
Abstract
Electrocatalytic urea synthesis is an emerging alternative technology to the traditional energy-intensive industrial urea synthesis protocol. Novel strategies are urgently needed to promote the electrocatalytic C–N coupling process and inhibit the side reactions. Here, we report a CuWO4 catalyst with native bimetallic sites that achieves a high urea production rate (98.5 ± 3.2 μg h−1 mg−1cat) for the co-reduction of CO2 and NO3− with a high Faradaic efficiency (70.1 ± 2.4%) at −0.2 V versus the reversible hydrogen electrode. Mechanistic studies demonstrated that the combination of stable intermediates of *NO2 and *CO increases the probability of C–N coupling and reduces the potential barrier, resulting in high Faradaic efficiency and low overpotential. This study provides a new perspective on achieving efficient urea electrosynthesis by stabilizing the key reaction intermediates, which may guide the design of other electrochemical systems for high-value C–N bond-containing chemicals.
Similar content being viewed by others


Introduction
Urea is used in nitrogen-based fertilizers and has supported a large proportion of the crop yield increase and assured the food supply of humanity1,2. In industry, urea is synthesized using the Bosch-Meiser process, where CO2 and liquid ammonia are mixed in urea through an ammonium carbamate intermediate under severe high-pressure conditions (150–250 bar) and elevated temperature (150–200 °C)3. Moreover, industrial ammonia manufacture (Haber-Bosch method) is highly energy-intensive, consumes considerable amounts of fossil fuels, and heavily emits CO2; thus, the current urea synthetic protocol is far from meeting the demands of society for sustainable development4. By contrast, electrosynthesis can convert feedstocks into high-value-added chemicals using renewable energy, being a more sustainable process and enabling the decarbonization of urea production2,5.
To date, direct activation of N2 coupled to CO2 for urea in ambient conditions is still challenging, because of the high overpotential requirements for the dissociation of highly stable C=O (806 kJ mol−1) and N≡N (941 kJ mol−1) bonds, as well as the strong competition of the parallel reactions6,7,8. Reactive nitrogen-oxygen bond-containing species, such as nitric oxide (NO) and nitrate/nitrite (NO3−/NO2−) ions, are more active nitrogen feedstocks. Among these species, NO3− is a nitrogen-containing reactant with a better intrinsic instability, which can be obtained from industrial wastewater; or by potentially sustainable nitrate generation technology in the future, such as non-thermal plasma activation of nitrogen9,10. Moreover, the lower dissociation energy of the nitrogen-oxygen bond (204 kJ mol−1) eases the coupling of NO3− reduction with CO2 reduction to accomplish urea electrosynthesis11. Thus, the NO3− to urea process is a suitable model reaction to study the electrochemical C–N bond formation; and a potential synthetic protocol for urea synthesis.
Since 1998, NO3− has been reported as a nitrogen-containing feedstock that can be coupled with CO2 for urea electrosynthesis12. However, the complex 16-electron reduction process has restricted studies on the development of NO3−-to-urea13; in particular, the comprehensive mechanistic understanding of electrocatalytic C–N bond formation and the structure design of catalysts remain fundamental challenges14,15. To date, urea electrosynthesis with NO3− and CO2 as feedstocks has reached an FE of up to 53% with a current density of 0.3 mA cm−216. Furthermore, the applied potentials for efficient urea electrosynthesis range from −0.6 to −1.5 V vs. reversible hydrogen electrode (RHE)15,16, which are far away from the thermodynamic potential for urea synthesis from CO2 and NO3− (0.48 V vs. RHE)17. Under such negative applied potentials, kinetically favorable competing side reactions, such as H2 generation, CO2 reduction, and NO2−, NH3 production, easily occur and reduce the selectivity of urea electrosynthesis18.
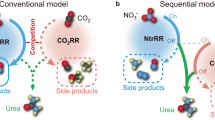
As the primary intermediate of NO3− reduction, the formation of *NO2 does not involve complex elementary reactions that may dissociate various by-products. Thus, *NO2 serves as a vital N-intermediate that reacts with intermediates of CO2 reduction, which may reduce the overpotential of urea electrosynthesis and reduce the probability of by-product generation. Cyanobacteria can utilize NO3− to photosynthetically synthesize organic nitrogen compounds with the help of nitrate and nitrite reductase metalloenzymes. In the first step of nitrate assimilation, nitrate reductase, a Mo-bis-molybdopterin guanine dinucleotide with a high-valence Mo4+-based reaction center, can convert NO3− into *NO2 intermediate and rapidly dissociate to NO2− at a low reduction potential19,20. A relatively long lifetime of N-related intermediates is expected to provide more opportunities for coupling with the intermediates of CO2 reduction, which the C–N bond formation can achieve urea electrosynthesis. The reaction of a high-valence metal center can decrease the electron density of the adsorbed species, resulting in a more positive reaction overpotential21,22. As tungsten is a homolog of molybdenum, high-valence tungsten-oxide derivatives are conducive to stabilizing *NO2 intermediate(s); such as WO3 could strongly adsorb NO2 molecules on the surface23,24. However, WO3 itself cannot trigger the CO2 reduction at low overpotentials, because *CO intermediate is challenging to be formed25. Contrarily, *CO is the common CO2 reduction intermediate on Cu-based catalysts, but continuous Cu sites enable the C–C coupling between adsorbed *CO and/or *CHO/*COH intermediates26. For urea electrosynthesis, a higher C–N coupling proportion is expected; separating the Cu sites could reduce the probability of C–C coupling by-products27.
Based on the considerations above, in this study, a Cu–W bimetallic oxide (CuWO4) catalyst with alternating bimetallic reaction sites was utilized for urea electrosynthesis with CO2 and NO3− as feedstocks (Fig. 1). A milliampere-level current of urea electrosynthesis could be realized at a remarkably operating potential with the highest FE reported to date. The reaction pathways and intermediates were systematically studied by in situ Raman spectroscopy and differential electrochemical mass spectrometry (DEMS), demonstrating that the rate-determining step of the urea generation from CO2 and NO3− is the *NO2 and *CO intermediates hydrogenation and coupling on CuWO4. Combined with thermodynamic adsorption energy analysis and theoretical calculation, the alternating bimetallic sites effectively improve the formation and coverage of the two intermediates on the surface of the catalyst, increasing the probability of C–N coupling and the selectivity for urea electrosynthesis.

The stabilization of the activated *CO and *NO2 intermediates at Cu and W bimetallic sites may play an important role in the high efficiency of C–N coupling.
