
Abstract
N-functionalized aziridines, which are both useful intermediates and important synthetic targets, can be envisioned as arising from the addition of nitrenes (i.e., NR fragments) to olefinic substrates. The exceptional reactivity of most nitrenes, in particular with respect to unimolecular decomposition, prevents general application of nitrene-transfer to the synthesis of N-functionalized aziridines. Here we demonstrate N-aryl aziridine synthesis via 1) olefin aziridination with N-aminopyridinium reagents to afford N-pyridinium aziridines followed by 2) Ni-catalyzed C–N cross-coupling of the N-pyridinium aziridines with aryl boronic acids. The N-pyridinium aziridine intermediates also participate in ring-opening chemistry with a variety of nucleophiles to afford 1,2-aminofunctionalization products. Mechanistic investigations indicate aziridine cross-coupling proceeds via a noncanonical mechanism involving initial aziridine opening promoted by the bromide counterion of the Ni catalyst, C–N cross-coupling, and finally aziridine reclosure. Together, these results provide new opportunities to achieve selective incorporation of generic aryl nitrene equivalents in organic molecules.
Similar content being viewed by others
Introduction
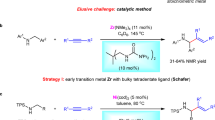
Aziridines, which are three-membered nitrogen-containing heterocycles, are attractive synthetic intermediates en route to 1,2-aminofunctionalization products and are present in various naturally occurring alkaloids and pharmacologically active compounds1,2,3,4. Retrosynthetically, aziridines can be envisioned as arising from the combination of a nitrene equivalent with an olefinic substrate. In practice, aziridination via nitrene transfer is severely limited by the promiscuous reactivity of unstabilized nitrenes5: For example, attempts to access N-phenylaziridines from phenylnitrene (generated by thermolysis or photolysis of phenyl azide) result in polymeric tars instead of the desired aziridine6. Since Evans’s report of Cu-catalyzed olefin aziridination7, myriad transition metal-catalyzed methods have been developed for nitrene transfer to olefins (Fig. 1a)8,9,10. Metal-catalyzed nitrene transfer catalysis typically requires electron-withdrawing groups, such as N-sulfonyl substituents, to activate the nitrogen equivalent for transfer11,12,13; there are limited reports of metal-catalyzed nitrene transfer from aryl azide precursors14, 15. The resulting N-protected aziridines can be challenging to utilize in downstream N-functionalization chemistry. For example, exposure of N-sulfonyl aziridines to metal-catalyzed cross-coupling conditions typically results in aziridine opening, not N-functionalization16,17,18,19,20,21,22,23,24. N–H aziridines can be accessed by either deprotection of N-protected aziridines25,26,27 or by direct synthesis of olefinic precursors (Fig. 1b)28,29,30,31,32. While derivatization of the N–H valence can provide access to some N-functionalization products, arylation of these compounds is not broadly developed33,34,35,36,37.

a Nitrene transfer to olefins provides access to aziridines but often requires the utilization of sulfonyl groups to activate the nitrogen. b N–H aziridines can be accessed directly from olefins and metal-catalyzed allylation methods enable functionalization of the N–H valence. c Here we advance N-pyridinium aziridines as platforms for C–N cross coupling to provide access to N-substituted aziridines.
N-aminopyridinium reagents represent a burgeoning class of bifunctional reagents38 which combine a nucleophilic N-amino group with a low-lying pyridinium-centered LUMO that enables access to N-centered radicals via reductive N–N cleavage (LUMO = lowest unoccupied molecular orbital)39. In the context of amination chemistry, N-sulfonylaminopyridiniums have been utilized in photoredox-promoted olefin difunctionalization40,41,42,43,44,45,46,47 and aromatic C–H amination reactions48,49,50,51,52,53,54. These reactions also rely of the presence of the electron withdrawing N-sulfonyl substituents to stabilize incipient N-centered radicals. Broad application of N-aminopyridiniums as bifunctional reagents in amination chemistry is stymied by the limited methods currently available to prepare N-functionalized aminopyridiniums, which are accessed by either addition of hydrazines to pyrylium salts or by sulfonylation of N-aminopyridiniums55, 56.
