
Abstract
Sea ice is a key factor for the functioning and services provided by polar marine ecosystems. However, ecosystem responses to sea-ice loss are largely unknown because time-series data are lacking. Here, we use shotgun metagenomics of marine sedimentary ancient DNA off Kamchatka (Western Bering Sea) covering the last ~20,000 years. We traced shifts from a sea ice-adapted late-glacial ecosystem, characterized by diatoms, copepods, and codfish to an ice-free Holocene characterized by cyanobacteria, salmon, and herring. By providing information about marine ecosystem dynamics across a broad taxonomic spectrum, our data show that ancient DNA will be an important new tool in identifying long-term ecosystem responses to climate transitions for improvements of ocean and cryosphere risk assessments. We conclude that continuing sea-ice decline on the northern Bering Sea shelf might impact on carbon export and disrupt benthic food supply and could allow for a northward expansion of salmon and Pacific herring.
Similar content being viewed by others


Introduction
The organismal composition in high-latitudinal oceans is highly vulnerable to anthropogenic global warming, which may alter ecosystem services (e.g., food supply, biological carbon pump) due to sea-ice loss and related effects on rising ocean temperature, increasing light transmission, stronger water column stratification and decreasing nutrient availability1. This will likely change the seasonal timing, biomass, and composition of algal blooms, which play a central role in trophic interactions by supporting food webs, including benthic communities and primary producers2.
Long-term trends in the Bering Sea show an ongoing decline of sea-ice duration which is projected to shorten further due to later freeze-up (by 20 to 30 days) and earlier break-up (by 10–20 days) until the middle of this century3. Extreme events, such as the lowest sea-ice extent in the Bering Sea over the last 5500 years4 in 2018 with persistent southerly warm winds, allow for assessments of immediate ecosystem responses to sea-ice loss. Cascading effects through the food web have been recorded, possibly resulting from reduced energy transfer from lower to upper trophic levels5: A late spring phytoplankton bloom and a scarcity of large, lipid-rich copepods led to decreasing abundance of young pollock and other forage fish that were later linked with seabird reproductive failures as well as seabird and seal mortality events in 2018 and 20196.
A better understanding of ecosystem changes during the transition from seasonal sea-ice to ice-free conditions due to ongoing climate warming is needed. Despite extensive monitoring efforts, we still have scarce knowledge on long-term ecosystem responses to climate transitions for many taxonomic groups, particularly zooplankton, fish, non-fossilizing algae, and benthic organisms such as tunicates, starfish, or macrophytes. Furthermore, the ability and constraints of ecosystems to adapt are uncertain, which limits ocean and cryosphere risk assessments, as emphasized in the IPCC report7. The short-term responses of the Bering Sea ecosystem to warmer and colder regimes have been well-documented2,5,8, yet long-term rearrangements of the organismal composition in pelagic and benthic communities are not clear and more data are needed to constrain the future ecosystem development. While pelagic communities are strongly linked to hydrographic factors and climate, benthic communities are strongly dependent on primary production and sinking particles from the water column above9. Past ecosystem responses to sea-ice loss at the Pleistocene–Holocene boundary in the southern part of the Bering Sea could possibly reveal such long-term rearrangements and serve as an analog for future changes in the Pacific Arctic.
Marine sediments are a natural archive of climate history from which sea-ice can be reconstructed via proxy records, such as from biomarkers10 or microfossil remains11. In the Bering Sea, a palaeoceanographic framework for past sea-ice coverage has been based on reconstructions using the highly branched isoprenoid alkane biomarker IP2512 (produced by diatoms bound to a life in seasonal sea-ice10) and microfossils (from diatoms)11. Alkenones, which are produced by haptophytes can add complementary information about sea-surface temperatures of the late summer/early fall season (SSTUk’37)13. Physical remains, such as diatom frustules11,14,15,16, provide evidence for past organismal responses to sea-ice variability. For many non-fossilizing organismal groups (e.g., many protists and zooplankton) or such whose fossil records remain underexplored (e.g., fish), analyses of sedimentary ancient DNA (sedaDNA) have the potential to fill the gap.
Within marine settings, sedaDNA metabarcoding (enrichment of a genetic marker sequence via polymerase chain reaction (PCR)) has revealed past responses of plankton to changes, for example, in water mass characteristics17 or sea-ice18,19,20. Recently, sedaDNA applications were expanded by metagenomic shotgun sequencing21,22,23, which increases the spectrum of taxonomic groups that can be analyzed in a single approach, facilitates authentication by profiling damage patterns, and circumvents typical PCR biases, arising, for example, from the mismatch between the ancient DNA fragment size distribution and the size of genetic markers22.
Correlation networks have been used in the interpretation of multidimensional data by means of co-occurrence networks and contributed to our understanding of ecosystems by identifying, for example, habitat preferences in aquatic bacterial communities24 or geographic co-occurrence patterns in European amphibians, reptiles, breeding birds, and mammals25. The sparsity of metagenomic datasets resulting from either true absences or insufficient sequencing depth can lead to spurious correlations26. Gaussian copula graphical models (GCGMs) are a novel statistical framework developed to separate environmental effects from intrinsic interactions among taxa27. They are suited to address the compositional structure of sequencing data, non-linear correlations, and to remove spurious correlations via mediator taxa (if a correlation between two taxa arises only because both are correlated to a third taxon) or via similar responses to environmental effects (covariance). Correlation networks have not yet been applied to sedaDNA data before. As sedaDNA samples contain averaged information on decadal to centennial time scales depending on sedimentation rates, correlation in the network should not be interpreted as ecological interactions between linked taxa or actual co-occurrence on a short-timescale. We here assume that positively correlated families show similar responses to environmental changes (i.e., sea-ice cover and SSTs).
Here, we explore the potential of sedaDNA metagenomic shotgun sequencing to reveal shifts in ecosystem composition in response to postglacial sea-ice loss using sediment core SO201-2-12KL, which was taken from the western continental slope of Kamchatka about 70 km from the coast of Kamchatka (Fig. 1). To identify taxa that show a similar response to sea-ice loss, we used co-occurrence networks and compared Spearman networks with GCGMs. The paleoenvironmental backbone is based on previous multi-proxy reconstructions from the same core, including diatom microfossils and the biomarker IP25. The proxies suggest that the coring site was covered by seasonal sea-ice during the Last Glacial Maximum (LGM), Heinrich Stadial 1, and the Younger Dryas, while the phases of the Bølling-Allerød and the Holocene were predominantly sea-ice free12,16. Our approach includes a broad spectrum of taxonomic and functional groups, particularly among eukaryotes, which we consider a major step towards deciphering the consequences of climate transitions for marine ecosystem structure.

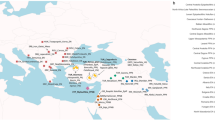
a The coring site (53.993°N, 162.37°E; water depth 2173 m) is located south of the modern sea-ice extent as shown by the median for March between 1981–2010103 (dotted line) and the mean modern sea-ice concentration for March in 2020104, 105 (color scale from blue to white). b Modern seasonal patterns of primary productivity are indicated by a low mean chlorophyll-a concentration in January compared to c higher mean chlorophyll-a concentrations in July from 2020 (E.U. Copernicus Marine Service Information; https://resources.marine.copernicus.eu/?option=com_csw&view=details&product_id=GLOBAL_ANALYSIS_FORECAST_BIO_001_028). The map was created with ArcGIS using the Global 30 Arc-Second Elevation (https://www.usgs.gov/centers/eros/science/usgs-eros-archive-digital-elevation-global-30-arc-second-elevation-gtopo30#overview).