Results
Catalyst synthesis and characterization
The CuWO4 catalyst was prepared by a hydrothermal synthesis approach using tungstate and Cu salts as raw materials28. The X-ray diffraction (XRD) pattern, Raman and Fourier-transform infrared spectrum of the synthesized sample could be assigned to the CuWO4 triclinic structure (Fig. 2a, Supplementary Fig. 1)29,30. The X-ray photoelectron spectrum (XPS) of Cu 2p (Fig. 2b) and W 4 f (Fig. 2c) exhibited two distinct split spin-orbit peaks located at 934.3 (Cu2+ 2p3/2), 954.2 (Cu2+ 2p1/2), 35.2 (W6+ 4f7/2), and 37.4 eV (W6+ 4f5/2), which indicated that Cu and W were in the II and VI oxidation states31, respectively. The lattice oxygen (O–Cu or W, 530.1 eV) was observed in O 1 s XPS (Supplementary Fig. 2)32. The morphology of the CuWO4 catalyst manifested as nanoparticles with a size of 40–60 nm by scanning electron microscopy (SEM, Fig. 2d) and cryogenic transmission electron microscopy (cryo-TEM, Fig. 2e). Cu, W, and O were homogeneously dispersed throughout the nanoparticle-like catalyst (Supplementary Fig. 3). The entire lattice exhibited highly ordered rectangular arrays with alternating bright and dark columns of atoms (Fig. 2e right), which was revealed by the high-angle, annular dark-field scanning transmission electron microscopy (HAADF-STEM). The transverse and longitudinal array spacings were 3.77 and 3.42 Å, corresponding to the interplanar spacing of (0−11) and (−101) facets, respectively33. The spacing between metal atoms in the same longitudinal array showed a weak periodic change. The triclinic CuWO4 crystal structure was constructed (Supplementary Fig. 4) and exhibited the dominant (111) facet (Fig. 2f), which is consistent with the atomic structure in the HAADF-STEM image. Therefore, the synthesized nanoparticles were highly ordered triclinic CuWO4 and did not contain other detectable impurities such as other derivatives of Cu or W. Pure CuO and WO3 catalysts were also prepared with similar morphology and valence state to CuWO4 for comparison; and were fully investigated by various characterization methods (Supplementary Figs. 5–9).

a XRD pattern. Inset: crystal structure of CuWO4. b, c XPS spectra of Cu 2p (b) and W 4 f (c). d SEM image. e TEM and atomic-resolution HAADF-STEM image with line profile of the corresponding area. f Atomic structure of CuWO4 (111) facet.
Electrochemical urea synthesis
Electrochemical measurements of urea synthesis were conducted to investigate the catalytic performance of CuWO4 and other samples (Methods, Supplementary Fig. 10). Figure 3a shows the linear sweep voltammetry (LSV) curves and I-V plots of CuWO4, CuO, and WO3. When switching the atmosphere of the electrolyzer from Ar to CO2, the reduction current of CuWO4 and CuO increased at the potential range from −0.1 to −0.4 V vs. RHE, indicating that additional CO2 reduction-related reactions may occur. For WO3, no significant difference between the two LSV curves was observed until to the more negative potential region (−0.7 to −0.9 V vs. RHE). For accurately measuring the urea synthesis efficiency of the catalysts at the corresponding potential, the chronoamperometry (CA) method was employed (Methods, Supplementary Fig. 11); and the production of urea was quantified by diacetylmonoxime-thiosemicarbazide (DAMO-TSC) (Supplementary Figs. 12–13) and nuclear magnetic resonance (NMR) methods. As the UV-visible spectral of DAMO-TSC method could be affected by the variation in the concentration of nitrite (Supplementary Figs. 14–17)34,35. Therefore, the urease decomposition method was further adopted and calibrated by two ammonium ion quantification methods (Supplementary Figs. 18–19). Other N-based by-products such as nitrite, ammonia, and hydrazine were quantitatively analyzed using ion chromatography, indophenol blue, and Watt and Christo’s methods, respectively (Methods)35,36. Gas-phase by-products were analyzed by gas chromatography (GC) (Supplementary Figs. 20–25)6,37.

a LSV curves of CuWO4, CuO, and WO3 in 0.1 M KNO3 with Ar or CO2 bubbling and I–V plots of CuWO4, CuO, and WO3 in 0.1 M KNO3 with CO2 at different potentials. b Yield rates and FE values of urea production for CuWO4 at different applied potentials in 0.1 M KNO3 with CO2 bubbling (20 mL min−1). c 1H NMR data of isotope calibration experiment in 0.1 M K15NO3 with CO2 bubbling (20 mL min−1) at different applied potentials. d 15N-urea yield rates and FEs via integrated peak area from NMR data. e FE values of all products for CuWO4 at different applied potentials in 0.1 M KNO3 with CO2 bubbling (20 mL min−1). f Nurea-selectivity and NO3−RR-FE for CuWO4 at different applied potentials in 0.1 M KNO3 with CO2 bubbling (20 mL min−1). g Urea FEs for CuWO4 at −0.2 V vs. RHE in 0.1 M KNO3 in different concentrations of KNO3 electrolyte with CO2 bubbling (20 mL min−1). h Stability test of urea synthesis during 10 h of electrolysis at −0.2 V vs. RHE in 0.1 M KNO3 with CO2 bubbling (20 mL min−1). i Comparison of the results of this work with state-of-art electrocatalytic synthesis urea catalysts in terms of operation potential and FE. b, d–g Error bars in accordance with the standard deviation of at least three independent measurements.
The corresponding FEs and average yield rates were calculated according to Eqs. 1–5. The optimal applied potential for urea synthesis of CuWO4 was the same as that of CuO (−0.2 V vs. RHE), which is more positive than that of WO3 (−0.8 V vs. RHE) (Fig. 3b). The peak urea yield rate of 99.5 ± 3.0 μg h−1 mg−1 with a remarkable FE as high as 70.9 ± 2.2% could be obtained for the CuWO4 catalyst at −0.2 V vs. RHE with a current density of nearly 1.0 mA cm−2. By contrast, the maximum urea yield rate of CuO was 63.9 ± 2.2 μg h−1 mg−1 and the maximum FE of 25.4 ± 2.7%. However, for WO3, a maximum urea yield rate of 61.7 ± 1.2 μg h−1 mg−1 and a urea FE of 24.7 ± 1.7% could be obtained only at a more negative operating potential (−0.8 V vs. RHE). The electrochemical active surface area (ECSA) was investigated and CuWO4 still had the highest urea yield after normalization (Supplementary Figs. 26–27). To verify the accuracy of the results, the isotope 15N-labeled potassium nitrate (K15NO3) was used as the electrolyte for CA measurements, and the yield rates of urea were quantified by 1H NMR spectroscopy (Fig. 3c, Supplementary Figs. 28–29)6. The urea yield rate calibrated by NMR spectra was 98.5 ± 3.2 μg h−1 mg−1 with a urea FE of 70.1 ± 2.4% (Fig. 3d), similar to that measured using the urease method. Other control experiments were also supplemented, and no urea was detected in the absence of CO2, NO3−, catalyst, or potential (Supplementary Figs. 30–31). Therefore, the generated urea was a result of the coupling of the feeding CO2 and NO3− via the electrocatalysis of CuWO4; and no other C–N coupling products were detected.
Compared with the complex process of coupling CO2 and NO3− to urea, side reactions such as NO3− reduction to NO2− or NH3 and HER could occur more easily; as a result, the generation of by-products leads to low FEs of urea for most systems18. Therefore, investigating the FE of the entire NO3−RR (Eq. 6) and the selectivity of products (Eq. 7) will help us understand the advantages of using CuWO4 (Fig. 3e, f, Supplementary Fig. 21). At the low overpotential region (−0.1 to −0.2 V vs. RHE), the FEs of the entire NO3−RR were close to 100%, most of the NO3− could react with CO2 to form urea, and the nitrogen selectivity for urea reached 76.2 ± 1.7% at −0.2 V vs. RHE. Both the urea yield rate (Fig. 3d) and the nitrogen selectivity (Fig. 3f) gradually decreased under more negative applied potentials, where kinetically favorable competitive reactions occur. Significantly, the CuO could effectively catalyze the reaction of NO3− to NO2−, which significantly decreased the FE of urea (Supplementary Figs. 16, 23); but WO3 can effectively avoid the NO2− generation (Supplementary Figs. 16, 25). With the assistance of high-valence W reaction center, the competitive reactions for urea synthesis catalyzed by CuWO4 are effectively suppressed.