In this work, we describe the first example of olefin aziridination with N-aminopyridinium reagents (Fig. 1c). Inspired by the Ni-catalyzed C–C coupling chemistry of N-alkylpyridinium electrophiles pioneered by Watson57,58,59,60 and others61,62,63,64,65,66, we demonstrate that the resulting N-pyridinium aziridines are competent electrophiles for C–N bond-forming cross-coupling with aryl boronic acids to afford N-aryl aziridines. Analogous chemistry is unknown for N-tosyl or N-phthalyl aziridines. Moreover, in contrast to classical methods for functionalization of N–H aziridines based on N-centered nucleophilicity, the described protocol leverages N-centered electrophilicity to provide access to the products of formal aryl nitrene transfer to olefinic substrates. Initial mechanistic experiments suggest that the cross-coupling proceeds via a non-canonical mechanism involving halide-promoted ring opening, C–N bond-forming cross coupling, and aziridine reclosure.
Results
We began the development of a formal nitrene transfer sequence by developing robust conditions for olefin aziridination with N-aminopyridinium reagents as nitrogen sources. Combination of styrene and N-aminopyridinium triflate in the presence of iodobenzene diacetate (PhI(OAc)2) and MgO resulted in 1-(2-phenylaziridin-1-yl)pyridin-1-ium in 64% yield. Aziridination could also be accomplished using N-amino-2,4,6-triphenylpyridinium tetrafluoroborate (2) as the nitrogen source under these conditions (42% yield of the corresponding pyridinium aziridine (3a)). During subsequent studies of C–N cross coupling (vide infra), the triphenyl derivative was found to provide superior results and thus we optimized the aziridination reaction with compound 2 as the pyridinium source. Examination of the impact of various catalysts, solvents, reaction temperatures, and additives (see Supplementary Information Section C.1 for details) identified optimized aziridination conditions based on iodide catalysis in the presence of 4 Å molecular sieves, which affords aziridine 3a in 71% yield (Fig. 2).

Condition a 1 (1.0 equiv), 2 (1.0 equiv), PhIO (1.0 equiv), TBAI (5 mol%); condition b 1 (1.0 equiv), 2 (1.6 equiv), PhIO (1.6 equiv), TBAI (20 mol%); condition c 1 (1.0 equiv), N-aminopyridinium triflate (1.0 equiv), PhIO (1.0 equiv), TBAI (5 mol%). X-ray structure of 3a presented as displacement ellipsoid plot (50% probability) with BF4– counterion removed for clarity. py* = N-2,4,6-triphenylpyridinium, py = N-pyridinium.
An array of 4-substituted vinyl arenes participate in aziridination with the optimized conditions: hydrocarbyl substituents (3b and 3c), electron-donating alkoxy and Boc-protected amines (3d and 3e), as well as various electron-withdrawing substituents (3f–3k) are all well tolerated. Both meta- and ortho-substituents (3l–3o and 3p–3r, respectively) are compatible with the developed aziridination conditions and 2-vinylnaphthalene and 2-vinylbenzothiophene undergoes aziridination to 3s and 3t in 79% and 60% yield, respectively. For electron-neutral and electron-rich substrates, 5 mol% [TBA]I is utilized; for electron deficient substrates we increased the catalyst loading to 20 mol% to achieve efficient aziridination. Consistent with previous reports of iodide-catalyzed aziridination that are proposed to proceed via radical mechanisms, functionalization of 1,2-disubstituted olefins is not stereospecific12: aziridination of trans-β-methylstyrene (trans-1u) affords a 45:55 cis:trans mixture of 3u; aziridination of cis-1u affords a 35:65 cis:trans mixture of 3u. For geometrically constrained 1,2-disubstituted olefins, such as indene (1v), aziridination provides a single diastereomer (e.g., 3v in 31% yield). The developed conditions are effective for the aziridination on more complex substrates, including 3w–3aa, which are derived from pharmaceutically relevant estrone, ibuprofen, tufnil, probenecid, and isatin. Application to aliphatic olefins is available, albeit in lower efficiency: Aziridination of cyclohexene and allylbenzene with N-aminopyridinium triflate afforded the corresponding aziridines 3ab and 3ac in 33% and 16% yield, respectively.
