
Abstract
Sulfonyl and sulfonimidoyl fluorides are versatile substrates in organic synthesis and medicinal chemistry. However, they have been exclusively used as S(VI)+ electrophiles for defluorinative ligations. Converting sulfonyl and sulfonimidoyl fluorides to S(VI) radicals is challenging and underexplored due to the strong bond dissociation energy of SVI−F and high reduction potentials, but once achieved would enable dramatically expanded synthetic utility and downstream applications. In this report, we disclose a general platform to address this issue through cooperative organosuperbase activation and photoredox catalysis. Vinyl sulfones and sulfoximines are obtained with excellent E selectivity under mild conditions by coupling reactions with alkenes. The synthetic utility of this method in the preparation of functional polymers and dyes is also demonstrated.
Similar content being viewed by others


Introduction
Compared to the chloride analogues, sulfonyl and sulfonimidoyl fluorides (SFs) are relatively inert due to the high reduction potential and bond strength of SVI−F (Fig. 1a)1,2. They have exhibited better reaction selectivity and improved stability to heat, hydrolysis, and reduction3,4,5,6. In addition, SFs are optically stable at the stereogenic sulfur(VI) centers, whereas the chloride analogues are liable to racemization7,8,9,10,11. These unique advantages have made SFs useful in synthesis12,13,14,15,16,17,18 and laid the foundation for the recent sulfur(VI) fluoride-exchange (SuFEx) click chemistry19,20,21,22.

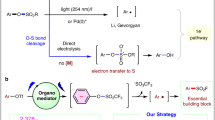
a The properties of sulfonyl and sulfonimidoyl halides (F, Cl). b Different mechanisms of activating SFs. c Our S(VI) radical ligation platform.
Activation of the SFs for fluoride-exchange ligations often requires stringent conditions23,24. Vorbrüggen and Gembus found that organosuperbases were able to promote nucleophilic substitution of sulfonyl fluorides by heteroatoms25,26. This strategy has been widely used and further developed by several groups27,28,29,30,31,32. Hydrogen bonding was another potential driving force for SVI−F activation33, especially during covalent capture of biomolecules under physiological conditions19,34,35,36,37,38. Strong Lewis acids could also abstract fluoride to induce Friedel-Craft-type sulfonylations39,40,41 and sulfoximidations42,43. However, almost all reported reactions were based on the heterolytic SVI−F cleavage mechanism, where SFs served as S(VI)+ electrophiles.
Generating S(VI) radicals from SFs is still underexplored to date, but once achieved would greatly expand their synthetic utility. The excellent balance between thermal stability and kinetic reactivity of SFs makes them superior to many reported S(VI) radical precursors, especially when dealing with complex and challenging systems. For example, sulfonyl chlorides are well known for radical sulfonylations (−1.30 V for PhSO2Cl, Fig. 1a)44,45,46,47, but are often accompanied by hydrolysis and unwanted chlorination products in many reactions (Fig. 1b)48,49. The use of sulfinates as radical S(VI) reagents is also limited because of their high sensitivity to oxidants. During the preparation of this manuscript, the Luo and Molander groups reported radical sulfonylations of aryl sulfonyl fluorides50,51. However, sulfonimidoyl fluorides were not discussed and a general method for radical ligation of a variety of SFs is still lacking.
Direct homolytic or reductive cleavage of SVI−F bonds to S(VI) radicals is challenging due to their high bond strength and reduction potentials (−1.74 V vs SCE for PhSO2F)2. In recent studies52,53,54,55,56, FSO2• radical was generated from FSO2Cl or related precursors by photocatalysis, but SVI−F cleavage to RSO2• was not observed from either the reagents or the products. Inspired by the emerging use of photoredox-based dual catalysis as an appealing approach for inert bond functionalization57,58,59,60,61,62, we envisioned that a cooperative strategy might be worth trying. We hypothesized that intermediates RSO(X)[B]+ derived from the organosuperbase activation of SFs could be converted in situ to related S(VI) radicals upon visible light irradiation in the presence of photocatalyst, which were then trapped by substrates to complete a reaction cycle (Fig. 1c)63,64. Herein, we report that this protocol is generally applicable for converting SFs electrophiles to S(VI) radicals in the reaction with alkenes.
Results
We initiated the study using phenyl sulfonyl fluoride 1a and styrene 2a as model substrates and Ru(bpy)3Cl2 as photocatalyst. And representative results were summarized in Fig. 2 (see Supplementary Information for details). Under the blue LED illumination with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as an additive in dry CH3CN, nearly quantitative sulfonylation product 3aa was obtained in absolute E configuration at room temperature (entry 1). Both visible light and the photocatalyst were required for this reaction (entry 2−3). Without or with less than 3.5 eq. of DBU, no or incomplete conversion of 1a was observed (entry 4−5). There was no reaction using inorganic bases such as Cs2CO3 (entry 6). Et3N was an effective sacrificial reduction reagent in promoting many photoredox reactions65,66,67. However, it did not work for this reaction (entry 7). Only organosuperbases with similar pKb values to DBU were effective68, including DBN, BTMG and MTBD (entry 8−10). Ru(bpy)3Cl2 was found optimal over other photocatalysts at a 1.5 mol% loading (entry 11−13). Considering the solvents, the use of DMSO also gave a quantitative yield of 3aa, while the less polar DCE resulted in no conversion (entry 14−15). CH3CN was used in the following studies because of its low boiling point and ease for handling. Sulfonyl chloride was investigated as a substitute for 1a, but decomposed completely to give only a trace amount of 3aa (entry 16). During the reaction optimization, the light on/off experiment was also performed with a time interval of 2 hours. We found that the reaction stopped completely when the light was turned off, but recovered when the blue LED was turned on (Fig. 2, bottom right). This result was helpful in elucidating the reaction mechanism and was further discussed in the mechanism study part of this manuscript.