The ratio of two reactants may impact the efficiency and selectivity of the reaction. CO2 with different flow rates was continuously fed, and the performance of CuWO4 was measured at −0.2 V vs. RHE (Supplementary Fig. 32). When the flow rate of CO2 was more than 5 mL min−1, the urea FEs remained at approximately 70%; this is because when the flow rates are higher than the consumption rate, the CO2 concentration remains roughly the same. The flow rate of CO2 was fixed at 20 mL min−1, and the concentration of KNO3 in the electrolyte was changed. As shown in Fig. 3g and Supplementary Fig. 33, the urea FEs had a volcano peak-shaped distribution, reaching a peak at the KNO3 concentration of 0.1 M. As the saturated concentration of CO2 (0.033 M) in aqueous solution at standard temperature and pressure, the ratios of CO2 and NO3− in different concentrations of KNO3 aqueous solution were 1:0, 1:0.75, 1:1.5, 1:2, 1:3, 1:4.5, and 1:6. Considering that the mass transfer efficiency of CO2 may be higher owing to the small amount of bubble transport, the actual ratio of CO2 and NO3− in 0.1 M KNO3 may be closer to the stoichiometric ratio of C and N atoms in urea, which is more conducive to a higher FE and the formation of urea. In addition, no other C–N coupling products (such as methylamine, ethylamine, formamide, acetamide, etc) were detected when the ratios of CO2 and NO3− changed.
The durability and reproducibility of CuWO4 toward urea electrosynthesis were also examined. As shown in Fig. 3h, the CuWO4/CP electrode maintained a relatively stable operation current density of approximately 1.0 mA cm−2 for 10 h at −0.2 V vs. RHE with an average urea FE of 68.0%. The longer-term continuous electrolysis was also performed, and after 20 h of electrolysis without electrolyte renewal, the Faradaic efficiency of urea was 56.4% (Supplementary Figs. 34–35). The final Faradaic efficiency remained above 50% after three 20 h long-term repeated tests, demonstrating excellent reproducibility. After the reaction, no appreciable topography and crystal structure changes were observed for CuWO4 (Supplementary Figs. 36–38). The only change detected is the binding energy of Cu on the surface of the CuWO4 after electrolysis immediately transfer to the XPS under the protection of Ar (Supplementary Fig. 39)38, indicating that the crystal structure of CuWO4 is stable, and the lower valence state Cu involved during the electrosynthesis. Moreover. no evident vibrational changes of CuWO4 (Supplementary Fig. 40) and WO3 (Supplementary Fig. 41) were found in in situ Raman spectra at operating potentials. By contrast, for CuO, multiple vibration peaks corresponding to Cu2O and other Cu derivatives appeared after applying the working potentials (Supplementary Fig. 42)39,40. And the valence state of Cu in CuO did not change completely back to Cu2+ after exposure to air, due to the structural transformation of CuO (Supplementary Figs. 42–43).
The FE of urea and the operating potential of this work were compared with those of previous reports (Fig. 3i and Supplementary Table 1)41,42,43,44,45; the FEs of urea in previous works were less than 53%, and the operating potentials were more negative than −0.4 V. The CuWO4 catalyst system significantly improved the urea FE (70.1 ± 2.4%) and reduced the operating potential (−0.2 V vs. RHE) with a current density of nearly 1.0 mA cm−2.
Unraveling the origin of C–N bond formation and reaction mechanism
As CuWO4 can obtain high FEs of urea at low operating potentials, an in-depth understanding of the C–N bond formation mechanisms of NO3− and CO2 co-reduction mechanism and structure–function relationship of CuWO4 is attractive, which may be helpful for the development of more advanced catalytic systems.
In situ Raman spectroscopy (Supplementary Fig. 44 shows the setup) was used to probe the stretching vibrations of generated intermediates on the surface of the CuWO4 under working conditions. Compared with the vibration peaks of NO3− (1048 and 1365 cm−1) under Ar-saturated conditions, new peaks at 1060 and 1351 cm−1 corresponding to *CO32− and *HCO3− species, respectively, emerged after CO2 bubbling (Fig. 4a) at the open circuit state46,47. At the working state (potential range from 0.1 to −0.3 V vs. RHE), the peak at 1979 cm−1 could be observed when the potential reached −0.1 V vs. RHE, which is assigned to the *CO on bridge sites48. Moreover, when the applied potential increased, the peak intensities of *CO32− and *HCO3− gradually decreased, indicating that *CO came from these derivatives of CO2. Interestingly, only one NO3− reduction species could be detected at 1428 cm−1 at the working state, corresponding to the υ(N=O) of *NO2 intermediate in bridging configuration49. Under the potential of −0.2 V vs. RHE, the maximum peak intensities of 1428 and 1979 cm−1 were reached, indicating that the highest coverage of *NO2 and *CO intermediates on the surface of CuWO4 can be received. It is in agreement with the best-applied potential (−0.2 V vs. RHE) for the highest rate and FE of urea formation. As reaction intermediates irrelevant to the rate-determining step, and the product of rate-determining step could be rapidly consumed, which are difficult to be observed by non-time-resolved characterization methods. Thus, the undissociated intermediates that serve as substrates for the rate-determining step can be observed in Raman spectra37. Therefore, assuming that *NO2 and *CO are the key intermediates in urea formation is reasonable, and the C–N bond formation between *NO2 and *CO coupling is very likely the rate-determining step of the urea electrosynthesis reaction on the surface of CuWO4. In contrast, various species from NO3− and CO2 reduction can be observed on the surface of the CuO (Supplementary Fig. 45). Stretching peaks of *CHO and *CH3O species at 1048 and 1110 cm−1 are observed in the range from −0.3 to −0.5 V vs. RHE, which is consistent with the increased yield rate of methane (Supplementary Fig. 23)50. A variety of nitrogenous intermediates with weak peak intensities are observed, indicating that the coverage of nitrogenous intermediates was low, which are in agreement with the low yield rate and FEs of urea for CuO. The WO3 exhibited a mismatch between the reduction potentials of CO2 and NO3− (Supplementary Fig. 46). The stretching peak at 2000 cm−1, corresponding to the *CO on atop sites48,51, could not be observed until the potential reached −0.6 V vs. RHE. As the applied potential changed, the intermediates of NO3− reaction on the surface of WO3 changed. In the range from −0.2 to −0.4 V vs. RHE, the main intermediate of NO3− reduction was *NO2, which is consistent with the observed for CuWO4. When the applied potential increased from −0.5 to −0.7 V vs. RHE, the vibration peaks at 1143, 1163, 1390, and 1533 cm−1 gradually appeared, corresponding to υ(N–O) and υ(N=O) of nitrito orientation, and υ(N=O) of nitroxyl for chelating nitrito49. These results explain the more negative potential requirement and low selectivity for WO3, which is because the generation of *CO intermediate requires a relatively negative applied potential; at such a relatively negative potential range, the NO3− reaction intermediate transformed from *NO2 to other intermediates.