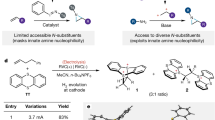
With conditions in hand to efficiently access N-pyridinium aziridines, we turned our attention to engaging these species as electrophiles in C–N coupling reactions. We envisioned a C–N cross coupling of N-pyridinium aziridines would (1) provide access to the products of formal nitrene transfer to olefins, (2) provide a rare example of an aziridine cross-coupling in which the aziridine ring remains intact, and (3) represent the first application of pyridinium electrophiles in C–N cross-coupling chemistry. We initiated our investigations by examining potential Ni-catalyzed cross coupling of N-pyridinium aziridine electrophiles with appropriate organometallic nucleophiles (i.e., Grignard reagents, organolithiums, organostannanes, and boronic acids). We identified that treatment of N-pyridinium aziridine 3a with tolyl boroxine and NiCl2(dme) in MeCN afforded N-arylaziridine 5b in 36% yield. The coupling efficiency is extremely sensitive to the Ni(II) counter anion: Under identical conditions, NiCl2 provided 5b in 36% yield while NiBr2 afforded 5b in 60% yield. Ni(OAc)2, Ni(acac)2, and NiSO4 salts were completely ineffective. Optimization of the cross-coupling reaction (see Supplementary Information Section C.2 for details) ultimately identified the use of NiBr2(phen) as catalyst in the presence of K3PO4 and 2,4,6-collidine provided N-tolylaziridine 5b in 79% yield (Fig. 3). The catalyst loading could be reduced to 10 mol% without significant loss of yield, but further reduction to 5 mol% resulted in substantial reduction in reaction efficiency. For comparison, attempted cross-coupling reactions with N-phthalyl or N-sulfonyl aziridines under our optimized conditions or with N–H aziridines using C–N cross-coupling conditions described in the literature were unsuccessful (see Supplementary Information sections C.8 and C.9)33, 34. We also examined potential Ni-catalyzed cross-electrophile-coupling and found inferior results: Combination of 3a with 4-bromotoluene in the presence of NiCl2 (30 mol%), 1,10-phenanthroline (30 mol%), and either Mn or Zn dust as terminal reductant afforded the product 5b at 50% and 11% NMR yield, respectively (see Table S7)60.

Cross-coupling of 3 with aryl boronic acids provides access to N-arylaziridines, which are the products of formal transfer of aryl nitrenes to olefins. a These reactions were carried out with 10 mol% Ni(phen)Br2. b For these substrates, K2CO3 was used in place of K3PO4. c Prepared from the unsubstituted N-pyridinium aziridine (i.e. 3aa and 3ab). Yields reported are based on isolated products (based on 1H NMR integration). Phen = 1,10-phenanthroline.
The developed cross-coupling conditions enable cross-coupling with simple arylboronic acids substituted in the 4-position (5a–5j) and in the 3-position (5k), but substitution in the 2-position (5l) and alkyl boronic acids (i.e., n-butylboronic acid) were not tolerated. Notably, electron-neutral and electron-rich aryl groups can be incorporated in the N-arylaziridine efficiently, which represent specific challenges in direct nitrene transfer strategies11, 15. Similarly, various substitutions of the pyridinium aziridine coupling partner were also tolerated (5m–5s). The developed aziridination reaction is compatible with both complex boronic acids, such as those derived from thioflavin T (5t), indomethacin (5u), loratadine (5v), and chloropyramine (5w), and with complex pyridinium aziridine partners, such as those derived from estrone (5x–5y), probenecid (5z), and ibuprofen (5aa–5ab). Fragment coupling reactions in which both complex boronic acids and complex pyridinium aziridine partners are directly linked via an aziridine ring were efficient (5ac–5ad). Coupling of aziridines derived from aliphatic olefins is less efficient than coupling of vinyl arene-derived aziridines: Cross-coupling of 3ab and 3ac with (4-(ethoxycarbonyl)phenyl)boronic acid (4j) afforded aziridines 5ae and 5af in 36% and 30% yield, respectively.
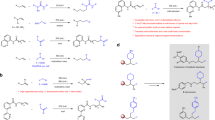
In addition to direct C–N coupling with boronic acids, the developed N-pyridinium aziridines participate in ring-opening chemistry to access 1,2-difunctionalization products (Fig. 4). Exposure of N-pyridinium aziridine 3a to halide sources (i.e. [TBA]Br, [TBA]Cl, or pyridine·HF) or H2O in the presence of BF3·OEt2 resulted in opening of the aziridine to afford 1,2-haloamine derivatives 6a–6c or 1,2-hydroxyamine 6d. Attempts to isolate 6a and 6b resulted in low isolated yields due to aziridine reclosure to N-pyridinium aziridine 3a (vide infra). A variety of other oxygen-, nitrogen-, and sulfur-based nucleophiles participate in aziridine opening to afford isolable aminopyridinium derivatives 6e–6j. Indole can serve as a carbon nucleophile to provide 6k in 37% yield. These ring-opened compounds could be isolated as analytically pure materials and participate in efficient Ni-catalyzed cross coupling to generate 1,2-aminofunctionalized compounds 7e–7k (the products of p-tolylboronic acid coupling), respectively. The ring-opened product 6g and 6j also participated in cross coupling with more complex boronic acids, as highlighted by the synthesis of 7l and 7m, which are derived from cross-coupling of ring-opened compounds with the boronic acid derived from indomethacin.

