
Abstract
Designing efficient catalyst for the oxygen evolution reaction (OER) is of importance for energy conversion devices. The anionic redox allows formation of O-O bonds and offers higher OER activity than the conventional metal sites. Here, we successfully prepare LiNiO2 with a dominant 3d8L configuration (L is a hole at O 2p) under high oxygen pressure, and achieve a double ligand holes 3d8L2 under OER since one electron removal occurs at O 2p orbitals for NiIII oxides. LiNiO2 exhibits super-efficient OER activity among LiMO2, RMO3 (M = transition metal, R = rare earth) and other unary 3d catalysts. Multiple in situ/operando spectroscopies reveal NiIII→NiIV transition together with Li-removal during OER. Our theory indicates that NiIV (3d8L2) leads to direct O-O coupling between lattice oxygen and *O intermediates accelerating the OER activity. These findings highlight a new way to design the lattice oxygen redox with enough ligand holes created in OER process.
Similar content being viewed by others

Introduction
The electrocatalysis of the oxygen evolution reaction (OER) is at the core of many energy conversion devices, such as water and CO2 electrolysers, and metal-air batteries1,2,3. Because of the sluggish kinetics of OER, the exploration of OER electrocatalysts with high intrinsic activity plays a decisive role in determining the device performance, and thus the fundamental understanding of OER mechanisms is essential4,5,6,7. The conventional adsorbate evolution mechanism, involving multiple adsorbed intermediates with highly correlated adsorption strengths, exhibits a minimum theoretical overpotential of about 0.37 V8,9. This limitation has been circumvented by a recently identified mechanism10,11—the lattice oxygen oxidation mechanism—wherein the oxygen ligands are electrochemically activated, coupled directly with *O intermediates, and released from the lattice matrix4. Lattice-oxygen-activated electrocatalysts, such as 3d late-transition-metal (TM) oxides with high metal oxidation states, universally feature a high TM-O covalency with a strong orbital hybridization between TM 3d and O 2p orbitals, which enables intramolecular electron transfer from the oxygen ligands to the TM cations, leaving ligand holes for lattice oxygen activation4,10. Such an anionic redox mechanism is well documented in lithium-ion-battery (LIB) cathodes12 and thermal oxidative catalysis13.
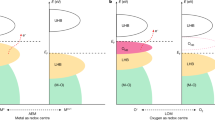
NiIII oxides are intrinsically anionic-redox-active. Here we use the notation NiIII rather than Ni3+ as a reminder that the formally trivalent Ni ion is in a highly covalent situation where holes in the oxygen 2p ligands play a significant role. NiIII oxides such as RNiO3, RSrNiO4 (R = rare earth element), and LiNiO2 are in the negative charge-transfer energy regime (Δ < 0, Fig. 1a), leading to 3d8L (L denotes a ligand hole located on a molecular orbital formed by the O 2p orbitals) as the dominant ground state configuration, instead of the conventional 3d7 (Δ > 0, Fig. 1b)14,15,16,17,18,19. It is well known that as the NiIII oxidized to NiIV, the electron removal in such charge transfer system should happen in the O 2p band rather than the Ni 3d band20, resulting in a 3d 8L2 configuration, namely double O 2p hole states (Fig. 1a).

a Δ < 0 and b Δ > 0 scenarios. Charge-transfer energy Δ: energy cost for electron transferring from the O 2p band to the Ni 3d band, and U: energy cost for electron transferring from the occupied 3d band to the unoccupied 3d band. The hybridization between Ni 3d and O 2p is neglected for simplicity. The Fermi level (EF) is shown as a vertical dash black line and the O 2p band is used as the energy reference considering the position of EF is arbitrary in the band gap.
Based on the lattice oxygen oxidation mechanism, we proposed that a catalyst featuring double ligand hole states would endow high OER activity. To test our prediction, LiNiO2 is employed as the precatalyst for the double ligand hole states considering that it has an edge-shared Ni-O-Ni network, which is more stable than the corner-shared network in RNiO321 and RSrNiO422. In addition, as a well-known cathode material of LIB, LiNiO2 can be easily delithiated and oxidized to form NiIV ions23,24, although this oxidizing process is rarely observed during OER in aqueous electrolytes. The stabilization of the highly active NiIV has been recently attributed to the high-entropy charge-glass-like state of LiNiO217. The electronic structure of LiNiO2 is found to have many nearly degenerate states composed of [NiO6] octahedra with 3d8, 3d8L and 3d8L2 configurations17. The LiNiO2 system can fluctuate between these states such that the hole states on the oxygen ligands are stabilized by the entropy term of the free energy.
In this study, a pristine Ni2+-free LiNiO2 sample was prepared using high-pressure O2, and its structure was comprehensively characterized using multiple complementary techniques, including microscopic techniques, X-ray diffraction (XRD), and X-ray absorption spectroscopy (XAS) at the Ni K, Ni L3,2, and O K edges. The electrocatalytic properties of LiNiO2 was robustly assessed using rotating ring disk electrodes (RRDE). The delithiation, oxidation and resulting structural transition of LiNiO2 during OER were probed using operando XRD, XAS and Raman spectroscopies. Combined analysis of the operando results, density function theory (DFT) calculation and in situ differential electrochemical mass spectrometry (DEMS) reveals that the enhanced OER activity of LiNiO2 is originated from the structurally stabilized NiIV with a 3d8L2 configuration.
Results
Structural characterization of LiNiO2
The crystalline phase and morphology of the as-prepared LiNiO2 were characterized using synchrotron X-ray diffraction (XRD) and transmission electron microscopy (TEM) (Fig. 2). The XRD pattern of LiNiO2 is well-matched with the standard pattern of R-3m LiNiO2 (JCPDS No. 09-0063) without any noticeable crystalline impurities (Fig. 2a and Supplementary Fig. 1). The sharp diffraction peaks, and the well separation of (006) and (10-2) and of (10-8) and (110), indicate a well-crystalized layered structure with a low degree of Li+/Ni2+ intermixing (Supplementary Fig. 1)23,25. Rietveld refinement was carried out to obtain the crystal structural parameters of the as-prepared LiNiO2 (Fig. 2a), yielding a good fit for the R-3m structure and ~3.9% Ni occupancy in the [LiO2] layers, demonstrating the high-purity of the sample. The single crystalline nature of the as-prepared LiNiO2 is suggested by TEM techniques (Fig. 2b–d). A typical selected-area electron diffraction (SAED) pattern of LiNiO2 shows clear diffraction spots that can be indexed to the R-3m structure of LiNiO2 along the [010] zone axis (Fig. 2b). High-resolution TEM (HRTEM) images of the thin parts of LiNiO2 display uniform lattice fringes of (003) and (104) of the R-3m structure, respectively (Fig. 2c and Supplementary Fig. 2). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were also recorded to directly visualize the Ni atomic arrangement along the edges, which well matches the (003) lattice plane of R-3m LiNiO2 (Fig. 2d). TEM, scanning electron microscopy (SEM) and atomic force microscopy (AFM) suggest that the general morphology of the as-prepared LiNiO2 is nanoflakes with 200 − 400 nm in the lateral size and ~10 nm in the layer thickness (Supplementary Fig. 3 and Fig. 2e). LiNiO2-raw, a LiNiO2 sample prepared without the second annealing step under high-pressure O2, shares a similar morphology with the LiNiO2 (Supplementary Fig. 4).

a A synchrotron XRD pattern (18 keV incident beam energy, λ = ~0.6888 Å) and a Rietveld refinement fit with the refined structural parameters listed in Supplementary Table 1, along with the structural model of R-3m LiNiO2 which consists of alternate [LiO2] and [NiO2] layers formed by edge-sharing [LiO6] or [NiO6] units. A set of extra diffraction peaks are from graphitic carbon of carbon paper and two broad diffraction peaks at 12° and 18° are from the sample holder. b A typical SAED pattern of LiNiO2 along a [010] zone axis of the R-3m structure, c a HRTEM image and its fast Fourier transform pattern, showing the (003) lattice fringes, and d an AC-HAADF-STEM image projected perpendicular to the (003) lattice plane. e An AFM image and the corresponding thickness profile across the horizontal red line.
