory (NEVPT2)18 corrects this state ordering. Near degeneracy of low- and high-spin states is an effect often seen in 3d systems, due to the similar magnitude of Coulomb interactions and various valence level splittings; in solid-state context, a well-known example is LaCoO3 (see19 for a quantum chemical investigation). Spin-crossover effects were also observed in DMFT calculations on GaV4S814. In contrast, for the Nb- (Fig. 2b) and Ta-based materials (Fig. 2c), the CASSCF(7e,12o) methodology already ensures a reasonably good description—the NEVPT2 scheme provides only minor corrections to the relative energies in the 4d and 5d systems (see Supplementary Tables 4 and 5).
While a 2T2 state is found as ground state in all compounds, the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) electronic configuration (where a1, e, and t1 are symmetry-adapted, molecular-like orbitals in Td point-group symmetry) contributes with weights well below 100% to the ground-state wave-functions; this aspect is discussed in detail in the following section. SOC further splits the degenerate 2T2 components into a jeff = 3/2 ground and a jeff = 1/2 excited state in all instances, with the magnitude of this splitting increasing from 12 meV (3d) to 97 meV (4d) and 345 meV (5d ions); the latter number, in particular, suggests that the origin of the peak found at ≈ 0.3 eV in resonant inelastic X-ray scattering (RIXS) measurements on GaTa4Se820 is spin-orbit splitting within the 2T2 (\({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\)) levels, different from the initial interpretation in terms of \({e}^{4}{t}_{2}^{1}\) → \({e}^{3}{t}_{2}^{2}\) transitions12,20.
It is seen that the separation between the ground 2T and the first excited 4T state also increases for heavier transition-metal species: 41 meV in GaV4S8, 475 meV in GaNb44Se8, and 598 meV in GaTa4Se8. A denser set of low-lying excited-state levels in the vanadate could explain the significant deviation from Curie-Weiss behavior seen at higher temperatures in susceptibility measurements21,22, although a quantitative analysis would require the inclusion of vibronic couplings and intersite magnetic interactions (see discussion in Section V of the Supplementary Information and in our previous work12). The latter then dictates the structure of the inelastic neutron scattering spectra23.
Ground-state correlations in cubic group-5 spinels
A major difference between how the single-tetrahedron electronic structure is presently depicted in the literature and in our quantum chemical results is the composition of the ground-state 2T2 term. Different from the 100% \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) ground state assumed so far on the basis of DFT computations for these materials, we find weights of 72% in GaTa4Se8, 65% in GaNb44Se8, and as little as 21% in GaV4S8 for the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) configuration. Other configurations contributing to the ground-state wave functions imply, for example, double excitations into higher-lying t1 and t2 levels, each of those configurations with a weight of a few percent or even less (see also Supplementary Table 6).
The much more pronounced multiconfigurational character of the vanadate ground-state wave function in the symmetry-adapted orbital basis is also reflected in the correlation index proposed by Ramos-Cordoba et al.15, which serves as a measure for the extent of near-degeneracy effects (also referred to as nondynamical correlation in quantum chemistry). Using the (7e,12o) CASSCF natural-orbital occupation numbers, we derived nondynamical correlation indices IND15 ranging from ≈2 in vanadates (1.96 for GaV4S8 and 2.00 for GaV4Se8) to ≈ 1.05 in the 4d variants (1.05 for GaNb4S8 and 1.07 for GaNb44Se8) and 0.92 in GaTa4Se8 (details are given in the Supplementary Table 7). To put this in perspective, along the H2 dissociation curve, IND evolves from less than 0.1 at equilibrium distance to 0.5 towards dissociation15, although a direct comparison of IND values between chemically different systems is not straightforward.
