
Abstract
Performing CO2 reduction in acidic conditions enables high single-pass CO2 conversion efficiency. However, a faster kinetics of the hydrogen evolution reaction compared to CO2 reduction limits the selectivity toward multicarbon products. Prior studies have shown that adsorbed hydroxide on the Cu surface promotes CO2 reduction in neutral and alkaline conditions. We posited that limited adsorbed hydroxide species in acidic CO2 reduction could contribute to a low selectivity to multicarbon products. Here we report an electrodeposited Cu catalyst that suppresses hydrogen formation and promotes selective CO2 reduction in acidic conditions. Using in situ time-resolved Raman spectroscopy, we show that a high concentration of CO and OH on the catalyst surface promotes C-C coupling, a finding that we correlate with evidence of increased CO residence time. The optimized electrodeposited Cu catalyst achieves a 60% faradaic efficiency for ethylene and 90% for multicarbon products. When deployed in a slim flow cell, the catalyst attains a 20% energy efficiency to ethylene, and 30% to multicarbon products.
Similar content being viewed by others


Introduction
The electrocatalysis community has demonstrated high selectivity toward multicarbon (C2+) products in the CO2 reduction (CO2R) reaction1,2,3,4,5. In particular, the faradaic efficiency (FE) to ethylene (C2H4) in CO2R has increased at an impressive rate in systems operating in neutral and alkaline conditions2,6,7,8,9,10,11. Unfortunately, the single-pass carbon efficiency (SPCE) (i.e. the utilization of CO2) has till now remained low12,13. A primary cause is the rapid conversion of CO2 to (bi)carbonate in these mid- to high-pH conditions. This imposes a substantial energy cost penalty for carbonate regeneration and CO2 (re)capture12,14.
Performing CO2R in acidic conditions provides a potential route to high SPCE of CO212,14,15. However, to date, the selectivity of CO2R in acidic conditions has been limited by the competing hydrogen evolution reaction (HER)12,14, which benefits from a faster kinetics and a lower reaction overpotential16. Consequently, the energy efficiency (EE) for producing C2+ chemicals in acidic CO2R has remained well below that in neutral and alkaline conditions.
The activity and selectivity of CO2R can be tuned by tailoring the interaction between reaction intermediates and the Cu surface12,17,18,19,20,21,22. Achieving a high coverage and long residence time of CO on the surface of the catalyst can help acidic CO2R compete successfully against HER22. Previous CO2R studies in neutral/alkaline conditions showed that adsorbed hydroxyl groups (OH*, * indicating surface site) form on the Cu surface during CO2R due to an increase in local alkalinity23,24,25. Such species have been shown to affect the adsorption energy of CO and the reaction rate of C–C coupling26. In acidic CO2R, the local pH at the electrode surface is near-neutral under moderate current densities14, a fact that will limit the concentration of surface OH* and lead to low C2+ selectivity in acidic CO2R.
Here we focus therefore on a strategy to increase the concentration of OH* on the catalyst surface during acidic CO2R. We report that it suppresses HER and enhances C–C coupling. Using density functional theory (DFT) calculations, we found an increase in the CO adsorption energy and a decrease in the barrier for C–C coupling when a sub-monolayer coverage of OH* exists on the Cu surface. By tailoring synthesis conditions, we synthesized catalysts that maintain a high concentration of co-adsorbed CO and OH during acidic CO2R. Using time-resolved in situ Raman spectroscopy, we find that the residence time of CO on the catalyst surface increases in the presence of OH, a finding we correlate with higher C2+ selectivity and suppressed HER activity. The method of catalyst synthesis involves electrodepositing a Cu catalyst in situ during CO2 reduction (EC–Cu). This leads to achieving >50% and 90% FE for C2H4 and C2+, respectively, with 10 h of operating stability. When we add poly(amino acid) promotors, the catalyst achieves a C2H4 FE of 60% and a C2H4 EE of 20%.
Results and discussion
DFT calculations
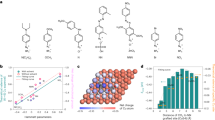
We first applied DFT calculations to investigate the free energy of CO adsorption (ΔGCO*) on Cu(100) as a function of the surface coverage of CO (θCO) and OH (θOH). We chose Cu(100) as the model surface due to its high activity for the C–C coupling reactions10,27,28,29 and investigated the dependence of ΔGCO* on the θCO and θOH up to 3/9 of a monolayer (ML) (Supplementary Fig. S1). We found that ΔGCO* is similar at different θCO without co-adsorbed OH (Fig. 1a). However, an increase in CO binding strength is observed for θCO between 1/9 and 2/9 ML when the θOH is at 2/9 ML, indicating that the ΔGCO* can be fine-tuned with co-adsorbed OH. Comparing the barrier for C–C coupling with and without OH* (Fig. 1b, Supplementary Figs. S2–5), we found a lower activation energy for CO dimerization and a lower free energy for OCCO (ΔGOCCO*) on Cu(100) with nearby adsorbed OH. A similar trend on the influence of OH* in CO dimerization is also observed for Cu(111) (Supplementary Figs. S6−10). DFT calculation results suggest that introducing OH* species in acidic CO2R could be a promising strategy to suppress HER and promote C2+ formation.