Results and discussion
Shotgun sequencing results
Sequencing (25 samples, five negative controls) resulted in 918,186,452 paired-end reads of which, on average, 70.76% of the reads passed the quality check within samples (Supplementary data file 1). Of those, we were able to classify 13,119,146 read pairs (merged and paired-read pairs together). About 98.5% of our sequences remained unclassified (based on the full NCBI nt database), which is in a similar range as reported for metagenomic shotgun sequencing of ancient cave sediments (79.1–96.1%)28 and ancient lake sediments (99.629, 8430,31, 97%32). Our results are not surprising since estimates from 2011 suggest that 91% of marine species have not yet been described33. Especially among protists, the sequence reference database is largely incomplete34, resulting in low confidence at genus and species level assignments35. Therefore, we limited our analyses to assignments on the family level. Most classified read pairs overlapped and were merged (72.5%). Among samples, this resulted in mean fragment lengths between 83 and 105 bp per sample, which matches the expectation that sedaDNA is highly fragmented36.
Pelagic taxonomic assignments are dominated by primary producers and fish
The novel sedaDNA metagenomics approach allowed for the successful identification of 167 families with 456,058 read pairs after filtering and resampling, representing functional and taxonomic groups (Supplementary data file 1) which are known from sea-ice or the pelagic or demersal zones (hereafter grouped as pelagic) of the subarctic North Pacific and the Bering Sea. Of all samples combined, high shares of sequences are assigned to fish (66.8%), phototrophic protists (10.2%), and phototrophic bacteria (12.8%) after resampling. Among phototrophic families (including mixotrophic taxa), the highest numbers of reads are assigned to pico-sized Cyanobacteria (11.5%), Chlorophyta (3.6%), and Chlorobi (0.8%), as well as to typically nano- to micro-sized diatoms (4.7%) and haptophytes (1.0%) (Fig. 2).

Only families represented in at least three samples and by at least 1.5% proportion of read counts after resampling are shown. The dendrogram (based on constrained hierarchical clustering) and IP25 concentrations from the same core98 and sea-surface temperature (SSTUk’37) reconstructions of the Northwest Pacific52 (light red) and Northeast Pacific97 (dark red) are added for comparison. Families which are significant, and positively correlated with IP25 or SSTUk’37 are highlighted in blue or red, respectively. The pelagic:benthic ratio shows the proportion of reads assigned to pelagic families in relation to reads assigned to benthic families. The ratio between reads assigned to phototrophic bacteria and reads assigned to phototrophic protists is given as a blue-green line. Black triangles next to the IP25 record mark radiocarbon-dated calibrated ages52. LGM last glacial maximum, HS1 Heinrich stadial 1, B/A Bølling/Allerød, YD Younger Dryas, and colors of the boxes refer to warm (red) and cold (blue) phases. Source data are provided as a Source Data file.
The proportions of pelagic resampled reads assigned to phototrophic primary producers exceed those of zooplankton, which comprises single-celled, heterotrophic protists (2.1%) and copepods (0.3%). The group of heterotrophic protists is phylogenetically diverse, and the highest read counts are found for Ciliophora (0.8%), colloidarian Polycystinea (0.3%), Discoba (0.3%), Choanoflagellata (0.2%), and Dinoflagellata (0.1%).
The low proportion of assigned reads likely reflects a strong grazing intensity on zooplankton in the upper water column, which leads to a low zooplankton DNA export towards the seafloor. Alternatively, this discrepancy between the proportions of assigned reads to zooplankton (multicellular organisms) and phytoplankton (unicellular organisms) could be explained by differential preservation or degradation due to different skeletal properties or large gaps in the sequence reference database regarding marine macrofauna for both barcoding genes and genomes37.
Among the analyzed metazoans, most reads were assigned to fish of the families Salmonidae (12.1%), Serranidae (8%), and Gadidae (6%), which are common in the subarctic North Pacific and its adjacent seas2,38. Furthermore, we found few, but notable read counts assigned to marine mammals (4.5%) such as whales (Delphinidae 0.3% and Balaeonopteridae 0.3%), seals (Otariidae 0.1% and Phocidae 0.4%), and sea otter (Mustelidae 2.9%), which are also common in this region.
Co-occurrence network-derived pelagic environment associations
We investigated the influence of environmental variables such as temperature and seasonal sea-ice cover using a co-occurrence network based on Spearman rank correlations (Fig. 3). The Spearman network (167 pelagic families kept after filtering for at least ten counts and in at least three samples and resampling) is composed of two modules containing 148 nodes (families) and 446 edges (positive correlations Spearman’s ρ > 0.4, adjusted p value <0.1) (Fig. 3a). The upper module contains 42 nodes, of which eight positively correlated (adjusted p value <0.1; Supplementary Table 4) with IP25. We, therefore, define it as the sea-ice module. The lower module contains 120 nodes, of which 13 are showing a trend or are positively correlated (adjusted p value <0.1; Supplementary Table 4) with higher SST. Therefore, we define it as the sea-ice-free module, hereafter.

Families (nodes) that are positively correlated (Spearman rank correlation coefficients >0.4, Benjamini–Hochberg adjusted p value < 0.1) with each other are connected by links (edges) after resampling. The node size is log-scaled according to taxa abundance, while node colors represent a the functional group to which the organisms belong, and b whether the node showed a positive trend (Spearman rank correlation coefficients >0.4, adjusted p value < 0.1) with the seasonal sea-ice biomarker IP2598 (blue) or SSTs97 (red). Node labels are given if a family exceeds the log-scaled threshold of 4. Source data are provided as a Source Data file.
As Spearman correlations cannot separate environmental effects from intrinsic interactions among taxa, we used ecoCopula to test how mediator taxa and environmental conditions (IP25 and SSTs used as covariates) affect associations between taxa.
The ecoCopula network is composed of 167 nodes and 474 edges, which is in a similar range compared to the edge density of the Spearman network. Two modules are dominated by a specific functional group: the green and red modules by diatoms and the violet module by fish, while the other modules are more mixed (Supplementary Fig. 13a). Despite removing the environmental effects of IP25 and SSTs for network generation, the green module is mainly composed of families that are positively correlated with IP25 in the Spearman network suggesting a strong co-occurrence pattern of these families based on biogeographic distribution patterns or other environmental factors not included here, such as nutrient availability, salinity, or light conditions. After accounting for the effects of IP25, SSTs, and mediator taxa, a high overlap of edges (34% of positive associations) can be found between the two network approaches, which is significantly different from the randomized null-model (Supplementary Fig. 13a, b).
Environmental preference of pelagic taxa inferred by network analysis
Nodes of the sea-ice module contain families that show higher proportions during the LGM, Heinrich Stadial 1, and Younger Dryas compared to warmer phases (Fig. 3a). Among algae, this pattern can be observed for Bacillariaceae (0.8% of pelagic counts), Stephanodiscaceae (0.6%), Thalassiosiraceae (1.1%), Triparmaceae (0.3%), and Phaeocystaceae (0.6%), which can be found in sea-ice or as a part of cryopelagic communities39. They co-occur with the chlorophytes Mamiellaceae (0.5%) and Bathycoccaceae (1.7%), which contain cold-adapted lineages that prevail in the marginal ice zone or in landfast sea-ice40,41. Closely linked to these primary producers are copepods (Calanidae 0.03%, Metridinidae 0.03%), which rely on sea-ice algae as a food source due to their high quantity of polyunsaturated fatty acids42. Among fish, Gadidae, for example, are linked with the above-mentioned algae and copepods. Reads assigned to this family can most likely be attributed to Pacific cod (Gadus macrocephalus), walleye pollock (Gadus chalcogrammus), or Polar cod (Boreogadus saida). While pollock and Pacific cod are generalist predators in the Bering Sea, they are strongly influenced by bottom-up effects through prey composition and availability38,43 compared to the Polar cod, which is a keystone species of the high Arctic with a strong linkage to sea-ice44.
The 8 families that are positively correlated with IP25 (adjusted p value <0.1) contain 109 links to 28 families. Of those links, 42.2% are shared with diatoms (15.6% centric and 26.6% pennate), which comprise overall some of the most connected families in the network. This emphasizes that diatoms are the central functional group among primary producers, particularly during phases characterized by seasonal sea-ice cover, and that sea-ice might act as a strong environmental filter favoring organisms with adapted functional traits.
