
Abstract
Homogenous transition-metal catalysts bearing a chiral ligand are widely used for asymmetric hydrogenation of unsaturated compounds such as olefins and ketones, providing efficient concise access to products with chiral carbon centers. However, distinguishing the re and si prochiral faces of a double bond bearing two substituents that are sterically and electronically similar is challenging for these catalysts. Herein, we report a relay strategy for constructing compounds with a chiral gem-diaryl carbon center by means of a combination of selective arene exchange between 1,1-diarylethylenes or benzophenones with (naphthalene)Cr(CO)3 and subsequent asymmetric hydrogenation. During the hydrogenation, the Cr(CO)3 unit facilitate differentiation of the two prochiral faces of the substrate double bond via formation of a three-dimensional complex with one of the aromatic rings by selective arene exchange. Density functional theory calculations reveal that during the hydrogenation, chromium coordination affected π–π stacking of the substrate and the catalyst ligand, leading to differentiation of the prochiral faces.
Similar content being viewed by others

Introduction
Chiral carbon centers are present in a wide variety of functional molecules, including natural products, pharmaceuticals, and agrochemicals1. Extensive study of asymmetric hydrogenation reactions over the last several decades has led to the establishment of a number of remarkable methods involving homogeneous transition-metal catalysis, providing reliable access to chiral compounds from abundant unsaturated substrates2,3,4,5,6,7,8,9,10,11,12,13,14,15. To achieve high enantioselectivity, researchers have designed chiral catalysts that can distinguish between the two prochiral faces of substrates on the basis of differences between the substituents on the double bonds16,17,18,19,20,21,22,23,24,25,26,27,28,29. However, general methods for discriminating between the re and si faces of double bonds bearing two different aryl groups, which would provide compounds with chiral gem-diaryl motifs30,31,32,33,34,35,36,37,38,39,40,41, are still under development owing to the small difference in electronic properties between the two aromatic rings and to their similar planar structures.
Depending on the positions of the substituents on the aromatic rings of 1,1-diaryl unsaturated compounds such as 1,1-diarylethylenes and benzophenones, they can be divided into two categories. High enantioselectivities have been achieved for category a, compounds in which one of the aromatic rings bears an ortho substituent (Fig. 1a)42,43,44,45,46,47,48,49,50,51,52. For example, Wang et al. reported rhodium/Duanphos-catalyzed asymmetric hydrogenation of 1,1-diarylethylenes bearing a ortho-hydroxy directing group44. In addition, Song et al. accomplished elegant syntheses of enantiomerically enriched 1,1-diarylethanes by means of iridium-catalyzed hydrogenation reactions assisted by an ortho carboxylic acid group45. Sterically bulky substituents without a chelating atom can also facilitate catalyst differentiation of the prochiral faces of double bonds. For example, Mazuela et al. accomplished asymmetric hydrogenation of 1,1-diarylethylenes by using an iridium phosphite–oxazoline catalyst42, and Chen et al. used a chiral oxazoline iminopyridine–cobalt catalyst for enantioselective hydrogenation of 1,1-diarylethylenes48. Ohkuma et al.49 and Touge et al.52 found that ruthenium-catalyzed asymmetric hydrogenation of benzophenones also requires an ortho substituent for high enantioselectivity. In addition, owing to the less difference of steric hinderance, chiral induction in hydrogenation of unsaturated compounds bearing two ortho-substituted aromatic rings is challenging; a highly enantioselective catalytic method has yet to be reported.

a Metal-catalyzed asymmetric hydrogenation of 1,1-diarylethylenes and benzophenones with ortho-substituent. b Metal-catalyzed asymmetric hydrogenation of 1,1-diarylethylenes and benzophenones without ortho-substituent.
In stark contrast, asymmetric hydrogenation of 1,1-diaryl-substituted double bonds without the assistance of a proximal substituent is fundamentally difficult, and enantioselectivities are generally low (Fig. 1b)53,54,55,56. Exceptionally, double bonds bearing highly electronically biased aromatic rings have been successfully hydrogenated with good enantioselectivities. For example, Bess et al. achieved asymmetric hydrogenation of 1,1-diarylalkenes bearing a 3,5-dimethoxylphenyl ring by using an iridium/phosphoramidite catalyst53, and Touge et al. observed high enantioselectivities in ruthenium/1,2-diphenyl-1,2-ethylenediamine-catalyzed transfer hydrogenation of benzophenones with electron-poor aromatic rings, such as a 3,5-dinitrophenyl ring52.