a CO adsorption energy on Cu(100) surface with different OH and CO coverage. b Free energy profiles of CO dimerization on Cu(100) with and without adsorbed OH. Different positions of OH* denoted as Cu-OH*-1, Cu-OH−-2, and Cu-OH*-3 were considered. The bottom right panel illustrates the OH* positions in DFT models.
In situ electrodeposition of catalysts during acidic CO2R
Previous work reported that Cu catalysts with a surface OH* are obtained at high pH by electrochemically reducing a pre-deposited Cu(OH)2 film26. We electrodeposited Cu catalysts (EC–Cu) in acidic conditions by reducing Cu2+ species during CO2R, which generates a locally alkaline condition (H3O+ mass transport limitation) necessary for the formation of Cu(OH)x pre-catalyst and provides a reductive potential needed to reduce the as-formed Cu(OH)x into Cu (Fig. 2a). The local formation of Cu(OH)x pre-catalyst was found to be necessary to obtain high performance of EC–Cu catalysts—an observation arrived at by comparing with control samples prepared under various deposition conditions (Supplementary Fig. S11). The EC–Cu catalyst produced by Cu(OH)x–mediated in situ deposition differs in morphology and structure from the electrodeposited Cu catalysts obtained at locally acidic conditions. It also demonstrates a higher C2+ selectivity (vide infra).

a Schematic of Cu(OH)x mediated in situ electrodeposition during acidic CO2R in a flow cell. b Gas-phase product distribution of sCu in acidic CO2R. The error bars correspond to the standard deviation of three independent measurements. c FEH2 and FEC2H4 of in situ formed EC–Cu at different time of electrodeposition. The CO2R was performed at 200 mA cm−2 in 0.05 M H2SO4 and 2.5 M KCl containing the copper precursor (5 mM). The FE of CO and CH4 were less than 1% and not listed. d Morphology evolution of EC–Cu. Scanning electron microscope (SEM) image of a series of EC–Cu catalysts at different times of electrodeposition. e SEM and transmission electron microscopy (TEM) images and corresponding EDX mapping of Cu. f High-resolution TEM. g Fourier-transformed operando XAS spectra of the formations of EC–Cu with respect to time at 200 mA cm−2 in 0.05 M H2SO4 and 2.5 M KCl containing the copper precursor (3 mM).
We performed electrodeposition of Cu on a sputtered Cu (sCu) covered polytetrafluoroethylene (PTFE) gas diffusion layer (sCu/PTFE) in a flow cell during acidic CO2R (Fig. 2a). The sCu catalyst, a control, showed a C2H4 FE of 25% and a H2 FE of 26% (Fig. 1b)9,11. During electrodeposition, the selectivity of EC–Cu towards C2H4 improved, reaching ~50% C2H4 FE, while the HER selectivity was reduced substantially (Fig. 2c). The FE to C1 products (CO and CH4) was <1%, lower than that of sCu. We also observed that deposition duration of >14 min has a negative effect on the FE of C2H4 with a concomitant increase in HER.
We relate the time-dependent CO2R activity to the morphology change of the deposited catalysts during synthesis. Scanning electron microscopy (SEM) images of EC–Cu show that dendritic structures gradually form on the sCu/PTFE surface with a longer deposition duration (Fig. 2d, e). A complete dendritic structure was formed at 14 min, corresponding to the best performance.
In a Cu2+ free electrolyte, the optimally deposited EC–Cu catalyst (at 14 min) maintained high CO2R selectivities, excluding CO2R catalysis by solvated Cu2+ ions (Supplementary Fig. S12). Transmission electron microscopy (TEM) shows that different Cu facets exist (Fig. 2f). This is consistent with OH− adsorption experiments showing a mixture of (100), (110), and (111) surface crystal orientations on EC–Cu, and the ratios of these facets differ greatly from that of sCu (Supplementary Fig. S13). Operando X-ray absorption spectroscopy (XAS) was used to track the oxidation state and local bonding configuration of Cu as a function of deposition duration (Fig. 2g, Supplementary Fig. S14). The EC–Cu catalysts were metallic and showed Cu coordination similar to that of sCu.