The ice-free ecosystem composition, which prevailed for most of the Holocene (Fig. 2), suggests a response of past biota to warmer SSTs with reads assigned to families known to occur in warmer surface waters from their modern spatial distribution (Hemiaulaceae45, Rhizosoleniaceae46, and Chloropicaceae47).
The 28 families in the network that are positively correlated with SSTs (adjusted p value <0.1) encompass 114 links to 51 families within 13 functional groups (Fig. 3). Hence, connections of SST-correlated families are more diverse compared to those of IP25-correlated families. Families, which are positively correlated with SSTs, have on average fewer neighbors (i.e., links to adjacent nodes) and, therefore, are less densely linked in comparison to IP25-correlated families (p value <0.001, Supplementary Fig. 11). Neighbors of SST-correlated families belong mostly to the functional groups of fish (43% of links), phototrophic bacteria (25.4%), Chlorophyta (11.4.3%), and centric diatoms (1.8%). We assume that predominantly sea-ice-free conditions, and thus a longer growing season, might have favored a broader spectrum of families.
Higher read counts assigned to Clupeidae (Pacific herring) and Salmonidae in warm phases, for example, the early Holocene (Fig. 2), are consistent with previous studies that have shown that warmer temperatures provide good conditions for fish spawning, quick hatching from eggs, and low mortality rates during early life stages48. These patterns support modern observations of increased herring distribution on the northern Bering Sea shelf in warmer than in colder years49 and of increasing population densities in pink salmon since the 1990s50. Rivers in Kamchatka and nearby Chukotka are spawning grounds for Salmonidae51 and are known as a diversity hotspot; for example, all species of Pacific salmon (Oncorhynchus) occur in the area. Summer temperature reconstructions from Chukotka show a similar trend to SST reconstructions from our core52 with lowest values pre-15 ka and an about 5 °C rise until the early-to-mid Holocene temperature maximum53.
The ice-free sub-network is mainly characterized by phototrophic bacteria and chlorophytes (Fig. 3a). Furthermore, we find indications of increased continental runoff reflected in the presence of different functional groups of freshwater organisms (e.g. Sarcinofilaceae (Sarcinofilum mucosum; chlorophyte54), Roseiflexceae (Roseiflexus; bacteria55), Acanthocerotaceae (Acanthoceras zachariasii; diatom56); Supplementary Fig. 17). While freshwater discharge is a source of organic carbon and nutrients that can enhance productivity, the lower salinity of surface waters increases vertical water column stratification57. Over the course of the productive season, nutrients may become depleted by the spring/early summer phytoplankton blooms, especially when the water column is strongly stratified by warm sea-surface temperatures and freshwater input. Under nutrient-depleted conditions, cyanobacteria are assumed to have several advantages, such as the small surface area-to-volume ratio of their cells and thus lower nutrient requirements58, the capability of nitrogen fixation (here limited to Nostocaceae), and their preference for inorganic nitrogen in the form of ammonium. Ammonium has an energetic advantage compared to nitrate, the preferred form by diatoms, which has to be transformed to ammonium within the cell at an energetic cost59.
Benthic sedaDNA composition and temporal patterns
Despite the vastness of deep-sea benthic ecosystems, the responses of its inhabitants to past climatic changes are still underexplored. From benthic foraminifers and ostracods, we know that deep-sea habitats have been influenced by glacial–interglacial cycles in the past60,61,62.
The overall lower number of reads attributed to benthic (263,193 originally, 45,975 after resampling) in comparison to pelagic (476,058 originally, 210,800 after resampling) organisms could suggest that upper-ocean productivity is higher than benthic productivity at the deep continental margin. However, the low assignment rate, particularly among benthic fauna, may, to an unknown extent, also relate to a large number of undescribed species and the sequence database gap of deep-sea biota in general63. Some typical deep-sea biota were detected, such as free-living nematodes (Oxystominidae, 0.1%), albeit only with a few reads (Supplementary data file 2).
The majority of resampled reads were assigned to families, which potentially represent the benthic composition of the shelf and upper continental slope64,65, such as bivalves (Pectinidae, 22.7% and Mytilidae, 1.5%), starfish (Asteriidae, 14.1%), priapulid worms (Priapulidae, 6.9%), corals (Nephtheidae,1%; Pocilloporidae, 2.8%), and sea urchins (Strongylocentrotidae, 4.5%) (Fig. 4). Storms can whirl up and redistribute benthic inhabitants66,67, and the very steep Kamchatka slope may foster downslope transport, which might also explain the considerable share of reads assigned to near-shore macrophytes such as seagrass (Zosteraceae, 0.8%) and brown (Phaeophyta, 2.9%) and red macroalgae (Rhodophyta, 10.9%) in our record. The ratio between counts assigned to pelagic and benthic families (pelagic:benthic ratio) is highest in samples dated to the late glacial and Younger Dryas compared to the warmer Bølling/Allerød and the Holocene (Figs. 2, 4). During phases with higher SSTs there is a general increase in read counts of pelagic and benthic taxa. This increase is proportionally stronger in the benthic compared to pelagic taxa resulting in a smaller difference between them under warmer SSTs and, thus a smaller pelagic:benthic ratio. Overall, the pelagic:benthic ratio shows a weak, negative correlation with SSTs (Spearman’s ρ = −0.39, p value = 0.052), indicating that more reads are assigned to benthic families in phases with warmer SSTs.

Only families represented in at least three samples and by at least 1.5% proportion of read counts after resampling are shown. The dendrogram (based on constrained hierarchical clustering) and IP25 concentrations from the same core16 and sea-surface temperature (SSTUk‘37) reconstructions of the Northwest Pacific52 (light red) and Northeast Pacific97 (dark red) are added for comparison. Zosteraceae, which are significant, and positively correlated with SSTUk’37, are highlighted in red. The pelagic:benthic ratio shows the proportion of reads assigned to pelagic families in relation to reads assigned to benthic families. The ratio between reads assigned to phototrophic bacteria and reads assigned to phototrophic protists is given as a blue-green line. Black triangles next to the IP25 record mark radiocarbon-dated calibrated ages52. LGM last glacial maximum, HS1 Heinrich stadial 1, B/A Bølling/Allerød, YD Younger Dryas, and colors of the boxes refer to warm (red) and cold (blue) phases. Source data are provided as a Source Data file.
In our dataset, Zosteraceae have higher read counts during the Holocene and are significantly, positively correlated with summer/early fall SSTs (Spearman’s ρ = 0.73, adjusted p value = 0.026; Fig. 4b). Laminariaceae (kelp) have higher read counts during the late glacial. Kelp form underwater forests on shallow, rocky, cold-water coasts with complex vertical structures that provide habitat for epiphytic algae, and a food source for sea urchins (Strongylocentrotidae)68. In addition, the occurrence of kelp in the North Atlantic is linked to the behavioral traits of cod69. The co-occurrence of kelp and Pacific cod in the assemblages of ice-rich periods (Figs. 2, 4) suggests functional links between benthic and pelagic taxa in our study. Seagrass rather grows in temperate waters with soft sediments, such as sheltered, shallow bays70. The modern distribution range in the north is restricted to circumboreal coasts, which is in line with our finding of low late-glacial abundance in our study area and that in response to warmer and ice-free conditions in the Holocene, a range expansion occurred. Important benthic-pelagic relationships are established between seagrass and salmon as well as Pacific herring, including foraging opportunities and spawning grounds71,72, which are reflected in our data by similar temporal co-occurrence patterns.