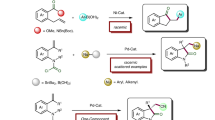
To provide a reliable method for asymmetric hydrogenation of 1,1-diaryl-substituted double bonds, we wondered a transition-metal unit would facilitate the differentiation of the two prochiral faces by ligating with one of the aromatic rings. Metal η6-benzene complexes, such as (benzene)Cr(CO)3, have different electronic properties than free benzene and have three-dimensional structures57,58,59,60,61,62,63,64,65,66. In 1995, Corey and co-workers conducted an elegant study of the electronic effects of remote substituents on aromatic rings during Corey-Bakshi-Shibata (CBS) reduction of benzophenones and found that Cr(CO)3-ligated p’-chlorobenzophenone synthesized from (benzene)Cr(CO)3 can be reduced to the corresponding alcohol with 94% ee (Fig. 2a)67,68,69,70. Given that the stability of a metal η6-arene complex varies with the ligated aromatic ring (for an example involving chromium, see Fig. 2b)71,72,73, we envisioned that combining selective arene coordination with a metal unit and transition-metal-catalyzed asymmetric hydrogenation would provide a reliable solution to the challenge posed by category b. Two questions needed to be answered: Could a metal unit distinguish and coordinate selectively to only one aromatic ring of a conjugated compound, and would the metal complexes be compatible with homogenous hydrogenation conditions74,75?

a Synthesis and Corey-Bakshi-Shibata (CBS) reduction of Cr(CO)3 benzophenone complex. b Relative stability of Cr(CO)3 arene complex. c This work: synthesis of compounds bearing a chiral gem-diaryl motif from either 1,1-diarylethylenes or benzophenones by a sequence involving selective arene exchange and stereoselective asymmetric hydrogenation.
Herein, we report a relay strategy for synthesis of compounds bearing a chiral gem-diaryl motif from either 1,1-diarylethylenes or benzophenones by a sequence involving selective arene exchange with (naphthalene)Cr(CO)3 (2) and subsequent ruthenium-catalyzed asymmetric hydrogenation (Fig. 2c).
Results
Reaction optimization
To explore the feasibility of our strategy, we began by investigating arene exchange reactions between different metal species and 4-chlorobenzophenone (1a) as a model substrate (see Supplementary Fig. 1). To our delight, upon reaction of 1a with (naphthalene)Cr(CO)3 (2)76,77,78,79,80 in dioxane at 80 °C, arene exchange took place selectively on the unsubstituted benzene ring (1a-Cr: 1a-Cr’ > 10:1).
The arene exchange conditions showed great generality, allowing for ligation of an array of benzophenones with generally high regioselectivities (Fig. 3). Specifically, the selectivity for the two aromatic rings was in line with the trend in the relative stabilities of the Cr(CO)3–arene complexes shown in Fig. 2b. In addition, we found that the selectivity varied somewhat with temperature. For example, after a mixture of Cr-ligated 4-methoxylbenzophenone (1b) was heated at 140 °C for 3 h, the 1b-Cr/1b-Cr’ ratio increased to 4.4:1 from the 1.5:1 ratio that was observed under our kinetically controlled conditions, and a complex bearing one Cr(CO)3 units on each aromatic ring was detected (see Supplementary Fig. 3). We attributed this result to arene exchange between complexes under thermodynamic conditions, and metal migration along the conjugated system may also have taken place81,82,83.

Arene exchange reactions between (naphthalene)Cr(CO)3 (2) and an array of benzophenones. Reaction conditions, unless otherwise stated: benzophenones (0.10 mmol), 2 (0.15 mmol), dioxane (1.0 mL), N2, 80 °C, 24 h. a2 (0.11 mmol). Ratios were determined by 1H NMR spectroscopy.
Next, we investigated the asymmetric hydrogenation of Cr(CO)3-ligated 1a as a model substrate by using various chiral Ru catalysts (see Supplementary Fig. 4a). To our delight, the Ru(II)-NHC-diamine catalyst developed by Glorius group84 afforded the corresponding reduced complex with high enantioselectivity (93% ee) upon reaction with H2 gas (5 atm) in the presence of NaOtBu. The free alcohol could be readily released from the chromium with no obvious loss of enantiomeric purity by irradiation of the crude reaction mixture at 440 nm under air85. In contrast, when 1a was directly subjected to the same hydrogenation conditions, the enantioselectivity was much lower (43% ee) (see Supplementary Fig. 4b). Beside Ru-1, we also found that the spiro diphosphines supported ruthenium catalyst developed by Zhou group86 also provided alcohol 4a with high enantioselectivity (91% ee) (see Supplementary Fig. 4a).
Substrate scope
Having optimized the conditions for the selective arene exchange and the asymmetric hydrogenation, we evaluated the scope of the sequence by applying it to an array of benzophenones 1 (Fig. 4). Monosubstituted benzophenones were suitable substrates (4a–4j), providing the desired alcohols in moderate to good yields with high enantioselectivities, regardless of whether the substituent was in the para or meta position. In addition, substrates bearing bisfunctionalized aromatic rings (4k–4u) and polyfunctionalized aromatic rings (4v, 4w), were suitable and gave high enantioselectivities (up to 99% ee). Notably, an array of functional groups, such as carboxylate (4h), amide (4i), piperonyl (4k), and siloxy (4q), were well tolerated under the reaction conditions.