Experimental mechanistic studies
We employed time-resolved in situ Raman spectroscopy to probe the surface-adsorbed CO—an important reaction intermediate in CO2R. When CO chemisorbs on the Cu catalysts, its characteristic stretching frequency (ν(COatop)) ranges from 1850−2100 cm−1. The values of ν(COatop) depend on multiple factors, including the adsorption site (atop vs. bridge sites) and surface CO coverage (θCO). Based on the peak positions, we assign the peaks at 2082 and 2042 cm−1 to CO molecules adsorbed at the atop sites of the Cu surface on sCu and EC–Cu, respectively (Fig. 3a). The high-frequency band (HFB) at 2082 cm−1 is assigned to COatop on isolated step sites, and the lower frequency band (LFB) at 2042 cm−1 is attributed to COatop on terrace sites17 (Fig. 3b, Supplementary Fig. S15). The lower ν(COatop) frequency on EC–Cu indicates stronger COatop binding on its surface than sCu. This result is consistent with a higher ν(Cu–CO) frequency observed for EC–Cu at 375 cm−1 compared to sCu at 360 cm−1 (see Supplementary Note 1 for additional discussion)30. A consequence of stronger COatop binding on EC–Cu is a longer residence time on the catalyst surface, which could benefit CO–CO coupling for C2+ product formation. We evaluate CO residence time on the Cu surface by monitoring the decrease in ν(COatop) peak intensity after changing the applied potential from high (−1.5 V) to low (−1.2 V) rate conditions. For sCu and EC–Cu, the ν(COatop) peak intensity decays exponentially with time constants (τ) of 0.33 and 0.61 s, respectively (Fig. 3d, e). Similar τ values are also observed for the corresponding Cu–CO peak at 360 and 378 cm−1 (Supplementary Fig. S16, S17). The larger τ values of the 2042 and 378 cm−1 peaks indicate a longer residence time of COatop on the surface of EC–Cu, which can promote C–C coupling in addition to having a high θCO. This effect is also in line with the higher C2+ FE for the EC–Cu (>90%) compared with sCu (<50%). Furthermore, the degraded EC–Cu samples (<65% C2+ FE) show a high-frequency ν(COatop) band with short τ, reaffirming the importance of longer residence time for adsorbed COatop (Fig. 3c, Supplementary Figs. S18, 19).

a Raman spectra of sCu and EC–Cu after different electrodeposition time. b Deconvolved spectra of the ν(COatop) region of (a). c Deconvolved spectra of the ν(COatop) region for EC–Cu catalyst after 5, 10, and 90 min of CO2 electrolysis. d Time-resolved in situ Raman spectra of EC–Cu under −1.2 and −1.5 V applied potentials. The top right panel summarizes the normalized intensity of the 2042 cm−1 peak as a function of time. The bottom right panel illustrates the time-dependent change of the normalized intensity for the 2042 cm−1 peak after a potential step to −1.2 V. The red curve is an exponential fit to the intensity decay. e Time-resolved in situ Raman spectra of sCu under −1.2 and −1.5 V applied potentials. The top right panel plots the normalized intensity of the 2082 cm−1 peak as a function of time. The bottom right panel illustrates the time-dependent change of the normalized intensity for the 2082 cm−1 peak after a potential step to −1.2 V. The red curve is an exponential fit to the intensity decay. In situ Raman spectra were collected using a 785 nm laser. The time-resolved spectra were collected every 250 ms.
The larger τ value for EC–Cu is closely associated with the surface OH* group. The greater intensity of the Cu-OH peak at 531 cm−1 for EC–Cu suggests a greater concentration of OH* at the surface compared to sCu under the same applied potential (Fig. 3a). DFT calculations show that the ΔGCO* can increase with the number of nearby adsorbed OH. Experimentally, we observed the interactions between co-adsorbed CO and OH based on a red shift in the ν(COatop) peak position to 2042 cm−1 and a lower frequency shoulder at 1972 cm−1 (Fig. 3a, Supplementary Note 1)26. Combining in situ characterization of surface OH and COatop, we propose that EC–Cu enables high surface coverage of co-adsorbed OH and CO, which increases the residence time of CO on the catalyst surface and promotes C–C coupling. Evidence from Raman spectroscopy indicates the presence of OH* on the surface of Cu catalysts. However, since we cannot observe and quantify the concentration of locally generated hydroxyl ions by Raman spectroscopy, we do not rule out the effect of this species on the CO2R selectivity (Supplementary Figs. S20, 21).