Implications for sea-ice decline in the northern Bering Sea
Our study combines metagenomic shotgun sequencing of sedaDNA with a correlation network analysis24 to explore changes in organismal composition and potential co-occurrence in the course of sea-ice change over the past ~20,000 years. During warmer, ice-free phases, namely the Bølling/Allerød and since the early Holocene, fewer sequences are assigned to diatoms, and sequences assigned to Cyanobacteria dominate among primary producers. Phytoplankton groups with large cell sizes, such as diatoms, are assumed to contribute more to carbon export than other pico-sized plankton, such as phototrophic bacteria73, which are presumed to be mostly recycled by the microbial loop in the water column74. A shift from larger phototrophic protists to smaller bacterioplankton in the course of sea-ice loss could therefore alter the amount and quality of food-sustaining benthic organisms75 and the efficiency of the biological carbon pump58. Similar observations have been made in the Arctic, where sea-surface freshening due to increased meltwater and runoff have led to a rise in cell abundance of upper-ocean bacterioplankton, while the abundance of nanophytoplankton decreased58. The latter study also found that despite the abundance shifts of the two plankton size classes, the chlorophyll-a content, which is often used as a proxy for biomass, remained stable. These results, combined with the data presented in this study, suggest that paleo-productivity reconstructions based on biogenic silica (produced e.g., by diatoms and silicoflagellates) or calcium carbonate (produced e.g., by haptophytes and foraminifers) can strongly underestimate past primary productivity estimations especially during climate transitions, because they do not consider changes in taxonomic composition among primary producers.
Although metagenomic shotgun sequencing is currently limited by the lack of reference databases to assess past diversity changes, our approach detected a variety of fish and marine mammals that are common in coastal areas of the Bering Sea and the subarctic North Pacific. So far, no studies have been published that target marine mammals in sediments using ancient DNA, and the only marine sedaDNA record targeting fish extends back 300 years76. By pushing back the detection limit of fish and mammals to almost 20,000 years, our approach paves the way to assess millennial-scale to glacial–interglacial changes in the distribution and natural dynamics of these key groups in future studies. Furthermore, our results suggest that in the future, sedaDNA holds promise in identifying the organismal sources of organic matter, which has been recognized as one of the key fundamental questions in blue-carbon science77.
Overall, our results indicate that sea-ice is a key factor for the structure and functioning of the near-coastal Western Bering Sea ecosystem on a millennial timescale. By analogy to the past, substantial shifts of pelagic and benthic compositions can be expected for subarctic regions that will likely become ice-free in the next decades, such as the northern Bering Sea shelf, which today is subject to frequent regime shifts between cold and warm conditions and related variation in sea-ice extent2. Along with changes in ecosystem composition, alterations in ecosystem services can be expected, particularly with respect to carbon sinks and fishing grounds.
This sedaDNA record implies that ongoing climate warming on the Bering Sea shelf might lead to a shift towards a pico-sized phytoplankton community composed mainly of cyanobacteria (Fig. 5) where previously, primary productivity was dominated by nano- and microphytoplankton. This is supported by observations that recent warming and increasing freshwater runoff is paralleled by increasing picoplankton abundance58. The dominance of picoplankton-derived DNA in our dataset indicates efficient carbon export and burial of Cyanobacteria, potentially facilitated by aggregate formation or inclusion in detritus. However, the lack of polyunsaturated fatty acids and their potential toxicity might limit the value of Cyanobacteria as a high-quality food source for benthic organisms78.

Representatives of functional groups of a the seasonal sea-ice ecosystem, which prevailed for most of the late glacial and b the ice-free ecosystem, which dominated during the Holocene.
Our data also suggest that climate warming and prolonged sea-ice-free conditions in coastal areas allow for the northward expansions of seagrass in protected, soft-sediment bays and kelp along shallow, rocky coastlines of the Russian and North American Arctic79. The expansion of seagrass meadows, in particular, could assist the northward expansions of Pacific herring and salmon, which our data suggests could be benefiting from climate warming on the northern Bering Sea shelf. However, high abundances of pink salmon have been connected with negative effects on other species, such as increased seabird mortalities, changes in nesting phenology, and a decrease in breeding success due to their competitive advantage over shared prey5,50. In contrast, warmer, ice-free conditions leave pollock and cod at a disadvantage. Their survival depends on the availability of large copepods, which are reduced in warm years, as suggested by our data and modern observations2. Our results thus confirm that ecosystem service changes related to future sea-ice loss is of major relevance not only for global climate but also for regional economies.
Methods
Sample material
In 2018 we collected and extracted samples from the archive half of a 9.05 m piston corer (SO201-2-12KL, Fig. 1) which was retrieved during RV Sonne cruise SO201 “KALMAR” in 200952 and since then stored at 4 °C. We applied the chronostratigraphy established by ref. 52. Its high sedimentation rates with 80 cm kyr−1 during the last deglaciation and 30 cm kyr−1 in the Holocene allows us to detect ecosystem changes with a temporal resolution of, on average, 780 years according to our sampling density. According to our age model, the one-centimeter thick samples contain, on average, a contribution of DNA deposition over 15.5 years during the late glacial and over 28.7 years during the Holocene. The sampling was carried out at GEOMAR, Kiel, in a sedimentology laboratory devoid of any molecular biology work. Details about the subsampling procedure and the opening of the cores can be found in the Supplementary Information.
DNA extraction
DNA extractions and subsequent library preparation were carried out in a dedicated paleogenetic facility devoid of any post-PCR laboratories, whereas the PCRs and downstream preparations for sequencing were carried out in the genetics laboratories located in a different building.
The DNeasy PowerMax Soil Kit (Qiagen, Germany) was used to extract total DNA from an average of 7.5 g sediment80. For a previous DNA metabarcoding analysis20 we extracted DNA for 63 samples in seven batches, where each batch contained up to nine samples and an extraction blank (in total, seven negative controls). From those, we chose the stock solutions of 25 DNA extracts and the corresponding extraction blanks, which had been kept continuously frozen since the extraction. The sample choice led to a mean temporal resolution of ~780 years, thereby ensuring that each millennium is represented by at least one and each climatic phase by at least two samples.
We measured the DNA concentration on a Qubit 4.0 fluorometer (Invitrogen, Carlsbad, CA, USA) using the Qubit dsDNA BR Assay Kit (Invitrogen, USA) and depending on the initial DNA concentrations, we concentrated a volume of 300 or 600 µL using the GeneJET PCR Purification Kit (Thermo Scientific, USA) and eluted twice with 25 µL elution buffer. Immediately afterward, we checked the DNA yield again with 2 µL concentrated DNA using the Qubit dsDNA BR Assay Kit and the DNA was diluted to 3 ng µL−1. The DNA extracts and aliquots were stored at −20 °C.
Single-stranded library preparation and sequencing
For metagenomic shotgun sequencing, we chose the method of single-stranded library preparation, which was specifically developed for highly degraded ancient DNA81,82 and used 15 ng DNA as a template. We prepared three batches of libraries for sequencing. The first batch contained a set of samples covering each major climatic phase of the core for a first assessment of the method. Therefore, five of the seven extraction blanks (50 µL per extraction blank were pooled and 5 µL of the pool used for the library preparation) were sequenced together with their corresponding samples in the first run, while the last two blanks were processed later in the second sequencing run. To monitor contamination during library preparation, we included one negative control for each batch of libraries (library blank).
The libraries were quantified using qPCR82: First, a pUC19 vector (New England BioLabs, Germany) was amplified by PCR. The PCR products were purified with the MinElute PCR Purification Kit (Qiagen, Germany) according to the manufacturer’s instructions and subsequently quantified using the Qubit dsDNA BR Assay Kit. Three independent tenfold dilution series (109 to 102 copies per µL) were prepared to calculate the standard curves. The qPCR setup contained 1x Maxima™ SYBR™ Green (Thermo Scientific, Germany), 0.2 µM IS7 and IS8, and 1 µL of the libraries diluted 1:20 with TET buffer in a final volume of 25 µL. The qPCR was run in the RotorGeneQ cycler (Qiagen, Germany) with the following settings: 95 °C for 10 min, followed by 40 cycles of 30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. The fluorescence was acquired after each cycle.