Looking for further insight, we re-expressed the many-body ground-state wave functions in terms of atomic-like, single-site orbitals. The orbital localization module available in ORCA was employed for this purpose; from the 12 symmetry-adapted orbitals (1 × a1, 1 × e, 1 × t1, 2 × t2) obtained in the (7e,12o) CASSCF we arrive then to 12 t2g-like functions (three per pseudo-octahedrally coordinated transition-metal ion, see Supplementary Figure 1). For both orbital bases, symmetry-adapted or site-centered, the weights of the leading configurations are illustrated in Fig. 3, for GaV4S8, GaV4Se8, GaNb4S8, GaNb4Se8, GaTa4Se8, and an A-site-substituted member of the family (see also Supplementary Table 6). Interestingly, a low weight of the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) configuration in the symmetry-adapted orbital basis is associated with large weight of the \({t}_{{{{{\mbox{2g}}}}}}^{1}{t}_{{{{{\mbox{2g}}}}}}^{2}{t}_{{{{{\mbox{2g}}}}}}^{2}{t}_{{{{{\mbox{2g}}}}}}^{2}\) (i.e., V4+V3+V3+V3+) configurations in the localized-orbital representation; for the AlV4S8, GaV4S8, and GaV4Se8 vanadates, in particular, 15–20% \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) translates into 85–90% configurations of \({t}_{{{{{\mbox{2g}}}}}}^{1}{t}_{{{{{\mbox{2g}}}}}}^{2}{t}_{{{{{\mbox{2g}}}}}}^{2}{t}_{{{{{\mbox{2g}}}}}}^{2}\) type. The remaining part stems mainly from triply-occupied transition-metal centers, i.e., excited-state configurations of \({t}_{{{{{\mbox{2g}}}}}}^{1}{t}_{{{{{\mbox{2g}}}}}}^{2}{t}_{{{{{\mbox{2g}}}}}}^{1}{t}_{{{{{\mbox{2g}}}}}}^{3}\) (V4+V3+V4+V2+) type. Weights of only ≈9% for the latter indicate much stronger correlations in the case of V-based lacunar spinels: intersite fluctuations are heavily suppressed, compared to the 4d and 5d compounds. In a Mott-Hubbard picture, stronger localization is the result of larger U/t ratio. In other words, correlations are moderate in the 4d and 5d compounds and strong in the vanadates—for the latter, an expansion in terms of four V4+V3+V3+V3+ resonant valence structures already provides a reasonably good description. For perspectives on valence bond theory, the reader is referred to e.g.24,25.

For each system, the analysis is performed in terms of ground-state optimized CASSCF(7e,12o) wave-functions.
Discussion
To analyze in detail how correlations evolve from 3d to 4d and 5d ions for the same type of leading ground-state configuration and in the same crystallographic setting is difficult. 3d5 (Mn2+, Fe3+) species, for example, tend to adopt a \({t}_{{{\mbox{2g}}}}^{3}{e}_{{{\mbox{g}}}}^{2}\) ground-state electron configuration, while 4d5 (Ru3+, Rh4+) and 5d5 (Ir4+) varieties display a \({t}_{{{\mbox{2g}}}}^{5}\) valence-orbital occupation. Thinking of lower d-shell filling, Mo5+ 4d1 and Os7+ 5d1 ions, for instance, can be found in double-perovskite fcc settings26, but that is not the case for Ti3+ or V4+ 3d1.
Here we individualize the group-5 lacunar spinels as a unique platform that makes it possible to illustrate how correlations shape many-body wave functions across a given group of the d block—3d to 4d and 5d, for the same kind of leading electron configuration and in the same crystallographic setting. In particular, by expressing the many-body M4-tetrahedron wave function in terms of localized single-ion t2g orbitals, we show that strong correlations yield a weight of 85–90% for the \({t}_{{{\mbox{2g}}}}^{1}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}\) (i.e., V4+V3+V3+V3+) configurations in the vanadates; ferromagnetic double exchange occurs in this setting and yields near degeneracy of the low-lying low- and high-spin states—the S = 1/2 doublet is obtained as single-tetrahedron ground-state term only through a more advanced many-body treatment. In contrast, smaller U/t ratios in the 4d (Nb) and 5d (Ta) systems take us away from the regime of strongly correlated electrons: in localized-orbital basis, we see that stronger charge fluctuations reduce the weight of \({t}_{{{\mbox{2g}}}}^{1}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}\) resonances to ~ 50%; in molecular-orbital representation, significantly larger hoppings (t) and consequently larger bonding-antibonding splittings make that the Aufbau principle is to first approximation usable, with weights in the range of 65-75% for the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) configuration.
This result suggests a physical picture for the nonmagnetic states found in the Nb- and Ta-based lacunar spinels: as the M4-cluster electrons are prone to stronger fluctuations with larger spatial spread, inter-cluster couplings are able to create “valence bond” spin singlets, as concluded from experiment27,28,29. The peculiar pseudospin structure must play a role in the superconductivity mechanism under pressure, which is speculated to be unconventional owing to closeness of magnetic states and spin fluctuations.
While it is clear that the on-site correlations affect the magnetic properties, they are only indirectly discernible in available experimental data, as already pointed out for susceptibility21,22 and inelastic neutron scattering measurements23 in in the previous section. More direct experimental insight into the details of the single-tetrahedron correlated electronic structure might be derived from resonant inelastic x-ray scattering experiments, as in the case of other clustered compounds, either d-30 or p-electron31,32,33 based.