The local coordination environment of transition metal ions can be obtained by extended X-ray absorption fine structure (EXAFS) spectra at the corresponding K edges26,27. The Ni local coordination of LiNiO2 resemble that of NiO and Ni(OH)2, with two dominant peaks at ~1.8 and ~2.8 Å, which are attributed to Ni–O and Ni–Ni scattering paths within and between the edge-sharing [NiO6] units, respectively (Fig. 3a and Supplementary Fig. 5). For these peaks, LiNiO2 shows a shorter radial distance than those of Ni(OH)2 and NiO, supporting the presence of a shorter Ni-O bond length due to the higher formal oxidation state of Ni.

a Fourier transforms of k3-weigted EXAFS spectra of LiNiO2, LiCoO2 (R-3m) and NiO, and b XANES spectra of LiNiO2 and Ni references (NiO, Ni(OH)2, and KNiIO6) at the Ni K edge. The amplitude of LiNiO2 EXAFS spectra is adjusted for a clear comparison, and the Fourier transforms were phase-corrected. TEY-XAS spectra of LiNiO2 c at the Ni L3 edge and d at the O K edge, along with NiO and γ-NiOOH as the Ni and O references.
From an ionic picture, low spin NiIII ions adopt a (t2g)6(eg)1 configuration in an octahedral crystal field and are Jahn-Teller (JT)-active, so that one can expect a strong local distortion of the [NiO6] octahedra to remove the degeneracy of the eg orbitals. There are local distortions of the [NiIIIO6] units here, as evidenced by the broader first coordination shell at 1.7 A and a larger disorder factor (σ2) of LiNiO2 by fitting R-space EXAFS spectra, as compared with that of the analogous R-3m LiCoO2 (CoIII, (t2g)6(eg)0, Fig. 3a, Supplementary Fig. 6, and Table 1). Two distortion mechanisms are proposed. One is the conventional local JT distortion, as suggested by previous EXAFS studies of LiNiO228,29, with four short Ni-O (~1.920 Å) and two long Ni-O (~2.069 Å) bonds in the [NiIIIO6] octahedra (Table 1 and Supplementary Fig. 7a). However, pure JT distortion fails to explain the neutron-pair distribution function and electronic structure of LiNiO2; therefore, a [NiO6]-size disproportionation (SD) mechanism was proposed, where the number of Ni−Oshort and Ni−Olong is equal17 (Table 1 and Supplementary Fig. 7b). Based on these two possible structural models, EXAFS fitting using two Ni−O coordination shells was performed. Both models gave comparable but better fits of the EXAFS data than that using only one Ni–O scattering path (Table 1 and Supplementary Fig. 7c), supporting the presence of distorted [NiIIIO6] in pristine LiNiO2. To distinguish JT distortion from SD, double cluster simulation would be needed to simulate the Ni-L2,3 XAS spectrum of LiNiO216, which is beyond the scope of this study.
The oxidation state of the as-prepared LiNiO2 was assessed by X-ray absorption near edge structure (XANES) spectroscopy (Fig. 3b). At the Ni K edge, the energy position of LiNiO2 locates in-between those of Ni2+ (NiO and Ni(OH)2) and NiIV references (KNiIO6), suggesting that the Ni of the LiNiO2 is in the intermediate oxidation state between Ni2+ and NiIV. Assuming that the formal oxidation state of Ni is proportional to the energy at 0.7 edge jump height, the average Ni oxidation state of LiNiO2 can be quantified as ~3.0 (Supplementary Fig. 9), in a good agreement with the nominal value.
The detailed electronic structure at the surface region can be obtained by total electron yield soft X-ray absorption spectra (TEY-sXAS) at the L3 edges. The multiplet spectral feature and the energy position at the L3 edges of 3d elements are highly sensitive valence state and local environments30,31,32,33. Figure 3c shows the Ni L3 edge sXAS of LiNiO2, along with those of NiO and γ-NiOOH as the Ni2+ and NiIII references, respectively. Both the multiplet spectral features and energy positions of the Ni-L3 edge are very similar to those of γ-NiOOH, indicating that the Ni of LiNiO2 is in the formal +3 oxidation state. The dominant peak for both γ-NiOOH and LiNiO2 locates at ~855 eV, much higher than that of NiO at ~853 eV, and further inspection reveals that the low-energy peak of LiNiO2 locates ~0.3 eV higher than the dominant peak of NiO in Fig. 3c. The low-energy peak is an intrinsic spectral feature of NiIII as found in theoretical and experimental XAS15,16 and high-quality single crystal LaNiO3 and NdSrNiO421,22 In addition, NiIII (3d8L) in our LiNiO2 is supported by theoretical simulation of the Ni L3,2 XAS in Supplementary Fig. 10a, where the LiNiO2 spectra can be nicely reproduced by the full multiplet cluster calculation with a nominal 3d8L configuration. Our LiNiO2 is free from Ni2+ impurity that often observed in the literature (Supplementary Fig. 10b).
The Ni valence state and Ni-O covalency were further studied by the O-K sXAS. The pre-edge peak below 533 eV can be assigned to the unoccupied 3d eg hybridised with O 2p orbitals. An increase in the valence state of the 3d element correlates with a shift of the pre-edge to lower energy and an enhancement of the spectral intensity due to the strong covalence21,34,35. The O-K sXAS of LiNiO2 is dominated by a strong peak at 528.2 eV without obvious contribution of a peak at 531.9 eV found from NiO (Fig. 3d). Thus, TEY-sXAS at both the Ni L3 edges and the O K edge suggest that the surface Ni oxidation state of LiNiO2 is NiIII and free from Ni2+ impurities.
Electrochemical measurements
The OER activity of LiNiO2 was studied using rotating ring disk electrodes (RRDE, disk: glassy carbon, ring: Pt) at 1600 rpm. The potential of the Pt ring was held at 0.42 V throughout the measurements, so that O2 once generated on the disk can be collected and electrochemically reduced on the ring (Fig. 4a and Supplementary Fig. 11), which enables the determination of OER onset potentials. The OER activity of LiNiO2 was estimated using linear sweep voltammetry (LSV) in Ar-saturated and Fe-free 1 M KOH and compared with the benchmark IrO2 (99.9%) catalyst and a NiOOH reference. The LiNiO2 shows the best OER geometric activity (Fig. 4b, current density normalized by disk electrode area) and intrinsic activity (Supplementary Fig. 13, current density normalized by electrochemical surface area), with the potential to achieve 10 mA cmgeo−2 (E@10 mA cmgeo−2, ~1.545 V) lower than those of IrO2 (~1.558 V) and NiOOH (~1.753 V). In addition, LiNiO2 shares a Tafel slope (53 mV dec−1) similar to IrO2 (54 mV dec−1) and distinguishes itself from conventional NiOOH (96 mV dec−1, Fig. 4c), indicating rapid OER kinetics of LiNiO2. Compared to the reported overpotential values of LiMO2 (M = Co and Fe), and RMO3 (R = Rare earth elements, M = Fe, Co and Ni) and other unary 3d transition metal catalysts, our LiNiO2 requires much less overpotential to achieve certain current density (Fig. 4d), demonstrating the unique electrochemical properties to facilitate OER. Amongst the previously reported LiNiO2 and even Fe-doped LiNiO2, the as-prepared sample exhibit superior E@10 mA cm−2 and Tafel slopes, highlighting the excellent activity of pristine LiNiO2 (Supplementary Table 2).

a Schematic illustration of a rotating ring disk electrode (RRDE, ring: Pt, disk: glassy carbon). b Linear sweep voltammograms and c Tafel plots of LiNiO2, NiO-derived NiOOH, IrO2 (99.9% metal basis, Adamas-beta) and carbon (Vulcan XC-72R) in Ar-saturated 1 M KOH solution, along with the corresponding Pt ring current shown in the inset (the onset potential of OER indicated by arrows). d OER activity comparison of the as-prepared LiNiO2 to LiMO2 (M = Co and Fe), and RMO3 (R = Rare earth elements, M = Fe, Co and Ni) and other unary 3d transition metal catalysts (Supplementary Table 3). e Stability test of LiNiO2 in 1 M KOH at 10 mA cm−2 (on carbon paper) and (the inset of e) linear sweep voltammograms of LiNiO2 before and after the stability test. f Window-opening cyclic voltammograms of LiNiO2 in Fe-free 1 M KOH with stepwise decreasing the lower potential limits, showing the evolution of redox peaks (magnified in the corresponding insets) and the deterioration of OER current. The voltammograms were collected under 1600 rpm with a sweep rate of 5 mV s−1, and IR drops of the voltammograms were determined by EIS prior to the measurements and compensated. g Operando Raman spectra of LiNiO2 at 1.56 V with and without being cycled through Ni2+/NiIII redox.