Synthesis of compounds bearing a chiral gem-diaryl motif from either 1,1-diarylethylenes or benzophenones by a sequence involving selective arene exchange and stereoselective asymmetric hydrogenation. Reaction conditions, unless otherwise stated: (1) 1 (0.2 mmol), 2 (0.3 mmol), dioxane (2 mL), 80 °C, 24 h. (2) Ru-1 (3 mol%), NaOtBu (15 mol%), toluene (1.5 mL), H2 (5 atm), 0 °C, 36 h; then irradiation with 440 nm LEDs for 2 h in air at room temperature. a(1) 2 (0.22 mmol). b(2) Room temperature. c(2) Hexane (1.5 mL), H2 (50 atm). Isolated yields were reported. The absolute configurations of 4a, 4e, 4 h, and 4 m were S, as determined by comparison of their optical rotations with reported data.
Next, we evaluated the utility of the sequence for asymmetric hydrogenation of several 1,1-diarylethylenes (Fig. 4). By using hexane as the solvent for hydrogenation instead of toluene, we could obtain 1,1-diarylethanes in good yields with moderate to high enantioselectivities (up to 99% ee); both mono- (5a–5c) and disubstituted (5d) 1,1-diarylethylenes were suitable substrates.
Complexation of an unsymmetrically 1,2- or 1,3-disubstituted arene ring to a metal via η6-coordination results in a molecule lacking symmetry elements60. To determine whether planar chirality affected the stereochemical control of the asymmetric hydrogenation, we subjected a 1:1 mixture of both enantiomers of Cr(CO)3 complexes of 2-methyl-4’-chlorobenzophenone to the ruthenium-catalyzed hydrogenation conditions (Fig. 5a). This reaction yielded two pair of diastereomers with > 20:1 dr and 2.9:1 dr respectively. After removing the Cr(CO)3 unit, we obtained the free alcohol (4x) in overall 98% yield and 74% ee, implying that chiral catalyst Ru-1 dominated the differentiation of the two prochiral faces during the hydrogenation. Interestingly, shortening the reaction time to 15 minutes resulted in kinetic resolution of 1x-Cr87, providing alcohol 4x in 92% ee.

a Synthesis of 4x by a sequence involving selective arene exchange and stereoselective asymmetric transfer hydrogenation. b Synthesis of 4b by a sequence involving selective arene exchange and stereoselective asymmetric transfer hydrogenation.
Our strategy was also applicable to substrates that formed chromium complexes less selectively (Fig. 5b). For example, reaction of 4-methoxybenzophenone (1b) with (naphthalene)Cr(CO)3 (2) gave a mixture of two isomers, 1b-Cr and 1b-Cr’, which could be separated. Each of the isomers was then converted to the same enantioenriched alcohol (4b) by reduction catalyzed by the catalyst Ru-1 or its enantiomer Ru-1’, respectively.
In addition, we tested the versatility of our relay strategy by replacing H2 gas with other easy-handle hydrogenation sources, and we found that asymmetric hydrogenation of Cr(CO)3-ligated 4-chlorobenzophenone catalyzed by a 1,2-diphenyl-1,2-ethylenediamine-derived ruthenium complex Ru-288,89,90 with sodium formate provided alcohol 4a with high enantioselectivity. Then we evaluated the substrate scope of the combination of arene exchange and asymmetric hydrogenation (Fig. 6). To our delight, an array of diaryl ketones were suitable for this alternative method, providing enantioenriched 1,1-diarylmethanols with HCO2Na as the hydrogen source instead of H2 gas. Notably, substrates with a vinyl group (4ac) were tolerated under the reaction conditions.

Synthesis of compounds bearing a chiral gem-diaryl motif from benzophenones by a sequence involving selective arene exchange and stereoselective asymmetric transfer hydrogenation. Reaction conditions, unless otherwise noted: (1) 1 (0.2 mmol), 2 (0.3 mmol), dioxane (2.0 mL), 80 °C, 24 h. (2) Ru-2 (7 mol%), NaCO2H (2 mmol), DMF (1.0 mL), H2O (0.1 mL), 35 °C, 36 h; then irradiation for 2 h with 440 nm LEDs. Isolated yields were reported. The absolute configurations of 4a, 4e, 4 h and 4 m were S, as determined by comparing their optical rotations with reported data.
Mechanistic studies
Next, we turned our attention to the origin of the high enantioselectivity achieved in the hydrogenation of Cr(CO)3-ligated substrates. The steric bulk of the Cr(CO)3 unit can be expected to have hindered rotation of the aromatic rings relative to the extent of rotation possible in the unbound precursor. Indeed, density functional theory calculations involving a relaxed scan of the dihedral angle formed by the carbonyl group and the aromatic ring connected to the carbonyl group were consistent with this expectation. The maximum barrier to rotation for each ring of 4-chlorobenzophenone (1a) was around 3 kcal/mol, and there was no obvious energetical difference between the para-Cl-substituted and unsubstituted rings (Fig. 7a). In contrast, the barrier to rotation of Cr(CO)3-ligated phenyl group of 1a-Cr was remarkably higher than that of the unbound phenyl ring, which may require a higher energy cost to adapt the benzophenone to the catalytic pocket (Fig. 7b).