Acidic CO2R performance
We prepared EC–Cu on different conductive substrates and found that sCu/PTFE resulted in the highest FE of C2H4 and C2+. The interaction between the 200 nm Cu film (sCu) and electrodeposited Cu species (EC–Cu) has little influence on the performance of the catalysts (Supplementary Fig. S22). The thickness of Cu in sCu also has a minimal impact on the FE of C2H4, but it significantly affects the stability of EC–Cu. Additionally, the substrates’ structure and composition can influence EC–Cu performance (Supplementary Fig. S23)31,32,33. A 200 nm Cu film deposited on a PTFE-coated gas diffusion layer was used as the substrate in all experiments unless otherwise specified. We found that the optimal deposition time of EC–Cu decreased with greater CuSO4 concentrations (Fig. 4a). We identified two deposition conditions that produce highly selective catalysts for C2H4 as EC–Cu-1 and EC–Cu-2 (optimal electrodeposition duration at 5 mM and 7 mM CuSO4). The EC–Cu catalysts synthesized using the second deposition condition achieved a maximum C2H4 FE of 56%. Over a long-time operation, the average FE for C2H4, C2+, and H2 was maintained at 53%, 90%, and 10%, respectively (Fig. 4b).

a FEC2H4 of EC–Cu obtained from different electrodeposition durations and Cu precursor concentrations. b Acidic CO2R product distribution of EC–Cu−1 (5 mM CuSO4, 14 min) and EC–Cu-2 (7 mM CuSO4, 10 min) in (a). c The gas-phase product distribution of EC–Cu (7 mM CuSO4, 10 min) and sCu in acidic CO2R at different current densities. d FEH2 and FEC2H4 of EC–Cu-2 when using different concentrations of H2SO4 in the catholyte. e Performance of different poly(amino acid)s modified EC–Cu-2. f SPC of acidic CO2R. g Cell voltage, FEC2H4 and FEH2 of acidic CO2R in a flow cell. h Full-cell EE, energy consumption, C2H4 FE, C2+ FE and C2H4 SPCE compared to the best prior results for acidic CO2R. All the error bars correspond to the standard deviation of three independent measurements.
In previous acidic CO2R works, high current densities were necessary to increase the local pH and promote C–C coupling on Cu catalysts, and a high K+ concentration was employed to suppress HER (Supplementary Fig. S24)12,14,34. This was observed for sCu, which showed a lower FE to C2H4 at lower current densities (Fig. 4c, <10% at 75 mA/cm2). In contrast, EC–Cu demonstrated >45% FE to C2H4 from 75 to 250 mA/cm2 and was able to achieve high C2H4 FE at lower K+ concentrations (Fig. 4c, d and Supplementary Figs. S25, 26). We attribute this high selectivity partially to a high surface OH* concentration during CO2R, which stabilizes CO and reduces the C–C coupling barrier.
The stability of EC–Cu was also improved compared with sCu, from less than 0.5 h to 10 h (Supplementary Figs. S27–29). We note that the degradation of dendritic structures in EC–Cu accompanies a performance decrease, which correlates with a decrease in surface OH, and a shorter CO residence time (Supplementary Figs. S18, 19). To further increase the FE of C2H4, we introduced amide-bearing polymers during the catalyst deposition process. We evaluated five poly(amino acids) containing abundant amide groups and different functional side chains and found that poly(Lys, Phe) showed a stable C2H4 FE of 60% (Fig. 4e, Supplementary Figs. S30, 31). A similar promoting effect of amine/amide functional groups toward the C2H4 selectivity in CO2R has been reported previously6,35. Deploying the poly(Lys, Phe)-modified EC–Cu catalyst, we obtained a SPCE of 70% towards C2+ products (Fig. 4f) and stable operation for 10 h with 55% FE of C2H4 in acidic conditions (Fig. 4g). Based on a full cell voltage of 3.4 V (Supplementary Fig. S32), the EE towards C2H4 is 20%, which is 1.6-fold greater than the previous acidic CO2-to-C2H4 record. Figure 3h summarizes the result in comparison with the previous best acidic CO2R performance (Supplementary Table 1–3). Based on the performance summary in Fig. 3h, we achieved CO2-to-C2H4 conversion with an energy requirement of ~266 GJ ton−1, lower than previous neutral and alkaline CO2R systems13. Despite the promising performance, practical implementation requires further improvements in stability and selectivity.