The stoichiometric structure of LiNiO2 is essential to achieve high OER activity. LiNiO2-raw was also characterized and compared with the pristine LiNiO2, as the high-pressure or high-flow-rate O2 during synthesis has been found to be essential for stabilizing NiIII and minimizing oxygen vacancies in LiNiO2 and LiNiO2-based cathode materials36,37. The LiNiO2-raw shares similar morphology with the LiNiO2 (Supplementary Fig. 4) but has ~4.6% oxygen vacancies (vs. ~0.4% in LiNiO2) as characterized by XRD refinement (Supplementary Fig. 14 and Supplementary Table 4), and a significant fraction of Ni2+ impurities as shown in Ni-K XAS (Supplementary Fig. 15) and surface sensitive TEY-sXAS at the Ni L3 and O K edges (Supplementary Fig. 16 and 17). We found that the off-stoichiometric structure of LiNiO2-raw yields reduced OER activity (Supplementary Fig. 18).
From the ring current of the RRDE, the OER onset potentials of LiNiO2, IrO2 and NiOOH were estimated as ~1.437, ~1.438 and ~1.495 V, respectively (Fig. 4b and Supplementary Fig. 11), indicating that LiNiO2 activates the oxidation of water at a much lower overpotential than NiOOH. The OER activity of NiOOH is consistent with a common understanding that Ni (oxy)hydroxides without Fe doping and without the presence of Fe in the electrolyte are poor OER electrocatalysts38,39,40. Thus, from perspectives of overpotentials, Tafel slopes and onset potentials, the electrocatalytic properties of LiNiO2 for OER is as good as the state-of-the-art IrO2 and outperforms the standard NiOOH. The stability of LiNiO2 was measured using chronopotentiometry at 10 mA cm−2 (Fig. 4e). LiNiO2 largely maintained its initial potential, which gradually increased by ~10 mV for the first 18 h, and then remained constant for the subsequent 30 h. The high stability of LiNiO2 is also indicated by the little activity deterioration in LSV before and after the 48 h stability test (the inset of Fig. 4e). One may be interested in the stability of LiNiO2 as an OER electrocatalyst, as LiNiO2 is known to suffer from severe capacity degradation as a cathode material for LIB. The fast capacity fading is generally observed in high voltage region >4.1 V vs. Li+/Li, resulting from a large content of delithiation in LixNiO2 (x < 0.3) and in turn structural collapse23,41,42. If the highest cut-off voltage would be limited to 4.1 V vs. Li+/Li, the capacity of LiNiO2 could retain >95% of its initial capacity after >100 cycles41,43. However, operation potentials for our OER are below 1.8 V vs. RHE which corresponds to only 4.03 V vs. Li+/Li in battery operation.
The interconversion of Ni2+-based (hydr)oxides to NiOOH is usually indicated by Ni2+/NiIII redox peaks in the potential window of 1.2–1.4 V (vs. RHE); therefore, the formation of NiOOH from LiNiO2 was investigated using window-opening cyclic voltammetry. As shown in Fig. 4f, the lower potential limit decreased stepwise from 1.41 V (used in Figs. 4a) to 1.11 V, close to the values generally used in previous studies44,45,46,47,48,49. When the lower potential limit was set to 1.41 V, the OER current is optimal and remains stable over multiple CV cycles. However, decreasing the limit to ~1.21 and ~1.11 V deteriorates the OER current significantly, accompanied by the appearance and gradual increase of redox peaks at ~1.36 (anodic) and ~1.26 V (cathodic), indicating the formation of redox-active but OER-inactive phases on the surface. A LiNiO2 sample, cycled through the Ni2+/NiIII redox, was characterized using operando Raman spectroscopy at 1.56 V (Fig. 4g). Its Raman spectrum shows clear differences in the position and relative intensity of the Raman bands and a considerable increase in the overall Raman scattering intensity, as compared to pristine LiNiO2 at the same potential (for more details, see the operando Raman section below). All these spectral features can be indexed to β-NiOOH50, indicating the structural transformation of OER-inactive NiOOH species from LiNiO2 after being cycled through Ni2+/NiIII redox. Window-opening voltammetry was also carried out in unpurified 1 M KOH (Supplementary Fig. 19), wherein, despite the appearance of the Ni2+/NiIII redox peaks in the close potential range, the OER current remains stable even when the lower potential limit reached ~1.11 V. This indicates that Fe impurities significantly improve the performance of NiOOH, in agreement with the common understanding of Ni-based OER electrocatlaysts38,39,40. Therefore, the voltammetric analysis shows that the OER activity and electrochemical properties of LiNiO2 are impacted by the lower potential limits and presence of Fe impurities in the electrolyte. When undergoing the Ni2+/NiIII redox, the surface structure of LiNiO2 transforms and degrades into NiOOH, which has a poor OER activity in the absence of Fe. In contrast, the pristine and structurally unaltered LiNiO2, whose real active species has been covered and neglected in previous LiNiO2 OER studies44,45,47,48,49, exhibits better intrinsic OER activity.
Operando XRD, XAS, and Raman studies
From the aforementioned structural and electrochemical measurements, the pristine nature of LiNiO2 has been characterised in terms of crystalline phase, local coordination environment, oxidation state and electronic structure, and the superior OER activity of LiNiO2 to the analogous NiOOH is confirmed and studied in detail. However, the active species of LiNiO2, which can no longer be attributed to NiOOH, remains unclear. In addition, LiNiO2, a typical cathode material for lithium-ion batteries, can be delithiated to increase the Ni oxidation state and induce structural transition;24,46 therefore, delithiation is expected to become comparable or even easier during the OER in KOH aqueous solution. To establish the relationship between the structure of LiNiO2 and its OER activity, operando synchrotron-based XRD and XAS, and Raman spectroscopy experiments were carried out.
Figure 5a shows the operando synchrotron XRD of LiNiO2 in 1 M KOH in the potential range of 1.41–1.81 V. The patterns consist of three parts: an amorphous background from cell body and window materials (~12° and 18°), diffraction peaks of graphitic carbon from carbon paper (indicated by arrows), and diffraction patterns of LiNiO2 and its delithiated derivatives. Using the carbon peak at ~11.8° as the internal reference for intensity and peak position (Supplementary Fig. 20), the overall LiNiO2 pattern largely remains intact under the applied potentials, and another set of pattern emerges with potentials in around 2θ values at the expense of LiNiO2 pattern intensity when the applied potential is greater than 1.46 V, coinciding with the onset of OER. Whilst (00 l) diffraction peaks of LiNiO2 show shoulders at lower 2θ values, the (h0l) peaks show shoulders at higher 2θ values, which suggests the formation of another hexagonal phase from the parent LiNiO2 with interlayer expansion and intralayer contraction for the [NiO2] layers24. Based on the peak position, the new phase can be indexed to layered LixNiO2 (x = 0.45–0.65, Supplementary Fig. 21)51, and other LixNiO2 polymorphs and NiOOH phases were not found. The LiNiO2 delithiation during OER is also supported by increased Li concentration of KOH electrolyte and Ni/Li molar ratio of the sample (Supplementary Table 5). Considering that both the delithiation and OER initiate from the surface, the delithiated LixNiO2 can be considered as the active species of OER.

a Operando XRD patterns and b Operando Ni K edge XANES spectra of LiNiO2 under applied potentials of 1.41–1.81 V in 1 M KOH solution. Diffraction peaks in low 2θ ranges and the pre-edge peak were magnified in the corresponding insets. A set of extra peaks are from graphitic carbon and broad diffraction peaks at 12° and 18° from the sample holder.