a In situ Raman spectra of CuWO4 in 0.1 M KNO3 with CO2 bubbling at different applied potentials or Ar bubbling at open circuit state. b NO2-temperature programmed gas desorption (TPD) of CuWO4, CuO, and WO3. c Online DEMS of CuWO4 in 0.1 M KNO3 with saturated CO2 at −0.2 V vs. RHE. d Online DEMS of CuWO4 in 0.1 M K15NO3 with saturated 13CO2 at −0.2 V vs. RHE. e Control experiment results of different carbon and nitrogen sources for speculating the urea synthesis mechanism.
To further verify the speculations above, NO2-temperature-programmed gas desorption (TPD) and CO-TPD were conducted to evaluate the thermodynamic adsorption energy of *NO2 and *CO on the surface of CuO, WO3, and CuWO4. As shown in Fig. 4b, no evident NO2 desorption peak was observed on the CuO NO2-TPD curve, which means that the CuO surface hardly adsorbed NO2. On the contrary, WO3 showed a good adsorption capacity for NO2 with a desorption peak in the low-temperature region (<200 °C) and multiple desorption peaks in the high-temperature region (200–400 °C), representing the physical and chemical adsorptions of NO2 on the WO3 surface, respectively52. The NO2-TPD curve of CuWO4 also shows a series of NO2 desorption peaks. However, the strength is weaker than that of WO3, indicating that CuWO4 has a moderate adsorption capacity for *NO2 intermediate by balancing the properties of CuO and WO3. It is also consistent with the results of nitrite formation of the three catalysts (Supplementary Fig. 16). For the CO-TPD measurement (Supplementary Fig. 47), the peak strength of CO desorption for WO3 in both low and high-temperature regions was higher, indicating that WO3 had a stronger CO-adsorption capacity. CuO also showed a certain degree of chemical adsorption for CO. With the influence of octahedral [WO6] clusters, the physical adsorption of CO by CuWO4 significantly increased; however, the CO chemical adsorption capability of CuWO4 was the same as that of CuO, suggesting that CuWO4 maintains the advantages of higher physical adsorption ability of the W sites, and the favorable chemical adsorption capability of Cu sites for the *CO intermediate. Therefore, as predicted, CuWO4 could provide appropriate adsorption and stabilization capability for both *NO2 and *CO intermediates compared to WO3 and CuO, which is consistent with the in situ Raman spectroscopy results.
Differential electrochemical mass spectrometry (DEMS) can detect the products of the dissociated electrochemical-generated intermediates on the surface of the catalyst (Supplementary Fig. 48)38. The mass-to-charge ratio (m/z) signals of 2, 17, 28, 30, 33, and 46 corresponding to H2, NH3, CO, NO, NH2OH, and NO2, respectively, were recorded (Fig. 4c). For CuWO4, the signals of NH3 (m/z = 17), CO (m/z = 28), NO (m/z = 30), and NO2 (m/z = 46) showed fluctuations consistent with the switching cycles of open circuit and working states (at −0.2 V vs. RHE). The high captured signal strengths of NH3 (m/z = 17) corroborate that NH3 is the main by-product for CuWO4 at the applied potential of −0.2 V vs. RHE. Considering that a small amount NO may come from the disproportionation reaction of NO2 and the reaction during NH3 production, the observation of NO2 and CO, the dissociated electrochemical-generated intermediates, suggests that *NO2 and *CO are the critical intermediates for the C–N bond formation on the CuWO4 surface, which also corroborates the in situ Raman measurements. To confirm these results, the reactants KNO3 and CO2 were replaced with K15NO3 and 13CO2. As shown in Fig. 4d, corresponding isotopic molecule signals of 13CO (m/z = 29), 15NO (m/z = 31), and 15NO2 (m/z = 47) could be detected, indicating that *CO and *NO2 intermediates were generated from the reduction of CO2 and NO3− rather than pollutants. The signals of NH3 (m/z = 17) and NO2 (m/z = 46) could be detected for WO3 (Supplementary Fig. 49a); however, CO (m/z = 28) could not be clearly observed, which confirms that WO3 can only reduce NO3− to produce NH3 rather than trigger urea generation at −0.2 V vs. RHE, and that *CO generation potential is mismatched with that of NO2 formation (Supplementary Fig. 46). For CuO (Supplementary Fig. 49b), relatively high signal strengths of NO2 (m/z = 46) and CO (m/z = 28) could be detected, which is identical to the results that urea generation could occur at −0.2 V vs. RHE. However, these signals displayed a pulse-like intensity decay, which may be attributed to the weaker adsorption capacity of intermediates or insufficient structural stability of CuO. At the same time, considerable CO, H2, and NH3 were observed, which is consistent with the lower selectivity of urea electrosynthesis for CuO (Supplementary Fig. 23c).
The C- or N-sources were further changed to screen the C–N coupling process. Derivatives of CO2 in the aqueous phase (CO3− and HCO3−) and the desorption species (CO, HCHO, and CH3OH) of the intermediates generated during the CO2 reduction were used as the C-source for urea electrosynthesis with NO3−. As shown in Fig. 4e and Supplementary Table 2, CO3−, HCO3−, and CO can be co-reactants with NO3− for urea electrosynthesis by CuWO4; HCHO and CH3OH, the deeper CO2 reduction products, cannot conduct the process for CuWO4; which confirms that the *CO should be the key CO2 reduction intermediate on CuWO4 before C–N coupling. By contrast, NO2, NO2−, NO, NH2OH, and NH3, possible desorption species of the intermediates for NO3−RR, were used as the N-source for urea electrosynthesis with CO2. The results show that only NO2 can serve as the co-reactant with CO2 to form urea for CuWO4, inferring that *NO2 should be the critical NO3− reduction intermediate in the process of C–N coupling for urea generation. Electrosynthesis of urea on the surface of CuWO4 could also occur when CO and NO2 were used as feedstocks, further proving that *CO and *NO2 are the critical intermediates in the reaction pathway of urea electrosynthesis on the CuWO4 surface.