In summary, we report an EC–Cu catalyst that selects for C2H4 and C2+ products in acidic CO2R. Preparation via in situ electrodeposition led to a higher surface coverage of OH, and we found that this species interacts with the CO intermediate and increases the CO binding energy. This observation is consistent with DFT calculations showing that sub-monolayer coverage of OH can increase the free energy of CO adsorption and reduce the barrier for C–C coupling. Implementing the EC–Cu catalyst in acidic CO2R achieves a high FE toward multicarbon products (60% for C2H4, 90% for C2+), a high SPCE of CO2 to C2+ products (70%), and a high EE (20% for C2H4, 30% for C2+) simultaneously.
Methods
Materials
A high-purity Cu target (99.99%, Kurt J. Lesker Company, EJTCUXX403A2) was used in the fabrication of sputtered Cu catalysts. Sulfuric acid (H2SO4, Fischer Chemicals, A300S), copper(II) sulfate pentahydrate (CuSO4 · 5H2O, Aldrich, 209198), and potassium chloride (KCl, Aldrich, P3911) were used in the preparation of supporting electrolytes. Polytetrafluoroethylene (PTFE) gas diffusion layers (GDL) with 450 nm pore size were purchased from Beijing Zhongxingweiye Instrument Co., Ltd. Nafion 117 membrane was purchased from the FuelCell Store. All poly(amino acid)s: Poly(l-Lys, l-Phe) 1:1 hydrobromide (P3150), Poly(l-Glu, l-Lys) 1:4 hydrobromide (P8619), Poly(l-His) (P9386), Poly(l-Lys) hydrobromide (P2636), Poly(l-Arg) hydrochloride (P4663) were purchased from Sigma Aldrich.
In situ electrodeposition
Magnetron sputtering (Angstron Engineering, Nextdep) was used to deposit 200 nm or 500 nm of Cu catalyst on a PTFE GDL at a rate of 1 Å/s in a high vacuum condition. EC–Cu samples were prepared by electrodeposition under acidic CO2R conditions. Catalysts were electrodeposited at a constant current density of −200 mA cm-2 for a certain duration on the sputtered Cu in a three-electrode flow cell. The catholyte consisted of 3~8 mM CuSO4, 0.05 M H2SO4, and 2.5 M KCl. 0.05 M H2SO4 was used as the anolyte. For electrodeposition containing poly(amino acid), each polymer was added to the catholyte at 0.1 mg/ml concentration. The catholyte and anolyte chambers were separated by a cation exchange membrane (CEM, Nafion 117®). An Ag/AgCl reference electrode (3 M KCl) and a platinum mesh (Fisher Scientific, AA41814FF) counter electrode were used. The in situ nature of the catalyst deposition allows us to monitor the performance evolution of the formed catalysts in real-time. In situ electrodeposition was performed under the same electrochemical conditions as acidic CO2R except for the addition of Cu precursor (CuSO4) to the catholyte. After the deposition time interval, a Cu2+-free catholyte (0.05 M H2SO4, 2.5 M KCl) was introduced to the reactor, which terminated the electrodeposition process, allowing continued evaluation of the in situ formed catalysts.
Catalyst characterization
Scanning electron microscopy (SEM, Hitachi S3500) and transmission electron microscopy (TEM, Hitachi HF3300) were used to characterize the crystallinity and morphology of the in situ formed Cu catalysts. The XAS measurements were performed with a modified flow cell in the Soft X-ray Micro-Characterization Beamline (SXRMB) at the Canadian Light Source (Saskatoon, Canada), which is equipped with a water-cooled Si (111) and InSn (111) double-crystal monochromator covering a photon energy range from 1.7 to 10.0 keV. The XAS data was processed with Demeter (v.0.9.26)36. In situ Raman spectroscopy (Renishaw Invia Raman) was performed to investigate CO speciation on sCu and EC–Cu during CO2R.
CO2 reduction in a flow cell
Three chambers constitute the flow cell setup: anolyte chamber, catholyte chamber, and gas flow chamber. Each compartment has an inlet and outlet. The electrolytes were circulated through the flow cell using peristaltic pumps. CO2 (Linde Gas) flowing through the gas flow chamber was controlled using a mass flow controller (Brooks Instrument, GF100). The CO2 flow rate was fixed at 40 standard cubic centimeters per minute (sccm) unless stated otherwise. During the CO2R reaction, the catalyst-coated working electrodes, Nafion 117 membrane, and a platinum mesh (Fisher Scientific, AA41814FF) counter electrode were sandwiched between the three chambers. The working electrode potential was measured against Ag/AgCl reference electrode (CH Instruments, CHI111P) that was inserted into the catholyte chamber.