The oxidation of LiNiO2 accompanying with delithiation was tracked using operando XANES measurements at the Ni K edge. No discernible change in XANES spectra was observed when the electrode was immersed in 1 M KOH (Supplementary Fig. 22). Figure 5b shows the XANES spectra in the potential range of 1.41–1.81 V. The rising edge shifts towards higher energy with the applied potentials, indicating that the oxidation of NiIII coincides with the onset of OER. Using the relationship between the energy at 0.7 edge jump height and the Ni formal oxidation state obtained from ex-situ data (Supplementary Fig. 9), the average Ni oxidation state of LiNiO2 was plotted as a function of the applied potentials (Fig. 6c). The oxidation state is ~3.0 at 1.41 V, sharply increases to ~3.3 at 1.56 V, gradually at potentials >1.56 V and reaches a plateau of ~3.4. In addition, the pre-edge peak intensity was found to increase with applied potentials (the inset of Fig. 5b), corresponding to the decreased symmetry of [NiO6] octahedra (see EXAFS analysis below). Combining the potential-dependent oxidation state of Ni with the electrochemistry of LiNiO2 reveals a positive correlation between the number of NiIV ions and OER current (Fig. 6c), suggesting that NiIV in delithiated LixNiO2 can be considered as (part of) the OER active centre.

a, b Fourier transforms of operando Ni K edge EXAFS spectra of LiNiO2 under applied potentials of 1.41–1.81 V in 1 M KOH solution, with the Ni–O region and the Ni–Ni region magnified to better show the potential-dependent behaviour. The Fourier transformation was carried out over a k range of 2.5 − 12.4 Å−1, and the Fourier transforms were phase-corrected. c Average oxidation state, N(Ni–O) and R(Ni–O) of LiNiO2 as a function of applied potentials. The error bars represent the uncertainty of the fitted parameters. d Operando Raman spectra of LiNiO2, showing the Eg and A1g vibration bands, where oxygen atoms of [NiO6] units move oppositely along adjacent O-layers and symmetrically along the c-axis, respectively.
The dominance of NiIV with double ligand hole state (3d8L2) is supported by simulation of the experimental sXAS at the Ni-L3,2 edge of LiNiO2 after OER taken with surface sensitive TEY mode (black circles) using the full multiplet cluster calculation which includes full intra-atomic multiplet interaction, crystal field interaction and covalence. We found the experimental Ni-L3,2 sXAS of LiNiO2 after OER can be well reproduced by the sum (black line) of 75% 3d8L2 (blue line) and 25% 3d8L (red line) configurations (Supplementary Fig. 23b) giving rise to Ni3.75+ valence state.
Potential-dependent changes of LiNiO2-raw in XANES are much weaker than those of pure LiNiO2 (Supplementary Fig. 24). The average oxidation state of Ni of LiNiO2-raw grows much slower with applied potentials than that of LiNiO2, and reaches only ~3.2 at 1.81 V. The impeded Ni oxidation in LiNiO2-raw implies that the formation of NiIV and double ligand hole state in LiNiO2-raw is limited. For the LiNiO2-raw sample, the XRD patterns show increasing fraction of NiOOH phase with applied potentials, while the R-3m structures is largely maintained in LiNiO2 without discernible formation of any NiOOH phases (Supplementary Fig. 25). It should be noted that the nanoscale thickness of our LiNiO2 is a prerequisite to observe significant spectral changes in operando XRD and operando XAS, because the OER usually occurs within several nanometres from the surface. Thus, our results indicates that observation of NiIV with double ligand holes state is directly related to both our pure and nano-sheet pristine LiNiO2 sample.
Potential-dependent changes in the local coordination environment of Ni were probed using operando EXAFS (Fig. 6a, b). Similar to the ex-situ data, the Ni coordination shell is dominated by Ni–O and Ni–Ni scattering paths. The amplitude of Ni–O at ~1.45 Å increases with the applied potentials from 1.41 to 1.81 V (Fig. 6b). Because the Ni first-shell coordination in pristine LiNiO2 is saturated with six oxygen atoms, the coordination numbers (CN) of Ni-O cannot increase further, which alternatively implies the Ni-O bonds become increasingly ordered with the applied potentials. The lowered Ni-O distortion is also suggested by the decreased Ni-O peak border at the high R with increased amplitude (Fig. 6b) and by the increased frequency of Ni-O oscillation in the real part of the Fourier transforms (Supplementary Fig. 26). Quantitative structural information was extracted via EXAFS fitting (Supplementary Fig. 27 and Supplementary Table 6), where the parameter σ2(Ni–O) represents the distortion in the [NiO6] octahedra. The value of σ2(Ni–O) decreases as a function of the applied potential from 0.012 Å−2 at 1.41 V to 0.008 Å−2 at 1.81 V, supporting that the LiNiO2 evolves from the oxidation of SD-active NiIII atoms (t2g6eg1) to SD-inactive NiIV atoms (t2g6) under OER. In addition, the CN(Ni–O) decreases from 5.8 ± 0.6 at 1.46 V to 5.0 ± 0.4 at 1.81 V, indicating the formation of oxygen vacancies during OER52,53,54, which have been calculated to greatly reduce the OER overpotential by enhancing intermediate adsorption on the Ni sites53,54. In addition, the increase in the relative spectral intensity of the weak pre-edge peak at 8335.4 eV in the Ni-K XANES as a function of applied potentials (the inset of Fig. 5b) implies a reduced local symmetry of Ni ion, which generally suggests a decrease in CN53. Moreover, our DEMS measurements and DFT calculation indicate that lattice oxygen involves in OER process (see below). In this lattice oxygen mechanism, oxygen vacancies should be created.
In addition to the amplitude, the contour maps of Ni–O shows a gradually increasing contribution in the corresponding lower R regions (Fig. 6b), corresponding to a shift toward lower R in the real part of FT-EXAFS (Supplementary Fig. 26). A shrinking Ni–O bond length from 1.955 Å at 1.41 V to 1.909 Å at 1.81 V was determined via EXAFS fitting (Fig. 6c, Supplementary Fig. 27 and Supplementary Table 6), supporting the presence of a high valence state of NiIV under OER conditions. To summarise, by combining the fast operando Ni-K XANES and EXAFS spectra with the OER onset potential, NiIV and oxygen vacancies in delithiated LixNiO2 were identified during OER, and attributed to the high OER activity.
Operando Raman spectroscopy was further employed to track the intermediate species of LiNiO2 under the high applied potentials larger than 1.4 V. Different from the above Raman experiments in Fig. 4g, the LiNiO2 now was not forced to undergo Ni2+/NiIII redox, but directly applied to 1.46 V. The spectral profile of LiNiO2 Raman in Fig. 6d only exhibits slightly red-shifted bands (Eg: from 472 cm−1 to 462 cm−1; A1g: from 546 cm−1 to 544 cm−1) with increasing applied potentials during the OER process, in agreement with the formation of delithiated LiNiO255. This supports the results from the above operando XRD and XAS. From comparison between Fig. 6d and Fig. 4g we can conclude that the crystal structure, intermediates and the OER activity (Fig. 4f) of LiNiO2 strongly depend on the lowest potential applied.
The presence and formation of γ-NiOOH have been regarded as the origin of OER activity in many Ni-based oxides46,53,56. However, according to our operando Raman spectra (Fig. 6d), the active species of LiNiO2 could not be assigned to γ-NiOOH as per the band position and relative intensity (for LiNiO2, A1g: ~546 cm−1 and I(Eg)/I(A1g)=0.5; for γ-NiOOH, A1g: 558 − 560 cm−1 and I(Eg)/I(A1g) = 1.5 − 1.7)40,57,58,59. In addition, a large expansion in the interlayer spacing between [NiO2] layers, caused by the transformation from LiNiO2 (4.72 Å) to γ-NiOOH (≥7 Å), was not observed in our operando XRD data. In addition, post-mortem XRD and XAS indicate that the structure of LixNiO2 remains mainly intact after OER (Supplementary Fig. 29 and 30). Moreover, the surface and bulk AC-STEM images were employed to study the structural transformation of LiNiO2 after OER60,61 (Supplementary Fig. 31), and both show the atomic arrangement perpendicular to the (018) lattice plane of LiNiO2, supporting the surface structure of LiNiO2 remains mainly intact after OER. Thus, using operando X-ray diffraction/absorption and Raman spectroscopies and post-mortem characterizations, the active species were identified as the delithiated yet structurally intact LiNiO2 with oxygen vacancies.