To further unravel the fundamental origins of the high selectivity of CuWO4, density functional theory (DFT) calculations (Methods) were conducted on the preferentially oriented in the (111) direction of CuWO4 crystal (Supplementary Fig. 50) corresponding to the XRD and TEM results. Figure 5a and Supplementary Fig. 51 show the detailed free-energy diagram of the corresponding structures with the lowest energy pathway from CO2 and NO3− to urea. The reaction started with the reduction of NO3− to *NO2 with strong adsorption-free energy of −1.35 eV. By contrast, CO2 was first physically adsorbed on the surface of CuWO4. The hydrogenation of *CO2 is a potential determining step with a 0.93 eV uphill in reaction-free energy; whereafter, the *COOH was spontaneously reduced to *CO thermodynamically. As shown in Supplementary Fig. 51, in the relevant atomic configurations with the lowest energy, the *NO2 intermediate was adsorbed as the bridging nitro with the N bonding to Cu and O bonding to W, suggesting that the W sites play an important role in stabilizing *NO2. Whereas, the *CO intermediate was located at the bridge site between two Cu atoms, consistent with the in situ Raman results. As the *CO was formed, the *NO2 intermediate was involved in the urea production. In contrast to the reaction-free energy for hydrogenation of *NO2 to *HNO2 (−0.32 eV), the lower reaction-free energy of −0.70 eV for *CONO2 formation (insert in Fig. 5a) on the surface of CuWO4 was beneficial for the direct C–N coupling at a very early stage. Simultaneously, the reaction-free energies of *CO to *CHO (0.17 eV) and *CO to *COH (2.12 eV) are higher compared to the C–N coupling process (−0.70 eV, Supplementary Fig. 52). As a result, the NH3-related or *CO hydrogenation side reactions could be significantly suppressed, which may explain the high selectivity of urea production by CuWO4. Although the free energy of C–N bonding between *NO2 and *CO is lower thermodynamically, an activation energy barrier as high as 0.87 eV for the *CONO2 formation process exists (Fig. 5b), making the coupling of *CO and *NO2 a slow kinetics process compared to the other elementary reactions during urea production, which is in agreement with the experimental results that the C–N bond formation between *NO2 and *CO is the rate-determining step. Further calculations of the charge density difference of *NO2 and *CO adsorbed states on the (111) facet of CuWO4 were investigated (Supplementary Fig. 53). The electrons on the CuWO4 could transfer to the *CO and *NO2, corresponding to the nature of the reduction reaction, which enables the improvement of their intrinsic activity and realizes the C–N coupling between the two intermediates16. Compared with the change of charge region before and after C–N bonding (Fig. 5c) an evident electron exchange between the *CO and *NO2 intermediates was observed; the electrons mainly flowed from the C atom of *CO to the N atom of *NO2, and completed the C–N coupling. The further hydrogenation of *CONO2 to *CONH2 intermediate can occur in an energetically favorable pathway, which has an advantage over the formation of *CONONO2, *CONHONO2, and *CONHNO2 in the reaction-free energy (Supplementary Fig. 54). Although the free energy of the coupling of *CONH2 with the second *NO2 increased slightly, it was not sufficient to prevent a thermodynamically spontaneous pathway of urea formation. These results theoretically confirm that the rate-determining step is the C–N coupling between the *CO and *NO2 intermediates on the surface of CuWO4 for urea electrosynthesis. Furthermore, the lower reaction-free energy of the C–N coupling between the *CO and *NO2 intermediates and the thermodynamically spontaneous formation pathway determine the extremely high selectivity of urea electrosynthesis on the surface of CuWO4, in line with the preceding experimental analysis.

a Free-energy diagram for urea production and NO3− reduction on the CuWO4 (111) facet. b Mechanism of C–N coupling between *CO and *NO2. The initial, transition, and final states during the *CONO2 formation are presented. Gray, blue, red, orange, cyan, and yellow balls represent C, N, lattice O, Cu, W, and adsorbate O, respectively. c Charge density difference of the co-adsorption state of *CO and *NO2 (left side), and adsorbate *CONO2 (right side) on the CuWO4 (111) facet. The iso-value is 0.002 e/Å3. Electron accumulation and depletion are represented by yellow and cyan sections, respectively.
Discussion
Inspired by the efficient nitrate assimilation process by the high-valence Mo-based reaction center of nitrate reductase in nature, CuWO4 catalyst with the high-valence W reaction centers was designed and employed for urea electrosynthesis with CO2 and NO3− as the feedstocks. The prepared CuWO4 could achieve highly efficient urea production with a 98.5 ± 3.2 μg h−1 mg−1cat urea yield at −0.2 V vs. RHE and high FE of 70.1 ± 2.4%. As evidenced by in situ Raman, TPD, DEMS, C/N-sources changing experiments, and theoretical calculation, early C–N coupling originating for urea electrosynthesis from *NO2 and *CO intermediates on the surface of CuWO4 was verified. Owing to the relative positive formation potential and appropriate adsorption ability of *NO2 and *CO intermediates on the alternating bimetallic W and Cu sites, CuWO4 optimized the coupling for these critical intermediate species. The lower energy barrier of direct coupling of *NO2 and *CO enabled the C–N coupling to easily occur. As primary intermediates of NO3− and CO2 reduction, the coupling of *NO2 and *CO decreased the possibility of intermediates desorption from complex elementary reactions before the C–N formation, which inhibits other side reactions. This study may also provide new inspiration for designing electrochemical synthesis systems to produce a broader range of high-value C–N bond compounds.
Methods
Preparation of catalysts
CuWO4 nanoparticles were synthesized by the hydrothermal method. Na2WO4·2H2O (0.33 g) and CuSO4 (0.16 g) were dissolved in 20 mL of ultrapure water, respectively. In sequence, the Na2WO4·solution was slowly dripped into the solution of CuSO4 under vigorous stirring. The mixture was transferred to a Teflon-lined stainless steel autoclave (50 mL), and maintained at 180 °C for 20 h in an oven. For CuO nanoparticles, 20 mL of 0.5 M Na2CO3 was slowly dripped into CuSO4 solution (0.16 g in 20 mL H2O) under vigorous stirring. For WO3 nanoparticles, HCl solution (0.1 M, 20 mL) was slowly dripped into the Na2WO4·solution (0.33 g in 20 mL H2O) under vigorous stirring. The hydrothermal synthesis procedures were the same as that of CuWO4. After hydrothermal synthesis, all samples were separated and washed by centrifugation. Ultimately, all samples were calcined at 500 °C in the air for 2 h to remove organic pollutants and improve the crystallinity.
Electrode preparation and electrochemical measurements
All electrochemical measurements were conducted on a CHI760E electrochemical instrument. A mass loading of 1 mg cm−2 was used on carbon paper (CP) working electrodes in a three-electrode H-cell electrolyzer, at room temperature and atmospheric pressure. 0.01 g of catalyst powders and 50 uL of Nafion solution were dissolved in 1 mL of ethanol as the ink. Subsequently, catalyst ink (100 μL) was coated on carbon paper (Toray 060) (geometric area 1 × 1 cm2), and the working electrode was obtained after drying (catalyst loading of 1 mg cm–2). A Nafion 117 membrane (Dupont) was used for separation. A ruthenium oxide-coated titanium sheet was used as the counter electrode. A leaking-free Ag/AgCl (saturated KCl) electrode was used as the reference electrode. KNO3 solutions (0.1 M as N-source) with or without high-purity CO2 (20 mL min−1 with bubbling, as C-source) were employed as the optimized electrolytes for the electrochemical measurements. Before tests, the corresponding gas was used to pre-saturate the electrolyte of the cathode part. In the electrolytic process, the gas flow rate was set as 20 mL min–1. The LSV measurements were performed with a negative scan direction and a scan speed of 10 mV/s. For each experiment, the applied potential was converted to scales of RHE according to \({E}_{\left({{{{{\rm{vs}}}}}}.{{{{{\rm{RHE}}}}}}\right)}={E}_{\left({{{{{\rm{vs}}}}}}.{{{{{\rm{Ag}}}}}}/{{{{{\rm{AgCl}}}}}}\right)}+{E}_{{{{{{\rm{Ag}}}}}}/{{{{{\rm{AgCl}}}}}}{{{{{\rm{vs}}}}}}.{{{{{\rm{RHE}}}}}}}\), where \({E}_{{{{{{\rm{Ag}}}}}}/{{{{{\rm{AgCl}}}}}}{{{{{\rm{vs}}}}}}.{{{{{\rm{RHE}}}}}}}\) is the potential difference between the Ag/AgCl electrode and a commercialized RHE (HydroFlex®) under corresponding conditions (such as saturated Ar or CO2). The same methods were adopted for isotope-labeling experiments and operando Raman measurements.