For samples, the subsequent double-indexing was performed by PCR in 10–14 cycles (for samples and blanks) depending on library concentration with indexed P5 (5′−3′: AATGATACGGCGACCACCGAGATCTACACNNNNNNNACACTCTTTCCCTACACGACGCTCTT; IDT, Germany) and P7 (5′−3′: CAAGCAGAAGACGGCATACGAGATNNNNNNNGTGACTGGAGTTCAGACGTGT, IDT, Germany) primers using AccuPrime Pfx polymerase (Life Technologies, Germany) and 24 µL of each library. Blanks were amplified with the same number of cycles, although the initial DNA concentration was 0 ng/µL. The indexing PCR products were purified using MinElute PCR Purification Kit (Qiagen, Germany). Then the library size distribution was checked on the Agilent TapeStation using the D1000 ScreenTape (Agilent Technologies, USA). The samples were pooled equimolarly, but with the negative controls in a 10:1 ratio to achieve a final molarity of 10 mM. Three paired-end sequencing (2 × 125 bp) runs were carried out with a modified sequencing primer CL7282 (5′−3′: ACACTCTTTCCCTACACGACGCTCTTCC, IDT, Germany) by the Fasteris SA sequencing service (Switzerland). The first library pool was sequenced on a single lane of an Illumina HiSeq 2500 instrument (high-output V4, total output of 40.6 GB), while the second and the third pools were sequenced on two lanes of a full SP flow cell (total output 198.1 GB) using the Illumina NovaSeq device.
Bioinformatic processing and sequence classification
We used FastQC v. 0.11.983 to check the quality of the raw data, followed by duplicate removal with FastUniq v. 1.184, which reduced the number of read pairs from 918,186,452 to 876,349,245 (Supplementary Data 1). We additionally applied clumpify (BBMap package v. 38.87; sourceforge.net/projects/bbmap/) to perform deduplication. Both approaches resulted in comparable duplication rates. The average of sample and blank duplication rate for FastUniq is 5.3 and 10.6% for clumpify. The average duplication rate within samples is FastUniq = 4.2% and clumpify = 7.2%, when considering blanks only, the approaches identified a duplication rate of FastUniq = 9.5% and clumpify = 22.5 %, which indicates a better performance of clumpify to remove duplicates in the blanks.
Afterward we used Fastp v. 0.20.085 for trimming and merging of overlapping reads in parallel, which further reduced the total number of read pairs to 742,117,607. The reads were trimmed for quality, length residual adapters, and stretches of low complexity using a sliding window (settings: length_required = 30, cut_front, cut_tail, cut_window_size = 4, cut_mean_quality = 10, n_base_limit = 5, unqualified_percent_limit = 40, complexity_threshold = 30, qualified_quality_phred = 15, low_complexity_filter). Nucleotide stretches of at least 10 poly-x were trimmed at the tail of the reads (-q 10, -x 10). Overlapping reads were merged and the quality information was used for base pair correction (settings: correction, overlap_len_require = 10, overlap_diff_limit = 5, overlap_diff_percent_limit = 20).
For the taxonomic classification of reads, we used the k-mer-based classifier Kraken2 v. 2.0.8-beta86 in single mode for merged and in the paired mode for unmerged reads (confidence threshold of 0.2). We downloaded the National Center for Biotechnology Information (NCBI) non-redundant nucleotide database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nt.gz; downloaded in June 2021) and the NCBI taxonomy (obtained using kraken2-build in June 2021) for taxonomic classification and built the Kraken2 database.
Using R v. 4.0.387, we retrieved all entries assigned on the family level and combined merged and unmerged paired-read classifications. Not all classified reads were kept for further analyses. The sediment core was kept at 4 °C since 2009; hence we expected prokaryotic and fungal composition to be biased and possibly reflect storage conditions. As we are interested in organismal responses to past environmental conditions, we kept only families belonging to phototrophic bacteria, photo- and heterotrophic protists, marine macrophytes, and Metazoa of likely regional and aquatic origin (among fish, we kept only those which occur in the subarctic North Pacific and the Bering Sea based on FishBase88). According to FishBase, the region is inhabited by marine Cyprinidae, yet it has previously been debated as contamination22 and was detected in our blanks, which is why we removed it from our analyses. Among protists, we excluded parasites, mostly belonging to Apicomplexans, Euglenids, Amoebozoa, and Myxozoa, although we acknowledge the important roles of parasites in marine environments. Euglenids contain many free-living bacterivorous flagellates89, yet the majority of sequences were assigned to the human pathogens Trypanosoma and Leishmania. Apicomplexans are highly diverse in marine ecosystems, but the NCBI reference database suffers from false annotations with widespread mistakes in human and animal parasites, like Theileria, Babesia, Cryptosporidium and Eimeriidae, which we predominantly recovered in our data90.
Finally, we kept only families that occurred in at least three samples with at least ten counts and resampled the dataset 500 times to a pelagic sample effort of 6593 counts and a benthic sample effort of 1839 counts per sample to circumvent bias arising from different sequencing depths across the samples91 to capture the main compositional trend and allow for stable correlation signals. This measure was effective in reducing the zero-inflation of our dataset for the correlation network analysis and the few affected taxa have no significant role within the network (Supplementary Fig. 12).
Negative controls
After deduplication, quality filtering, and trimming of DNA reads in the blanks (see details in Supplementary Data 1), only 0.37 and 2.14% of the original read pairs were left. In reads classified to family level and lower, the negative controls showed a negligible number of read counts. Between 111 and 540 reads were assigned to blanks, summing up to 1519 reads (0.1%) in total (Supplementary Table 1). The majority of reads in blanks were assigned to Hominidae (28.1% of blanks) and bacteria (Sphingomonadaceae 10.5%, Alteromonadaceae 5.6%, Vibrionaceae 5.4%) (Supplementary Figs. 4, 5). A total of 98 families occurred with 1 read count in blanks and 38 families have up to 2 reads (Supplementary Fig. 5).
Damage pattern analysis
Damage pattern analysis applied by the automated computational screening pipeline HOPS Version 0.3492 supports the ancient origin of the analyzed DNA pool (Supplementary Fig. 6). HOPS can either run for merged or unmerged data, but it cannot combine both. For the analyses of ancient patterns, only those reads were considered which had at least one mismatch in the first five base pairs. According to this filter, we first analyzed ancient patterns for key taxonomic groups (eukaryotic and prokaryotic algae, pelagic fish, benthic crustacean) and excluded taxa (freshwater fish and marine bacteria) using the nt (non-redundant nucleotide) database (see Supplementary Information). The frequency of C to T substitutions of the reads in the first ten and the last ten positions are given in Supplementary Fig. 6a–c.
Statistical analysis
All statistical analyses have been performed in R v. 4.0.387. After resampling, we tested each of the pelagic and benthic taxa for temporal autocorrelation using acf from the stats package and concluded that our trends and correlation networks are not influenced by temporal autocorrelation (Supplementary Figs. 8, 9).
We calculated pairwise Spearman rank correlation coefficients (ρ) using corr.test including Benjamini–Hochberg correction93 for multiple testing from the psych package v. 2.0.1294, and those exceeding at least 0.4 (adjusted p value <0.1) were used for undirected network generation by the igraph packagev. 1.2.695 without self-loops and isolated nodes (Supplementary Data 2). Networks with more and less stringent p-value thresholds are shown in Supplementary Fig. 16.
We included only positive correlations, as negative correlations were rare and did not affect the structure of the network. Furthermore, the samples are temporally not sufficiently resolved to infer direct negative interactions such as exclusions, as usually, samples represent a time average of several years. For the scale-free, force-directed network layout, we used the Fruchtermann–Reingold method (layout_with_fr)96.