aStandard reaction conditions: 1a (0.4 mmol), 2a (0.2 mmol), Ru(bpy)3Cl2 (1.5 mol%), DBU (0.7 mmol), dry CH3CN (2.0 mL), blue LED (5 W), RT, N2 atmosphere. bDetermined by 1H NMR. cisolated yield.
Using the established conditions, the scope of aryl sulfonyl fluorides was investigated (Fig. 3). Substrates bearing either electron-donating or withdrawing substituents on arenes para to the sulfonyl fluoride gave sulfonylation products in high yields (3aa–3ja). However, the reaction rate was significantly accelerated by electron-deficient substituents, allowing the reaction to be completed within minutes (3da, 3fa, 3ia). Substrates bearing meta or ortho substituents also gave satisfactory yields (3ka–3oa). The mild conditions tolerated functional groups such as halides (3ha, 3ma), cyanide (3ia), and carboxylic esters (3ja) that allowed for post-functionalization.

a1x (0.4 mmol), 2y (0.2 mmol), Ru(bpy)3Cl2 (1.5 mol%), DBU (0.7 mmol), CH3CN (2.0 mL), blue LED (5 W), RT, under N2 atmosphere. bLinear internal alkene 3fo’ in 8% yield.
Heterocycles were ubiquitous and important building blocks in natural and synthetic molecules. The heteroaryl sulfonyl chlorides were often highly reactive and susceptible to decomposition, whereas their fluoride analogues were much more stable for storage and use. In Fig. 3, we highlighted that this method was applicable to a variety of heteroaryl compounds, including coumaran, benzofuran, thiophene, benzothiozole, quinoline, pyridine, 4-quinazolinone, and indazole (3na–3wa). Further functionalization of drug-related molecules has also been demonstrated, such as the Celecoxib and Neratinib derivatives 3xa and 3ya. In all these reactions, vinyl sulfones were obtained in absolute E configuration, as confirmed by X-ray analysis of the product 3aa (CCDC: 2180194).
The scope of alkenes was next investigated with two model sulfonyl fluorides of opposed electron properties (1a and 1f, Fig. 3). One limitation of this method at this stage is that only styrene derivatives work. Aliphatic alkenes were unreactive, probably due to the mismatched redox potentials of related intermediates in the catalytic cycle. Again, the reactions were very fast with the electron-deficient substrate 1f, while it usually took hours to reach full conversions with the electron-rich substrate 1a. A standard reaction time of 14 hours was used for the remaining studies for consistency. Yields were good regardless of the electron properties and steric effects of the styrene derivatives (3fb–3fm). The 1,1-disubstituted alkene (3fn) and the 1,2-disubstituted alkene (3ap) worked equally well. Notably, a terminal alkene 3fo was the major product instead of the intramolecular isomer 3fo’, probably because that it was kinetically favored in the DBU-deprotonation step (see below). The modification of functional molecules and their derivatives was also demonstrated, including Menthol, Thymol, and Galactose (3fr–3ft).
Divinyl sulfones had unique utility as dyes69, building blocks70,71, anti-inflammatory agents, and tumor cell growth inhibitors72,73. But there was no reliable synthetic access to them, especially for the unsymmetrical ones. Reported routes installed the vinyl double bond by using aldehyde-based Wittig-Horner reactions, which often gave E/Z mixtures69, or by using multi-step sequences under harsh reaction conditions71,74. In a related study, vinyl sulfonyl chlorides were treated with styrene derivatives to afford atom transfer radical addition adducts. An additional step was required to afford divinyl sulfones at high temperatures, accompanied by uncontrollable desulfonylation side reactions75. The vinyl sulfonyl fluorides were good Michael acceptors and SuFEx electrophiles. Methods for their preparation and applications were readily available52,76,77,78. Encouraged by the radical aryl sulfonylation, we then became interested in extending the chemistry to vinyl sulfonyl fluorides. In this context, unsymmetrical divinyl sulfones could be prepared.
To our delight, the standard condition was applicable without further optimization (Fig. 4). Divinyl sulfones were obtained in exclusive E configuration. A variety of functional groups were tolerated, such as halides (5bj–5dj, 5lj, 5mj), thiol ether (5fj), carboxylic esters (5jj, 5oj), and cyanide (5rd). Multi-substituted vinyl sulfonyl fluorides also gave the desired products (5pa, 5qa). Styrene derivatives worked equally well, including heteroaryl alkenes (5sm, 5sp) and disubstituted alkene (5su). Complex skeletons could be constructed by taking advantage of the distinctive reactivity of multiple SuFEx handles. For instance, a fluorosulfate −OSO2F handle allowed the connection of Norestrone to 4-iodophenol via nucleophilic substitution. The resulting product was converted to vinyl sulfonyl fluoride and further enabled the preparation of 5ta with our method. The reaction could be extended to aliphatic sulfonyl fluorides, including the primary and secondary alkyl-substituted substrates (6aa, 6ba), but only ~10% yields of products were obtained. This was probably due to the deprotonation of α-methylene by DBU, which induced the decomposition of the sulfonyl fluorides.