Quantitative analysis and identification of urea, ammonia, nitrite, and hydrazine
The content of urea, ammonia, and hydrazine was determined by urease, diacetylmonooxime, indophenol blue, and Watt and Christo, respectively6,15. For the urease decomposition method, 0.5 mL of urease solution (urease: 5 mg mL−1) was added into 4.5 mL of electrolyte, and then reacted at 40 °C for 40 min. The urease solution also contained 0.1 g ethylenediaminetetraacetic acid disodium salt and 0.49 g K2HPO4 per 100 mL. Then, the NH3 concentrations of electrolyte before and after decomposition were detected by ion chromatograph and indophenol blue method. The urea yield was calculated according to the NH3 concentrations before and after decomposition. For the diacetylmonoxime method, two chromogenic solutions, A and B, were prepared. For solution A, 10 mL of phosphoric acid, 30 mL of concentrated sulfuric acid, and 10 mg of ferric chloride were added to 60 mL of deionized water; finally, deionized water was added such that the volume reached 100 mL. For solution B, 0.5 g of diacetylmonoxime and 10 mg of thiosemicarbazide were dissolved in 100 mL of deionized water. 1 mL of the post-test electrolyte was mixed with 2 mL of solution A and 1 mL of solution B and heated at 100 °C for 15 min. After cooling, the absorbance of the solution at 525 nm was measured by a UV-vis spectrophotometer (Thermo One Plus). The calibration was performed using a standard concentration-absorbance curve of urea solution, including urea concentrations in the electrocatalytic test (Supplementary Fig. 12). For the indophenol blue method, 10 ml of electrolyte was added to 2 mL of salicylic acid solution (5 wt%), 1 mL of sodium hypochlorite solution (0.05 mol L−1), and 0.2 mL of sodium nitrosoferricyanide solution (1 wt%). After uniform mixing, the sample solution was placed for 45 min and tested with a UV-vis spectrophotometer (at 655 nm). The calibration was performed using a standard concentration-absorbance curve of ammonia solution, including ammonia concentrations in the electrocatalytic test (Supplementary Fig. 18). The concentration of ammonium ion in the electrolyte was also detected by ion chromatography (Thermo Scientific Dionex Aquion). The calibration was performed using a standard chromatographic curve of ammonium solution, including ammonium concentrations in the electrocatalytic test (Supplementary Fig. 19). For Watt and Christo’s method, 5 mL of a reagent consisting of a mixture of p-dimethylaminobenzaldehyde (5.99 g), concentrated hydrochloric acid (30 mL), and ethanol (300 mL) was added to 5 mL of electrolyte. The absorbance of the resulting solution was measured at 455 nm. The concentration of nitrite ions in the electrolyte was detected by ion chromatography (Thermo Scientific Dionex Aquion). The calibration was performed using a standard chromatographic curve of nitrite solution, including nitrite concentrations in the electrocatalytic test (Supplementary Fig. 14).
The FE is the ratio of the number of electrons transferred between the formation of products and the total current flowing through the circuit. As 16 electrons are required to form a urea molecule, and 8 electrons are required to form an NH3 molecule, the FE of urea and NH3 can be calculated, respectively, as follows:
where \({c}_{{{{{{\rm{urea}}}}}}}\) and \({c}_{{{{{{{\rm{NH}}}}}}}_{3}}\) (μg/mL) are the measured urea and NH3 concentrations, respectively; V (mL) is the total volume of the electrolyte, F is the Faraday constant (96,485.3 C mol−1); and Q (C) is the total charge passed through the working electrode.
The average yield rates of urea and NH3 were calculated according to the following equation.
where t is the time (h) for electrocatalysis and m is the catalyst loading (mg).
The NO3−RR-FE was calculated as follows:
where \({Q}_{{{NO}}_{3}^{-}{RR}}\) represents the charge consumed by the products involved in nitrate reduction.
The Nurea-selectivity was calculated as follows:
where \({n}_{{{{{{\rm{urea}}}}}}}\) is the number of moles of nitrogen in as-produced urea, and \({{{n}}}_{{{{{{\rm{total}}}}}}}\) is the total number of moles of N atoms in the products from NO3−RR.
Isotope-labeling product quantification and identification
The isotope-labeling experiments for the identification of products were conducted using a solution of 0.1 M K15NO3 as the electrolyte. The concentrations of 15N-urea and 15N-ammonia were both quantified by NMR6,37. For NMR sample preparation, the test solution was mixed with dimethyl sulfoxide at a ratio of 9:1 and shaken evenly. The NMR solution used to detect ammonia was adjusted to a pH of 3 by adding an appropriate amount of 1.0 M HCl solution. The NMR test was conducted under the water suppression mode with a scanning circle of 420 times. 15N-urea and 15NH4Cl were used to prepare the standard solution, and the standard linear curves between the NMR signal and the product concentration were established (Supplementary Fig. 28). The yield rates and FEs were calculated by reference to the calculation formula in the spectrophotometer method.
Quantitative analysis of other gas and liquid-phase products
The yield rates of CO, CH4, H2, and N2 in gas products were determined by an online gas chromatograph (GC-2014, Shimadzu) with thermal conductivity and flame ionization detectors. Other liquid-phase products were identified by 1H NMR.
NO2 and CO temperature program desorption
The NO2- and CO-TPD measurements were completed by AutoChem 2920 temperature program instrument and Hiden QIC-20 mass spectrometer. First, the catalyst was pretreated, and the temperature was raised to 200 °C with Ar flow for 0.5 h. In sequence, the catalyst was cooled down to 25 °C and exposed to NO2 or CO flow of 30 mL min−1 for 30 min. Before desorption, the sample was flushed in Ar gas for 10 min. Subsequently, NO2 or CO desorption was performed in the range of 25–600 °C at a heating rate of 10 °C min−1 under an Ar flow of 30 mL min−1. The mass-to-charge ratio (m/z) signals of 28, 44, and 46 corresponding to CO, CO2, and NO2, respectively, were recorded.
In-situ Raman spectroscopy measurements
In-situ Raman spectra were obtained from a homemade H-type three-electrode spectroelectrochemical cell with an embedded quartz window under the same working conditions of activity measurements (Supplementary Fig. 44 shows the experimental setup). The electrochemical workstation provided the corresponding applied potential. Before electrolysis, Ar or CO2 was injected into the electrolyte for 20 mins to purge, and the gas flow rate was maintained at 20 mL min-1 during the electrolysis. The Raman data was recorded after 15 mins of electrolysis to ensure that enough intermediates had accumulated on the surface of the catalyst. The laser intensity, the data recording circle, and the sweep speed were kept consistent.
XPS measurement after electrolysis
The CuWO4 and CuO electrodes were electrolyzed for an hour under at −0.2 V vs. RHE in 0.1 M KNO3 with CO2 bubbling (20 mL min−1). After the test, the electrodes were treated instantly with deionized water and vacuum drying (20 °C). The Ar-protected samples were transferred to an Ar-filled sealed chamber for storage. The samples, which were not protected by Ar, were exposed to air for an hour. Then, the two types of samples were tested together by XPS.
DEMS measurements
Differential electrochemical mass spectrometry (DEMS) test employed a high precision three-stage filter quadrupole mass analyzer with softer ionization (Ionic energy: 4–150 eV) (Supplementary Fig. 48a). Hiden QMS has a unique soft ionization technology. By optimizing the tuning of the electron energy of the EI source, the fragmentation peak can be reduced and the prominent molecular ion peak can be strengthened, which achieves the purpose of reducing interference. The DEMS cell was a dual thin-layer flow cell customized by Hiden, which was reported in previous work (Supplementary Fig. 48b)53. The working electrode was a custom-made glass carbon electrode uniformly coated with 50 μL catalyst ink (in Electrochemical measurements). The counter electrode was a ruthenium oxide electrode. Nafion 117 membrane was used as the separation membrane. The reference electrode was an Ag/AgCl electrode calibrated by a commercial RHE (HydroFlex). The electrolyte was 0.1 M KNO3 solution, and the cathode electrolyte was saturated with CO2. A microinjector ensured electrolyte flow in the DEMS cell at a flow rate of 200 μL min−1. The electrochemical workstation provided the corresponding applied potential. In isotope-labeling experiments, 0.1 M K15NO3 and 13CO2 were used as feedstocks.