DFT calculation and DEMS for OER mechanisms
First-principles DFT calculations were employed to investigate the underlying mechanism of the enhanced OER activity of delithiated LiNiO2 with respect to the reaction steps. Similar to our previous work11,62,63, three scenarios were considered for the OER mechanisms involving four proton-electron transfer steps: metal-site adsorbate evolution (MAE) mechanism, lattice-oxygen-vacancy-site (LOV) mechanism, and metal-and lattice-oxygen-vacancy-site (MLOV) mechanism. In these mechanisms, the adsorbate sites of the reaction steps are different: only the Ni sites in MAE, oxygen sites in LOV, and both in MLOV (Fig. 7a). We built models for perfect LiNiO2 and delithiated LixNiO2 to investigate the effects of the unsaturated NiIV state generated during OER. The (102) plane of LiNiO2 with a 50% lithium vacancy was used to model the surface reaction pathways on delithiated LixNiO2 (x = 0.5, Supplementary Fig. 32), as the delithiation and oxidation of LiNiO2 and the concomitant OER all start from the edge25,52.

a Schematic 4-step reaction pathways of MAE, LOV, and MLOV OER mechanisms. Free energy of each OER step of b Li0.5NiO2 and LiNiO2 at URHE = 1.23 V. The estimated thermodynamic overpotentials of each potential-determining step are labelled. c MSCV and faradic CV curves recorded in 18O-enriched 0.1 M KOH electrolyte prepared using H218O (99% 18O), showing the mass ion current (iMS) of 18O2, 16O18O and 16O2 along with the faradaic current (iF) and the applied potential E from the first CV scan. (The inset of c) Comparison of the measured iMS(16O18O) curve and the theoretically expected iMS(16O18O) curve purely from electrolyte oxidation, based on the 16O abundance of 1.18% in the electrolyte (Supplementary Equation S27). d The evolution of measured 18O abundance of evolved oxygen (from total DEMS charge) for 18O-labled LiNiO2 with CV cycles in H216O-based 0.1 M KOH electrolyte. The expected 18O fractions are based on the natural isotope abundance and on the measured value from DEMS experiment of unlabelled LiNiO2 in the 16O-based electrolyte.
The Gibbs free energy differences (ΔG) of each OER step via the three OER mechanisms for Li0.5NiO2 and LiNiO2 are shown in Fig. 7b, respectively. First, regardless of the OER pathway, the potential-limiting step of Li0.5NiO2 possess lower overpotential than that of LiNiO2, in agreement with the operando measurement results indicating delithiated LixNiO2 as the actual active phase for OER. Second, among the three OER scenarios for Li0.5NiO2 the MLOV route shows the lowest overpotential (0.69 V), suggesting that the OER via MLOV prevails on the delithiated Li0.5NiO2, and that the potential-limiting step is the adsorption of *OH (step 3). Third, for the OER on LiNiO2, the MLOV route also prevails, but the potential-limiting step switches to the formation of *OO intermediates and oxygen vacancies (step 1).
The transition of the potential-limiting step of LiNiO2 before and after delithiation can be attributed to the double ligand hole state induced by the presence of NiIV ions in Li0.5NiO2. In the electronic structure of octahedral-symmetric [NiO6] system, the charge-transfer energy from the O 2p to Ni 3d orbitals decreases with the increased valence state of the Ni ion4,20,52. The LiNiO2 system is already in the negative charge-transfer regime, and consequently, the Ni-O interaction is highly covalent14,17, in which NiIII adopts an average configuration close to 3d8L (rather than the formal 3d7), leaving a ligand hole residing on the O 2p orbitals. The covalency increases further when the NiIII is (partly) oxidised to NiIV via LiNiO2 delithiation, and the resulting NiIV was endowed with an average configuration of 3d8L2, that is, a double ligand hole state in O 2p. The high unoccupied density state with the O 2p ligand character in LixNiO2 activates lattice oxygen, favouring direct coupling with oxygenated intermediates and the formation of oxygen vacancies (i.e. step 1 of the MLOV route), which are the potential-limiting step of LiNiO2. The prediction of oxygen vacancies formation during OER is in good agreement with the experimental observation of operando XAS (Fig. 6c). Thus, density-functional theory indicates that the double ligand holes created under OER condition easily activates lattice oxygen, accelerating the formation of O-O binding and oxygen vacancies, and the presence of double ligand hole states is closely correlated to the high OER activity of the delithiated LixNiO2.
To confirm the participation of lattice oxygen during OER, we performed 18O-isotope labelling experiments using in situ differential electrochemical mass spectrometry (DEMS). The experiments were first carried out on unlabelled LiNiO2 in an 18O-enriched KOH electrolyte (prepared using 99% H218O) over six consecutive CV cycles from 1.4 V to 1.8 V. Figure 7c shows the first CV and simultaneous mass spectrometric cyclic voltammogram (MSCV) in the time domain. In the MSCV curves, the mass signal m/z = 34 of 16O18O, which is mainly originated from the coupling of 16O of the LiNiO2 lattice and 18O of the electrolyte, shows significant ion current, with 36O2 as the dominant molecular oxygen isotope and 32O2 within the noise level. The measured 16O18O mass signal was also compared with the theoretically expected profile of the oxidation of the 18O-enriched electrolyte (1.18% 16O abundance, the inset of Fig. 7c), revealing a significant excess of the 16O18O ion charge. This evidences that the lattice oxygen plays a direct role in forming O2 during OER4,10,64. Furthermore, after the lattice 16O was partially replaced by 18O from the electrolyte, the resulting 18O-labelled LiNiO2 was cycled in a H216O-based KOH electrolyte for another six consecutive cycles to study the reverse oxygen isotope replacement (Supplementary Fig. 33). The first-cycle data reveal a nearly two-fold 18O abundance than the expected 18O fractions from the natural isotope abundance and from the measured value of unlabelled LiNiO2 in the same O16-based electrolyte (Supplementary Fig. 34 and Supplementary Equation S28). The 18O isotope excess gradually decreases with the number of cycles Fig. 7d), indicating the consumption of 18O on the labelled LiNiO2 in forming O2. Therefore, the two-way isotope labelling experiments confirm the participation of LiNiO2 lattice oxygen in the OER, supporting the predicted MLOV mechanism and activated lattice-oxygen in the presence of double ligand holes.
Fe3+ impurity effects
It is widely accepted that the incorporation of Fe3+ into Ni (oxy)hydroxides promote OER activity by forming synergetic active centres of oxygen-bridged Ni and Fe sites56,65, and the activity can be further enhanced via structural ordering, as recently proposed by Lee et al.58. Structurally intact LixNiO2 with stabilised double ligand holes could be a better platform for forming such synergetic centres than the usually amorphous NiOOH.
Figure 8a compares the linear sweep voltammograms of LiNiO2 titrated with Fe3+ in purified 1 M KOH. Similar to the reported Ni (oxy)hydroxides, the OER activity of LiNiO2 is strongly correlated with the Fe3+ concentration in terms of E(@10 mA cm−2) and Tafel slopes (Fig. 8a–c): (1) E(@10 mA cm−2) decreases with the Fe3+ concentration and saturated at 1.500 V in the presence of 100 µM Fe3+ (45 mV lower than that in the Fe-free electrolyte, Figs. 8a), and (2) the Tafel slope plummets from 57 to 43 mV dec−1 in the presence of only 1 µM Fe3+, and largely remains constant up to a Fe3+ concentration of 200 µM (Fig. 8b). The decreased Tafel slope at such a low Fe3+ concentration indicates that OER is switched to a Fe-involving electrocatalytic site with higher activity, and the number of such new active sites increases with Fe3+ concentration, resulting in Fe-concentration-dependent E(@10 mA cm−2). For comparison, NiOOH was also measured in 1 M KOH containing 100 µM Fe3+. Although the overall activity is highly promoted as expected (Supplementary Fig. 35), NiOOH-Fe3+ shows 36 mV higher than LiNiO2-Fe3+ in E(@10 mA cm−2) and 25 mV in OER onset potential (Fig. 8d), proving LiNiO2 an excellent platform for forming Ni-Fe active sites. In addition, the presence of high-valent Ni in Ni-Fe active sites is also found to promote electrocatalytic durability30. The promotional mechanism and underlying role of double ligand holes in Ni-Fe lithiated oxide systems are also of interest and related studies are currently underway.