a4x (0.4 mmol), 2y (0.2 mmol), Ru(bpy)3Cl2 (1.5 mol%), DBU (0.7 mmol), CH3CN (2.0 mL), blue LED (5 W), RT, under N2 atmosphere. b4x (0.2 mmol), Ru(bpy)3Cl2 (1.5 mol%), DBU (0.7 mmol), CH3CN (2.0 mL), blue LED (5 W), RT, under N2 atmosphere.
During the study of vinyl sulfonyl fluorides, we observed an unexpected reaction pathway. Namely, vinyl sulfonyl fluorides underwent self-condensation in the absence of styrene to give symmetrical divinyl sulfones in low yields (5aa, 5bb, 5hh, Fig. 4, bottom section). Initially, we thought a styrene intermediate might be generated in situ from the vinyl sulfonyl radical by releasing one molecule of SO2. But styrene was not found in the large-scale synthesis. And the deuterated product 5aa was also not observed when the reaction of 4a was carried out in CD3CN. Therefore, styrene was ruled out as a potential intermediate. It was likely that the vinyl sulfonyl radical could be directly added the double bond of another vinyl sulfonyl fluoride, resulting in the subsequent ejection of an FSO2• fragment79 and the final product.
Sulfoximines are useful building blocks in many fields80,81,82,83,84. But sulfoximine synthesis via the sulfonimidoyl radical was still rare. To date, only two protocols have been reported, both using sulfonimidoyl chlorides85,86. We found that our cooperative activation model was also able to convert sulfonimidoyl fluorides to the corresponding radicals after minor modification of the reaction conditions (see Supplementary Information). As depicted in Fig. 5, imine groups had an important influence on the reaction. Substrates with N-pivaloyl (Piv) group gave the best yield (8aa). Their analogues also afforded good yields (8ab–8ae). However, other N-functional groups such as the tosyl (Ts) were less effective (8af), giving mainly the reduction and hydrolysis products. The N-alkyl and N-aryl sulfonimidoyl fluorides were also not good substrates because of their poor electrophilicity42,43. The reaction was not sensitive to different electron or steric properties of styrene derivatives. Variations of the aryl group on sulfonimidoyl fluorides were also investigated and worked quite well. In general, this method is a good complement to the synthesis of vinyl sulfoximines87,88,89.

a4x (0.4 mmol), 2y (0.2 mmol), Ru(bpy)3Cl2 (1.0 mol%), DBU (0.7 mmol), DMF (2.0 mL), blue LED (24 W), RT, under N2 atmosphere.
Control experiments were subsequently carried out to gain deeper insight into the mechanism (Fig. 6a). The reaction was inhibited by radical scavengers, such as TEMPO and butylated hydroxytoluene (BHT). Treatment of 1f with the radical probe cyclopropylstyrene 2t under standard reaction conditions gave the ring expansion product 9. Together with the light on/off experiment in Fig. 2, these results supported a photoredox single electron transfer (SET) process, but excluded a radical chain reaction pathway. Besides, sulfonate ArSO3− and sulfinate ArSO2− salts were excluded as active intermediates, because they did not give any conversion under the standard reaction conditions. The essential cooperative effect of DBU for SVI−F activation was further confirmed through a modified experiment based on a previous report53. The vinyl sulfonyl fluoride 4a was obtained from the fluorosulfonylation of styrene under the photoredox reaction conditions. 4a did not undergo further SVI−F reaction in the absence of DBU. However, the addition of DBU to the same reaction mixture allowed the spontaneous conversion of 4a to 5aa by further reaction with styrene (Fig. 6a, last equation).

a Investigation of radical intermediates. b Hammett analysis performed with the styrene substrates. c 1H NMR study of the reaction progress in DMSO-d6. (Spectrum c-1: mixture of 0.1 mmol of 1f and 0.1 mmol of 1,3,5-trifluorobenzene as internal standard. Spectrum c-2: 15 min after the addition of 0.2 mmol of DBU. Spectrum c-3: 5 min after the reaction with 0.1 mmol of styrene in the presence of 1.5 mol% Ru(bpy)3Cl2 and blue LED illumination. Spectrum c-4: purified 3fa). d Proposed reaction mechanism.
A Hammett analysis of the relative reaction rate of the para-substituted styrenes indicated that the electron-rich substrates underwent faster reactions than the electron-deficient ones (Fig. 6b). Taken together with the olefin migration observed in product 3fo (Fig. 3), a benzylic cation intermediate should be involved in the late stage of the reaction cycle. In addition, we learned from Figs. 3 and 4 that the electron-rich sulfonyl fluorides reacted much more slowly than the electron-deficient ones (14 hours for 1a vs 5 mins for 1f).