Nucleophilic opening of aziridine 3a provides access to 1,2-aminofunctionalization products 6. Ni-catalyzed cross-coupling of compounds 6 provides the opportunity to elaborate the resulting acyclic N-aminopyridinium derivatives to generate anilines 7. *Yields determined by 1H NMR due to instability of these compounds towards intramolecular elimination.
Metal-catalyzed cross-coupling of aziridine often results in ring opening products16,17,18,19,20,21,22,23,24. In contrast, we observed Ni-catalyzed C–N coupling to generate N-arylaziridines in which the aziridine ring is conserved in the product. To better understand this unusual reaction outcome, we were interested in evaluating the mechanism of C–N bond-forming chemistry that leaves the three-membered aziridine ring intact. These investigations were guided by (1) a desire to understand the bromide-specific activity noted in our original catalyst optimization studies and (2) the observation that while treatment of 3a with [TBA]Br and BF3·OEt2 resulted in ring opening, attempts to isolate the resulting benzyl bromide 6a resulted in re-isolation of 3a, which suggested that ring-opening with bromide is reversible. This observation suggested the possibility that aziridine opening, followed by cross-coupling of an open-chain aminopyridinium intermediates, and finally aziridine reclosure may be operative (Fig. 5a). Consistent with this hypothesis, treatment of aziridine 3a with NiBr2(dme) (with or without added phenanthroline) results in the observation of ring-opened compound 6a by 1H NMR (Fig. 5b). While BF3·OEt2 was required for ring opening with [TBA]Br, spontaneous ring-opening is observed upon addition of NiBr2, which suggest that under these conditions Ni2+ is serves as a cooperative Lewis acid activator and bromide source for aziridine opening. Finally, exposure of a sample of compound 6a to Ni(OTf)2 or Ni(BF4)2 and p-ethoxylcarbonylphenylboronic acid results in the formation of N-arylaziridine 5j, which demonstrates the viability of cross-coupling and aziridine reclosure (Fig. 5c). For further discussion of the impact of added bromide on cross-coupling efficiency, see Section C.7 of the Supplementary Information.

a Reversible halide-promoted aziridine opening, cross coupling, and aziridine reclosure are proposed to mediate C–N cross coupling of N-pyridinium aziridines. Consistent with this mechanism. b NiBr2 reacts with N-pyridinium aziridine 3a to generate ring-opened 6a and c exposure of ring-opened 6a to Ni(OTf)2 or Ni(BF4)2 affords arylated aziridine 5j. *Yields determined by 1H NMR spectroscopy.
Discussion
In summary, we report a strategy for the synthesis of N-aryl aziridines, which are the formal products of aryl nitrene addition to olefins. This method overcomes the inherent instability of free nitrene fragments by harnessing N-pyridinium aziridine intermediates that participate in Ni-catalyzed C–N cross-coupling. By decoupling the aziridination from installation of the N-substituent, this strategy overcomes the common requirement for difficult-to-remove N-substituents in aziridination chemistry. The observed C–N cross-coupling chemistry contrasts the typical reactivity pattern of N-sulfonylaziridine cross-coupling, which typically participate in ring-opening C–N activation, by taking advantage of a unique reversible ring opening/reclosure mechanism. These studies not only provide strategies to access products of formal nitrene transfer to olefins but significantly expand the synthetic scope of nitrene transfer by demonstrating N-aminopyridinium to be a bifunctional amination reagent.
Methods
General procedure for olefin aziridination
In an N2-filled dry box, a 20-mL scintillation vial was charged with 1-amino-2,4,6-triphenylpyridin-1-ium tetrafluoroborate (2, 82.0 mg, 0.200 mmol, 1.00 equiv), tetrabutylammonium iodide (3.7 mg, 0.010 mmol, 5 mol%), 4 Å molecular sieves, vinyl arene (1, 0.200 mmol, 1.00 equiv), and iodosylbenzene (44.0 mg, 0.200 mmol, 1.00 equiv). Acetonitrile (1.0 mL) was added, the vial was removed from the dry box, and the reaction mixture was stirred for 18 h. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by silica gel flash chromatography (2:1 ethyl acetate:hexanes) to afford compound 3.