a LSVs and b Tafel plots of LiNiO2 in Fe-free 1 M KOH with the addition of Fe3+ solutions (prepared from ultrapure water and Fe(NO3)3 ∙ 9H2O, 99.999%-Fe, PURATREM, Strem). c Potentials at 10 mA cm−2 and Tafel slopes as a function of Fe3+ concentrations. d Comparison of the Fe3+ promotional effects on LiNiO2 and NiOOH. Note that 100 µM Fe3+ reaches the saturated concentration of Fe(OH)4- (the predominant Fe3+ species in alkaline solution)70.
In conclusion, a comprehensive picture correlating electrochemical properties and operando spectroscopies is present to uncover the pristine activity, actual active species, and reaction centres of high-entropy LiNiO2 for OER. Pristine LiNiO2 with single-crystalline R-3m structure and with pure NiIII was prepared and characterized unambiguously using electron microscopy, XRD, and XAS. The best and real OER activity of LiNiO2 is achieved when the electrochemical redox of Ni2+/NiIII and the introduction of Fe impurities are prevented, and the activity is found to be comparable with that of the state-of-the-art IrO2 and much higher than that of the conventional NiOOH in terms of overpotentials, Tafel slopes and OER onset potentials., Operando XRD, XAS, and Raman spectra were combined to probe the active species and reaction centre. The in situ delithiation, NiIII oxidation and oxygen-vacancy formation in LiNiO2 under OER were found to coincide with OER for the first time, and the active species was then identified as delithiated yet structurally intact LixNiO2 with high-valent NiIV and double ligand holes, which is also supported by the prevalence of the MLOV OER mechanism from DFT calculation and by the activated lattice oxygen from in situ DEMS measurements. This unique LixNiO2 active sites were also found to be superior to those of NiOOH for the formation of a synergetic electrocatalyst with Fe3+. This study elucidates the electrocatalysis of lithiated metal oxides in OER and highlights the importance of considerate electrochemical measurements and complementary operando techniques in understanding the pristine electrochemistry and actual active species.
Methods
Preparation of LiNiO2
The pristine LiNiO2 was synthesized using a solid-state reaction method with two annealing steps. NiO and LiOH were grinded and mixed by a mortar and pestle for 30 min, and transferred into a tubular furnace. A 15% excess of the Li precursor was added to compensate for the evaporation of Li2O and/or Li2O2 during annealing. After the furnace was purged with O2 (100 sccm) for 15 min, under such O2 flow the temperature was increased to 750 °C with a ramping rate of 5 °C min−1 and held at this temperature for 72 h. The second annealing step was carried out at ~100 bar O2 pressure with cooling naturally to room temperature.
Structural characterisation
Lab-based X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance diffractometer with Cu Kα radiation (λ = 1.5418 Å) and a LYNXEYE detector, and diffraction peaks from Cu Kα2 was stripped off using Jade 6 software. Transmission electron microscopy (TEM) measurements, including low-magnification TEM, high-resolution TEM (HRTEM) and selected area electron diffraction (SAED), were carried out on a FEI Tecnai G2 F20 S-TWIN electron microscope. Scanning electron microscopy (SEM) measurements were performed on a Zeiss Crossbeam 540. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were recorded using an aberration-corrected FEI Themis Z STEM. The microscope was operated with an accelerating voltage of 60 kV and the HAADF detector angle range was 50–200 mrad. Note that the accelerating voltage of STEM was selected as 60 kV to prevent beam damage, which is reported to induce Ni displacement and oxygen release on LiNiO2 after long exposure to electron irradiation66. X-ray absorption spectra (XAS) at the Ni K edge were measured in the transmission mode at TPS 44 A beamline of National Synchrotron Radiation Research Centre (NSRRC, Hsinchu), and using an in-house lab-based X-ray absorption spectrometer (Supplementary Fig. 36). The electron storage ring of the TPS was operated at 3.0 GeV with a ring current of ~500 mA. The measured spectra were processed and analysed using ATHENA and ARTEMIS (Demeter software package)67. Soft XAS (sXAS) at the Ni L3,2 edge and the O K edge were collected at TLS 11 A and 20 A beamlines of the NSRRC. The Ni L edge data of γ-NiOOH and KNiIO6 were measured in fluorescence mode and inverse partial fluorescence yield mode, respectively, to reduce the spectral distortion of surface Ni2+.
Electrochemical measurements
Electrochemical measurements were performed on a standard three-electrode setup connected to a PGSTAT302N potentiostat (Metrohm Autolab, equipped with FRA32M and BA modules). A Pt gauze was used as the counter electrode, and a Hg|HgO electrode (in 1 M KOH) as the reference electrode. The potential measured using the Hg|HgO reference electrode, E(V vs. Hg|HgO, 1 M KOH), was converted into RHE scale in 1 M KOH, E(V vs. RHE), using the following equation,
E(V vs. RHE) = E(V vs. Hg|HgO, 1 M KOH) + 0.0977 V + 0.05916 pH (V) (1), where the pH of the purified 1 M KOH is measured as 13.73 at 25 °C. The values of potential in the text are thus referenced against RHE, unless stated otherwise. The contact and solution resistance were determined using electrochemical impedance spectroscopy under open circuit potential (OCP) before each voltammetry measurement, and the voltammograms were corrected by IR drops.
The working electrode was prepared by drop-casting 10 μL of a catalyst ink onto a polished rotating ring-disk electrode (RRDE, with a glassy carbon disk and a Pt ring) and drying under ambient condition to obtain an even film. The catalyst ink was prepared by dispersing 5 mg of each sample and 5 mg carbon (Vulcan XC-72R) into 1 mL isopropanol alcohol (IPA) aqueous solution (25 vol% of IPA) containing 40 μL 5 wt% Nafion solution.
Linear sweep voltammograms and cyclic voltammograms (CV) were collected using a RRDE configuration with 5 mV s−1 in Ar-saturated 1 M purified KOH solution at 25 °C. The voltammograms were collected at 1600 rpm to promptly remove the generated O2, and the disk potential was swept from the corresponding OCP of each sample, while the ring potential was held at −0.5 V (vs. Hg|HgO, 1 M KOH) to electrochemically reduce and detect the O2 evolved from the disk. For the long-term stability test, the working electrode was prepared using carbon paper (Toray TGP-H-060) with the same catalyst loading and ink recipe as the RRDE, and the stability was evaluated using chronopotentiometry at 10 mA cm−2 for 48 h, before and after which LSV curves were measured with 5 mV s−1. The OER activity of NiOOH and IrO2 (99.9%, Adamas-beta) was measured for comparison. The NiOOH was prepared by potential cycling NiO nanopowder (50 nm, 99.9% metal basis, Aladdin) in 1 M KOH between 1.01 V and 1.66 V for at least 40 cycles until the Ni2+/NiIII redox couple at ~1.32 V was fully developed (Supplementary Fig. 37).
To avoid the inference of Fe impurities on the measured OER activity, the electrochemical cell was cleaned by concentrated H2SO4 overnight and boiled with ultrapure H2O (18.2 MΩ cm) for at least 3 times to remove residual H2SO4 and other impurities, and the KOH solution was purified using Trotochaud’s method38. Briefly, 1 M KOH solution was first prepared from semiconductor grade KOH (99.9996% metal basis, 4 ppm total metal impurity excluding Na+, Aladdin) and ultrapure water. Into a H2SO4-rinsed 50 mL polypropylene centrifuge tube, ~2 g of Ni(NO3)2·6H2O (99.9985%, PURATREM, STREM) were dissolved in ~4 mL ultrapure H2O and mixed with 20 mL 1 M KOH. The resulting Ni(OH)2 suspension was shaken and centrifuged, and the supernatant was decanted. The Ni(OH)2 was then washed 3 times with ~20 mL ultrapure H2O and ~2 mL 1 M KOH, and transferred to a H2SO4-rinsed 100 mL polypropylene bottle. The bottle was filled with 75 mL 1 M KOH for purification, and the freshly prepared and washed Ni(OH)2 was redispersed and shaken for 10 min, followed by resting overnight.