The reaction of sulfonyl fluoride 1f with styrene was monitored by 1H and 19F NMR in deuterated CH3CN and DMSO (Supplementary Information, Page 66-71). A typical example was depicted in Fig. 6c. Substrate 1f was partially hydrolyzed to sulfonic acid after 15 min of incubation with DBU in DMSO-d6 at room temperature. This was because a trace amount of water could not be avoided in the deuterated solvents (Fig. 6c, c−2). A set of newly formed peaks Int probably belonged to the proposed intermediate RSO2[DBU]+, which was confirmed by the mass spectrum but could not be isolated due to its high activity (Supplementary Information, Page 69). Upon light irradiation in the presence of styrene and photocatalyst, the remaining sulfonyl fluorides were fully consumed and 3fa was obtained in 5 min. The Int species was also consumed (Fig. 6c, c−3). DBU was quantitatively recovered after the reaction, suggesting that it was not a sacrificial reagent for the photoredox cycle.
In the luminescence quenching experiments, DBU was able to quench the excited Ru(bpy)3Cl2, while other components were not (Supplementary Information, Page 84). Although this suggested that DBU might be directly involved in the early stage of the photoredox cycle, we still thought it unlikely that the reaction would proceed through a reductive quenching cycle for the following reasons. The reduction potential of Ru(II)*/Ru(I) was +0.77 V (vs SCE)59, which was known for reductive quenching by aliphatic amines such as Et3N (+0.83 V vs SCE) and i-Pr2NEt65,66,67. But we did not observe any reaction when Et3N and i-Pr2NEt were used during reaction optimization. The potential of DBU was even higher (+1.28 V vs SCE)90. Therefore, the reductive quenching of Ru(II)* by DBU did not make sense in this reaction. If this reductive quenching with DBU occurred for whatever reason, it remained challenging for Ru(I) (EII/I = −1.33 V vs SCE) to reduce sulfonyl fluorides. Because the reduction potential of PhSO2F was −1.74 V vs SCE. On the other hand, oxidative quenching of Ru(II)* by sulfonyl fluorides was also unlikely, because the reduction potential of Ru(II)*/Ru(III) was −0.83 V vs SCE. Hydrogen bonding between the sulfonyl fluoride and the in situ generated DBU•H+ was not observed by 1H and 19F NMR when the two reagents were incubated in CD3CN at a 1:2 ratio (ArSO2F vs DBU, Supplementary Information, Page 73−80). And considering that the experimental reaction rate of the electron-poor aryl sulfonyl fluoride is much faster than that of the electron-rich one, a reaction mechanism based on the hydrogen bonding activation of SFs seemed unlikely. However, we could not rule out other potential hydrogen bonding species as the active intermediate in our reaction.
A plausible reaction mechanism is proposed in Fig. 6d. Nucleophilic activation of sulfonyl fluoride by DBU was very fast at room temperature, especially with the electron-deficient substrates, giving the intermediate RSO2[DBU]+ by expelling a fluoride anion. This species was highly active and often ended up with hydrolysis to sulfonic acid in the presence of water. But under blue LED irradiation, it was reduced by the exited photocatalyst Ru(II)*. And a sulfonyl radical RSO2• was subsequently released via intramolecular fragmentation, then added to styrene that gave a benzylic radical. The radical was further oxidized to carbocation by Ru(III), which yielded vinyl sulfone after deprotonation by DBU. As mentioned above, only those organosuperbases that are effective in catalyzing the nucleophilic substitutions of sulfonyl fluorides could promote this photoredox reaction, such as DBU, BTMG, and MTBD.
We were curious about which ligation pathway the sulfonyl fluoride would be primarily involved when both the heteroatom nucleophile and the alkene were incubated in one reaction flask under the current reaction conditions, the radical sulfonylation or the conventional DBU-promoted nucleophilic substitution. We found the results varied depending on which competing nucleophile was used (Fig. 7a). Nucleophilic substitution dominated in the presence of phenol, giving sulfate in 90% yield. In contrast, radical sulfonylation became the main reaction when tert-butanol, morpholine, and aniline were used as nucleophiles. Both the nucleophilic and radical products were present in trace amounts using TMSN3 as an additive. These results were helpful for evaluating the relative rates of the two ligation pathways and their functional group tolerance.

a Competition reaction between the radical and nucleophilic substitution. b Gram-scale synthesis and post-modification. (Reaction conditions: atrimethylsulfoxonium iodide, NaOH, rt. btris(trimethylsilyl)-silane, AIBN, reflux. cethyl isocyanoacetate, NaOH, rt. dDPPO, KOH, O2, rt. ebenzyl mercaptan, Et3N, rt. fpyrrolidine, rt. gdimethyl malonate, triton-B, 70 oC. hcyclopentanone, triton-B, 70 oC. in-butylamine, 70 oC). c Divinyl sulfone as dyes. d New polymer synthesis.
In Fig. 7b, we demonstrated that our method could be scaled up to prepare grams of vinyl sulfone 3ad and divinyl sulfone 5da. The vinyl sulfones were useful in organic synthesis for divergent post-modifications. For instance, cyclopropanation at the double bond of 3ad yielded 11a. The sulfonyl motif was also transformable by silylation (11b), pyrrolation (11c), and phosphinoylation (11d). Vinyl sulfones were excellent Michael acceptors for nucleophilic addition by thiols and amines (11e, 11f). It had important applications in the covalent modification of biomolecules in vitro or in vivo. Chemical probes and enzyme inhibitors based on the vinyl sulfone skeleton have been reported91.
