
Abstract
As isomers of the regular porphyrins, N-confused porphyrins have attracted extensive attention of chemists because of their unique chemical structures, chemical reactivities, and physical properties, which result in their promising applications in the fields of catalytic chemistry, biochemistry and material science. Typically, N-confused porphyrins are synthesized via acid catalyzed condensation and following oxidation during which lactams are often formed as the byproducts. Here we report doubly N-confused and ring-contracted [24]hexaphyrin(1.1.0.1.1.0) mono- and bis-Pd-complexes as stable antiaromatic N-confused expanded porphyrins, which are synthesized through Pd-catalyzed Suzuki-Miyaura coupling of 1,14-dibromotripyrrin. These macrocycles show a paratropic ring currents, an ill-defined Soret band, a red-shifted weak absorption tail, and a small HOMO-LUMO gap. NBS bromination of the bis Pd-complex give its mono- and dibromides regioselectively, which are effectively used to synthesize a [24]hexaphyrin dimer and a NiII porphyrin-[24]hexaphyrin-NiII porphyrin triad, respectively.
Similar content being viewed by others
Introduction
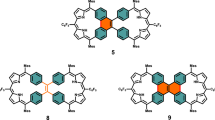
In the chemistry of porphyrinoids, expanded porphyrins1,2,3,4,5 and N-confused porphyrins (NCPs)6,7,8,9 are two important newcomers, which have contributed to highlighting the glorious potentials of porphyrinoids. In 2003, Furuta et al. have combined these two porphyrinoids by synthesizing expanded NCPs such as doubly N-confused [26]dioxohexaphyrins(1.1.1.1.1.1) 1 and doubly N-confused and ring-contracted [26]dioxohexaphyrins(1.1.1.1.1.0) 2 (Fig. 1)10,11,12,13,14. These molecules were synthesized by acid-catalyzed condensation reactions and the lactam units were formed via facile oxidation of the N-confused pyrroles. The oxidized N-confused pyrrole units in 1 and 2 have been shown to be quite effective for the coordination of transition metal ions, allowing for the synthesis of various metal complexes. On the other hand, attempts to synthesize doubly N-confused non-oxo [26]hexaphyrin metal complexes ended in very low yields15. In addition, these synthetic methods provided only thermodynamically stable aromatic products. As a rare example, Furuta et al. reported that electrochemical oxidation of 2Cu led to the generation of 24π-antiaromatic species14, but antiaromatic expanded NCP has been never isolated before. In this work, we report the serendipitous synthesis of doubly N-confused and ring-contracted (DNCRC) [24]hexaphyrin(1.1.0.1.1.0) PdII complexes 8 and 9 by Pd-catalyzed self-coupling reaction of α,α-dibromotripyrrin 3b.

Structures of doubly N-confused [26]dioxohexaphyrins(1.1.1.1.1.1) 1, doubly N-confused and singly ring-contracted [26]dioxohexaphyrins(1.1.1.1.1.0) 2, and its CuII complex 2Cu.
Results
With the aim to synthesize m-benziporphyrin(1.1.0.0) 5, we attempted the cyclization reaction of α,α’-diboryltripyrrane 3a with 1,3-dibromo-4-methoxybenzene under the usual Suzuki–Miyaura coupling conditions (Pd2(dba)3, X-phos, Cs2CO3, CsF, and toluene/DMF). In addition to the target 516 which was actually obtained in 5% yield, we isolated dark green product 6 in ca. 1% (Fig. 2). Fortunately, the structure of 6 has been revealed by X-ray analysis to be a symmetric and planar [18]triphyrin(5.1.1) with a small mean-plane deviation (MPD) of 0.26 Å (Fig. 3a, b). The carbonyl oxygen atom is hydrogen bonded with the two adjacent pyrrolic NH groups and the C=O bond length is 1.288(7) Å, being distinctly longer than those of usual ketones (1.23 Å)17, 18. This structural feature suggests a significant contribution of a dipolar resonance state. The 1H NMR spectrum of 6 shows a singlet at 9.74 ppm due to the vinylic proton, two doublets at 8.36 and 8.08 ppm (J = 4.0 Hz) and a singlet at 8.31 ppm due to the pyrrolic protons, and signals due to the NH protons at 3.89 (2H) and −4.13 (1H) ppm, suggesting a distinct diatropic ring current, reflecting its 18π-electronic circuit. The absorption spectrum of 6 shows a split Soret-like band at 419 and 444 nm and vibronic-structured Q-like bands as characteristic features of aromatic porphyrinoids (Fig. 4). Further, 6 displays vibronic-structured fluorescence at 652 and 721 nm with a high quantum yield of ΦF = 0.48. These data strongly indicate that 6 is an aromatic tripyrrolic porphyrinoid. Actually, the calculated NICS values are negative (−12.15 ppm) inside the macrocycle.

Dba = dibenzylideneacetone, Mes = 2,4,6-trimethylphenyl, Bpin = pinacolatoboryl, DDQ = 2,3-dicyano-5,6-dichlorobenzoquinone.

a Top view of 6, b side view of 6, c top view of 7, d side view of 7. Ellipsoids are drawn at the 30% probability level. All hydrogen atoms except those connected to N and O atoms are omitted for clarity. Carbon atom, black ellipsoid; nitrogen atom, blue; palladium atom, orange; oxygen atom, red; hydrogen atom small black ball (These instructions are omitted in the following figures for clarity).

Solid line, absorption spectrum; dash line, fluorescence spectrum.