CO2 reduction in a full cell
A slim flow cell was used to measure the full cell performance. The slim flow cell also contains anolyte, catholyte, and gas flow compartments. The catholyte chamber is made of PEEK and has a total thickness of 1/16”. The reduced dimension is intended to lower ohmic losses between the cathode and the anode. In the full cell measurement, oxygen evolution reaction takes place on the anode and it was catalyzed by iridium oxide catalyst supported on titanium felt (IrOx–Ti)37. The IrOx–Ti anode electrodes were fabricated according to the following steps: (1) immersing the high porosity titanium fiber felts (Fuel Cell Store, porosity approximately 70-73%) into an ink of 2-propanol, iridium (IV) chloride dehydrate (Premion®, 99.99%, metal basis, Ir 73%, Alfa Aesar), and 0.1 M hydrochloric acid (ACS reagent; 37 vol%), (2) drying at 100 °C for 15 min and sintering at 500 °C for 15 min, and (3) repeating the first two steps until a final Ir mass loading of 1.5 mg/cm2 is achieved.
Electrochemical measurement
All electrochemical measurements were performed using a potentiostat (Autolab PGSTAT302N). The CO2R performance was evaluated in a flow cell (three-electrode setup) or a slim flow cell (full cell, two-electrode setup) under galvanostatic modes. Gas-phase products were quantified using gas chromatography (GC, Shimadzu, GC-2014) equipped with a thermal conductivity detector (TCD) for the detection of H2, O2, N2, and CO, and a flame ionization detector (FID). Helium (Linde, 99.999%) was used as the carrier gas. Liquid products were analyzed using 1H NMR spectroscopy (600 MHz, Agilent DD2 NMR Spectrometer) with water suppression. Dimethyl sulfoxide (DMSO) was used as the internal reference and deuterium oxide (D2O) as the lock solvent. The Faradaic efficiency (FE) of the gas product was calculated based on the following equation:
where n is the number of electrons transferred, F is the Faraday constant, v is the CO2 flow rate, r is the concentration of the gas product in parts-per-million (ppm), i is the total current and Vm is the unit molar volume of gas.
The energy efficiency (EE) for the formation of ethylene is calculated as follows:
where FEC2H4 denotes the FE of ethylene (C2H4), Eoa and Eoc are the standard reduction potentials for the anode and cathode (CO2-to-ethylene) reactions, respectively.
The single-pass carbon efficiency (SPCE) of CO2 for a particular product is determined as follows:
where j is the partial current density of a specific product, n is the number of electrons transferred for every molecule of the product
In situ Raman spectroscopy
A custom-made cell was used to carry out in situ Raman spectroscopy. In an epi-illumination configuration, a 785 nm laser was used as the excitation source. The laser power was kept lower than 0.20 mW in all experiments to minimize sample damage. The scattered Raman light was collected by a water immersion objective (Leica, 63×, NA 0.9). Raman spectrometer calibration was done with a Si standard.
Electrochemical OH− adsorption
Electrochemical OH− adsorption was performed in an N2-saturated 1 M KOH electrolyte. Linear sweep voltammetry was carried out between −0.2 and +0.6 V versus RHE with a sweep rate of 100 mV s−1. Before the experiment, all copper catalysts were reduced at −0.6 V versus RHE for 3 min.
DFT Calculations
All density functional theory (DFT) calculations were carried out using the Vienna ab initio simulation package (VASP)38,39,40,41. The generalized gradient approximation was used with the Perdew–Burke–Ernzerhof exchange-correlation functional42. The projector-augmented wave method43,44 was used to describe the electron-ion interactions and the cut-of energy for the plane wave basis set was 450 eV. To illustrate the long-range dispersion interactions between the adsorbates and catalysts, the D3 correction method was employed45,46. Brillouin zone integration was accomplished using a 3 × 3 × 1 and 2 × 3 × 1 Monkhorst-Pack k-point mesh for CO adsorption energy calculations and C–C coupling calculations. A vacuum region of more than 15 Å thickness was included along the perpendicular direction to avoid artificial interactions.
A periodic six-layer model and p(3 × 3) or p(6 × 3) super cell were chosen. A monolayer of charged water molecules was included in C–C coupling calculations. Adsorption geometries of the different states were optimized by a force-based conjugate gradient algorithm, whereas the transition states were located using the climbing image-nudged elastic band method47. During the calculations, the three lower layers were fixed and the three upper layers together with water molecules and adsorbates were allowed to relax. The Gibbs free energy (∆G) was calculated by converting the electronic energy using the equation: ∆G = ∆E + ∆ZPE + ∫ ∆CpdT − T∆S, where ∆E, ∆ZPE, ∆Cp, and ∆S are the differences in electronic energy, zero-point energy, heat capacity and entropy, respectively, and T was set to room temperature (298.15 K).