Divinyl sulfones bearing a push-pull structure were useful chromophores with large Stokes shift69. Our method provided a straightforward protocol for their synthesis. Compound 5uv was a typical example (Fig. 7c). On the other hand, the mono- or di-functionalizations of divinyl sulfones was controllable (11f−11i). The reaction with aliphatic amine was particularly efficient that gave the disubstituted product in near quantitative yield (11i). This reactivity could be utilized for the preparation of new functional polymers92. The condensation of 5da with diamines yielded polyamines with high molecular weight and low dispersity (Mn = 21.5 KDa, PDI = 1.09, Fig. 7d). A systematic study of this polymerization reaction and related applications is underway.
Discussion
In summary, sulfonyl and sulfonimidoyl fluorides are advantageous over their analogues in synthesis, which have been exclusively used as electrophiles for fluoride-exchange chemical ligations. A general platform for their conversion to radical ligation reagents has been achieved in this manuscript, by combining photoredox catalysis and organosuperbase activation under mild conditions. The reaction with alkenes affords vinyl sulfones and sulfoximines. It also allows for the preparation of new polymeric materials and chromophores. Their synthetic utility has been largely expanded in linkage chemistry and beyond.
Methods
Under the N2 atmosphere, a 10 mL dry Schlenk tube equipped with a magnetic stirring bar was charged with sulfonyl fluoride, [Ru(bpy)3]Cl2, anhydrous solvent, alkene, and DBU. The reaction mixture was stirred at room temperature for 14 hours under blue LED illumination. The resulting mixture was then transferred to a 50 mL round-bottom flask. After removing the solvent, the residue was purified by flash column chromatography to give vinyl sulfones.
Data availability
The crystallographic data reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2180194 (3aa). They are free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the article and Supplementary Information files, and are available from the corresponding author upon request.
References
Takacs, G. A. Heats of formation and bond dissociation energies of some simple sulfur- and halogen-containing molecules. J. Chem. Eng. Data. 23, 174–175 (1978).
Salaudeen, B. Electrochemical reduction of arene sulfonyl fluorides and arene sulfonyl succinimides and their potential use as precursors for surface modification. (University of Guelph, 2020).
Parsons, J. S. Analysis of sulfonic acids by forming sulfonyl fluoride derivatives. J. Chromatogr. Sci. 5, 254–256 (1967).
Ciuffarin, E., Senatore, L. & Isola, M. Nucleophilic substitution at four-co-ordinate sulphur. Mobility of the leaving group. J. Chem. Soc., Perkin Trans. 2 4, 468–471 (1972).
Kice, J. L. & Lunney, E. A. Catalysis of the hydrolysis of aryl sulfonyl fluorides by acetate ion and triethylamine. J. Org. Chem. 40, 2125–2127 (1975).
Bellale, E. V., Chaudhari, M. K. & Akamanchi, K. G. A simple, fast and chemoselective method for the preparation of arylthiols. Synthesis 19, 3211–3213 (2009).
Johnson, C. R., Jonsson, E. U. & Wambsgans, A. Nucleophilic substitution at sulfur in sulfonimidoyl compounds: synthesis of sulfoximines. J. Org. Chem. 44, 2061–2065 (1979).
Johnson, C. R. et al. Preparation and reactions of sulfonimidoyl fluorides. J. Org. Chem. 48, 1–3 (1983).
Liang, D.-D. et al. Silicon-free SuFEx reactions of sulfonimidoyl fluorides: scope, enantioselectivity, and mechanism. Angew. Chem. Int. Ed. 59, 7494–7500 (2020).
Greed, S. et al. Synthesis of highly enantioenriched sulfonimidoyl fluorides and sulfonimidamides by stereospecific sulfur-fluorine exchange (SuFEx) reaction. Chem. Eur. J. 26, 12533–12538 (2020).
Greed, S., Symes, O. & Bull, J. A. Stereospecific reaction of sulfonimidoyl fluorides with grignard reagents for the synthesis of enantioenriched sulfoximines. Chem. Commun. 58, 5387–5390 (2022).
Suter, C. M. The organic chemistry of sulfur, tetracovalent sulfur compounds. (John Wiley & Sons, Ltd, 1944).
Chinthakindi, P. K. & Arvidsson, P. I. Sulfonyl fluorides (SFs): more than click reagents? Eur. J. Org. Chem. 27–28, 3648–3666 (2018).
Lou, T. S. B. & Willis, M. C. Sulfonyl fluorides as targets and substrates in the development of new synthetic methods. Nat. Rev. Chem. 6, 146–162 (2022).
Magre, M., Ni, S. & Cornella, J. (Hetero)aryl-S (VI) fluorides: synthetic development and opportunities. Angew. Chem. Int. Ed. 61, e202200904 (2022).
Bizet, V., Kowalczyk, R. & Bolm, C. Fluorinated sulfoximines: syntheses, properties and applications. Chem. Soc. Rev. 43, 2426–2438 (2014).