Overall weights of ≳50% for the \({t}_{{{\mbox{2g}}}}^{1}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}{t}_{{{\mbox{2g}}}}^{2}\) resonant valence structures suggest the t-U-V model (or U-\(t{t}^{{\prime} }\)-\(V{V}^{{\prime} }\) variants, where the primes denote inter-tetrahedral hopping matrix elements and Coulomb repulsion integrals) as means to explore correlation-induced symmetry breaking. Such numerical investigations could provide qualitative insights into the different types of low-temperature lattice symmetries realized in lacunar spinels and also into the polar properties of these materials. An analysis in terms of only three molecular-like t2 orbitals and one electron as in e.g., ref. 34 does not seem promising: according to our data, especially in the vanadates, the (\({a}_{1}^{2}{e}^{4}\))\({t}_{2}^{1}\) description is not appropriate—the M4 tetrahedron t2 electron cannot be separated from the other six d-ion valence electrons, symmetry breaking should be rather described in terms of resonating holes in localized-orbital basis (i.e., V4+ ‘holes’ in V3+ ‘background’).
In sum, our quantum chemical data provide unparalleled specifics as concerns the correlated electronic structure of the group-5 lacunar spinels, well beyond the featureless \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) picture circulated so far in the literature. Stronger correlations in the vanadates imply substantially less weight for the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) configuration as compared to the Nb and Ta compounds and render the molecular-orbital picture8,9,35 inappropriate. Remarkably, spin-orbit coupling is still effective, even for the vanadates—the predicted fine structure with a splitting of ≈10 meV between the j ≈ 3/2 and j ≈ 1/2 terms should be detectable experimentally. The stronger intersite fluctuations (M3+M3+→M2+M4+) and the larger weight (65–75%) of the \({a}_{1}^{2}{e}^{4}{t}_{2}^{1}\) molecular-orbital configuration in the Nb and Ta spinels indicate that the 4d and 5d systems are closer to the Hartree-Fock limit. The different nature of valence-space charge fluctuations across the group-5 family of lacunar spinels should be relevant to inter-tetramer exchange; on-site charge fluctuations, for example, were shown to strongly renormalize intersite exchange in cuprate superconductors36. Assessing cooperative effects in group-5 lacunar spinels through the calculation of inter-cluster couplings will require approaches able to incorporate the correlated nature of the M4-cluster ground states.
Methods
The basic building block in the lacunar-spinel structure was described by a [M4X28Ga6]25− embedded cluster model (M = V, Nb, Ta; X = S, Se) (see Fig. 1). Experimentally determined high-temperature lattice parameters were adopted, as reported by Stefancic et al.22 for GaV4S8 and by Pocha et al. for GaNb4Se8 and GaTa4Se87. The influence of the surrounding bulk atoms was modeled by a finite point charge field (PCF) generated through the EWALD program37,38. A buffer region of 60 capped effective core potentials (cECPs) was set up between the quantum cluster and PCF (indicated by the smaller atoms in Fig. 1) (for details, see Section I of the Supplementary Information and Supplementary Dataset 1).
As initial step in our study, quasi-restricted orbitals (QROs39) were generated from an unrestricted Kohn-Sham B3LYP calculation for a single-configuration S = 5/2 state with initial-guess orbitals from Hueckel theory. The Hueckel guess ensures that the QROs fulfill Td point-group symmetry. Subsequently, 12 [M4]13+ molecular orbitals around the HOMO-LUMO gap were identified from the QROs and used as a starting point for CASSCF17 calculations. Major convergence problems as encountered in earlier quantum chemical studies11 are circumvented in this way. The valence-space multiplet structure was derived from state averaged (SA) CASSCF optimizations with those 12 orbitals and seven valence electrons defining the active space (denoted in quantum chemistry as (7e,12o) CASSCF), consequently corrected for dynamical correlation by N-electron valence 2nd order perturbation theory (NEVPT2)18. Both methods were accelerated by the resolution of identity (RI40) and chain-of-spheres (COS41) approximations for Coulomb and exchange integrals with automatically generated auxiliary basis sets42. All calculations were done using the program package ORCA, v5.0.343.
Data availability
The quantum chemical data (coordinates of quantum clusters and point charge fields, ORCA outputs, and magnetic susceptibility simulations) generated in this study have been deposited in the RADAR database under the https://doi.org/10.22000/1655.