The relative Gibbs free energies are given in kcal/mol. a Energy diagram of a relaxed scan of the dihedral angle between the carbonyl group and the aromatic ring in 1a. b Energy diagram of a relaxed scan of the dihedral angle between the carbonyl group and the aromatic ring in 1a-Cr.
To gain more insight into the mechanism of the stereoselectivity of the ruthenium-catalyzed hydrogenation, we performed additional density functional theory calculations. On the basis of previous studies52,84, we began by investigating the reaction of noncoordinated 4-chlorobenzophenone (Fig. 8, left part), and we found that the transition states for the different stereoselectivities were quite similar in structure; there was only a tiny energy difference (0.3 kcal/mol) between them, which is consistent with the observed absolute stereochemical outcome and the low enantioselectivity (43% ee). Moreover, similar weak interactions (the green area, see IGMH analysis) between the aromatic substituents of the catalyst and the 4-chlorobenzophenone ring in transition states TS-1 and TS-2 lead to quite close in energy.

The relative Gibbs free energies are given in kcal/mol. Left part, computational studies of transition states for asymmetric hydrogenation of 1a. Right part, computational studies of transition states for asymmetric hydrogenation of 1a-Cr.
In contrast, calculations on the asymmetric hydrogenation of Cr(CO)3-ligated 4-chlorobenzophenone revealed relatively lower energy barriers to the formation of both enantiomeric products, which can probably be attributed to the electron-withdrawing effect of the Cr(CO)3 unit (for atomic charge population and conceptual density functional analysis see Supplementary Tables 2 and 3). Furthermore, transition state TS-4, formed by reduction from the re face, was favored by 2.1 kcal/mol over TS-3, formed by reduction from the si face (Fig. 8, right part). Specifically, in TS-3, the chromium-ligated benzene ring was located in a rigid pocket formed by a phenyl group of the diamine ligand and a naphthyl group of the carbene ligand, leading not only to greater steric repulsion (for activation strain model, see Supplementary Table 2), but also to a weaker π–π stacking interaction between the phenyl ring of the diamine ligand and the chromium-ligated benzene ring (the corresponding green area in TS-3 was smaller than that in TS-4, see IGMH analysis) because rotation of the latter was hindered, as indicated by the clearly smaller | ∠O1-C1C2-C3 | angle (27.7°) relative to other transition states TS-1 (39.2°), TS-2 (39.1°), and TS-4 (41.9°).
In conclusion, we have developed a relay strategy for accessing compounds with a chiral carbon center bearing two aromatic substituents by means of a combination of selective arene exchange with (naphthalene)Cr(CO)3 and subsequent enantioselective hydrogenation mediated by a chiral ruthenium catalyst. Density functional theory calculations revealed that the chromium markedly affected the energy of the transition state for addition of the ruthenium hydride species to the carbonyl group. Work aimed at extending this strategy to related transformations is currently underway in our laboratory.
Methods
General procedure for synthesis of Cr complexes
In a glovebox, a solution of Cr(CO)3(naphthalene) 2 (0.3 mmol, 1.5 equiv.), 1 or 3 (0.2 mmol, 1.0 equiv.) in 1,4-dioxane (2 mL) in a 4 ml glass vial was stirred at room temperature for 2 min, then the sealed reaction vial was taken out of the glovebox and the reaction mixture was stirred at 80 °C in the dark for 24 h. After cooling to room temperature, the solution was evaporated under reduced pressure. The residue was purified by column chromatography eluting with Ethyl acetate/Petroleum ether, affording complex 1-Cr or 3-Cr.
General procedure for asymmetric hydrogenation
(Figure 4) To a 5 ml glass tube, Ru-1 (3 mol%), NaOtBu (15 mol%), 1-Cr or 3-Cr (1.0 equiv.), and toluene or hexane (0.10 M) were added in a glovebox. The glass tube was taken out of the glovebox and placed in a pre-cooled (0 °C) stainless steel autoclave, then the autoclave and pressurized and depressurized with hydrogen gas five times before the indicated pressure (5 atm or 50 atm) was set. The reaction mixture was stirred at 0 °C for 36 h. After the autoclave was carefully depressurized, the tube was irradiated under 440 nm LEDs (20 W) for 2 hours at room temperature in air atmosphere. Then the product was purified by flash column chromatography on silica gel. The enantiomeric excess was determined by SFC or HPLC analysis using chiral column. (Fig. 6) To a 4 ml glass vial, Ru-2 (7 mol%), HCO2Na (10 equiv.), 1-Cr (1.0 equiv.), and DMF/H2O (10:1, 0.18 M) were added. The reaction mixture was stirred at 35 °C for 36 h. Then the vial was irradiated under 440 nm LEDs (20 W) for 2 hours at room temperature in air atmosphere. Then the product was purified by flash column chromatography on silica gel. The enantiomeric excess was determined by SFC or HPLC analysis using chiral column.