Since 6 can be regarded as a cyclized product of 3a and dibenzalacetone, we examined the reaction of only 3a under the same conditions, which actually gave 6 in 5–8% yield. In the next step, we examined the reaction of 3a with dibenzalacetone and found that 6 was obtained in 8.8% under conditions (3 eq. dibenzalacetone, 0.4 equiv PPh3, 2 mol% Pd2(dba)3, 2 eq. Cs2CO3, CsF). In continuation of this study, we examined the reaction of 1,14-dibromotripyrrin 3b under the similar Suzuki–Miyaura coupling conditions (X-phos, Cs2CO3, CsF, Pd2(dba)3, toluene, DMF, reflux, 48 h), and found quite unexpectedly that [24]hexaphyrin Pd-complexes 8 (9%) and 9 (10%) were obtained along with 6 (1%) and tripyrrolic compound 7 (2%) (Fig. 5). The structure of 7 has been revealed by X-ray analysis (Fig. 3c, d) to be a coupling product of 3 and dibenzalacetone.

Reaction conditions: a X-phos, Cs2CO3, CsF, Pd2(dba)3, toluene, DMF, reflux, 48 h; b M(OAc)2, NaOAc, CHCl3, MeOH, reflux.
The parent ion peaks of 8 and 9 were observed at m/z = 1120.2568 (calcd for (C64H54N6Pd2)+ = 1120.2504 ([M]+)) and at m/z = 1030.3687 (calcd for (C64H56N2Pd)+ = 1030.3566 ([M]+)) by high-resolution MALDI-TOF mass measurement. Both the structures of 8 and 9 have been revealed to be doubly N-confused and ring-contracted (DNCRC) [24]hexaphyrins(1.1.0.1.1.0) by X-ray analysis (Fig. 6). Product 8 is a nonoxo DNCRC [24]hexaphyrin, displaying a very planar structure with a small MPD (0.04 Å), in which the two Pd metals are coordinated with the three pyrrolic nitrogen atoms and one α-carbon of the N-confused pyrrole with a bond distance of 2.002(4) Å for Pd1–N1, 2.027(5) Å for Pd1–N2, 1.959(5) Å for Pd1–N3, and 1.972(5) Å for Pd1–C. As compared with the previously reported PdII complexes of NCPs that showed bond distances of Pd–C (2.002–2.003 Å) and Pd–N (2.003 Å)19,20,21, Pd1–N2 and Pd1–C bond lengths are distinctly shorter. The 1H NMR spectrum of 8 shows signals due to the β-pyrrolic protons as a singlet at 4.01 ppm and two doublets at 4.85 and 5.10 ppm (J = 4.5 Hz) and at 5.25 and 5.30 ppm (J = 4.5 Hz), indicating a weak but distinct paratropic ring current. The absorption spectrum of 8 is ill-defined, showing peaks at 396, 502, 618, 687, 763, 1015, and 1176 nm. The HOMA value was calculated to be 0.51 based on the crystallographic data, and the NICS value at the center of the molecule was calculated to be 5.41, indicating the weak antiaromatic character of 8. In addition, the ACID plot (see Supplementary Fig. 58) of complex 8 displayed a continuous paratropic ring current, being consistent with the NICS(0) value.

a Top view of 8, b side view of 8, c top view of 9, d side view of 9, e top view of 9Pd, f side view of 9Pd. Ellipsoids are drawn at the 30% probability level. All hydrogen atoms except those connected to N atoms are omitted for clarity.
The structure of 9 has been determined by X-ray diffraction analysis to be a DNCRC keto tautomer, in which one α-position of the N-confused pyrrole is oxidized. Unfortunately, the serious disorder impeded a detailed analysis of the structure. The 1H NMR spectrum of 9 displays two signals due to the inner NH protons at 21.89 and 19.85 ppm and those in the range of 5.22–3.79 ppm due to the pyrrolic β-protons, clearly indicating its paratropic ring current and thus antiaromatic character. Judging from these chemical shifts, it is conceivable that the antiaromatic character of 9 is slightly larger as compared with 8. The NICS value of 9 was calculated to be 10.43, which is larger than that of 8. Importantly, 8 and 9 are stable antiaromatic N-confused expanded porphyrins.
Hexaphyrin 9 has been demonstrated to be a nice platform to form bis-metalated complexes. Actually, complexes 9Pd, 9Ni, and 9Cu were readily obtained in 80%, 95%, and 85% upon complexation with PdII, NiII, and CuII ions, respectively. The 1H NMR spectra of 9Pd and 9Ni show signals due to the peripheral β-protons in the range of 5.40–4.09 and 5.20–3.77 ppm, respectively, revealing paratropic rings current comparable to that in 9. The structure of 9Pd has been revealed by X-ray analysis, displaying a slightly curved structure with a large MPD (0.36 Å). Pd1 is coordinated with the three pyrrolic nitrogen atoms and one α-carbon of the N-confused pyrrole with a bond length of 2.034(6) Å for Pd1–N1, 2.029(5) Å for Pd1–N2, 1.956(5) Å for Pd1–N3, and 2.073(6) Å for Pd1–C. Pd2 is coordinated with the three pyrrolic nitrogen atoms and one oxygen atom with a bond length of 1.967(5) Å for Pd2–N4, 1.962(5) Å for Pd2–N5, 1.968(5) Å for Pd2–N6, and 2.002(5) Å for Pd2–O1.
The absorption spectra of 8, 9, 9Pd, 9Ni, and 9Cu are shown in Fig. 7a. Characteristically, all these DNCRC [24]hexaphyrin complexes show ill-defined Soret bands and weak bands in the range of 800–1400 nm in line with their antiaromatic properties. Judging from the most red-shifted bands, the optical HOMO–LUMO gaps have been estimated to be 1.05, 1.15, 1.12, 1.05, and 1.08 eV for 8, 9, 9Pd, 9Ni, and 9Cu, respectively.