Guo, J. K. et al. Rapid deoxyfluorination of alcohols with N-tosyl-4-chlorobenzenesulfonimidoyl fluoride (sulfoxfluor) at room temperature. Chem. Eur. J. 25, 7259–7264 (2019).
Liang, D. D., Pujari, S. P., Subramaniam, M., Besten, M. & Zuilhof, H. Configurationally chiral SuFEx‐based polymers. Angew. Chem. Int. Ed. 61, e202116158 (2022).
Dong, J., Krasnova, L., Finn, M. G. & Sharpless, K. B. Sulfur (VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chem. Int. Ed. 53, 9430–9448 (2014).
Barrow, A. S. et al. The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 48, 4731–4758 (2019).
Zeng, D. M., Deng, W.-P. & Jiang, X. F. Linkage chemistry of S (VI) fluorides. Chem. Eur. J. 29, e202300536 (2023).
Zeng, D. M., Deng, W. P. & Jiang, X.-F. Advances in the construction of diverse SuFEx linkers. Natl. Sci. Rev. 10, nwad123 (2023).
Lee, C. et al. The emerging applications of sulfur (VI) fluorides in catalysis. ACS Catal. 11, 6578–6589 (2021).
Frye, L. L., Sullivan, E. L., Cusack, K. P. & Funaro, J. M. Sulfonylation of organometallic reagents with arenesulfonyl fluorides: a simple one-step synthesis of sulfones. J. Org. Chem. 57, 697–701 (1992).
Bennua-Skalmowski, B. & Vorbrüggen, H. A facile conversion of primary or secondary alcohols with n-perfluorobutane-sulfonyl fluoride/1,8-diazabicycl [5.4.0]-undec -7-ene into their corresponding fluorides. Tetrahedron Lett. 36, 2611–2614 (1995).
Gembus, V., Marsais, F. & Levacher, V. An efficient organocatalyzed interconversion of silyl ethers to tosylates using DBU and p-toluenesulfonyl fluoride. Synlett 10, 1463–1466 (2008).
Nielsen, M. K., Ugaz, C. R., Li, W. & Doyle, A. G. Pyfluor: a low-cost, stable, and selective deoxyfluorination reagent. J. Am. Chem. Soc. 137, 9571–9574 (2015).
Jones, C. S., Bull, S. D. & Williams, J. M. J. DBN hexafluorophosphate salts as convenient sulfonylating and phosphonylating agents. Org. Biomol. Chem. 14, 8452–8456 (2016).
Ungureanu, A., Levens, A., Candish, L. & Lupton, D. W. N-heterocyclic carbene catalyzed synthesis of δ-sultones via α, β-unsaturated sulfonyl azolium intermediates. Angew. Chem. Int. Ed. 54, 11780–11784 (2015).
Yatvin, J., Brooks, K. & Locklin, J. SuFEx on the surface: a flexible platform for postpolymerization modification of polymer brushes. Angew. Chem. Int. Ed. 54, 13370–13373 (2015).
Wei, M. et al. A broad-spectrum catalytic amidation of sulfonyl fluorides and fluorosulfates. Angew. Chem. Int. Ed. 60, 7397–7404 (2021).
Smedley, C. J. et al. Accelerated SuFEx click chemistry for modular synthesis. Angew. Chem. Int. Ed. 61, e202112375 (2022).
Emsley, J. Very strong hydrogen bonding. Chem. Soc. Rev. 9, 91–124 (1980).
Fahrney, D. E. & Gold, A. M. Sulfonyl fluorides as inhibitors of esterases. i. rates of reaction with acetylcholinesterase, α-chymotrypsin, and trypsin. J. Am. Chem. Soc. 85, 997–1000 (1963).
Narayanan, A. & Jones, L. H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 6, 2650–2659 (2015).
Jones, L. H. & Kelly, J. W. Structure-based design and analysis of SuFEx chemical probes. RSC Med. Chem. 11, 10–17 (2020).
Brighty, G. J. et al. Using sulfuramidimidoyl fluorides that undergo sulfur (VI) fluoride exchange for inverse drug discovery. Nat. Chem. 12, 906–913 (2020).
Zheng, Q. et al. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. 116, 18808–18814 (2019).
Hyatt, J. A. & White, A. W. Synthesis of aryl alkyl and aryl vinyl sulfones via friedel-crafts reactions of sulfonyl fluorides. Synthesis 3, 214–217 (1984).
Mukherjee, P. et al. Sulfonamide synthesis via calcium triflimide activation of sulfonyl fluorides. Org. Lett. 20, 3943–3947 (2018).
Mahapatra, S. et al. SuFEx activation with Ca(NTf2)2: a unified strategy to access sulfamides, sulfamates, and sulfonamides from S (VI) fluorides. Org. Lett. 22, 4389–4394 (2020).
Zeng, D., Ma, Y., Deng, W. P., Wang, M. & Jiang, X. F. The linkage of sulfonimidoyl fluorides and unactivated alkenes via hydrosulfonimidoylation. Angew. Chem. Int. Ed. 61, e202207100 (2022).
Zeng, D., Ma, Y., Deng, W. P., Wang, M. & Jiang, X. F. Divergent sulfur (VI) fluoride exchange linkage of sulfonimidoyl fluorides and alkynes. Nat. Synth. 1, 455–463 (2022).