General procedure for cross-coupling of pyridinium aziridines
A 20-mL scintillation vial was charged with Ni(Phen)Br2 (20 mol%), base (2.8 equiv), aryl boronic acid (4, 2.4 equiv), pyridinium aziridine (3, 1 equiv) and a magnetic stir bar. In an N2 filled dry box, a solution of 2,4,6-collidine in acetonitrile (0.08 M, 1.0 equiv) was added to the scintillation vial with the rest of the reaction components. The reaction vial was heated at 65 °C for 36 h. After cooling to 23 °C, the reaction mixture was transferred to a centrifuge tube and centrifuged at 3220×g (6000 rpm) for 10 min. The supernatant was decanted. The residue was washed with CH2Cl2 and the combined supernatants were concentrated under reduced pressure and the crude mixture was purified as indicated in the Supplementary Information to afford the aziridine compound 5.
General procedure for nucleophilic opening of pyridinium aziridines
In an N2-filled dry box, a 20-mL scintillation vial was charged with 2,4,6-triphenyl-1-(2-phenylaziridin-1-yl)pyridinium tetrafluoroborate (3, 300 mg, 0.586 mmol, 1.00 equiv), BF3·OEt2 (0.075 mL, 0.59 mmol, 1.0 equiv), and CH2Cl2 (5 mL). A 25-mL Schlenk tube is charged with the appropriate nucleophile (0.703 mmol, 1.20 equiv). The CH2Cl2 solution of 3 and BF3·OEt2 was added to the Schlenk flask that contained the nucleophile. The resulting reaction mixture was allowed to stir at indicated temperature for indicated time. Solvent was removed under reduced pressure and the residue was purified by SiO2 gel chromatography (eluent 2:1 ethyl acetate:hexane) to afford the compound 6.
General procedure for cross-coupling of N-pyridinium amines 6
In an N2-filled dry box, a 20-mL scintillation vial was charged with compound 6 (0.100 mmol, 1.00 equiv) and CH3CN (1 mL). A separate 20-mL was charged with NiBr2(dme) (3.1 mg, 0.010 mmol, 10 mol%), 1,10-phenanthroline (2.4 mg, 0.014 mmol, 0.14 equiv), K3PO4 (53.1 mg, 0.250 mmol, 2.50 equiv), p-toluyl boronic acid (20.4 mg, 0.150 mmol, 1.50 equiv). The CH3CN solution of 6 was added to the vial containing NiBr2·DME. The reaction mixture was stirred at 65 °C for 18 h. The reaction mixture was cooled to 23 °C. The reaction mixture was centrifuged at 3220×g (6000 rpm) for 10 min and the supernatant was decanted. Solvent was removed under reduced pressure and the residue was purified by alumina gel column chromatography to obtain compound 7.
Data availability
All data generated in this study are provided in the Supplementary Information. Crystallography data for the structure reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2159504 (3a). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
References
Ismail, F. M. D., Levitsky, D. O. & Dembitsky, V. M. Aziridine alkaloids as potential therapeutic agents. Eur. J. Med. Chem. 44, 3373–3387 (2009).
Degennaro, L., Trinchera, P. & Luisi, R. Recent advances in the stereoselective synthesis of aziridines. Chem. Rev. 114, 7881–7929 (2014).
Sweeney, J. B. Aziridines: epoxides’ ugly cousins? Chem. Soc. Rev. 31, 247–258 (2002).
Schneider, C. Catalytic, enantioselective ring opening of aziridines. Angew. Chem. Int. Ed. 48, 2082–2084 (2009).
Wentrup, C. Carbenes and nitrenes: recent developments in fundamental chemistry. Angew. Chem. Int. Ed. 57, 11508–11521 (2018).
Gritsan, N. & Platz, M. Photochemistry of azides: the azide/nitrene interface. In Organic Azides: Syntheses and Applications (eds Bräse, S. & Banert, K.) 311–372 (John Wiley & Sons, Inc., 2009).
Evans, D. A., Faul, M. M. & Bilodeau, M. T. Copper-catalyzed aziridination of olefins by (N-(p-toluenesulfonyl)imino)phenyliodinane. J. Org. Chem. 56, 6744–6746 (1991).