a UV–Vis absorption spectra of 8, 9, 9Pd, 9Ni and 9Cu in CH2Cl2; b UV–Vis absorption spectra of 8, 10 and 12 in CH2Cl2.
Cyclic voltammetry (CV) and differential-pulse voltammetry (DPV) experiments were conducted and the redox potentials are summarized in Table 1.
The electrochemical properties were investigated by cyclic voltammetry and differential pulse voltammetry. Complex 8 showed reversible oxidation waves at 0.65 and 0.18 V and reduction waves at −1.05, −1.52, and −1.79 V, indicating the HOMO–LUMO gap to be 1.23 eV. Complexes 9, 9Pd, 9Ni, and 9Cu showed HOMO–LUMO gaps of 1.12, 1.19, 1.20, and 1.15 eV, respectively, roughly matching with the optical HOMO-LUMO gaps.
Further fabrications of 8 were attempted. Monobromide 8a was obtained in 78% yield by treating 8 with 1 equiv. NBS at 0 °C (Fig. 8). The parent ion peak of 8a was observed at m/z = 1196.1615 (calcd for (C64H53BrN6Pd2)+ = 1196.1601 ([M]+)) and its 1H NMR spectrum showed nine peaks at 5.23–4.05 ppm, indicating an asymmetric substitution. Subsequently, directly β-to-β linked DNCRC [24]hexaphyrin dimer 10 was successfully obtained by reductive coupling of 8a with Ni(cod)2 in 61% yield. The parent ion peak of 10 was observed at m/z = 2234.4843 (calcd for (C128H106N12Pd4)+ = 2234.4862 ([M]+)). The 1H NMR spectrum of 10 exhibited three singlets at 4.30, 3.91, and 3.74 ppm and three pairs of doublets in the range of 5.13–4.69 ppm. The structure of 10 has been confirmed by X-ray analysis as shown in Fig. 9. The dihedral angle between the two hexaphyrins is 54.2° and the bond length of the connecting C–C is 1.48 (1) Å. The first oxidation potentials of 10 were split at 0.31 and 0.15 V, indicating the electronic interaction between the two DNCRC [24]hexaphyrin units.

Reaction conditions: a NBS (1.0 equiv or 2.0 equiv.), 0 °C, CHCl3; b Ni(cod)2 (1.0 equiv), 2,2’-bipyridine (1.0 equiv), reflux, 24 h; c SPhos Pd G2 (10 mol %), K3PO4 (5.0 equiv.), THF/H2O = 20:1, reflux, 36 h.

a Top view of 10, b side view of 10, c top view of 12, and d side view of 12. Ellipsoids are drawn at the 30% probability level. All hydrogen atoms are omitted for clarity. Meso-substituents of porphyrin parts are omitted for clarity. Nickel atom, green.
Dibromide 8b was obtained in 86% yield by treating 8 with 2 equiv. NBS at 0 °C. The parent ion peak of 8b was observed at m/z = 1274.0660 (calcd for (C64H52Br2N6Pd2)+ = 1274.0701 ([M]+)) and its 1H NMR spectrum showed two doublets at 5.22 and 4.84 ppm (J = 4.7 Hz) and two singlets at 5.20 and 4.23 ppm, indicating a symmetric substitution. The single-crystal structure of 8b was also determined by X-ray diffraction analysis, which shows its centrosymmetric dibromide feature. Finally, the Suzuki–Miyaura coupling reaction between 8b and 5-boryl-10,15,20-triarylporphyrinato NiII 11 gave NiII porphyrin-DNCRC [24hexaphyrin-NiII porphyrin triad 12, in 43% yield. The parent ion peak of 12 was observed at m/z = 2975.2621 (calcd for (C188H194N14Ni2Pd2)+ = 2975.2429 ([M]+)). The 1H NMR spectrum showed two singlets at 5.94 and 5.58 ppm and a pair of doublets at 5.19 and 4.70 ppm, revealing its symmetric structure. The dihedral angle between the hexaphyrin and NiII porphyrin is 58.9° and the newly formed C–C bonds are 1.47 (1) Å long.
The absorption spectra of 8, 10, and 12 are shown in Fig. 7b. Dimer 10 shows bands at 395, 503, 635, 712, and 796 nm along with a weak tail. The three bands observed at longer wavelengths are considerably red-shifted in comparison to those of 8. Triad 12 displays a Soret band of the NiII porphyrins at 416 nm. A red-shifted band of the DNCRC [24]hexaphyrin observed at 792 nm suggests its substantial electronic interaction with the NiII porphyrin.
In summary, 1,14-dibromotripyrrin 3b underwent a self-coupling reaction under Pd-catalyzed Suzuki–Miyaura coupling conditions, giving DNCRC [24]hexaphyrin(1.1.0.1.1.0) PdII complexes 8 and 9, along with 1,14-dibromotripyrrin–benzalacetone coupled products 6 and 7. While triphyrin 6 is an aromatic [18] triphyrin(5.1.1), DNCRC [24]hexaphyrins 8 and 9 are stable antiaromatic expanded NCP showing paratropic ring currents, ill-defined absorption spectra with red-shifted weak absorption bands, and small HOMO–LUMO gaps. Monobromide 8a was reductively dimerized to give 10 and dibromide 8b was coupled with 11 to give NiII porphyrin-DNCRC [24hexaphyrin-NiII porphyrin triad 12.