Pan, X. Q., Zou, J. P., Yi, W. B. & Zhang, W. Recent advances in sulfur- and phosphorous-centered radical reactions for the formation of S-C and P-C bonds. Tetrahedron 71, 7481–7529 (2015).
Zhang, R., Cai, Y., Sun, D., Xu, S. & Zhou, Q. Eosin Y-sensitized photocatalytic reaction of tertiary aliphatic amines with arenesulfonyl chlorides under visible-light irradiation. Synlett. 28, 1630–1635 (2017).
Muralirajan, K., Kancherla, R. & Rueping, M. Dehydrogenative aromatization and sulfonylation of pyrrolidines: orthogonal reactivity in photoredox catalysis. Angew. Chem. Int. Ed. 57, 14787–14791 (2018).
Hell, S. M. et al. Silyl radical-mediated activation of sulfamoyl chlorides enables direct access to aliphatic sulfonamides from alkenes. J. Am. Chem. Soc. 142, 720–725 (2020).
Hakimelahi, G. & Hosein Just, G. Trifluoromethanesulfonyl chloride, a mild chlorinating agent. Tetrahedron Lett. 20, 3643–3644 (1979).
Corrêa, C. M. M., da Silva & Oliveira, M. A. B. C. S. Reaction of arenesulphonyl halides with free radicals. J. Chem. Soc. Perkin Trans. 2 6, 711–715 (1983).
Zhen, J. et al. Sulfonylation of aryl boronic acids by sulfonyl fluorides in water under visible-light irradiation. Org. Chem. Front. 10, 404–409 (2023).
Shreiber, S. T. & Molander, G. A. Alkyl (het)arylsulfones from SuFEx reagents via photochemical S-F bond activation. Org. Lett. 25, 2084–2087 (2023).
Nie, X. et al. Radical fluorosulfonylation: accessing alkenyl sulfonyl fluorides from alkenes. Angew. Chem. Int. Ed. 60, 3956–3960 (2021).
Wang, P., Zhang, H., Nie, X., Xu, T. & Liao, S. Photoredox catalytic radical fluorosulfonylation of olefins enabled by a bench-stable redox-active fluorosulfonyl radical precursor. Nat. Commun. 13, 3370 (2022).
Wang, P. et al. Radical hydro-fluorosulfonylation of unactivated alkenes and alkynes. Angew. Chem. Int. Ed. 61, e202207684 (2022).
Zhang, W. et al. A practical fluorosulfonylating platform via photocatalytic imidazolium-based SO2F radical reagent. Nat. Commun. 13, 3515 (2022).
Erchinger, J. E. et al. EnT-mediated N-S bond homolysis of a bifunctional reagent leading to aliphatic sulfonyl fluorides. J. Am. Chem. Soc. 145, 2364 (2023).
Yoon, T. P., Ischay, M. A. & Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2, 527–532 (2010).
Narayanam, J. M. R. & Stephenson, C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 40, 102–113 (2011).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Chan, A. Y. et al. Metallaphotoredox: the merger of photoredox and transition metal catalysis. Chem. Rev. 122, 1485–1542 (2022).
Skubi, K. L., Blum, T. R. & Yoon, T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 10035–10074 (2016).
Lu, F. D. et al. Recent advances in transition-metal-catalysed asym-metric coupling reactions with light intervention. Chem. Soc. Rev. 50, 12808–12827 (2021).
Schweitzer-Chaput, B., Horwitz, M. A., de Pedro Beato, E. & Melchiorre, P. Photochemical generation of radicals from alkyl electrophiles using a nucleophilic organic catalyst. Nat. Chem. 11, 129–135 (2019).
Meng, Q. Y., Döben, N. & Studer, A. Cooperative NHC and photoredox catalysis for the synthesis of β-trifluoromethyl-ated alkyl aryl ketones. Angew. Chem. Int. Ed. 59, 19956–19960 (2020).
DeLaive, P. J. et al. Photoinduced redox reactions of hydrophobic ruthenium (II) complexes. J. Am. Chem. Soc. 99, 7094–7097 (1977).
Hu, J., Wang, J., Nguyen, T. H. & Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 9, 1977–2001 (2013).
Beatty, J. W. & Stephenson, C. R. J. Amine functionalization via oxidative photoredox catalysis: methodology development and complex molecule synthesis. Acc. Chem. Res. 48, 1474–1484 (2015).
Ishikawa, T. Superbases for organic synthesis: guanidines, amidines, phosphazenes and related organocatalysts (John Wiley & Sons, Ltd, 2009).
Monçalves, M., Rampon, D. D. S., Schneider, P. H., Rodembusch, F. S. & Silveira, C. D. C. Divinyl sulfides/ sulfones-based D-π-A-π-D dyes as efficient non-aromatic bridges for π-conjugated compounds. Dyes Pigm. 102, 71–78 (2014).
Padmavathi, V., Venkata Subbaiah, D. R. C. & Reddy, B. J. M. Double Michael addition reactions to tetrakis sulfonyl activated olefins. J. Heterocycl. Chem. 42, 255–258 (2005).
Pal, T. K., Dey, S. & Pathak, T. A general route to mono- and disubstituted divinyl sulfones: acyclic Michael acceptors for the synthesis of polyfunctionalized cyclic sulfones. J. Org. Chem. 76, 3034–3041 (2011).