Chang, J. W. W., Ton, T. M. U. & Chan, P. W. H. Transition-metal-catalyzed aminations and aziridinations of C–H and C–C bonds with iminoiodinanes. Chem. Rec. 11, 331–357 (2011).
Shimbayashi, T., Sasakura, K., Eguchi, A., Okamoto, K. & Ohe, K. Recent progress on cyclic nitrenoid precursors in transition-metal-catalyzed nitrene-transfer reactions. Chem. Eur. J. 25, 3156–3180 (2019).
Driver, T. G. Recent advances in transition metal-catalyzed N-atom transfer reactions of azides. Org. Biomol. Chem. 8, 3831–3846 (2010).
Deng, T. et al. Rh2(II)-catalyzed intermolecular N-aryl aziridination of olefins using nonactivated N atom precursors. J. Am. Chem. Soc. 143, 19149–19159 (2021).
Kiyokawa, K., Kosaka, T. & Minakata, S. Metal-free aziridination of styrene derivatives with iminoiodinane catalyzed by a combination of iodine and ammonium iodide. Org. Lett. 15, 4858–4861 (2013).
Siu, T. & Yudin, A. K. Practical olefin aziridination with a broad substrate scope. J. Am. Chem. Soc. 124, 530–531 (2002).
Fantauzzi, S. et al. The (porphyrin)ruthenium-catalyzed aziridination of olefins using aryl azides as nitrogen sources. Eur. J. Org. Chem. 2007, 6053–6059 (2007).
Jin, L.-M. et al. Effective synthesis of chiral N-fluoroaryl aziridines through enantioselective aziridination of alkenes with fluoroaryl azides. Angew. Chem. Int. Ed. 52, 5309–5313 (2013).
Lin, B. L., Clough, C. R. & Hillhouse, G. L. Interactions of aziridines with nickel complexes: oxidative-addition and reductive-elimination reactions that break and make C−N bonds. J. Am. Chem. Soc. 124, 2890–2891 (2002).
Huang, C.-Y. & Doyle, A. G. Nickel-catalyzed Negishi alkylations of styrenyl aziridines. J. Am. Chem. Soc. 134, 9541–9544 (2012).
Nielsen, D. K., Huang, C.-Y. & Doyle, A. G. Directed nickel-catalyzed Negishi cross coupling of alkyl aziridines. J. Am. Chem. Soc. 135, 13605–13609 (2013).
Duda, M. L. & Michael, F. E. Palladium-catalyzed cross-coupling of N-sulfonylaziridines with boronic acids. J. Am. Chem. Soc. 135, 18347–18349 (2013).
Takeda, Y., Ikeda, Y., Kuroda, A., Tanaka, S. & Minakata, S. Pd/NHC-catalyzed enantiospecific and regioselective Suzuki–Miyaura arylation of 2-arylaziridines: Synthesis of enantioenriched 2-arylphenethylamine derivatives. J. Am. Chem. Soc. 136, 8544–8547 (2014).
Jensen, K. L., Standley, E. A. & Jamison, T. F. Highly regioselective nickel-catalyzed cross-coupling of N-tosylaziridines and alkylzinc reagents. J. Am. Chem. Soc. 136, 11145–11152 (2014).
Takeda, Y., Kuroda, A., Sameera, W. M. C., Morokuma, K. & Minakata, S. Palladium-catalyzed regioselective and stereo-invertive ring-opening borylation of 2-arylaziridines with bis(pinacolato)diboron: experimental and computational studies. Chem. Sci. 7, 6141–6152 (2016).
Woods, B. P., Orlandi, M., Huang, C.-Y., Sigman, M. S. & Doyle, A. G. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J. Am. Chem. Soc. 139, 5688–5691 (2017).
Davies, J. et al. Ni-catalyzed carboxylation of aziridines en route to β-amino acids. J. Am. Chem. Soc. 143, 4949–4954 (2021).
Ankner, T. & Hilmersson, G. Instantaneous deprotection of tosylamides and esters with SmI2/amine/water. Org. Lett. 11, 503–506 (2009).
Dauban, P. & Dodd, R. H. Phinses: a new iminoiodinane reagent for the copper-catalyzed aziridination of olefins. J. Org. Chem. 64, 5304–5307 (1999).
Nandi, P. et al. Alkali metals in silica gel (M-SG): a new reagent for desulfonation of amines. Org. Lett. 10, 5441–5444 (2008).
Varszegi, C. et al. A micellar iodide-catalyzed synthesis of unprotected aziridines from styrenes and ammonia. Angew. Chem. Int. Ed. 47, 1477–1480 (2008).