DFT calculations
All the DFT calculations in this work were conducted using the Vienna ab initio simulation program (VASP)54,55. The core-valence interactions were described by the projector-augmented wave (PAW) method56,57, and the cut-off energy of plane-wave basis expansion was set to 450 eV. All spin-polarized calculations were performed by the generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) for the exchange and association of functions58. The lattice constants were calculated to be a = 4.681 Å, b = 5.867 Å, and c = 4.898 Å, consistent with previous results, including calculated constants and experiment values58,59,60,61. For the surface construction, the CuWO4 surface was cleaved along a (111) direction, which is the dominant surface observed in XRD. A p(2 × 2) CuWO4 (111) surface with six atomic layers was modeled. The two atomic layers at the bottom remain fixed to mimic the bulk phase, while the other layers were fully relaxed. A ~15 Å vacuum layer was employed to eliminate the interaction of adjacent slabs. The free molecules of HNO3, H2O, H2, and CO2 were placed in a (15 × 15 × 15) Å3 cubic box to diminish the interplay between neighboring molecules. A 2 × 2 × 1 Monkhorst–Pack k-point mesh sampling was utilized for all optimizations. When the forces on the relaxed atoms became less than 0.05 eV/Å, and the energies in the self-consistent iterations reached 10−5 eV, the optimized structures converged. The van der Waal (vdW) interaction was described using the DFT-D3 method62,63. The climbing image nudged elastic band (CI-NEB) method was used for locating the transition states (TSs)64,65.
The Gibbs free energy can be expressed as:
where \(\triangle E\) is the reaction energy calculated by the DFT methods. \(\triangle {ZPE}\) and \(T\cdot \triangle S\) are the thermodynamic corrections of zero-point-energy (ZPE) and entropy (S) derived from the vibrational partition function at 298.15 K, respectively. Gaussian 03 software package was used for gas-phase species to calculate the thermodynamic corrections for the ideal gas approximation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data that support the findings of this study are present in the paper and the Supplementary Information. Additional data related to the study are available from the corresponding author upon reasonable request.
References
McKie, D. Wöhler’s ‘synthetic’ urea and the rejection of vitalism: a chemical legend. Nature 153, 608–610 (1944).
Li, J., Zhang, Y., Kuruvinashetti, K. & Kornienko, N. Construction of C–N bonds from small-molecule precursors through heterogeneous electrocatalysis. Nat. Rev. Chem. 6, 303–319 (2022).
Lan, R., Tao, S. & Irvine, J. T. S. A direct urea fuel cell-power from fertiliser and waste. Energy Environ. Sci. 3, 438–441 (2010).
MacFarlane, D. R. et al. A roadmap to the ammonia economy. Joule 4, 1186–1205 (2020).
Wu, Y., Jiang, Z., Lin, Z., Liang, Y. & Wang, H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat. Sustain. 4, 725–730 (2021).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Yang, H. B. et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 3, 140–147 (2018).
Yuan, M. et al. Unveiling electrochemical urea synthesis by co-activation of CO2 and N2 with Mott–Schottky heterostructure. Catalysts. Angew. Chem. Int. Ed. 60, 10910–10918 (2021).
Wang, Y., Wang, C., Li, M., Yu, Y. & Zhang, B. Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720–6733 (2021).
Li, L. et al. Efficient nitrogen fixation to ammonia through integration of plasma oxidation with electrocatalytic reduction. Angew. Chem. Int. Ed. 60, 14131–14137 (2021).
Saravanakumar, D., Song, J., Lee, S., Hur, N. H. & Shin, W. Electrocatalytic conversion of carbon dioxide and nitrate ions to urea by a titania–nafion composite electrode. ChemSusChem 10, 3999–4003 (2017).
Shibata, M., Yoshida, K. & Furuya, N. Electrochemical synthesis of urea at gas-diffusion electrodes: III. Simultaneous reduction of carbon dioxide and nitrite ions with various metal catalysts. J. Electrochem. Soc. 145, 595–600 (1998).
Huang, Y. et al. Direct electrosynthesis of urea from carbon dioxide and nitric oxide. ACS Energy Lett. 7, 284–291 (2022).
Leverett, J. et al. Tuning the coordination structure of Cu-N-C single atom catalysts for simultaneous electrochemical reduction of CO2 and NO3– to urea. Adv. Energy Mater. 12, 2201500 (2022).
Zhang, X. et al. Identifying and tailoring C–N coupling site for efficient urea synthesis over diatomic Fe–Ni catalyst. Nat. Commun. 13, 5337 (2022).
Lv, C. et al. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 4, 868–876 (2021).
Huang, Y., Wang, Y., Wu, Y., Yu, Y. & Zhang, B. Electrocatalytic construction of the C-N bond from the derivates of CO2 and N2. Sci. China Chem. 65, 204–206 (2022).
Liu, X., Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S.-Z. Mechanism of C-N bonds formation in electrocatalytic urea production revealed by ab initio molecular dynamics simulation. Nat. Commun. 13, 5471 (2022).
Campbell, W. H. Nitrate reductase structure, function and regulation: bridging the gap between biochemistry and physiology. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 277–303 (1999).
Hageman, R. H. & Hucklesby, D. P. In: Methods Enzymol. 23, 491–503 (Academic Press, 1971).
Wu, Y. et al. Electron density modulation of NiCo2S4 nanowires by nitrogen incorporation for highly efficient hydrogen evolution catalysis. Nat. Commun. 9, 1425 (2018).
Zhao, Z. et al. Molecular engineering of photocathodes based on polythiophene organic semiconductors for photoelectrochemical hydrogen generation. ACS Appl. Mater. Interfaces 13, 40602–40611 (2021).
Bai, S. et al. Synthesis mechanism and gas-sensing application of nanosheet-assembled tungsten oxide microspheres. J. Mater. Chem. A 2, 7927–7934 (2014).
Qin, Y., Liu, M. & Ye, Z. A DFT study on WO3 nanowires with different orientations for NO2 sensing application. J. Mol. Struct. 1076, 546–553 (2014).
Mendieta-Reyes, N. E., Díaz-García, A. K. & Gómez, R. Simultaneous electrocatalytic CO2 reduction and enhanced electrochromic effect at WO3 nanostructured electrodes in acetonitrile. ACS Catal. 8, 1903–1912 (2018).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Yang, F. et al. Scalable strategy to fabricate single Cu atoms coordinated carbons for efficient electroreduction of CO2 to CO. Carbon 168, 528–535 (2020).
Thiruppathi, M., Vinoth Kumar, J., Vahini, M., Ramalingan, C. & Nagarajan, E. R. A study on divergent functional properties of sphere-like CuWO4 anchored on 2D graphene oxide sheets towards the photocatalysis of ciprofloxacin and electrocatalysis of methanol. J. Mater. Sci. Mater. Electron. 30, 10172–10182 (2019).