Energy assessment for the acidic CO2R electrolyzer
Energy assessment of the acidic CO2R electrolyzer was performed using an energy evaluation model similar to that reported in refs. 13,48. This section provides an overview of the model used to obtain the energy intensity of ethylene production in an acidic CO2R electrolyzer. To calculate the energy intensity, we utilized the experimentally achieved performance metrics under various operating conditions (for example, under various CO2 input flow rates). The metrics inputted to the energy model include Faradaic efficiency, single-pass conversion efficiency, full-cell voltage, and current density—all towards ethylene (which is the major product of the acidic CO2R system). The model considers the presence of hydrogen (the product of competing HER) in addition to ethylene and CO2 at the cathode gas stream. The model also considers oxygen (the product of oxygen evolution) as the only product in the anode gas stream. The model considers a pressure swing adsorption (PSA) gas separation unit at the cathodic downstream to recover ethylene from the hydrogen side product and unreacted CO2. The CO2 recovered from the downstream of the cathode is considered to be returned to the cathode inlet for utilization in CO2R. An electrolyte requirement of 100 L per m2 of the active electrode geometric area is considered. This consideration is based on an estimation of the electrolyte requirement for a lab-scale electrolyzer with a geometric flow field area of 5 cm2. The electrolyte is considered to be circulating through a closed loop and used for 1 year without replacement. An example calculation for the energy intensity associated with the electrolyzer electricity and cathode separation is provided in Supplementary Note 2.
Data availability
All the data supporting the findings of this study are available within the article and its Supplementary Information. All other relevant source data are available from the corresponding authors upon reasonable request.
References
Choi, C. et al. Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4. Nat. Catal. 3, 804–812 (2020).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Wang, X. et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 5, 478–486 (2020).
Wang, X. et al. Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag–Ru–Cu catalyst. Nat. Energy 7, 170–176 (2022).
Ji, Y. et al. Selective CO-to-acetate electroreduction via intermediate adsorption tuning on ordered Cu–Pd sites. Nat. Catal. 5, 251–258 (2022).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2020).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Liu, W. et al. Electrochemical CO2 reduction to ethylene by ultrathin CuO nanoplate arrays. Nat. Commun 13, 1877 (2022).
Wakerley, D. et al. Bio-inspired hydrophobicity promotes CO2 reduction on a Cu surface. Nat. Mater. 18, 1222–1227 (2019).
Wang, Y. H. et al. Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis. Nat. Catal. 3, 98–106 (2020).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Ozden, A. et al. Carbon-efficient carbon dioxide electrolysers. Nat. Sustain. 5, 563–573 (2022).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
An, H. et al. Sub‐second time‐resolved surface‐enhanced raman spectroscopy reveals dynamic CO intermediates during electrochemical CO reduction on copper. Angew. Chem. 133, 16712–16720 (2021).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Jiang, K. et al. Metal ion cycling of Cu foil for selective C–C coupling in electrochemical CO2 reduction. Nat. Catal. 1, 111–119 (2018).
Cheng, T., Xiao, H. & Goddard, W. A. Nature of the active sites for CO reduction on copper nanoparticles; suggestions for optimizing performance. J. Am. Chem. Soc. 139, 11642–11645 (2017).
Fan, L. et al. Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2+ products. Sci. Adv. 6, eaay3111 (2020).
Chang, X. et al. Understanding the complementarities of surface-enhanced infrared and Raman spectroscopies in CO adsorption and electrochemical reduction. Nat. Commun. 13, 2656 (2022).
Zhao, Y. et al. Speciation of Cu surfaces during the electrochemical CO reduction reaction. J. Am. Chem. Soc. 142, 9735–9743 (2020).
Zhang, Z. et al. pH matters when reducing CO2 in an electrochemical flow cell. ACS Energy Lett. 5, 3101–3107 (2020).
Yang, K., Kas, R. & Smith, W. A. In situ infrared spectroscopy reveals persistent alkalinity near electrode surfaces during CO2 electroreduction. J. Am. Chem. Soc. 141, 15891–15900 (2019).
Iijima, G., Inomata, T., Yamaguchi, H., Ito, M. & Masuda, H. Role of a hydroxide layer on Cu electrodes in electrochemical CO2 reduction. ACS Catal. 9, 6305–6319 (2019).
Sandberg, R. B., Montoya, J. H., Chan, K. & Norskov, J. K. CO-CO coupling on Cu facets: coverage, strain and field effects. Surf. Sci. 654, 56–62 (2016).