Jat, J. L. et al. Direct stereospecific synthesis of unprotected N–H and N–Me aziridines from olefins. Science 343, 61–65 (2014).
Legnani, L., Prina-Cerai, G., Delcaillau, T., Willems, S. & Morandi, B. Efficient access to unprotected primary amines by iron-catalyzed aminochlorination of alkenes. Science 362, 434–439 (2018).
Cheng, Q.-Q. et al. Organocatalytic nitrogen transfer to unactivated olefins via transient oxaziridines. Nat. Catal. 3, 386–392 (2020).
Holst, D. E., Wang, D. J., Kim, M. J., Guzei, I. A. & Wickens, Z. K. Aziridine synthesis by coupling amines and alkenes via an electrogenerated dication. Nature 596, 74–79 (2021).
Sasaki, M., Dalili, S. & Yudin, A. K. N-arylation of aziridines. J. Org. Chem. 68, 2045–2047 (2003).
Witulski, B., Senft, S., Bonet, J. & Jost, O. Palladium-catalyzed N-arylation reactions with aziridine and azetidine. Synthesis 2007, 243–250 (2007).
Watson, I. D. G., Styler, S. A. & Yudin, A. K. Unusual selectivity of unprotected aziridines in palladium-catalyzed allylic amination enables facile preparation of branched aziridines. J. Am. Chem. Soc. 126, 5086–5087 (2004).
Dalili, S. & Yudin, A. K. Transition metal-catalyzed synthesis and reactivity of N-alkenyl aziridines. Org. Lett. 7, 1161–1164 (2005).
Watson, I. D. G. & Yudin, A. K. New insights into the mechanism of palladium-catalyzed allylic amination. J. Am. Chem. Soc. 127, 17516–17529 (2005).
Huang, H.-M., Bellotti, P., Ma, J., Dalton, T. & Glorius, F. Bifunctional reagents in organic synthesis. Nat. Rev. Chem. 5, 301–321 (2021).
Rössler, S. L. et al. Pyridinium salts as redox-active functional group transfer reagents. Angew. Chem. Int. Ed. 59, 9264–9280 (2020).
Chen, J.-Q., Yu, W.-L., Wei, Y.-L., Li, T.-H. & Xu, P.-F. Photoredox-induced functionalization of alkenes for the synthesis of substituted imidazolines and oxazolidines. J. Org. Chem. 82, 243–249 (2017).
Zhao, Y., Shi, C., Su, X. & Xia, W. Synthesis of isoquinolones by visible-light-induced deaminative [4+2] annulation reactions. Chem. Commun. 56, 5259–5262 (2020).
Mo, J.-N., Yu, W.-L., Chen, J.-Q., Hu, X.-Q. & Xu, P.-F. Regiospecific three-component aminofluorination of olefins via photoredox catalysis. Org. Lett. 20, 4471–4474 (2018).
Miyazawa, K., Koike, T. & Akita, M. Regiospecific intermolecular aminohydroxylation of olefins by photoredox catalysis. Chem. Eur. J. 21, 11677–11680 (2015).
Miyazawa, K., Koike, T. & Akita, M. Aminohydroxylation of olefins with iminopyridinium ylides by dual ir photocatalysis and Sc(OTf)3 catalysis. Tetrahedron 72, 7813–7820 (2016).
Guo, W., Wang, Q. & Zhu, J. Selective 1,2-aminoisothiocyanation of 1,3-dienes under visible-light photoredox catalysis. Angew. Chem. Int. Ed. 60, 4085–4089 (2021).
Yu, W.-L., Chen, J.-Q., Wei, Y.-L., Wang, Z.-Y. & Xu, P.-F. Alkene functionalization for the stereospecific synthesis of substituted aziridines by visible-light photoredox catalysis. Chem. Commun. 54, 1948–1951 (2018).
Moon, Y. et al. Visible light induced alkene aminopyridylation using N-aminopyridinium salts as bifunctional reagents. Nat. Commun. 10, 4117 (2019).
Kim, N., Lee, C., Kim, T. & Hong, S. Visible-light-induced remote C(sp3)–H pyridylation of sulfonamides and carboxamides. Org. Lett. 21, 9719–9723 (2019).
Jung, S., Lee, H., Moon, Y., Jung, H.-Y. & Hong, S. Site-selective C–H acylation of pyridinium derivatives by photoredox catalysis. ACS Catal. 9, 9891–9896 (2019).