Fang, S. H. et al. Biological evaluation of sulfone derivatives as anti-inflammatory and tumor cells growth inhibitory agents. Int. Immunopharmacol. 6, 1699–1705 (2006).
Vicik, R., Busemann, M., Baumann, K. & Schirmeister, T. Inhibitors of cysteine proteases. Curr. Top. Med. Chem. 6, 331–353 (2006).
Ramana Reddy, M. V., Reddy, S., Reddy, D. B. & Padmavathi, V. Thioglycolates in the synthesis of bis(styryl)-sulfones. Synth. Commun. 19, 1101–1107 (1989).
Kamigata, N., Ozaki, J. & Kobayashi, M. Reaction of alkenesulfonyl chlorides with olefins catalyzed by a ruthenium (II) complex. a novel method for synthesis of (E,E)-1,4-diaryl-1,3-butadienes. J. Org. Chem. 50, 5045–5050 (1985).
Nie, X. et al. Introducing a new class of sulfonyl fluoride hubs via radical chloro-fluorosulfonylation of alkynes. Angew. Chem. Int. Ed. 60, 22035–22042 (2021).
Zha, G. F. et al. Palladium-catalyzed fluorosulfonylvinylation of organic iodides. Angew. Chem. Int. Ed. 56, 4849–4852 (2017).
Lou, T. S. B., Bagley, S. W. & Willis, M. C. Cyclic alkenylsulfonyl fluorides: palladium-catalyzed synthesis and functionalization of compact multifunctional reagents. Angew. Chem. Int. Ed. 58, 18859–18863 (2019).
Frye, N. L., Daniliuc, C. G. & Studer, A. Radical fluorosulfonyl-2-alkynylation of unactivated alkenes. Angew. Chem. Int. Ed. 61, e202115593 (2022).
Lücking, U. Sulfoximines: a neglected opportunity in medicinal chemistry. Angew. Chem. Int. Ed. 52, 9399–9408 (2013).
Frings, M., Bolm, C., Blum, A. & Gnamm, C. Sulfoximines from a medicinal chemist’s perspective: physicochemical and in vitro parameters relevant for drug discovery. Eur. J. Med. Chem. 126, 225–245 (2017).
Mäder, P. & Kattner, L. Sulfoximines as rising stars in modern drug discovery? current status and perspective on an emerging functional group in medicinal chemistry. J. Med. Chem. 63, 14243–14275 (2020).
Chinthakindi, P. K. et al. Sulfonimidamides in medicinal and agricultural chemistry. Angew. Chem. Int. Ed. 56, 4100–4109 (2017).
Okamura, H. & Bolm, C. Sulfoximines: synthesis and catalytic applications. Chem. Lett. 33, 482–487 (2004).
Shi, P. et al. Regio- and stereoselective chloro sulfoximidations of terminal aryl alkynes enabled by copper catalysis and visible light. Adv. Synth. Catal. 363, 2552–2556 (2021).
Shi, P., Tu, Y., Wang, C., Ma, D. & Bolm, C. Visible light-promoted synthesis of β-keto sulfoximines from N-tosyl-protected sulfoximidoyl chlorides. J. Org. Chem. 87, 3817–3824 (2022).
Craven, G. B. et al. Synthesis and configurational assignment of vinyl sulfoximines and sulfonimidamides. J. Org. Chem. 86, 7403–7424 (2021).
Davies, T. Q. et al. Harnessing sulfinyl nitrenes: a unified one-Pot synthesis of sulfoximines and sulfonimidamides. J. Am. Chem. Soc. 142, 15445–15453 (2020).
Erdelmeier, I. & Gais, H. J. Stereoselective synthesis of enan-tiomerically pure 1-(E)-alkenylsulfoximines. Tetrahedron Lett. 26, 4359–4362 (1985).
Roth, H. G., Romero, N. A. & Nicewicz, D. A. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 27, 714–723 (2016).
Fang, Y., Luo, Z. & Xu, X. Recent advances in the synthesis of vinyl sulfones. RSC Adv. 6, 59661–59676 (2016).
Gao, B. et al. Bifluoride-catalysed sulfur (VI) fluoride exchange reaction for the synthesis of polysulfates and polysulfonates. Nat. Chem. 9, 1083–1088 (2017).
Acknowledgements
Financial support was provided by National Natural Science Foundation of China (22001065), the Science and Technology Foundation of Hunan Province (2021JJ30090). We thank professor Dr. K. Barry Sharples at Scripps Research for his helpful discussion, and thank professor Lin Yuan at Hunan University for instrumental support.
Author information
Authors and Affiliations
Contributions
B.G. conceived and directed the project. X.W. and W.Z. performed the synthetic experiments and analyzed the data. X.S. and Y.H. provided support for the cyclic voltammetry experiment. G.S. and X.Z. provided a helpful discussion on reaction development. B.G. wrote the manuscript. X.W. and W.Z. prepared the Supplementary Information and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xuefeng Jiang, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, X., Zhang, W., Sun, G. et al. Turning sulfonyl and sulfonimidoyl fluoride electrophiles into sulfur(VI) radicals for alkene ligation. Nat Commun 14, 5168 (2023). https://doi.org/10.1038/s41467-023-40615-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40615-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