Data availability
Experimental procedures, characterization data, copies of NMR spectra and computational details are available in the Supplementary Information. Supplementary Data 1 contains the cartesian coordinates of the optimized structures. Supplementary Data 2 contains the detailed data of electronic energy of relaxed scan of dihedral angles. Data is available from the corresponding authors upon request.
References
Nicolaou, K. C. & Montagnon, T. Molecules that changed the World (Wiley-VCH, 2008).
Knowles, W. S. Asymmetric hydrogenation. Acc. Chem. Res. 16, 106–112 (1983).
Knowles, W. S. Asymmetric Hydrogenations (Nobel Lecture). Angew. Chem. Int. Ed. 41, 1998–2007 (2002).
Tang, W. & Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3069 (2003).
Kitamura, M. & Noyori, R. Hydrogenation and transfer hydrogenation (Wiley-VCH, 2005).
Johnson, N. B., Lennon, I. C., Moran, P. H. & Ramsden, J. A. Industrial-scale synthesis and applications of asymmetric hydrogenation catalysts. Acc. Chem. Res. 40, 1291–1299 (2007).
Shimizu, H., Nagasaki, I., Matsumura, K., Sayo, N. & Saito, T. Developments in asymmetric hydrogenation from an industrial perspective. Acc. Chem. Res. 40, 1385–1393 (2007).
Roseblade, S. J. & Pfaltz, A. Iridium-catalyzed asymmetric hydrogenation of olefins. Acc. Chem. Res. 40, 1402–1411 (2007).
Xie, J.-H., Zhu, S.-F. & Zhou, Q.-L. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 111, 1713–1760 (2011).
Wang, D.-S., Chen, Q.-A., Lu, S.-M. & Zhou, Y.-G. Asymmetric hydrogenation of heteroarenes and arenes. Chem. Rev. 112, 2557–2590 (2012).
Verendel, J. J., Pàmies, O., Diéguez, M. & Andersson, P. G. Asymmetric hydrogenation of olefins using chiral Crabtree-type catalysts: scope and limitations. Chem. Rev. 114, 2130–2169 (2014).
Zhao, D., Candish, L., Paul, D. & Glorius, F. N-heterocyclic carbenes in asymmetric hydrogenation. ACS Catal 6, 5978–5988 (2016).
Zhang, Z., Butt, N. A. & Zhang, W. Asymmetric hydrogenation of nonaromatic cyclic substrates. Chem. Rev. 116, 14769–14827 (2016).
Seo, C. S. G. & Morris, R. H. Catalytic homogeneous asymmetric hydrogenation: successes and opportunities. Organometallics 38, 47–65 (2019).
Wang, H., Wen, J. & Zhang, X. Chiral tridentate ligands in transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 121, 7530–7567 (2021).
Noyori, R. & Takaya, H. BINAP: an efficient chiral element for asymmetric catalysis. Acc. Chem. Res. 23, 345–350 (2002).
Chen, Y., Yekta, S. & Yudin, A. K. Modified BINOL ligands in asymmetric catalysis. Chem. Rev. 103, 3155–3211 (2003).
McManus, H. A. & Guiry, P. J. Recent developments in the application of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev. 104, 4151–4202 (2004).
Desimoni, G., Faita, G. & Jorgensen, K. A. C2-symmetric chiral bis(oxazoline) ligands in asymmetric catalysis. Chem. Rev. 106, 3561–3651 (2006).
Xie, J.-H. & Zhou, Q.-L. Chiral diphosphine and monodentate phosphorus ligands on a spiro scaffold for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res. 41, 581–593 (2008).
Teichert, J. F. & Feringa, B. L. Phosphoramidites: privileged ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 49, 2486–2528 (2010).
Liao, S., Sun, X.-L. & Tang, Y. Side arm strategy for catalyst design: modifying bisoxazolines for remote control of enantioselection and related. Acc. Chem. Res. 47, 2260–2272 (2014).
Ye, B. & Cramer, N. Chiral cyclopentadienyls: enabling ligands for asymmetric Rh(III)-catalyzed C−H functionalizations. Acc. Chem. Res. 48, 1308–1318 (2015).
Janssen-Muller, D., Schlepphorst, C. & Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 46, 4845–4854 (2017).
Liu, X., Zheng, H., Xia, Y., Lin, L. & Feng, X. Asymmetric cycloaddition and cyclization reactions catalyzed by Chiral N,N’-dioxide−metal complexes. Acc. Chem. Res. 50, 2621–2631 (2017).
Zhang, L. & Meggers, E. Steering asymmetric lewis acid catalysis exclusively with octahedral metal-centered chirality. Acc. Chem. Res. 50, 320–330 (2017).
Xu, G., Senanayake, C. H. & Tang, W. P-chiral phosphorus ligands based on a 2,3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 52, 1101–1112 (2019).
Mas-Roselló, J., Herraiz, A. G., Audic, B., Laverny, A. & Cramer, N. Chiral cyclopentadienyl ligands: design, syntheses, and applications in asymmetric catalysis. Angew. Chem. Int. Ed. 60, 13198–13224 (2021).