Schouten, K. J. P., Gallent, E. P. & Koper, M. T. M. The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes. J. Electroanal. Chem. 716, 53–57 (2014).
Perez-Gallent, E., Marcandalli, G., Figueiredo, M. C., Calle-Vallejo, F. & Koper, M. T. M. Structure- and potential-dependent cation effects on CO reduction at copper single-crystal electrodes. J. Am. Chem. Soc. 139, 16412–16419 (2017).
Zhan, C. et al. Revealing the CO coverage-driven C-C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via operando Raman spectroscopy. ACS Catal. 11, 7694–7701 (2021).
Monteiro, M. C. O. et al. Probing the local activity of CO(2) reduction on gold gas diffusion electrodes: effect of the catalyst loading and CO(2) pressure. Chem. Sci. 12, 15682–15690 (2021).
Jaster, T. et al. Electrochemical CO(2) reduction toward multicarbon alcohols—the microscopic world of catalysts & process conditions. iScience 25, 104010 (2022).
Lees, E. W. et al. Linking gas diffusion electrode composition to CO2 reduction in a flow cell. J. Mater. Chem. A 8, 19493–19501 (2020).
Monteiro, M. C. O. et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654–662 (2021).
Ahn, S. et al. Poly-amide modified copper foam electrodes for enhanced electrochemical reduction of carbon dioxide. ACS Catal. 8, 4132–4142 (2018).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Radiat 12, 537–541 (2005).
Gabardo, C. M. et al. Continuous carbon dioxide electroreduction to concentrated multi-carbon products using a membrane electrode assembly. Joule 3, 2777–2791 (2019).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B: Condens. Matter 49, 14251–14269 (1994).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B: Condens. Matter 47, 558–561 (1993).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Michaelides, A. et al. Identification of general linear relationships between activation energies and enthalpy changes for dissociation reactions at surfaces. J. Am. Chem. Soc. 125, 3704–3705 (2003).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Ozden, A. et al. Cascade CO2 electroreduction enables efficient carbonate-free production of ethylene. Joule 5, 706–719 (2021).
Acknowledgements
The authors thank H. Xiao and X. Li for discussions. Funding: this work was financially supported by the Ontario Research Foundation: Research Excellence Program, the Natural Sciences and Engineering Research Council (NSERC) of Canada, TotalEnergies SE, the Australian Research Council (project number DE200100477), and the National Key Research and Development Program of China (2021YFC2102800). Synchrotron experiments were carried out at SXRMB beamline at the Canadian Light Source (CLS). We acknowledge the help from Dr. Qunfeng Xiao, Dr. Mohsen Shakouri, and Dr. Alisa Paterson for their kind technical assistance. All DFT computations were carried out at the Shenzhen Bay Laboratory Supercomputing Center. We acknowledge the Ontario Centre for the Characterization of Advanced Materials (OCCAM) for characterization facilities. Y.C. gratefully acknowledges a scholarship from China Scholarship Council (CSC) under Grant CSC N202006210266. W.N. gratefully acknowledges financial support from the Swiss National Science Foundation (SNSF) Postdoctoral Mobility Fellowship (No. P500PN_202906).
Author information
Authors and Affiliations
Contributions
E.H.S., J.G., and D.S. supervised the project. Y.C. conceived the idea, prepared catalysts, designed, and performed electrochemical experiments. P.O. supervised and Y.C. performed DFT calculations. Z.C. carried out Raman spectroscopy measurements and data analysis. P.L. and J.Z assisted in electrochemical measurements. W.N., J.A., and E.S. performed XAS measurements and data analysis. A.O. performed energy assessments. Y.C., Z.C., and E.H.S. co-wrote the manuscript. All authors discussed the results and assisted with the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
Provisional patent applications based on the electrodeposited copper catalysts described in this work have been filed: Y.C., Z.C. and E.H.S. “Electrodeposited catalysts promoting CO2 and/or CO electroreduction to carbon products in acidic conditions”. US Patent Application No. 63/489,035. Luxembourg Patent Application No. LU503602. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, Y., Chen, Z., Li, P. et al. Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions. Nat Commun 14, 2387 (2023). https://doi.org/10.1038/s41467-023-37898-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37898-8
This article is cited by
-
Acidic media enables oxygen-tolerant electrosynthesis of multicarbon products from simulated flue gas
Nature Communications (2024)
-
Electrochemical Carbon Dioxide Reduction in Acidic Media
Electrochemical Energy Reviews (2024)
-
Electrifying the future: the advances and opportunities of electrocatalytic carbon dioxide reduction in acid
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