Methods
Materials and characterization
1H NMR spectra (500 MHz) were taken on a Bruker ADVANCE-500 spectrometer, and chemical shifts were reported as the delta scale in ppm relative to CHCl3 (δ = 7.260 ppm) as an internal reference. UV/Vis absorption spectra were recorded on a Shimadzu UV-3600 spectrometer. MALDI-TOF mass spectra were obtained with a Bruker ultrafleXtreme MALDI-TOF/TOF spectrometer with trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as a matrix. X-ray data were taken on an Agilent Supernova X-ray diffractometer equipped with a large area CCD detector. Redox potentials were measured by cyclic voltammetry on a CHI900 scanning electrochemical microscope. Toluene and THF were distilled after refluxing with Na and benzophenone ketyl indicator under an argon atmosphere and stored over 3 Å molecular sieves in a glovebox for at least 12 h prior to use. DMF was refluxed with CaH2 under an argon atmosphere for at least 3 h and distilled prior to use. Unless otherwise noted, materials obtained from commercial suppliers were used without further purification.
Synthesis of 5 and 6
A suspension of 1,14-diboryltripyrrane 3a (214.0 mg, 0.30 mmol), 2,4-dibromo-1-methoxybenzene (53.2 mg, 0.20 mmol), Pd2(dba)3 (18.3 mg, 0.02 mmol), X-Phos (38.1 mg, 0.08 mmol), Cs2CO3 (130.3 mg, 0.40 mmol), and CsF (60.7 mg, 0.40 mmol) in toluene/DMF suspension (4 mL/2 mL) was degassed through three freeze–pump–thaw cycles, and the reaction flask was purged with argon. The resulting mixture was refluxed for 48 h. The reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate. Evaporation of the solvent followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:4, v/v) and recrystallization with CH2Cl2/MeOH gave 5 as green solids (5.6 mg, 0.010 mmol, 5% yield) and 6 as dark green solids (1.4 mg, 0.0020 mmol, 1% yield). 5: 1H NMR (500 MHz, CDCl3) δ = 17.85 (br, 1H, N-H), 12.50 (d, 1H, J = 2.0 Hz, Ph-H), 7.36 (dd, 1H, Ph-H), 6.87 (s, 4H, Mes-m-H), 6.69 (d, 1H, J = 4.5 Hz, β-H), 6.66 (d, 1H, J = 4.5 Hz, Ph-H), 6.58 (d, 1H, J = 4.5 Hz, β-H), 6.48 (d, 1H, J = 4.5 Hz, β-H), 6.46 (d, 1H, J = 4.5 Hz, β-H), 5.57 (m, 2H, β-H), 2.87 (s, 3H, OMe-H), 2.30 (s, 6H, Me-H) and 2.16 (s, 12H, Me-H) ppm. HR-MS (MALDI-TOF-MS): m/z = 561.2794, calcd for (C39H35N3O)+ = 561.2775 ([M]+).
6: 1H NMR (500 MHz, CDCl3) δ = 9.74 (s, 2H, vinyl-H), 8.36 (d, J = 4.0 Hz, 2H, β-H), 8.31 (s, 2H, β-H), 8.20 (d, J = 7.2 Hz, 4H, Ph-H), 8.08 (dd, J = 4.0, 1.5 Hz, 2H, β-H), 7.73 (t, 4H, Ph-H), 7.67 (t, J = 6.0 Hz, 2H, Ph-H), 7.28 (s, 4H, Mes-m-H), 3.87 (s, 2H, N-H), 2.63 (br, 6H, Me-H), 1.89 (br, 12H, Me-H) and −4.13(s, 1H, N-H) ppm. 13C NMR (126 MHz, CDCl3) δ = 166.7, 145.4, 139.8, 137.8, 137.6, 137.1, 134.8, 134.6, 132.7, 132.6, 128.2, 128.0, 127.9, 127.5, 127.1, 123.8, 123.4, 122.0, 106.8, 21.6 and 21.1 ppm. UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 419 (21,000), 444 (25,000), and 652(6300) nm. HR-MS (MALDI-TOF-MS): m/z = 689.3441, calcd for (C49H43N3O)+ = 689.3401 ([M]+).
Synthesis of 6, 7, 8 and 9
A suspension of 1,14-dibromotripyrrin (100.0 mg, 0.16 mmol), Pd2(dba)3 (50.0 mg, 0.05 mmol), X-Phos (38.1 mg, 0.08 mmol), Cs2CO3 (130.3 mg, 0.40 mmol), and CsF (60.7 mg, 0.40 mmol) in toluene/DMF (4 mL/2 mL) was degassed through three freeze–pump–thaw cycles, and the reaction flask was purged with argon. The resulting mixture was stirred at reflux for 48 h. The reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate. Evaporation of the solvent followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:3, v/v) and recrystallization with CH2Cl2/MeOH gave 6 as dark green solids (1.10 mg, 0.0016 mmol, 1% yield), 7 as blue solids (2.4 mg, 0.003 mmol, 2% yield), 8 as gray-green solids (8.4 mg, 0.0075 mmol, 9% yield), and 9 as emerald green solids (5.2 mg, 0.005 mmol, 10% yield).