Moon, Y., Lee, W. & Hong, S. Visible-light-enabled ortho-selective aminopyridylation of alkenes with N-aminopyridinium ylides. J. Am. Chem. Soc. 142, 12420–12429 (2020).
Greulich, T. W., Daniliuc, C. G. & Studer, A. N-aminopyridinium salts as precursors for N-centered radicals – direct amidation of arenes and heteroarenes. Org. Lett. 17, 254–257 (2015).
Rössler, S. L. et al. Pyridyl radical cation for C−H amination of arenes. Angew. Chem. Int. Ed. 58, 526–531 (2019).
Ham, W. S., Hillenbrand, J., Jacq, J., Genicot, C. & Ritter, T. Divergent late-stage (hetero)aryl C−H amination by the pyridinium radical cation. Angew. Chem. Int. Ed. 58, 532–536 (2019).
Hillenbrand, J., Ham, W. S. & Ritter, T. C–H pyridonation of (hetero-)arenes by pyridinium radical cations. Org. Lett. 21, 5363–5367 (2019).
He, F.-S., Ye, S. & Wu, J. Recent advances in pyridinium salts as radical reservoirs in organic synthesis. ACS Catal. 9, 8943–8960 (2019).
Pang, Y., Moser, D. & Cornella, J. Pyrylium salts: Selective reagents for the activation of primary amino groups in organic synthesis. Synthesis 52, 489–503 (2020).
Basch, C. H., Liao, J., Xu, J., Piane, J. J. & Watson, M. P. Harnessing alkyl amines as electrophiles for nickel-catalyzed cross couplings via C–N bond activation. J. Am. Chem. Soc. 139, 5313–5316 (2017).
Hoerrner, M. E., Baker, K. M., Basch, C. H., Bampo, E. M. & Watson, M. P. Deaminative arylation of amino acid-derived pyridinium salts. Org. Lett. 21, 7356–7360 (2019).
Plunkett, S., Basch, C. H., Santana, S. O. & Watson, M. P. Harnessing alkylpyridinium salts as electrophiles in deaminative alkyl–alkyl cross-couplings. J. Am. Chem. Soc. 141, 2257–2262 (2019).
Liao, J. et al. Deaminative reductive cross-electrophile couplings of alkylpyridinium salts and aryl bromides. Org. Lett. 21, 2941–2946 (2019).
Martin-Montero, R., Yatham, V. R., Yin, H., Davies, J. & Martin, R. Ni-catalyzed reductive deaminative arylation at sp3 carbon centers. Org. Lett. 21, 2947–2951 (2019).
Sun, S.-Z., Romano, C. & Martin, R. Site-selective catalytic deaminative alkylation of unactivated olefins. J. Am. Chem. Soc. 141, 16197–16201 (2019).
Yue, H. et al. Nickel-catalyzed C–N bond activation: activated primary amines as alkylating reagents in reductive cross-coupling. Chem. Sci. 10, 4430–4435 (2019).
Yi, J., Badir, S. O., Kammer, L. M., Ribagorda, M. & Molander, G. A. Deaminative reductive arylation enabled by nickel/photoredox dual catalysis. Org. Lett. 21, 3346–3351 (2019).
Ni, S. et al. Ni-catalyzed deaminative cross-electrophile coupling of katritzky salts with halides via C─N bond activation. Sci. Adv. 5, eaaw9516 (2019).
Zhu, Z.-F., Tu, J.-L. & Liu, F. Ni-catalyzed deaminative hydroalkylation of internal alkynes. Chem. Commun. 55, 11478–11481 (2019).
Acknowledgements
The authors gratefully acknowledge financial support from the National Institutes of Health R35GM138114 (D.C.P.), the Welch Foundation A-1907 (D.C.P.), and an Alfred P. Sloan Fellowship (D.C.P.). Andy Thomas and Danniel Arriaga are acknowledged for help with HPLC purification of selected compounds. Richard Thompson is acknowledged for assistance with X-ray crystallography experiments.
Author information
Authors and Affiliations
Contributions
H.T., S.S., and D.C.P. conceived of the project. H.T., S.S., A.M. and P.R. carried out experimental work. All authors participated in data analysis, manuscript writing, and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hegui Gong, and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, H., Samanta, S., Maity, A. et al. N-Aminopyridinium reagents as traceless activating groups in the synthesis of N-Aryl aziridines. Nat Commun 13, 3341 (2022). https://doi.org/10.1038/s41467-022-31032-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-31032-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