References
Kitaev, A. Anyons in an exactly solved model and beyond. Ann. Phys. 321, 2–111 (2006).
Takagi, H., Takayama, T., Jackeli, G., Khaliullin, G. & Nagler, S. E. Concept and realization of Kitaev quantum spin liquids. Nat. Rev. Phys. 1, 264–280 (2019).
Kézsmárki, I. et al. Néel-type skyrmion lattice with confined orientation in the polar magnetic semiconductor GaV4S8. Nat. Mater. 14, 1116–1122 (2015).
Geirhos, K. et al. Cooperative cluster Jahn-Teller effect as a possible route to antiferroelectricity. Phys. Rev. Lett. 126, 187601 (2021).
Geirhos, K. et al. Optical, dielectric, and magnetoelectric properties of ferroelectric and antiferroelectric lacunar spinels. Phys. Status Solidi B 259, 2100160 (2021).
Abd-Elmeguid, M. M. et al. Transition from mott insulator to superconductor in GaNb4Se8 and GaTa4Se8 under high pressure. Phys. Rev. Lett. 93, 126403 (2004).
Pocha, R., Johrendt, D., Ni, B. & Abd-Elmeguid, M. M. Crystal structures, electronic properties, and pressure-induced superconductivity of the tetrahedral cluster compounds GaNb4S8, GaNb4Se8, and GaTa4Se8. J. Amer. Chem. Soc. 127, 8732–8740 (2005).
Pocha, R., Johrendt, D. & Pöttgen, R. Electronic and Structural Instabilities in GaV4S8 and GaMo4S8. Chem. Mater. 12, 2882–2887 (2000).
Müller, H., Kockelmann, W. & Johrendt, D. The magnetic structure and electronic ground states of mott insulators GeV4S8 and GaV4S8. Chem. Mater. 18, 2174–2180 (2006).
Kim, H.-S., Im, J., Han, M. J. & Jun, H. Spin-orbital entangled molecular jeff states in lacunar spinel compounds. Nat. Commun. 5, 3988 (2014).
Hozoi, L., Eldeeb, M. S. & Rößler, U. K. V4 tetrahedral units in AV4X8 lacunar spinels: near degeneracy, charge fluctuations, and configurational mixing within a valence space of up to 21 d orbitals. Phys. Rev. Res. 2, 022017 (2020).
Petersen, T. et al. How correlations and spin-orbit coupling work within extended orbitals of transition-metal tetrahedra of 4d/5d lacunar spinels. J. Phys. Chem. Lett. 13, 1681–1686 (2022).
Petersen, T. et al. Dressed jeff-1/2 objects in mixed-valence lacunar spinel molybdates. Sci. Rep. 13, 2411 (2023).
Kim, H.-S., Haule, K. & Vanderbilt, D. Molecular Mott state in the deficient spinel GaV4S8. Phys. Rev. B 102, 081105 (2020).
Ramos-Cordoba, E., Salvador, P. & Matito, E. Separation of dynamic and nondynamic correlation. Phys. Chem. Chem. Phys. 18, 24015–24023 (2016).
Izsák, R., Ivanov, A. V., Blunt, N. S., Holzmann, N. & Neese, F. Measuring electron correlation: the impact of symmetry and orbital transformations. J. Chem. Theor. Comput. 19, 2703–2720 (2023).
Roos, B.O. The complete active space self-consistent field method and its applications in electronic structure calculations, pp. 399–445. John Wiley & Sons, Ltd. Advances in Chemical Physics https://doi.org/10.1002/9780470142943.ch7 (1987).
Angeli, C., Cimiraglia, R., Evangelisti, S., Leininger, T. & Malrieu, J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 114, 10252–10264 (2001).
Hozoi, L., Birkenheuer, U., Stoll, H. & Fulde, P. Spin-state transition and spin-polaron physics in cobalt oxide perovskites: ab initio approach based on quantum chemical methods. New J. Phys. 11, 023023 (2009).
Jeong, M. Y. et al. Direct experimental observation of the molecular Jeff = 3/2 ground state in the lacunar spinel GaTa4Se8. Nat. Commun. 8, 782 (2017).
Widmann, S. et al. On the multiferroic skyrmion-host GaV4S8. Philos. Mag. 97, 3428–3445 (2017).
Štefančič, A. et al. Establishing magneto-structural relationships in the solid solutions of the skyrmion hosting family of materials: GaV4S8−ySey. Sci. Rep. 10, 9813 (2020).