7: 1H NMR (500 MHz, CDCl3) δ = 17.17 (br, 1H, OH), 7.29 (d, J = 7.5 Hz, 2H, Ph-o-H), 7.13-7.08 (m, 5H, Ph-H), 7.04 (t, J = 7.1 Hz, 1H, Ph-p-H), 6.99 (d, 2H, J = 7.5 Hz, Ph-o-H), 6.91 (s, 1H, Mes-m-H), 6.89 (s, 1H, Mes-m-H), 6.88 (s, 1H, Mes-m-H), 6.87 (s, 1H, Mes-m-H), 6.60–6.59 (m, 2H, β-H), 6.51 (d, J = 4.5 Hz, 1H, β-H), 6.26 (d, J = 4.3 Hz, 1H, β-H), 6.24 (d, J = 5.0 Hz, 1H, β-H), 5.91 (d, J = 4.3 Hz, 1H, β-H), 5.68 (d, J = 3.0 Hz, 1H, sp3C-H), 4.23 (d, J = 7.7 Hz, 1H, sp3C-H), 3.25 (dd, J = 16.5, 7.8 Hz, 1H, sp3C-H), 2.63 (dd, J = 16.5, 3.1 Hz, 1H, sp3C-H), 2.34 (s, 3H, Me-H), 2.32 (s, 3H, Me-H), 2.18 (s, 3H, Me-H), 2.09 (s, 3H, Me-H), 2.05 (s, 3H, Me-H) and 1.96 (s, 3H, Me-H) ppm. 13C NMR (126 MHz, CDCl3) δ = 179.7, 179.0, 146.9, 144.6, 141.2, 141.0, 139.7, 139.5, 137.7, 137.6, 137.54, 137.47, 137.0, 136.9, 134.2, 133.8, 133.6, 131.7, 130.0, 128.51, 128.46, 128.2, 128.1, 128.0, 127.8, 126.8, 126.4, 122.2, 121.9, 118.7, 64.4, 54.6, 54.2, 45.9, 21.3, 21.2, 20.7, 20.4(2C) and 20.2 ppm. (The absence of some peaks in 13C NMR may be due to overlapping.) UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 355 (21,100), 400 (26,500), and 667 (6600) nm. HR-MS (MALDI-TOF-MS): m/z = 811.2408, calcd for (C49H43N3O2Pd)+ = 811.2402 ([M]+).
8: 1H NMR (500 MHz, CDCl3) δ = 6.72 (s, 4H, Mes-m-H), 6.68 (s, 4H, Mes-m-H), 5.20 (d, J = 4.5 Hz, 2H, β-H), 5.18 (d, J = 4.5 Hz, 2H, β-H), 5.12 (d, J = 4.5 Hz, 2H, β-H), 4.80 (d, J = 4.5 Hz, 2H, β-H), 4.02 (s, 2H, β-H), 2.25 (s, 12H, Me-H), and 2.18–2.16 (m, 24H, Me-H). 13C NMR (126 MHz, CDCl3) δ = 223.8, 161.5, 153.1, 152.0, 149.5, 148.3, 143.9, 138.5, 137.3, 137.1, 136.1, 132.5, 131.9, 131.8, 131.6, 128.5, 127.9, 127.8, 121.2, 120.4, 114.3, 29.8, 21.0, 19.9, and 19.4 ppm. (The absence of some peaks in 13C NMR may be due to overlapping). UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 340 (25,200), 396 (44,100), 502 (28,800), 618 (35,300), 690 (7100), 763 (7600), and 1082 (100) nm. HR-MS (MALDI-TOF-MS): m/z = 1120.2568, calcd for (C64H54N6Pd2)+ = 1120.2504 ([M]+).
9: 1H NMR (500 MHz, CDCl3) δ = 21.89 (s, 1H, N-H), 19.85 (s, 1H, N-H), 6.68-6.64 (m, 8H, Mes-m-H), 5.22 (d, J = 4.5 Hz, 1H, β-H), 5.13 (d, J = 4.5 Hz, 1H, β-H), 5.10 (dd, J = 4.5, 2.0 Hz, 1H, β-H), 5.07 (dd, J = 4.5, 2.0 Hz, 1H, β-H), 5.02 (d, J = 4.5 Hz, 1H, β-H), 4.88 (d, J = 4.5 Hz, 1H, β-H), 4.73 (d, J = 4.5 Hz, 1H, β-H), 4.62 (d, J = 4.5 Hz, 1H, β-H), 4.30 (s, 1H, β-H), 3.79 (s,1H, β-H) and 2.22-2.12 (m, 36H, Me-H). 13C NMR (126 MHz, CDCl3) δ = 215.8, 187.1, 166.2, 157.6, 155.8, 155.4, 151.3, 149.7, 140.3, 137.64, 137.56, 137.1, 137.0, 135.9, 135.7, 132.8, 132.1, 131.9, 131.4, 130.7, 128.4, 128.0, 127.9, 126.7, 125.7, 124.4, 123.8, 123.0, 120.3, 116.0, 113.2, 110.7, 100.0, 29.7, 21.0, 20.0, 19.5, and 19.1 ppm. (The absence of some peaks in 13C NMR may be due to overlapping.) UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 364 (29,400), 487 (26,700), 628 (21,200), 987 (200), and 1119 (200) nm. HR-MS (MALDI-TOF-MS): m/z = 1030.3687, calcd for (C64H56N6OPd)+ = 1030.3566 ([M]+).
Synthesis of 8a
A solution of NBS (3.2 mg, 0.018 mmol) in CHCl3 (10 mL) was added to a solution of 8 (20.0 mg, 0.018 mmol) in CHCl3 (10 mL) dropwise at 0 °C. After the consumption of 8 was confirmed by TLC monitoring, the reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate, evaporation of the solvent was followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:4, v/v) and recrystallization with CH2Cl2/MeOH. 8a was obtained as grass-green solids (16.7 mg, 0.014 mmol, 78% yield).