The GCGM network was built using the package ecoCopula v. 1.0.227. The stackedsdm function was used to build the stacked species regression model with interpolated SSTs and IP25 values as covariates. Then a Gaussian copula graphical lasso was fitted to the co-occurrence data with the function cgr and a lambda of 0.51, which was determined empirically (Supplementary Fig. 13). To test the similarity between the Spearman network and the ecoCopula network, we computed the Jaccard-index over the edges in the networks and compared the results with a randomized null-model. The null-model was generated via double-edge swaps on the generated ecoCopula networks, which was performed ten times for each lambda value (shrinking parameter: with increasing lambda, the networks become less dense) between 0.1 and 1.0. Co-occurrence networks with negative associations for both Spearman and ecoCopula are presented in Supplementary Figs. 14, 15, respectively.
The IP25 record was available for this core, whereas we used an independent stack of northeast Pacific SST records that covers the full interval of this study but could be influenced by regional processes, such as ice-sheet sourced meltwater pulses97. SST97 and IP2598 data were interpolated to our sample ages using the R function approx87. As IP25 data could not be extrapolated for the last three samples, we calculated the mean of the last three interpolated values and replaced the missing data based on regional sea-ice reconstructions for the LGM12 (Supplementary Fig. 10). Then the interpolated variables were added to the sample-by-taxa matrix, and pairwise Spearman rank correlations were calculated as described above (Supplementary Table 3). Yet, we advise caution because of our limited sample size.
Furthermore, we highlight that both proxies contain biases. We, therefore, applied Spearman rank correlations following Djurhuus et al.35 to identify SST- or IP25-correlated families as described above (Supplementary Table 4). Then, we filtered out families which are linked with either SST- or IP25-correlated families (neighboring nodes) using the adjacent_vertices function of igraph and the table function to build a contingency table with the abundance of each family or functional group as a neighboring node (Supplementary Fig. 11 and Supplementary Data 2). This was followed by a two-sided t-test using the t.test function of the stats package v. 4.0.387. The stratigraphic diagrams, including the CONISS dendrogram, were prepared using packages rioja v. 0.9-2699 and vegan v. 2.7-7100.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited at the European Nucleotide Archive (ENA) under Bioproject number PRJEB46821. The metadata and taxonomic count data on the family level used in this study are available at PANGAEA101. The cores are stored at the GEOMAR core repository in Kiel. For access, please contact co-author Dirk Nürnberg. Source data are provided with this paper. The sea-ice data of March 2020 were retrieved from https://www.meereisportal.de (Funding: REKLIM-2013-04). The Global 30 Arc-Second Elevation (GTOPO30) was retrieved from https://www.usgs.gov/centers/eros/science/usgs-eros-archive-digital-elevation-global-30-arc-second-elevation-gtopo30#overview102. Source data are provided with this paper.
Code availability
The code used to process, filter, and analyse the sequencing data were available at https://github.com/ZimmermannHH/BeringSea_shotgun_sequencing/.
References
Lannuzel, D. et al. The future of Arctic sea-ice biogeochemistry and ice-associated ecosystems. Nat. Clim. Change 10, 983–992 (2020).
Duffy-Anderson, J. T. et al. Return of warm conditions in the southeastern Bering Sea: Phytoplankton—Fish. PLoS ONE 12, e0178955 (2017).
Wang, M., Yang, Q., Overland, J. E. & Stabeno, P. Sea-ice cover timing in the Pacific Arctic: the present and projections to mid-century by selected CMIP5 models. Deep Sea Res. Part II Top. Stud. Oceanogr. 152, 22–34 (2018).
Stabeno, P. J. & Bell, S. W. Extreme conditions in the Bering Sea (2017–2018): record-breaking low sea-ice extent. Geophys. Res. Lett. 46, 8952–8959 (2019).
Duffy‐Anderson, J. T. et al. Responses of the northern Bering Sea and southeastern Bering Sea pelagic ecosystems following record-breaking low winter sea ice. Geophys. Res. Lett. 46, 9833–9842 (2019).
Siddon, E. C., Zador, S. G. & Hunt, G. L. Ecological responses to climate perturbations and minimal sea ice in the northern Bering Sea. Deep Sea Res. Part Ii Top. Stud. Oceanogr. 181–182, 104914 (2020).
Intergovernmental Panel on Climate Change (IPCC). The Ocean and Cryosphere in a Changing Climate: Special Report of the Intergovernmental Panel on Climate Change. (Cambridge Univ. Press, 2022).
Grebmeier, J. M. et al. A major ecosystem shift in the Northern Bering Sea. Science 311, 1461–1464 (2006).
Grebmeier, J. M. & Barry, J. P. The influence of oceanographic processes on pelagic-benthic coupling in polar regions: a benthic perspective. J. Mar. Syst. 2, 495–518 (1991).
Belt, S. T. & Müller, J. The Arctic sea ice biomarker IP25: a review of current understanding, recommendations for future research and applications in palaeo sea ice reconstructions. Quat. Sci. Rev. 79, 9–25 (2013).
Matul, A. G. Probable limits of sea ice extent in the northwestern Subarctic Pacific during the last glacial maximum. Oceanology 57, 700–706 (2017).
Méheust, M., Stein, R., Fahl, K. & Gersonde, R. Sea-ice variability in the subarctic North Pacific and adjacent Bering Sea during the past 25 ka: new insights from IP25 and Uk′37 proxy records. Arktos 4, 8 (2018).
Harada, N., Shin, K. H., Murata, A., Uchida, M. & Nakatani, T. Characteristics of alkenones synthesized by a bloom of Emiliania huxleyi in the Bering Sea. Geochim. Cosmochim. Acta 67, 1507–1519 (2003).
Smirnova, M. A., Kazarina, G. K. H., Matul, A. G. & Max, L. Diatom evidence for paleoclimate changes in the northwestern Pacific during the last 20000 years. Oceanology 55, 383–389 (2015).
Caissie, B. E., Brigham‐Grette, J., Lawrence, K. T., Herbert, T. D. & Cook, M. S. Last glacial maximum to Holocene sea surface conditions at Umnak Plateau, Bering Sea, as inferred from diatom, alkenone, and stable isotope records. Paleoceanography 25, PA1206 (2010).
Méheust, M., Stein, R., Fahl, K., Max, L. & Riethdorf, J.-R. High-resolution IP25-based reconstruction of sea-ice variability in the western North Pacific and Bering Sea during the past 18,000 years. Geo-Mar. Lett. 36, 101–111 (2016).
Pawłowska, J., Łącka, M., Kucharska, M., Pawlowski, J. & Zajączkowski, M. Multiproxy evidence of the neoglacial expansion of Atlantic water to eastern Svalbard. Clim. Past 16, 487–501 (2020).
De Schepper, S. et al. The potential of sedimentary ancient DNA for reconstructing past sea ice evolution. ISME J. https://doi.org/10.1038/s41396-019-0457-1 (2019).
Zimmermann, H. H. et al. Changes in the composition of marine and sea-ice diatoms derived from sedimentary ancient DNA of the eastern Fram Strait over the past 30 000 years. Ocean Sci. 16, 1017–1032 (2020).
Zimmermann, H. H. et al. Sedimentary ancient DNA from the subarctic North Pacific: how sea ice, salinity, and insolation dynamics have shaped diatom composition and richness over the past 20,000 years. Paleoceanogr. Paleoclimatol. 36, e2020PA004091 (2021).
Armbrecht, L., Hallegraeff, G., Bolch, C. J. S., Woodward, C. & Cooper, A. Hybridisation capture allows DNA damage analysis of ancient marine eukaryotes. Sci. Rep. 11, 3220 (2021).
Armbrecht, L. et al. Paleo-diatom composition from Santa Barbara Basin deep-sea sediments: a comparison of 18S-V9 and diat-rbcL metabarcoding vs shotgun metagenomics. ISME Commun. 1, 1–10 (2021).
More, K. D., Giosan, L., Grice, K. & Coolen, M. J. L. Holocene paleodepositional changes reflected in the sedimentary microbiome of the Black Sea. Geobiology 17, 436–448 (2019).