Operando and online characterisation
Operando synchrotron XRD patterns were collected at TLS 01C2 beamlines of the NSRRC. The XRD experiments were performed at an incident beam energy of 18 keV using a mar345 image plate area detector. The diffraction intensity and the peak shape were calibrated with a CeO2 reference sample (SRM 674b). Operando Quick-XAS measurements at Ni K were carried out in the transmission mode at TPS 44 A beamline of the NSRRC, and 120 XAS spectra collected within 1 min were merged to achieve high data quality. Operando Raman spectra were collected using a Horiba LabRAM HR Evolution confocal Raman microscope with an excitation wavelength of 473 nm. The spectra resolution is ~1 cm−1, and the spectral shifts were calibrated routinely using silicon wafer (520.7 cm−1). The operando Raman experiments were performed in a homemade spectro-electrochemistry cell, consisting of a Au disk working electrode (5 mm), a Pt wire counter electrode and a Ag|AgCl (saturated KCl) reference electrode. Online differential electrochemical mass spectrometry (DEMS) measurements were carried out using a QAS-100 equipped with a PrismaPro quadrupole mass spectrometer (Linglu Instruments). The O18-labeled LiNiO2 was prepared by cycling the unlabelled one to the OER range (e.g., 1.8 V) in 18O-enriched electrolyte, and before the experiment in the 16O-based electrolyte, rinsing and drying steps were conducted to remove free 18O species.
Computation details
The present calculations employ the Vienna ab initio Simulation Package (VASP) implementation of density functional theory (DFT) in conjunction with the projector augmented wave (PAW) formalism68. The H 1s1, Li 2s1, O 2s22p4, and Ni 4s23d8 states are treated as valence electrons. The electronic wave functions were expanded in plane waves using an energy cutoff of 520 eV, and the force and energy convergence criteria were set to 0.02 eV Å−1 and 10−5 eV respectively. To overcome the self-interaction error of the exchange correlation functional, we employed the Hubbard U model for describing the strong correlation of the localized Ni 3d states, and set the value of Ueff = (U − J) to 6.40 eV according to previous work46. The reciprocal space was sampled using a 4×4×1 Monkhorst–Pack k-point mesh for DFT + U calculations. To simulate the valence states of Ni in real catalytic environment, we built the Li0.5NiO2 system containing 50% lithium vacancy. The periodic slab models of LiNiO2 and Li0.5NiO2 system with 9 Ni sites per surface, with two layers at the bottom fixed during the relaxation. To prevent spurious interactions, the thickness of vacuum spacing was ~15 Å in the z-direction. We calculated the Gibbs free-energy differences (∆G) using the computational hydrogen electrode model under standard conditions69, with the voltage applied URHE = 0 and 1.23 V. The calculation details regarding ∆G can be found in the Supplementary Information.
Data availability
The data that support the findings of this study are available either from the Supplementary Information and the Source data or from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Yu, M., Budiyanto, E. & Tuysuz, H. Principles of water electrolysis and recent progress in cobalt-, nickel-, and iron-based oxides for the oxygen evolution reaction. Angew. Chem., Int. Ed. 61, e202103824 (2022).
Vass, A., Kormanyos, A., Koszo, Z., Endrodi, B. & Janaky, C. Anode catalysts in CO2 electrolysis: challenges and untapped opportunities. ACS Catal. 12, 1037–1051 (2022).
Suen, N.-T. et al. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 46, 337–365 (2017).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Yuan, Y. et al. Co3Mo3N-An efficient multifunctional electrocatalyst. Innovation 2, 100096 (2021).
Jing, C., et al. Electrocatalyst with dynamic formation of the dual-active site from the dual pathway observed by in situ Raman spectroscopy. ACS Catal. 12, 10276–10284 (2022).
Hu, Y. et al. Dynamic structural transformation induced by defects in nano-rod FeOOH during electrochemical water splitting. J. Mater. Chem. A 10, 602–610 (2022).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196–2214 (2020).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Zhou, J. et al. Voltage- and time-dependent valence state transition in cobalt oxide catalysts during the oxygen evolution reaction. Nat. Commun. 11, 1984 (2020).
Li, M., Bi, X., Amine, K. & Lu, J. Oxygen-based anion redox for lithium batteries. Acc. Chem. Res. 53, 1436–1444 (2020).
Kwon, S., Deshlahra, P. & Iglesia, E. Dioxygen activation routes in Mars-van Krevelen redox cycles catalyzed by metal oxides. J. Catal. 364, 228–247 (2018).
Kuiper, P., Kruizinga, G., Ghijsen, J., Sawatzky, G. A. & Verweij, H. Character of holes in LixNi1-xO and their magnetic behavior. Phys. Rev. Lett. 62, 221–224 (1989).
Bisogni, V. et al. Ground-state oxygen holes and the metal-insulator transition in the negative charge-transfer rare-earth nickelates. Nat. Commun. 7, 13017 (2016).
Green, R. J., Haverkort, M. W. & Sawatzky, G. A. Bond disproportionation and dynamical charge fluctuations in the perovskite rare-earth nickelates. Phys. Rev. B 94, 195127 (2016).
Foyevtsova, K., Elfimov, I., Rottler, J. & Sawatzky, G. A. LiNiO2 as a high-entropy charge- and bond-disproportionated glass. Phys. Rev. B 100, 165104 (2019).
Chen, H., Freeman, C. L. & Harding, J. H. Charge disproportionation and Jahn-Teller distortion in LiNiO2 and NaNiO2: A density functional theory study. Phys. Rev. B 84, 085108 (2011).
Zaanen, J., Sawatzky, G. A. & Allen, J. W. Band gaps and electronic structure of transition-metal compounds. Phys. Rev. Lett. 55, 418–421 (1985).
Bocquet, A. E., Mizokawa, T., Saitoh, T., Namatame, H. & Fujimori, A. Electronic structure of 3d-transition-metal compounds by analysis of the 2p core-level photoemission spectra. Phys. Rev. B 46, 3771–3784 (1992).
Guo, H. et al. Antiferromagnetic correlations in the metallic strongly correlated transition metal oxide LaNiO3. Nat. Commun. 9, 43 (2018).
Guo, H. et al. Charge disproportionation and nano phase separation in RSrNiO4. Sci. Rep. 10, 18012 (2020).
Bianchini, M., Roca-Ayats, M., Hartmann, P., Brezesinski, T. & Janek, J. There and back again-the journey of LiNiO2 as a cathode active material. Angew. Chem., Int. Ed. 58, 10434–10458 (2019).
Balasubramanian, M., Sun, X., Yang, X. Q. & McBreen, J. In situ X-ray diffraction and X-ray absorption studies of high-rate lithium-ion batteries. J. Power Sources. 92, 1–8 (2001).
Li, N. et al. Unraveling the cationic and anionic redox reactions in a conventional layered oxide cathode. ACS Energy Lett. 4, 2836–2842 (2019).
Liu, H. et al. Insight into the role of metal–oxygen bond and O 2p hole in high-voltage cathode LiNixMn2–xO4. J. Phys. Chem. C 121, 16079–16087 (2017).
Guan, D. et al. Utilizing ion leaching effects for achieving high oxygen-evolving performance on hybrid nanocomposite with self-optimized behaviors. Nat. Commun. 11, 3376 (2020).
Demourgues, A., Gautier, L., Chadwick, A. V. & Delmas, C. EXAFS study of the Jahn-Teller distortion in layered nickel oxyhydroxide. Nucl. Instrum. Methods Phys. Res., Sect. B 133, 39–44 (1997).
Rougier, A., Delmas, C. & Chadwick, A. V. Non-cooperative Jahn-Teller effect in LiNiO2: an EXAFS study. Solid State Commun. 94, 123–127 (1995).
Chen, G. et al. An amorphous nickel-iron-based electrocatalyst with unusual local structures for ultrafast oxygen evolution reaction. Adv. Mater. 31, e1900883 (2019).
Vasiliev, A. N. et al. Valence states and metamagnetic phase transition in partially B-site-disordered perovskite EuMn0.5Co0.5O3. Phys. Rev. 77, 104442 (2008).