8a: 1H NMR (500 MHz, CDCl3) δ = 6.71 (s, 4H, Mes-m-H), 6.67 (s, 4H, Mes-m-H), 5.23 (d, J = 4.5 Hz, 1H, β-H), 5.21 (d, J = 4.5 Hz, 1H, β-H), 5.15 (s, 1H, β-H), 5.14 (s, 1H, β-H), 5.13 (s, 1H, β-H), 4.81-4.80 (m, 2H, β-H), 4.17 (s, 1H, β-H), 4.05 (s, 1H, β-H), 2.22 (br, 6H, Me-H), 2.21 (br, 6H, Me-H), and 2.18-2.15 (m 24H, Me-H). HR-MS (MALDI-TOF-MS): m/z = 1196.1615, calcd for (C64H53BrN6Pd2)+ = 1196.1601 ([M]+).
Synthesis of 8b
A solution of NBS (6.4 mg, 0.036 mmol) CHCl3 (10 mL) was added to a solution of 8 (20.0 mg, 0.018 mmol) in CHCl3 (20 mL) dropwise at 0 °C. After the consumption of 8 and 8a was confirmed by TLC monitoring, the reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate, Evaporation of the solvent was followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:4, v/v) and recrystallization with CH2Cl2/MeOH gave 8b as grass green solids (19.7 mg, 0.016 mmol, 86% yield).
8b: 1H NMR (500 MHz, CDCl3) δ = 6.72 (s, 4H, Mes-m-H), 6.68 (s, 4H, Mes-m-H), 5.21 (d, J = 4.5 Hz, 2H, β-H), 5.20 (s, 2H, β-H), 4.84 (d, J = 4.5 Hz, 2H, β-H), 4.22 (s, 2H, β-H), 2.22 (br, 12H, Me-H), 2.18 (br, 6H, Me-H), and 2.16 (br, 18H, Me-H). HR-MS (MALDI-TOF-MS): m/z = 1274.0660, calcd for (C64H52Br2N6Pd2)+ = 1274.0701 ([M]+).
Synthesis of 9Cu
A suspension of 9 (20.0 mg, 0.019 mmol), Cu(OAc)2 (37.8 mg, 0.19 mmol), and NaOAc (15.6 mg, 0.19 mmol) in CHCl3/MeOH (20 mL/10 mL) was stirred at 65 °C overnight. The solvent was removed under reduced pressure. The product was separated by silica-gel column chromatography (eluent:CH2Cl2/n-hexane = 1:1, v/v) and recrystallization with CH2Cl2/MeOH gave 9Cu as brown crystals (17.6 mg, 0.016 mmol, 85% yield). UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 368 (38,200), 472 (33,300), 630 (24,000), 994 (300) and 1166 (200) nm. HR-MS (MALDI-TOF-MS): m/z = 1091.2730, calcd for (C64H54CuN6OPd)+ = 1091.2702([M + H]+).
Synthesis of 9Pd and 9Ni
By following the method used for the synthesis of 9Cu—9Pd and 9Ni were synthesized upon treatment with Pd(OAc)2 and Ni(OAc)2·4H2O in 80% and 95% yields. 9Pd: 1H NMR (500 MHz, CDCl3) δ = 6.90 (br, 1H, Mes-m-H), 6.76 (br, 1H, Mes-m-H), 6.73 (br, 1H, Mes-m-H), 6.72 (br, 2H, Mes-m-H), 6.69 (br, 1H, Mes-m-H), 6.65 (br, 2H, Mes-m-H), 5.40 (d, J = 4.6 Hz, 1H, β-H), 5.38–5.33 (m, 4H, β-H), 5.10 (d, J = 4.6 Hz, 1H, β-H), 5.00 (d, J = 4.6 Hz, 1H, β-H), 4.86–4.83 (m, 2H, β-H), 4.09 (s, 1H, β-H), 2.85 (s, 3H, Me-H), 2.37 (s, 3H, Me-H), 2.31 (s, 3H, Me-H), 2.22 (s, 3H, Me-H), 2.20 (s, 3H, Me-H), 2.19 (s, 9H, Me-H), 2.16 (s, 3H, Me-H), 2.02 (s, 3H, Me-H), 1.99 (s, 3H, Me-H), and 1.65 (s, 3H, Me-H). 13C NMR (126 MHz, CDCl3) δ = 212.4, 184.4, 159.0, 158.0, 154.0, 150.4, 150.2, 150.0, 147.7, 145.9, 142.8, 141.7, 137.7, 137.4, 137.3, 137.2, 136.8, 136.5, 136.3, 136.3, 136.1, 136.1, 136.0, 136.0, 132.2, 131.6, 131.2, 131.2, 129.9, 129.9, 129.3, 128.2, 128.0, 127.8, 127.8, 127.7, 124.5, 121.5, 120.3, 118.0, 116.8, 113.2, 21.0, 21.0, 20.8, 20.2, 19.7, 19.6 and 19.4 ppm. (The absence of some peaks in 13C NMR may be due to overlapping.) UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 411 (26,600), 316 (5700) and 490 (38,000) nm. HR-MS (MALDI-TOF-MS): m/z = 1136.2426, calcd for (C64H54N6OPd2)+ = 1136.2453 ([M + H]+).