Comte, J., Lovejoy, C., Crevecoeur, S. & Vincent, W. F. Co-occurrence patterns in aquatic bacterial communities across changing permafrost landscapes. Biogeosciences 13, 175–190 (2016).
Araújo, M. B., Rozenfeld, A., Rahbek, C. & Marquet, P. A. Using species co-occurrence networks to assess the impacts of climate change. Ecography 34, 897–908 (2011).
Matchado, M. S. et al. Network analysis methods for studying microbial communities: a mini review. Comput. Struct. Biotechnol. J. 19, 2687–2698 (2021).
Popovic, G. C., Warton, D. I., Thomson, F. J., Hui, F. K. C. & Moles, A. T. Untangling direct species associations from indirect mediator species effects with graphical models. Methods Ecol. Evol. 10, 1571–1583 (2019).
Slon, V. et al. Neandertal and denisovan DNA from pleistocene sediments. Science https://doi.org/10.1126/science.aam9695 (2017).
Pedersen, M. W. et al. Postglacial viability and colonization in North America’s ice-free corridor. Nature 537, 45–49 (2016).
Parducci, L. et al. Shotgun environmental DNA, pollen, and macrofossil analysis of lateglacial lake sediments from southern Sweden. Front. Ecol. Evol. 7, 189 (2019).
Ahmed, E. et al. Archaeal community changes in Lateglacial lake sediments: evidence from ancient DNA. Quat. Sci. Rev. 181, 19–29 (2018).
Schulte, L. et al. Hybridization capture of larch (Larix Mill.) chloroplast genomes from sedimentary ancient DNA reveals past changes of Siberian forest. Mol. Ecol. Resour. 21, 801–815 (2021).
Mora, C., Tittensor, D. P., Adl, S., Simpson, A. G. B. & Worm, B. How many species are there on Earth and in the Ocean? PLoS Biol. 9, e1001127 (2011).
Sibbald, S. J. & Archibald, J. M. More protist genomes needed. Nat. Ecol. Evol. 1, 1–3 (2017).
Djurhuus, A. et al. Environmental DNA reveals seasonal shifts and potential interactions in a marine community. Nat. Commun. 11, 254 (2020).
Pääbo, S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl Acad. Sci. USA 86, 1939–1943 (1989).
Hestetun, J. T. et al. Significant taxon sampling gaps in DNA databases limit the operational use of marine macrofauna metabarcoding. Mar. Biodivers. 50, 70 (2020).
Aydin, K., Lapko, V. V., Radchenko, V. & Livingston, P. A Comparison of the Eastern Bering and Western Bering Sea Shelf and Slope Ecosystems through the Use of Mass-balance Food Web Models (U.S. Department of Commerce, National Oceanic and Atmospheric Administration, National Marine Fisheries Service, Alaska Fisheries Science Center, 2002).
von Quillfeldt, C. H., Ambrose, W. G. & Clough, L. M. High number of diatom species in first-year ice from the Chukchi Sea. Polar Biol. 26, 806–818 (2003).
Joli, N., Monier, A., Logares, R. & Lovejoy, C. Seasonal patterns in Arctic prasinophytes and inferred ecology of Bathycoccus unveiled in an Arctic winter metagenome. ISME J. 11, 1372–1385 (2017).
Belevich, T. A., Ilyash, L. V., Milyutina, I. A., Logacheva, M. D. & Troitsky, A. V. Metagenomics of Bolidophyceae in plankton and ice of the White Sea. Biochemistry 82, 1538–1548 (2017).
Durbin, E. G. & Casas, M. C. Early reproduction by Calanus glacialis in the Northern Bering Sea: the role of ice algae as revealed by molecular analysis. J. Plankton Res. 36, 523–541 (2014).
Aydin, K. & Mueter, F. The Bering Sea—A dynamic food web perspective. Deep Sea Res. Part II Top. Stud. Oceanogr. 54, 2501–2525 (2007).
Kohlbach, D. et al. Strong linkage of polar cod (Boreogadus saida) to sea ice algae-produced carbon: evidence from stomach content, fatty acid and stable isotope analyses. Prog. Oceanogr. 152, 62–74 (2017).
Aizawa, C., Tanimoto, M. & Jordan, R. W. Living diatom assemblages from North Pacific and Bering Sea surface waters during summer 1999. Deep Sea Res. Part II Top. Stud. Oceanogr. 52, 2186–2205 (2005).
Ren, J., Gersonde, R., Esper, O. & Sancetta, C. Diatom distributions in northern North Pacific surface sediments and their relationship to modern environmental variables. Palaeogeogr. Palaeoclimatol. Palaeoecol. 402, 81–103 (2014).
Shiozaki, T. et al. Eukaryotic phytoplankton contributing to a seasonal bloom and carbon export revealed by tracking sequence variants in the western North Pacific. Front. Microbiol. 10, 2722 (2019).
Williams, E. H. & Quinn, T. J. II Pacific herring, Clupea pallasi, recruitment in the Bering Sea and north-east Pacific Ocean, II: relationships to environmental variables and implications for forecasting. Fish. Oceanogr. 9, 300–315 (2000).
Andrews, A. G., Strasburger, W. W., Farley, E. V., Murphy, J. M. & Coyle, K. O. Effects of warm and cold climate conditions on capelin (Mallotus villosus) and Pacific herring (Clupea pallasii) in the eastern Bering Sea. Deep Sea Res. Part II Top. Stud. Oceanogr. 134, 235–246 (2016).
Springer, A. M. & Vliet, G. B. V. Climate change, pink salmon, and the nexus between bottom-up and top-down forcing in the subarctic Pacific Ocean and Bering Sea. Proc. Natl Acad. Sci. USA 111, E1880–E1888 (2014).
Jones, V. & Solomina, O. The geography of Kamchatka. Glob. Planet. Change 134, 3–9 (2015).
Max, L. et al. Sea surface temperature variability and sea-ice extent in the subarctic northwest Pacific during the past 15,000 years. Paleoceanography 27, PA3213 (2012).
Andreev, A. A. et al. Late Pleistocene to Holocene vegetation and climate changes in northwestern Chukotka (Far East Russia) deduced from lakes Ilirney and Rauchuagytgyn pollen records. Boreas 50, 652–670 (2021).
Darienko, T. & Pröschold, T. Toward a monograph of non-marine Ulvophyceae using an integrative approach (Molecular phylogeny and systematics of terrestrial Ulvophyceae II.). Phytotaxa 324, 1–41 (2017).
Burgess, E. A., Unrine, J. M., Mills, G. L., Romanek, C. S. & Wiegel, J. Comparative geochemical and microbiological characterization of two thermal pools in the Uzon Caldera, Kamchatka, Russia. Microb. Ecol. 63, 471–489 (2012).
Medvedeva, L., Nikulina, T. & Genkal, S. Centric diatoms (Coscinodiscophyceae) of fresh and brackish water bodies of the southern part of the Russian Far East. Oceanol. Hydrobiol. Stud. 38, 139–164 (2009).
Heikkilä, M., Ribeiro, S., Weckström, K. & Pieńkowski, A. J. Predicting the future of coastal marine ecosystems in the rapidly changing Arctic: the potential of palaeoenvironmental records. Anthropocene https://doi.org/10.1016/j.ancene.2021.100319 (2021).
Li, W. K. W., McLaughlin, F. A., Lovejoy, C. & Carmack, E. C. Smallest algae thrive as the Arctic Ocean freshens. Science 326, 539–539 (2009).
Glibert, P. M. et al. Pluses and minuses of ammonium and nitrate uptake and assimilation by phytoplankton and implications for productivity and community composition, with emphasis on nitrogen-enriched conditions. Limnol. Oceanogr. 61, 165–197 (2016).
Sweetman, A. K. et al. Major impacts of climate change on deep-sea benthic ecosystems. Elem. Sci. Anth. 5, 4 (2017).