Burnus, T. et al. Local electronic structure and magnetic properties of LaMn0.5Co0.5O3 studied by x-ray absorption and magnetic circular dichroism spectroscopy. Phys. Rev. B 77, 125124 (2008).
Zhou, J. et al. In situ exploring of the origin of the enhanced oxygen evolution reaction ffficiency of metal(Co/Fe)–organic framework catalysts via postprocessing. ACS Catal. 12, 3138–3148 (2022).
Chin, Y. Y. et al. Spin-orbit coupling and crystal-field distortions for a low-spin 3d5 state in BaCoO3. Phys. Rev. B 100, 205139 (2019).
Tjeng, L. H., Chen, C. T. & Cheong, S. W. Comparative soft-x-ray resonant-photoemission study on Bi2Sr2CaCu2O8, CuO, and Cu2O. Phys. Rev. B 45, 8205–8208 (1992).
Xiao, P. et al. Effects of Oxygen Pressurization on Li+/Ni2+ Cation Mixing and the Oxygen Vacancies of LiNi0.8Co0.15Al0.05O2 Cathode Materials. ACS Appl. Mater. Interfaces.14, 31851–31861 (2022).
Li, L. et al. Role of Residual Li and Oxygen Vacancies in Ni-rich Cathode Materials. ACS Appl. Mater. Interfaces.13, 42554–42563 (2021).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel-iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014).
Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction. Nat. Energy. 5, 222–230 (2020).
Klaus, S., Cai, Y., Louie, M. W., Trotochaud, L. & Bell, A. T. Effects of Fe electrolyte impurities on Ni(OH)2/NiOOH structure and oxygen evolution activity. J. Phys. Chem. C 119, 7243–7254 (2015).
Yoon, C. S., Jun, D.-W., Myung, S.-T. & Sun, Y.-K. Structural Stability of LiNiO2 Cycled above 4.2 V. ACS Energy Lett. 2, 1150–1155 (2017).
Ikeda, N., Konuma, I., Rajendra, H. B., Aida, T. & Yabuuchi, N. Why is the O3 to O1 phase transition hindered in LiNiO2 on full delithiation? J. Mater. Chem. A 9, 15963–15967 (2021).
Sasaki, T. et al. Capacity-Fading Mechanisms of LiNiO2-Based Lithium-Ion Batteries. J. Electrochem. Soc. 156, A289 (2009).
Zhu, K. et al. Layered Fe-substituted LiNiO2 electrocatalysts for high-efficiency oxygen evolution reaction. ACS Energy Lett. 2, 1654–1660 (2017).
Gupta, A., Chemelewski, W. D., Buddie Mullins, C. & Goodenough, J. B. High-rate oxygen evolution reaction on Al-doped LiNiO2. Adv. Mater. 27, 6063–6067 (2015).
Ren, X. et al. Constructing an adaptive heterojunction as a highly active catalyst for the oxygen evolution reaction. Adv. Mater. 32, e2001292 (2020).
Wu, J. et al. Electrochemical water splitting by pseudo-spinel, disordered and layered lithium nickel oxides: correlation between structural motifs and catalytic properties. ChemCatChem 10, 2551–2557 (2018).
Ren, Y. et al. The effect of cation mixing in LiNiO2 toward the oxygen evolution reaction. ChemElectroChem 8, 70–76 (2020).
Fu, G. et al. Tuning the electronic structure of NiO via Li doping for the fast oxygen evolution reaction. Chem. Mater. 31, 419–428 (2018).
Lo, Y. L. & Hwang, B. J. In situ Raman studies on cathodically deposited nickel hydroxide films and electroless Ni−P electrodes in 1 M KOH solution. Langmuir 14, 944–950 (1998).
Hirano, A. Relationship between non-stoichiometry and physical properties in LiNiO2. Solid State Ion. 78, 123–131 (1995).
Kong, F. et al. Kinetic stability of bulk LiNiO2 and surface degradation by oxygen evolution in LiNiO2-based cathode materials. Adv. Energy Mater. 9, 1802586 (2019).
Zhao, S. et al. Structural transformation of highly active metal–organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy. 5, 881–890 (2020).
Wang, X. P. et al. Strain stabilized nickel hydroxide nanoribbons for efficient water splitting. Energy Environ. Sci. 13, 229–237 (2020).
Flores, E., Novák, P. & Berg, E. J. In situ and operando Raman spectroscopy of layered transition metal oxides for Li-ion battery cathodes. Front. Energy Res. 6, 82 (2018).
Dionigi, F. et al. In-situ structure and catalytic mechanism of NiFe and CoFe layered double hydroxides during oxygen evolution. Nat. Commun. 11, 2522 (2020).
Yeo, B. S. & Bell, A. T. In situ Raman study of nickel oxide and gold-supported nickel oxide catalysts for the electrochemical evolution of oxygen. J. Phys. Chem. C 116, 8394–8400 (2012).
Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel-iron layered double hydroxide. Angew. Chem., Int. Ed. 59, 8072–8077 (2020).
Garcia, A. C., Touzalin, T., Nieuwland, C., Perini, N. & Koper, M. T. M. Enhancement of oxygen evolution activity of nickel oxyhydroxide by electrolyte alkali cations. Angew. Chem., Int. Ed. 58, 12999–13003 (2019).
Maiyalagan, T., Jarvis, K. A., Therese, S., Ferreira, P. J. & Manthiram, A. Spinel-type lithium cobalt oxide as a bifunctional electrocatalyst for the oxygen evolution and oxygen reduction reactions. Nat. Commun. 5, 3949 (2014).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212–222 (2021).
Li, L. et al. In Situ/Operando Capturing Unusual Ir6+ Facilitating Ultrafast Electrocatalytic Water Oxidation. Adv. Funct. Mater. 31, 2104746 (2021).
Lin, X. et al. 5f covalency synergistically boosting oxygen evolution of UCoO4 catalyst. J. Am. Chem. Soc. 144, 416–423 (2022).
Ferreira de Araujo, J., Dionigi, F., Merzdorf, T., Oh, H. S. & Strasser, P. Evidence of Mars-Van-Krevelen mechanism in the electrochemical oxygen evolution on Ni-based catalysts. Angew. Chem., Int. Ed. 60, 14981–14988 (2021).
Anantharaj, S., Kundu, S. & Noda, S. “The Fe Effect”: A review unveiling the critical roles of Fe in enhancing OER activity of Ni and Co based catalysts. Nano Energy. 80, 105514 (2021).
Shim, J. H., Kang, H., Kim, Y. M. & Lee, S. In situ observation of the effect of accelerating voltage on electron beam damage of layered cathode materials for lithium-ion batteries. ACS Appl. Mater. Interfaces. 11, 44293–44299 (2019).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Bajdich, M., Garcia-Mota, M., Vojvodic, A., Norskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521–13530 (2013).
Furcas, F. E. et al. Solubility and speciation of iron in cementitious systems. Cem. Concr. Res. 151, 106620 (2022).
Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1502400), the “Transformational Technologies for Clean Energy and Demonstration,” Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA2100000), National Science Foundation of China (Grant No. 21876183 and No. 22179141), DNL Cooperation Fund, CAS (No. DNL202008), and the Young Potential Program of Shanghai Institute of Applied Physics, Chinese Academy of Sciences. We acknowledge support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials.
Author information
Authors and Affiliations
Contributions
L.Z., Z.H., and J.-Q.W. conceived the project and designed the experiments. H.H. performed the electrochemical measurements and data analysis. Y.O. and A.C.K conducted the material synthesis. Y.-C.C., Y-C.H., C.-W.P., T.-S.C., H.-J.L. C.-L.D., and Z.H. performed the operando synchrotron experiments. Y.W., and J.Z. performed the structural characterization. J.-M.C., S,-C.H., and C.-T.C. performed the sXAS measurements. L.L. and J.Z. carried out the DFT calculations. H.H. and L. Z. wrote the manuscript. Z.H. and L.H.T. provided interpretation of XAS and modified the manuscript. C.-Y.K. performed sXAS calculation. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jongwoo Lim and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, H., Chang, YC., Huang, YC. et al. Unusual double ligand holes as catalytic active sites in LiNiO2. Nat Commun 14, 2112 (2023). https://doi.org/10.1038/s41467-023-37775-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37775-4
This article is cited by
-
Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