Pokharel, G. et al. Spin dynamics in the skyrmion-host lacunar spinel GaV4S8. Phys. Rev. B 104, 224425 (2021).
Hoffmann, R., Shaik, S. & Hiberty, P. C. A conversation on VB vs MO theory: A never-ending rivalry? Acc. Chem. Res. 36, 750–756 (2003).
Truhlar, D. G. Valence bond theory for chemical dynamics. J. Comput. Chem. 28, 73 (2007).
Chen, G., Pereira, R. & Balents, L. Exotic phases induced by strong spin-orbit coupling in ordered double perovskites. Phys. Rev. B 82, 174440 (2010).
Waki, T. et al. Spin-singlet state formation in the cluster Mott insulator GaNb4S8 studied by μ-SR and NMR spectroscopy. Phys. Rev. B 81, 020401 (2010).
Ishikawa, H., Yajima, T., Matsuo, A., Ihara, Y. & Kindo, K. Nonmagnetic ground states and a possible quadrupolar phase in 4d and 5d lacunar spinel selenides GaM4Se8 (M = Nb, Ta). Phys. Rev. Lett. 124, 227202 (2020).
Yang, T.-H. et al. Bond ordering and molecular spin-orbital fluctuations in the cluster Mott insulator GaTa4Se8. Phys. Rev. Res. 4, 033123 (2022).
Gu, Y. et al. Site-specific electronic and magnetic excitations of the skyrmion material Cu2OSeO3. Commun. Phys. 5, 156 (2022).
Denlinger, J. D. et al. Bulk band gaps in divalent hexaborides. Phys. Rev. Lett. 89, 157601 (2002).
Denlinger, J. D., Gweon, G.-H., Allen, J. W., Bianchi, A. D. & Fisk, Z. Bulk band gaps in divalent hexaborides: a soft X-ray emission study. Surf. Rev. Lett. 9, 1309–1313 (2002).
Petersen, T., Rößler, U. K. & Hozoi, L. Quantum chemical insights into hexaboride electronic structures: correlations within the boron p-orbital subsystem. Commun. Phys. 5, 214 (2022).
Dally, R. L. et al. Magnetic phase transitions and spin density distribution in the molecular multiferroic system GaV4S8. Phys. Rev. B 102, 014410 (2020).
Browne, A. J. & Attfield, J. P. Orbital molecules in vanadium oxide spinels. Phys. Rev. B 101, 024112 (2020).
Bogdanov, N. A., Manni, G. L., Sharma, S., Gunnarsson, O. & Alavi, A. Enhancement of superexchange due to synergetic breathing and hopping in corner-sharing cuprates. Nat. Phys. 18, 190–195 (2022).
Klintenberg, M., Derenzo, S. E. & Weber, M. J. Accurate crystal fields for embedded cluster calculations. Comp. Phys. Commun. 131, 120–128 (2000).
Derenzo, S. E., Klintenberg, M. K. & Weber, M. J. Determining point charge arrays that produce accurate ionic crystal fields for atomic cluster calculations. J. Chem. Phys. 112, 2074–2081 (2000).
Neese, F. Importance of direct spin-spin coupling and spin-flip excitations for the zero-field splittings of transition metal complexes: a case study. J. Amer. Chem. Soc. 128, 10213–10222 (2006).
Neese, F. An improvement of the resolution of the identity approximation for the formation of the coulomb matrix. J. Comput. Chem. 24, 1740–1747 (2003).
Neese, F., Wennmohs, F., Hansen, A. & Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. a ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 356, 98–109 (2009).
Stoychev, G. L., Auer, A. A. & Neese, F. Automatic generation of auxiliary basis sets. J. Chem. Theor. Comput. 13, 554–562 (2017).
Neese, F. Software update: the ORCA program system–Version 5.0. WIREs Comput. Mol. Sci. 12, 1606 (2022).
Acknowledgements
We thank I. Kézsmárki, P. Fulde, and R. C. Morrow for discussions and U. Nitzsche for technical assistance. This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), Project No. 437124857. P.B. acknowledges funding from the DFG, Project No. 441216021.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
T.P. carried out the quantum chemistry calculations with assistance from P. B., U.K.R., and L.H. All authors were involved in writing the manuscript. U.K.R. and L.H. planned the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petersen, T., Bhattacharyya, P., Rößler, U.K. et al. Resonating holes vs molecular spin-orbit coupled states in group-5 lacunar spinels. Nat Commun 14, 5218 (2023). https://doi.org/10.1038/s41467-023-40811-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40811-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