Ye, F., Xu, Z. & Xu, L.-W. The discovery of multifunctional chiral P ligands for the catalytic construction of quaternary carbon/silicon and multiple stereogenic centers. Acc. Chem. Res. 54, 452–470 (2021).
Pu, L. & Yu, H.-B. Catalytic asymmetric organozinc additions to carbonyl compounds. Chem. Rev. 101, 757–824 (2001).
Schwab, R. S., Narayanaperumal, S., Teixeira, W. K. O. & de Albuquerque, D. Y. Recent advances in the synthesis of enantiomerically enriched diaryl, aryl heteroaryl, and diheteroaryl alcohols through addition of organometallic reagents to carbonyl compounds. Synthesis 52, 1855–1873 (2020).
Grélaud, S., Cooper, P., Feron, L. J. & Bower, J. F. Branch-selective and enantioselective iridium-catalyzed alkene hydroarylation via anilide-directed C−H oxidative addition. J. Am. Chem. Soc. 140, 9351–9356 (2018).
Friis, S. D., Pirnot, M. T. & Buchwald, S. L. Asymmetric hydroarylation of vinylarenes using a synergistic combination of CuH and Pd catalysis. J. Am. Chem. Soc. 138, 8372–8375 (2016).
Podhajsky, S. M., Iwai, Y., Cook-Sneathen, A. & Sigman, M. S. Asymmetric palladium-catalyzed hydroarylation of styrenes and dienes. Tetrahedron 67, 4435–4441 (2011).
Chen, Y.-G. et al. Nickel-catalyzed enantioselective hydroarylation and hydroalkenylation of styrenes. J. Am. Chem. Soc. 141, 3395–3399 (2019).
Lv, X.-Y., Fan, C., Xiao, L.-J., Xie, J.-H. & Zhou, Q.-L. Ligand-enabled Ni-catalyzed enantioselective hydroarylation of styrenes and 1,3-dienes with arylboronic acids. CCS Chem 1, 328–334 (2019).
Tran, H. N., Burgett, R. W. & Stanley, L. M. Nickel-catalyzed asymmetric hydroarylation of vinylarenes: direct enantioselective synthesis of chiral 1,1-diarylethanes. J. Org. Chem. 86, 3836–3849 (2021).
He, Y., Liu, C., Yu, L. & Zhu, S. Enantio- and regioselective NiH-catalyzed reductive hydroarylation of vinylarenes with Aryl iodides. Angew. Chem. Int. Ed. 59, 21530–21534 (2020).
Zhang, S. et al. Design and synthesis of tunable chiral 2,2’-bipyridine ligands: application to the enantioselective nickel-catalyzed reductive arylation of aldehydes. Angew. Chem. Int. Ed. 61, e202117843 (2022).
Zhu, Z., Xiao, J., Li, M. & Shi, Z. Nickel-catalyzed intermolecular asymmetric addition of aryl iodides across aldehydes. Angew. Chem. Int. Ed. 61, e202201370 (2022).
Jiang, X. et al. Photoassisted cobalt-catalyzed asymmetric reductive grignard-type addition of aryl iodides. J. Am. Chem. Soc. 144, 8347–8354 (2022).
Mazuela, J. et al. Iridium phosphite-oxazoline catalysts for the highly enantioselective hydrogenation of terminal alkenes. J. Am. Chem. Soc. 131, 12344–12353 (2009).
Mazuela, J., Norrby, P. O., Andersson, P. G., Pamies, O. & Dieguez, M. Pyranoside phosphite−oxazoline ligands for the highly versatile and enantioselective Ir-catalyzed hydrogenation of minimally functionalized olefins. A combined theoretical and experimental study. J. Am. Chem. Soc. 133, 13634–13645 (2011).
Wang, X. et al. Highly enantioselective hydrogenation of styrenes directed by 2’-hydroxyl groups. Org. Lett. 13, 1881–1883 (2011).
Song, S., Zhu, S.-F., Yu, Y.-B. & Zhou, Q.-L. Carboxy-directed asymmetric hydrogenation of 1,1-diarylethenes and 1,1-dialkylethenes. Angew. Chem. Int. Ed. 52, 1556–1559 (2013).
Besset, T., Gramage-Doria, R. & Reek, J. N. Remotely controlled iridium-catalyzed asymmetric hydrogenation of terminal 1,1-diaryl alkenes. Angew. Chem. Int. Ed. 52, 8795–8797 (2013).
Wang, Z. et al. Organocatalytic asymmetric synthesis of 1,1-diarylethanes by transfer hydrogenation. J. Am. Chem. Soc. 137, 383–389 (2015).
Chen, J., Chen, C., Ji, C. & Lu, Z. Cobalt-catalyzed asymmetric hydrogenation of 1,1-diarylethenes. Org. Lett. 18, 1594–1597 (2016).
Ohkuma, T., Koizumi, M., Ikehira, H., Yokozawa, T. & Noyori, R. Selective hydrogenation of benzophenones to benzhydrols. Asymmetric synthesis of unsymmetrical diarylmethanols. Org. Lett 2, 659–662 (2000).