Moffitt, S. E., Hill, T. M., Roopnarine, P. D. & Kennett, J. P. Response of seafloor ecosystems to abrupt global climate change. Proc. Natl Acad. Sci. USA 112, 4684–4689 (2015).
Yasuhara, M. et al. Climatic forcing of quaternary deep-sea benthic communities in the North Pacific Ocean. Paleobiology 38, 162–179 (2012).
Sinniger, F. et al. Worldwide analysis of sedimentary DNA reveals major gaps in taxonomic knowledge of deep-sea benthos. Front. Mar. Sci. 3, 92 (2016).
Gorbatenko, K. M. & Nadtochy, V. A. The biochemical composition and caloric content of the macrozoobenthos of the Western Kamchatka Shelf. Russ. J. Mar. Biol. 44, 283–291 (2018).
Selivanova, O. N. The Dynamical Processes of Biodiversity—Case Studies of Evolution and Spatial Distribution (InTech, 2011).
Corte, G. N. et al. Storm effects on intertidal invertebrates: increased beta diversity of few individuals and species. PeerJ 5, e3360 (2017).
Ortega, A. et al. Important contribution of macroalgae to oceanic carbon sequestration. Nat. Geosci. 12, 748–754 (2019).
Teagle, H., Hawkins, S. J., Moore, P. J. & Smale, D. A. The role of kelp species as biogenic habitat formers in coastal marine ecosystems. J. Exp. Mar. Biol. Ecol. 492, 81–98 (2017).
Cote, D., Ollerhead, L. M. N., Gregory, R. S., Scruton, D. A. & McKinley, R. S. Activity patterns of juvenile Atlantic cod (Gadus morhua) in Buckley Cove, Newfoundland. Hydrobiologia 483, 121–127 (2002).
McRoy, C. The distribution and biogeography of Zostera marina (eelgrass) in Alaska. Pacific Science 22, 507–513 (1968).
Kennedy, L. A., Juanes, F. & El‐Sabaawi, R. Eelgrass as valuable nearshore foraging habitat for juvenile Pacific salmon in the early marine period. Mar. Coast. Fish. 10, 190–203 (2018).
Levin, P. S., Francis, T. B. & Taylor, N. G. Thirty-two essential questions for understanding the social–ecological system of forage fish: the case of Pacific Herring. Ecosyst. Health Sustain. 2, e01213 (2016).
Basu, S. & Mackey, K. R. M. Phytoplankton as key mediators of the biological carbon pump: their responses to a changing climate. Sustainability 10, 869 (2018).
Legendre, L. & Le Fèvre, J. Microbial food webs and the export of biogenic carbon in oceans. Aquat. Microb. Ecol. 09, 69–77 (1995).
Kędra, M. et al. Status and trends in the structure of Arctic benthic food webs. Polar Res. 34, 23775 (2015).
Kuwae, M. et al. Sedimentary DNA tracks decadal-centennial changes in fish abundance. Commun. Biol. 3, 1–12 (2020).
Macreadie, P. I. et al. The future of Blue Carbon science. Nat. Commun. 10, 3998 (2019).
Nascimento, F. J. A., Karlson, A. M. L., Näslund, J. & Gorokhova, E. Settling cyanobacterial blooms do not improve growth conditions for soft bottom meiofauna. J. Exp. Mar. Biol. Ecol. 368, 138–146 (2009).
Krause-Jensen, D. et al. Imprint of climate change on pan-Arctic marine vegetation. Front. Mar. Sci. 7, 617324 (2020).
Zimmermann, H. H. et al. Sedimentary ancient DNA and pollen reveal the composition of plant organic matter in Late Quaternary permafrost sediments of the Buor Khaya Peninsula (north-eastern Siberia). Biogeosciences 14, 575–596 (2017).
Gansauge, M.-T. et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45, e79 (2017).
Gansauge, M.-T. & Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 8, 737–748 (2013).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. (2010).
Xu, H. et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS ONE 7, e52249 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
R Core Team. R: a language and environment for statistical computing. (R Foundation for Statistical Computing, 2018).
Froese, R. & Pauly, D. FishBase. https://www.fishbase.org (2020).
Ohtsuka, S., Suzaki, T., Horiguchi, T., Suzuki, N. & Not, F. (eds) Marine Protists: Diversity and Dynamics (Springer, 2015).
del Campo, J. et al. Assessing the diversity and distribution of apicomplexans in host and free-living environments using high-throughput amplicon data and a phylogenetically informed reference framework. Front. Microbiol. 10, 2373 (2019).
Kruse, S. R code for resampling and thus normalizing of count data to the minimum number of counts across a set of samples (e.g. sedaDNA sequence/pollen taxa counts per sample along a sediment core). (2019).
Hübler, R. et al. HOPS: automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol. 20, 280 (2019).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Revelle, W. psych: Procedures for psychological, psychometric, and personality research. (2020).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 1695, 1–9 (2006).
Fruchterman, T. M. J. & Reingold, E. M. Graph drawing by force-directed placement. Software Pract. Exper. 21, 1129–1164 (1991).
Praetorius, S. K. et al. The role of Northeast Pacific meltwater events in deglacial climate change. Sci. Adv. 6, eaay2915 (2020).
Méheust, M., Stein, R., Fahl, K., Max, L. & Riethdorf, J.-R. High-resolution IP25 of sediment core SO201-2-12. In supplement to: Méheust, M et al. (2015): High-resolution IP25-based reconstruction of sea-ice variability in the western North Pacific and Bering Sea during the past 18,000 years. Geo-Mar. Lett. 10.1007/s00367-015-0432-4 (2015).
Juggins, S. rioja: analysis of quaternary science data. R package version 0, 9–26 (2020).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2, 5–7 (2020).
Zimmermann, H. H. et al. Count data of classified sequences from sedimentary ancient DNA shotgun metagenomics of core SO201-2-12KL. https://doi.org/10.1594/PANGAEA.934664 (2021).
Earth Resources Observation And Science (EROS) Center. Global 30 arc-second elevation (GTOPO30). https://doi.org/10.5066/F7DF6PQS (2017).
Fetterer, F., Knowles, K., Meier, W. N., Savoie, M. & Windnagel, A. K. Sea Ice Index, Version 3. Monthly and daily GIS compatible shapefiles of median ice extent. National Snow & Ice Data Center https://doi.org/10.7265/n5k072f8 (2017).
Grosfeld, K. et al. Online sea-ice knowledge and data platform. Polarforschung 85, 143–145 (2016).
Spreen, G., Kaleschke, L. & Heygster, G. Sea ice remote sensing using AMSR-E 89-GHz channels. J. Geophys. Res. 113, C02S03 (2008).
Acknowledgements
We would like to thank Stefan Niehaus for bioinformatic support, Thomas Böhmer for generating the map, Janine Klimke for guidance with the library preparation, and Cathy Jenks for English proofreading and editing.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
H.H.Z.: conducting lab work, formal analysis, visualizations, and writing of the initial draft, K.R.S.-L.: formal analysis of damage patterns and conceptualization; V.D.: network similarity and ecoCopula networks; L.H.: software and formal analysis of damage patterns, L.S.: Methodology, M.-T.H.: supervision of network analyses, D.N.: Resources, R.T.: Resources, U.H.: conceptualization, study design, supervision, co-writing of the initial draft, and resources. All authors commented on the initial draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Linda Armbrecht, Alexandra Rouillard and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zimmermann, H.H., Stoof-Leichsenring, K.R., Dinkel, V. et al. Marine ecosystem shifts with deglacial sea-ice loss inferred from ancient DNA shotgun sequencing. Nat Commun 14, 1650 (2023). https://doi.org/10.1038/s41467-023-36845-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36845-x
This article is cited by
-
Millennial-scale variations in Arctic sea ice are recorded in sedimentary ancient DNA of the microalga Polarella glacialis
Communications Earth & Environment (2024)
-
Multi-omics for studying and understanding polar life
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