9Ni: 1H NMR (500 MHz, CDCl3) δ = 6.89 (br, 1H, Mes-m-H), 6.70 (br, 1H, Mes-m-H), 6.68 (br, 1H, Mes-m-H), 6.68 (br, 1H, Mes-m-H), 6.65 (br, 1H, Mes-m-H), 6.63 (br, 1H, Mes-m-H), 6.60 (br, 2H, Mes-m-H), 5.20 (d, 1H, J = 4.6 Hz, β-H), 5.14-5.12 (m, 2H, β-H), 5.10 (d, 1H, J = 4.6 Hz, β-H), 5.07 (d, 1H, J = 4.6 Hz, β-H), 4.90 (d, 1H, J = 4.6 Hz, β-H), 4.83 (d, 1H, J = 4.6 Hz, β-H), 4.61-4.60 (m, 2H, β-H), 3.77 (s, β-H), 2.95 (s, 3H, Me-H), 2.23 (s, 3H, Me-H), 2.22 (s, 3H, Me-H), 2.20 (s, 3H, Me-H), 2.20 (s, 3H, Me-H), 2.18 (s, 3H, Me-H), 2.16 (s, 3H, Me-H), 2.15 (s, 3H, Me-H), 2.12 (s, 3H, Me-H), 2.10 (s, 3H, Me-H), 2.08 (s, 3H, Me-H) and 1.62 (s, 3H, Me-H). 13C NMR (126 MHz, CDCl3) δ = 217.9, 187.1, 160.2, 158.7, 154.42, 154.39, 152.30, 152.0, 151.3, 147.6, 146.6, 144.1, 143.39, 139.8, 137.5, 137.3, 137.2, 136.0, 135.9, 132.0, 131.6, 131.4, 131.0, 130.2, 128.5, 128.2, 128.0, 127.9, 127.6, 122.6, 121.1, 119.9, 118.5, 113.2, 29.7, 21.0, 20.9, 20.8, 20.0, 19.8, 19.5, 19.4, 19.3 and 19.2 ppm. (The absence of some peaks in 13C NMR may be due to overlapping.) UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 393 (19,000), 836 (4000) and 931 (4600) nm. HR-MS (MALDI-TOF-MS): m/z = 1086.2635, calcd for (C64H54NiN6OPd)+ = 1086.2758 ([M]+).
Synthesis of 10
A THF solution (5 mL) of 8a (20.0 mg, 0.017 mmol), Ni(cod)2 (4.6 mg, 0.017 mmol), and 2,2’-bipyridine (2.7 mg, 0.017 mmol) in a 50 mL Schlenk tube was purged with argon. The mixture was refluxed for 24 h. The reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate. Evaporation of the solvent followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:3, v/v) and recrystallization with CH2Cl2/MeOH gave 10 as green solids (11.6 mg, 0.005 mmol, 61% yield).
10: 1H NMR (500 MHz, CDCl3) δ = 6.81 (s, 2H, Mes-m-H), 6.70-6.69 (m, 8H, Mes-m-H), 6.62 (s, 2H, Mes-m-H), 6.59 (s, 2H, Mes-m-H), 5.13 (d, 2H, J = 4.5 Hz, β-H), 5.12 (d, 2H, J = 4.5 Hz, β-H), 5.08 (d, 2H, J = 4.5 Hz, β-H), 4.95 (d, 2H, J = 4.0 Hz, β-H), 4.70 (d, 2H, J = 4.5 Hz, β-H), 4.69 (d, 2H, J = 4.0 Hz, β-H), 4.30 (s, 2H, β-H), 3.91 (s, 2H, β-H), 3.74 (s, 2H, β-H), 2.59 (s, 6H, Me-H), 2.37 (s, 6H, Me-H), 2.23 (br, 18H, Me-H), 2.19 (br, 9H, Me-H), 2.17 (br, 9H, Me-H), 2.13 (br, 9H, Me-H), 2.12 (br, 9H, Me-H), 2.05 (s, 6H, Me-H) and 2.02 (s, 6H, Me-H). UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 395 (72600), 503 (48,400), 635 (55,200), 712 (18,700), and 796 (30,700) nm. HR-MS (MALDI-TOF-MS): m/z = 2234.4843, calcd for (C128H106N12Pd4)+ = 2234.4862 ([M]+).
Synthesis of 12
A THF–H2O suspension (2 mL/0.1 mL) of 8b (20.0 mg, 0.016 mmol), 11 (34.0 mg, 0.032 mmol), Sphos Pd G2 (1.2 mg, 0.0016 mmol) and K3PO4 (6.5 mg, 0.027 mmol) in a 50 mL Schlenk tube was purged with argon. The mixture was refluxed for 36 h. The reaction mixture was diluted with CHCl3. The organic layer was separated and washed with water, and dried over anhydrous sodium sulfate. Evaporation of the solvent followed by silica-gel column chromatography (eluent: CH2Cl2/n-hexane = 1:3, v/v) and recrystallization with CH2Cl2/MeOH gave 12 as dark green solids (20.5 mg, 0.007 mmol, 43% yield).
1H NMR (500 MHz, CDCl3) δ = 8.89 (d, J = 4.9 Hz, 4H, Por-β-H), 8.75 (dt, J = 8.6, 4.9 Hz, 12H, Por-β-H), 7.76 (s, 4H, Por-Ar-H), 7.71 (s, 2H, Por-Ar-H), 6.74 (s, 4H, Mes-m-H), 5.94 (s, 2H, β-H), 5.58 (s, 2H, β-H), 5.19 (d, J = 4.5 Hz, 2H, β-H), 4.70 (d, J = 4.5 Hz, 2H, β-H), 2.45 (s, 12H, Me-H), 2.14 (s, 6H, Me-H), 1.70 (s, 18H, Me-H) and 1.60 (s, 108H, t-Bu-H). UV/Vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 416 (115,600), 636 (18,300), 714 (9300) and 792 (11,200) nm. HR-MS (MALDI-TOF-MS): m/z = 2975.2621, calcd for (C188H194N14Ni2Pd2)+ = 2975.2429 ([M]+).
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2239867, 2239868, 2239869, 2239872, 2239870, 2239871, 2278452, 2239873, 2239874 (6, 7, 8, 8a, 8b, 9, 9Pd, 10, 12). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Original 1H NMR and 13C NMR spectra, UV/vis absorption spectra, X-ray crystal data, electrochemical data, HR-MS Spectra, and DFT calculation results of the compounds generated in this study are provided in the Supplementary Information file.