Kokura, A., Tanaka, S., Ikeno, T. & Yamada, T. Catalytic enantioselective borohydride reduction of ortho-fluorinated benzophenones. Org. Lett. 8, 3025–3027 (2006).
Sui, Y.-Z. et al. CuII-catalyzed asymmetric hydrosilylation of diaryl- and aryl heteroaryl ketones: application in the enantioselective synthesis of orphenadrine and neobenodine. Chem. Eur. J. 18, 7486–7492 (2012).
Touge, T., Nara, H., Fujiwhara, M., Kayaki, Y. & Ikariya, T. Efficient access to chiral benzhydrols via asymmetric transfer hydrogenation of unsymmetrical benzophenones with bifunctional oxo-tethered ruthenium catalysts. J. Am. Chem. Soc. 138, 10084–10087 (2016).
Bess, E. N. & Sigman, M. S. Distinctive meta-directing group effect for iridium-catalyzed 1,1-diarylalkene enantioselective hydrogenation. Org. Lett. 15, 646–649 (2013).
Margarita, C. & Andersson, P. G. Evolution and prospects of the asymmetric hydrogenation of unfunctionalized olefins. J. Am. Chem. Soc. 139, 1346–1356 (2017).
Wang, B., Zhou, H., Lu, G., Liu, Q. & Jiang, X. Bifunctional oxo-tethered ruthenium complex catalyzed asymmetric transfer hydrogenation of Aryl N-heteroaryl ketones. Org. Lett. 19, 2094–2097 (2017).
Chen, F. et al. Chirality-economy catalysis: asymmetric transfer hydrogenation of ketones by Ru-catalysts of minimal stereogenicity. ACS Catal 9, 5562–5566 (2019).
Fischer, E. O. & Hafner, W. Di-benzol-chrom. Z. Naturforschg 10 b, 665–668 (1955).
Simion, D. V. & Sorensen, T. S. A theoretical computation of the aromaticity of (Benzene)Cr(CO)3 compared to benzene using the exaltation of magnetic susceptibility criterion and a comparison of calculated and experimental nmr chemical shifts in these compounds. J. Am. Chem. Soc. 118, 7345–7352 (1996).
Polunin, K. E. & Schmalz, H.-G. Application of chromium-arene complexes in the organic synthesis. efficient synthesis of stilbene phytoalexins. Russ. J. Coord. Chem. 30, 252–261 (2004).
Rosillo, M., Domínguez, G. & Pérez-Castells, J. Chromium arene complexes in organic synthesis. Chem. Soc. Rev. 36, 1589–1604 (2007).
Kündig, E. P. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis (Topics in Organometallic Chemistry 7, Springer, 2004).
McGrew, G. I., Temaismithi, J., Carroll, P. J. & Walsh, P. J. Synthesis of polyarylated methanes through cross-coupling of tricarbonylchromium-activated benzyllithiums. Angew. Chem. Int. Ed 49, 5541–5544 (2010).
McGrew, G. I. et al. Asymmetric cross-coupling of aryl triflates to the benzylic position of benzylamines. Angew. Chem. Int. Ed. 51, 11510–11513 (2012).
Zhang, J. et al. Palladium-catalyzed allylic substitution with (η6-arene–CH2Z)Cr(CO)3-based nucleophiles. J. Am. Chem. Soc. 133, 20552–20560 (2011).
Ricci, P., Krämer, K., Cambeiro, X. C. & Larrosa, I. Arene–metal π-complexation as a traceless reactivity enhancer for C–H arylation. J. Am. Chem. Soc. 135, 13258–13261 (2013).
Ricci, P., Kramer, K. & Larrosa, I. Tuning reactivity and site selectivity of simple arenes in C−H activation: ortho-arylation of anisoles via arene−metal π-complexation. J. Am. Chem. Soc. 136, 18082–18086 (2014).
Corey, E. J. & Helal, C. J. Novel electronic effects of remote substituents on the oxazaborolidine-catalyzed enantioselective reduction of ketones. Tetrahedron Lett 36, 9153–9156 (1995).
Corey, E. J. & Helal, C. J. Catalytic enantioselective synthesis of the second generation histamine antagonist cetirizine hydrochloride. Tetrahedron Lett 37, 4837–4840 (1996).
Corey, E. J. & Helal, C. J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Ed 37, 1986–2012 (1998).
Kloetzing, R. J., Krasovskiy, A. & Knochel, P. The Mg-oppenauer oxidation as a mild method for the synthesis of aryl and metallocenyl ketones. Chem. Eur. J. 13, 215–227 (2007).
Mahaffy, C. A. L. & Pauson, P. L. Exchange-reactions of arenetricarbonylchromium complexes with arenes. J. Chem. Res., Synop. 4, 126–127 (1979).
Traylor, T. G., Stewart, K. & Goldberg, M. Arene exchange reactions of (arene)tricarbonylchromium complexes. J. Am. Chem. Soc. 106, 4445–4454 (1984).