Ruiz-Fuertes, J., Pellicer-Porres, J., Segura, A., Rodríguez-Hernández, P. & Muñoz, A. Lattice and electronic contributions to the refractive index of CuWO4. J. Appl. Phys. 116, 103706 (2014).
Souza, E. L. S. et al. Structural evolution, growth mechanism and photoluminescence properties of CuWO4 nanocrystals. Ultrason. Sonochem. 38, 256–270 (2017).
Yourey, J. E. & Bartlett, B. M. Electrochemical deposition and photoelectrochemistry of CuWO4, a promising photoanode for water oxidation. J. Mater. Chem. 21, 7651–7660 (2011).
Gupta, R., Boruah, B., Modak, J. M. & Madras, G. Kinetic study of Z-scheme C3N4/CuWO4 photocatalyst towards solar light inactivation of mixed populated bacteria. J. Photochem. Photobiol. 372, 108–121 (2019).
Wang, T. et al. Self-assembled urchin-like CuWO4/WO3 heterojunction nanoarrays as photoanodes for photoelectrochemical water splitting. ChemElectroChem 8, 125–134 (2021).
Wei, X. et al. Oxygen vacancy-mediated selective C–N coupling toward electrocatalytic urea synthesis. J. Am. Chem. Soc. 144, 11530–11535 (2022).
Huang, Y. et al. Unveiling the quantification minefield in electrocatalytic urea synthesis. Chem. Eng. J. 453, 139836 (2023).
Li, D. et al. A reliable and precise protocol for urea quantification in photo/electrocatalysis. Small Methods 6, 2200561 (2022).
Zhao, Y. et al. Identification of M-NH2-NH2 intermediate and rate determining step for nitrogen reduction with bioinspired sulfur-bonded FeW catalyst. Angew. Chem. Int. Ed. 60, 20331–20341 (2021).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Chen, X. et al. Controlling speciation during CO2 reduction on Cu-alloy electrodes. ACS Catal. 10, 672–682 (2020).
Yang, P.-P. et al. Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J. Am. Chem. Soc. 142, 6400–6408 (2020).
Meng, N., Huang, Y., Liu, Y., Yu, Y. & Zhang, B. Electrosynthesis of urea from nitrite and CO2 over oxygen vacancy-rich ZnO porous nanosheets. Cell Rep. Phys. Sci. 2, 100378 (2021).
Shibata, M. & Furuya, N. Electrochemical synthesis of urea at gas-diffusion electrodes: Part VI. Simultaneous reduction of carbon dioxide and nitrite ions with various metallophthalocyanine catalysts. J. Electroanal. Chem. 507, 177–184 (2001).
Cao, N. et al. Oxygen vacancies enhanced cooperative electrocatalytic reduction of carbon dioxide and nitrite ions to urea. J. Colloid Interface Sci. 577, 109–114 (2020).
Shibata, M., Yoshida, K. & Furuya, N. Electrochemical synthesis of urea at gas‐diffusion electrodes: iv. simultaneous reduction of carbon dioxide and nitrate ions with various metal catalysts. J. Electrochem. Soc. 145, 2348–2353 (1998).
Liu, S. et al. AuCu nanofibers for electrosynthesis of urea from carbon dioxide and nitrite. Cell Rep. Phys. Sci. 3, 100869 (2022).
Bae, S.-E., Stewart, K. L. & Gewirth, A. A. Nitrate adsorption and reduction on Cu(100) in acidic solution. J. Am. Chem. Soc. 129, 10171–10180 (2007).
Chernyshova, I. V., Somasundaran, P. & Ponnurangam, S. On the origin of the elusive first intermediate of CO2 electroreduction. Proc. Natl Acad. Sci. USA 115, E9261–E9270 (2018).
Chang, X. et al. Understanding the complementarities of surface-enhanced infrared and Raman spectroscopies in CO adsorption and electrochemical reduction. Nat. Commun. 13, 2656 (2022).
Butcher, D. P. & Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: effect of oxide and Cl−. Nano Energy 29, 457–465 (2016).
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Gao, J. et al. Selective C–C coupling in carbon dioxide electroreduction via efficient spillover of intermediates as supported by operando raman spectroscopy. J. Am. Chem. Soc. 141, 18704–18714 (2019).
Yang, Y. et al. The crucial role of charge accumulation and spin polarization in activating carbon-based catalysts for electrocatalytic nitrogen reduction. Angew. Chem. Int. Ed. 59, 4525–4531 (2020).
Clark, E. L., Singh, M. R., Kwon, Y. & Bell, A. T. Differential electrochemical mass spectrometer cell design for online quantification of products produced during electrochemical reduction of CO2. Anal. Chem. 87, 8013–8020 (2015).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Xie, X. et al. Efficient photo-degradation of dyes using CuWO4 nanoparticles with electron sacrificial agents: a combination of experimental and theoretical exploration. RSC Adv. 6, 953–959 (2016).
Forsyth, J. B., Wilkinson, C. & Zvyagin, A. I. The antiferromagnetic structure of copper tungstate, CuWO4. J. Phys. Condens. Matter 3, 8433–8440 (1991).
Doumerc, J. P., Hejtmanek, J., Chaminade, J. P., Pouchard, M. & Krussanova, M. A photoelectrochemical study of CuWO4 single crystals. Phys. Status Solidi 82, 285–294 (1984).
Tian, C. M. et al. Elucidating the electronic structure of CuWO4 thin films for enhanced photoelectrochemical water splitting. J. Mater. Chem. A 7, 11895–11907 (2019).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work was conducted by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), financially supported by the National Key R&D Program of China (2022YFA0911904), the National Natural Science Foundation of China (NSFC) (Grant nos. 22172011 and 22088102), the Fundamental Research Funds for the Central Universities (DUT22LK06 and DUT22QN213), the Key Laboratory of Bio-based Chemicals of Liaoning Province of China, and the starting grant of Westlake University and the Kunpeng program of Zhejiang province.
Author information
Authors and Affiliations
Contributions
Y.Z., L.S., and Fu.L. conceived the project design and initiated the project. Y.Z. performed catalyst synthesis, most of the structural characterization, and electrochemical measurements. All spectra and electrochemical kinetics data were analyzed and interpreted by Y.Z. and Fu.L. Y.D. performed DFT calculations. W.L. and C.L. performed operando Raman spectra measurements and data analysis. Y.L. performed the SEM and TEM measurements. Z.Z. and Fe.L. performed partial product analysis of electrochemical measurements. Y.S. performed the XPS measurements and data analysis. All authors contributed to the discussions. Y.Z. wrote the paper with inputs from the other authors, and Fu.L., Fe.L., and L.S. revised the manuscript. All authors reviewed the paper. Fu.L. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Ding, Y., Li, W. et al. Efficient urea electrosynthesis from carbon dioxide and nitrate via alternating Cu–W bimetallic C–N coupling sites. Nat Commun 14, 4491 (2023). https://doi.org/10.1038/s41467-023-40273-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40273-2
This article is cited by
-
Pulsed co-electrolysis of carbon dioxide and nitrate for sustainable urea synthesis
Nature Sustainability (2024)
-
Upgrading waste NOx into amino acids via electrocatalysis on Co nanoparticles encapsulated in hollow carbon nanofibers
Science China Chemistry (2024)
-
Synergistic coupling of CO2 and NO3− for efficient electrosynthesis of urea using oxygen vacancy-rich Ru-doped CeO2 nanorods
Science China Materials (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