Change history
31 August 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41467-023-41072-5
References
Sessler, J. L. & Seidel, D. Synthetic expanded porphyrin chemistry. Angew. Chem. Int. Ed. 42, 5134 (2003).
Saito, S. & Osuka, A. Expanded porphyrins: intriguing structures, electronic properties, and reactivities. Angew. Chem. Int. Ed. 50, 4342 (2011).
Stępień, M., Sprutta, N. & Latos-Grażyński, L. Figure eights, Möbius bands, and more: conformation and aromaticity of porphyrinoids. Angew. Chem. Int. Ed. 50, 4288 (2011).
Tanaka, T. & Osuka, A. Chemistry of meso-aryl-substituted expanded porphyrins: aromaticity and molecular twist. Chem. Rev. 117, 2584 (2017).
Szyszko, B., Bialek, M. J., Pacholska-Dudziak, E. & Latos-Grażyński, L. Flexible porphyrinoids. Chem. Rev. 117, 2839 (2017).
Furuta, H., Asano, T. & Ogawa, T. “N-confused porphyrin”: a new isomer of tetraphenylporphyrin. J. Am. Chem. Soc. 116, 767 (1994).
Chmielewski, P. J., Latos-Grażyński, L., Rachlewicz, K. & Glowiak, T. Tetra-p-tolylporphyrin with an inverted pyrrole ring: a novel isomer of porphyrin. Angew. Chem. Int. Ed. 33, 779 (1994).
Furuta, H., Maeda, H. & Osuka, A. Confusion, inversion, and creation—a new spring from porphyrin chemistry. Chem. Commun. 1795–1804 (2002).
Toganoh, M. & Furuta, H. Creation from confusion and fusion in the porphyrin world-the last three decades of N-confused porphyrinoid chemistry. Chem. Rev. 122, 8313 (2022).
Srinivasan, A., Ishizuka, T., Osuka, A. & Furuta, H. Doubly N-confused hexaphyrin: a novel aromatic expanded porphyrin that complexes bis-metals in the core. J. Am. Chem. Soc. 125, 878 (2003).
Srinivasan, A., Ishizuka, T. & Furuta, H. Doubly N-fused pentaphyrin. Angew. Chem. Int. Ed. 43, 876 (2004).
Suzuki, M. et al. A new entry to doubly N-confused [26]hexaphyrins(1.1.1.1.1.1) from normal [26]hexaphyrins(1.1.1.1.1.1) through an unprecedented double pyrrolic rearrangement. Chem. Eur. J. 12, 1754 (2006).
Hisamune, Y. et al. Stable π radical from a contracted doubly N-confused hexaphyrin by double palladium metalation. Angew. Chem. Int. Ed. 54, 7323 (2015).
Yamasumi, K. et al. Bis-copper(II)/π-radical multi-heterospin system with non-innocent doubly N-confused dioxohexaphyrin(1.1.1.1.1.0) ligand. Chem. Eur. J. 23, 15322 (2017).
Wang, Y. et al. Near-infrared-III-absorbing and -emitting dyes: energy-gap engineering of expanded porphyrinoids via metallation. Angew. Chem. Int. Ed. 59, 16161 (2020).
Liu, L. et al. m-Benziporphyrin(1.1.0.0)s as a rare example of ring-contracted carbaporphyrins with metal-coordination ability: distorted coordination structures and small HOMO–LUMO gaps. Chem. Eur. J. 29, e202203517 (2023).
Lash, T. D. & Ferrence, G. M. Metalation and selective oxidation of diphenyl-23-oxa-, -thia-, and -selena-21-carbaporphyrins. Inorg. Chem. 56, 11426 (2017).
Berlicka, A. et al. 21-Carba-23-oxaporphyrinoids and 21-oxo-21-carba-23-oxaporphyrinoids: macrocyclic π-conjugation involving the carbonyl moiety. Org. Chem. Front. 9, 5440 (2022).
Furuta, H. et al. N-confused double-decker porphyrins. Inorg. Chem. 39, 5424 (2000).
Maeda, H. et al. N-confused porphyrin-bearing meso-perfluorophenyl groups: a potential agent that forms stable square-planar complexes with Cu(II) and Ag(II). Org. Lett. 5, 1293 (2003).
Ishizuka, T., Yamasaki, H., Osuka, A. & Furuta, H. Syntheses of aryl- and arylethynyl-substituted N-confused porphyrins. Tetrahedron 63, 513 (2007).
Acknowledgements
The work is supported by the National Natural Science Foundation of China (Grant Nos. 22071052 (J.S.), 21772036 (J.S.), 22271091 (L.X.), 22201072 (Y.R.)), Science and Technology Planning Project of Hunan Province (2018TP1017 (J.S.)), and Science and Technology Innovation Program of Hunan Province (2021RC4059 (J.S.), 2022WZ1019 (M.Z.)).
Author information
Authors and Affiliations
Contributions
J.S. designed and conducted the project. F.L., L.L., and H.W. performed the synthesis and characterization and measured the optical and electrochemical properties. L.X., M.Z., and Y.R. performed X-ray diffraction analysis and DFT calculations. A.O. and J.S. prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ablikim Kerim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, F., Liu, L., Wu, H. et al. Doubly N-confused and ring-contracted [24]hexaphyrin Pd-complexes as stable antiaromatic N-confused expanded porphyrins. Nat Commun 14, 5028 (2023). https://doi.org/10.1038/s41467-023-40700-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40700-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