Semmelhack, M. F., Chlenov, A. & Ho, D. M. Accelerated arene ligand exchange in the (Arene)Cr(CO)2L series. J. Am. Chem. Soc. 127, 7759–7773 (2005).
Frankel, E. N., Selke, E. & Glass, C. A. Homogeneous 1,4-addition of hydrogen catalyzed by tricarbonyl(arene)chromium complexes. J. Am. Chem. Soc. 90, 2446–2448 (1968).
Cais, M., Frankel, E. N. & Rejoan, A. Organometallic studies, XXIV. Selective hydrogenation of conjugated olefins catalyzed by arene chromium tricarbonyl complexes. Tetrahedron Lett. 9, 1919–1923 (1968).
Kündig, E. P., Perret, C., Spichiger, S. & Bernardinelli, G. Naphthalene complexes: V. Arene exchange reactions in naphthalenechromium complexes. J. Organomet. Chem. 286, 183–200 (1985).
Howell, J. A. S., Dixon, D. T., Kola, J. C. & Ashford, N. F. A kinetic investigation of arene exchange and substitution in (naphthalene)Cr(CO)3. J. Organomet. Chem. 294, C1–C4 (1985).
Howell, J. A. S. et al. The arene-exchange reaction in naphthalene– and pyrene–Cr(CO)3. Organometallics 10, 1852–1864 (1991).
Schmalz, H.-G., Millies, B., Bats, J. W. & Dürner, G. Diastereoselective complexation of temporarily chirally modified ligands: enantioselective preparation and configurational assignment of synthetically valuable η6-Tricarbonylchromium-1-tetralone Derivatives. Angew. Chem. Int. Ed. 31, 631–633 (1992).
Alexakis, A. et al. Resolution and asymmetric synthesis of ortho-substituted (benzaldehyde)tricarbonylchromium complexes. J. Am. Chem. Soc. 114, 8288–8290 (2002).
Cunningham, S. D., Öfele, K. & Willeford, B. R. Migration of tricarbonylchromium groups in phenylanthracenes. J. Am. Chem. Soc. 105, 3724–3725 (1983).
Kirss, R. U. & Treichel, P. M. Haptotropic rearrangements in naphthalene-chromium tricarbonyl complexes. J. Am. Chem. Soc. 108, 853–855 (1986).
Czerwinski, C. J., Fetisov, E. O., Gloriozov, I. P. & Oprunenko, Y. F. DFT study of intramolecular interring η6,η6-haptotropic rearrangements in tricarbonylchromium complexes of 2-aminobiphenyl and 4-aminobiphenyl. Dalton Trans 42, 10487–10494 (2013).
Li, W. et al. Design of Ru(II)-NHC-diamine precatalysts directed by ligand cooperation: applications and mechanistic investigations for asymmetric hydrogenation. J. Am. Chem. Soc. 142, 7100–7107 (2020).
Kündig, E. P. Synthesis of transition metal η6-arene complexes. Top. Organomet. Chem 7, 3–20 (2004).
Xie, J.-H. et al. Synthesis of spiro diphosphines and their application in asymmetric hydrogenation of ketones. J. Am. Chem. Soc. 125, 4404–4405 (2003).
Ursini, C. V., Dias, G. H. M. & Rodrigues, J. A. R. Ruthenium-catalyzed reduction of racemic tricarbonyl(η6-aryl ketone)chromium complexes using transfer hydrogenation: A simple alternative to the resolution of planar chiral organometallics. J. Organomet. Chem. 690, 3176–3186 (2005).
Dub, P. A., Wang, H., Watanabe, M., Gridnev, I. D. & Ikariya, T. A practical asymmetric conjugate addition to cyclic enones with chiral bifunctional Ru amido catalysts. Tetrahedron Lett 53, 3452–3455 (2012).
Dub, P. A. et al. C–F bond breaking through aromatic nucleophilic substitution with a hydroxo ligand mediated via water bifunctional activation. Bull. Chem. Soc. Jpn. 86, 557–568 (2013).
Dub, P. A., Matsunami, A., Kuwata, S. & Kayaki, Y. Cleavage of N−H bond of ammonia via metal−ligand cooperation enables rational design of a conceptually new Noyori−Ikariya catalyst. J. Am. Chem. Soc. 141, 2661–2677 (2019).
Acknowledgements
We thank the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2022SDXHDX0006), the National Natural Science Foundation of China (22071198, 22271235), and the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2020R01004) for research support. We thank Westlake university instrumentation and service center for molecular sciences and supercomputer center for the facility support and technical assistance.
Author information
Authors and Affiliations
Contributions
K.L. and W.-Q.W. contributed equally to this work. H.S. directed the project and wrote the manuscript. K.L. and W.-Q.W. performed the experiments and analysed the data. Y.L. performed density functional theory calculations. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Wu, WQ., Lin, Y. et al. Asymmetric hydrogenation of 1,1-diarylethylenes and benzophenones through a relay strategy. Nat Commun 14, 2170 (2023). https://doi.org/10.1038/s41467-023-37882-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37882-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
