
Abstract
Sequential double hydrofunctionalizationalization of alkynes is a powerful method to construct useful vicinal compounds. Herein, we report a cobalt-catalyzed sequential hydrosilylation/hydrohydrazidation of alkynes to afford 1,2-N,Si compounds via ligand relay catalysis. A phenomenon of ligand relay is found that the tridentate anionic N-ligand (OPAQ) could capture the cobalt ion from bidentate neutral P-ligand (Xantphos) cobalt complex. This protocol uses three abundant chemical feedstocks, alkynes, silanes, and diazo compounds, and also features operationally simple, mild conditions, low catalyst loading (1 mol%), and excellent functional group tolerance. The 1,2-N,Si compounds can be easily further derivatized to afford various substituted silane derivatives via Si-H functionalization, alcohols via Fleming-Tamao oxidation, free amines and amides via N-N bond cleavage and protection. The asymmetric reaction could also be carried out to afford chiral products with up to 86% ee. The ligand relay has been supported by control experiments and absorption spectra.
Similar content being viewed by others

Introduction
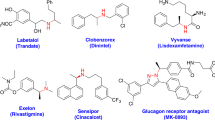
Organosilicon compounds have been widely used in materials science1 as well as agrochemistry and organic synthesis2,3, also play a growing role in medicinal chemistry as silasubstitution or bioisosteres of carbon or other moieties due to slight physical and electronic alterations4,5. Vicinal amino and silyl (1,2-N,Si) compounds as an important subclass of organosilicon compounds simultaneously bearing amino and silyl groups have presented in various biologically active compounds such as sila-haloperidol and sila-venlafaxine (Fig. 1)6,7,8.

Cyclic and acyclic 1,2-N,Si compounds in pharmaceuticals.
Sequential double hydrofunctionalization of readily available alkynes could atom- and pot-economically install two functional groups9,10. For two transformations with same catalytic species, one metal catalyst could achieve the sequential reactions11,12,13,14,15,16,17,18,19,20. For two mechanistically distinctive transformations, a strategy of bimetallic catalysis in which two different metal catalysts respectively play an individual role has been usually employed, however, compatibility issues of two metal catalysts used to limit their utility (Fig. 2a). Ligand exchange is a well-known fundamental elementary reaction in organometallic chemistry21. To avoid compatibility issues and also achieve metal economy, a strategy of ligand exchange by using mono metal and two ligands could be proposed in which the former ligand metal complex for catalyzing the first-step reaction would undergo ligand exchange after adding the latter ligand to catalyze the second-step reaction (Fig. 2a)22,23,24,25,26,27,28,29,30,31,32,33. Two reactions would not or slightly interrupt with each other. This one-way ligand exchange for catalyzing two mechanistically distinctive transformations could be called as ligand relay catalysis. However, to the best of our knowledge, this ligand relay catalysis has not been used in sequential double hydrofunctionalization of alkynes.

a Strategies for 1,2-double hydrofunctionalization of alkynes. b Cobalt-catalyzed sequential hydrosilylation/hydrohydrazidation of terminal alkynes via ligand relay catalysis.
Sequential double hydrofunctionalization of alkynes could be potentially used to access 1,2-N,Si compounds. Nevertheless, sequential hydrosilylation/hydrohydrazidation of alkynes to deliver 1-amino-2-silylalkanes has not yet been reported. There are several challenges: (1) inhibiting the side- reactions in sequential transformations, including dihydrosilylation11,12,13 and hydrogenation14,15; (2) controlling the compatibility of two reactions in one pot as well as one product bearing both N-H bond and Si-H bond; (3) achieving high regioselectivity to reduce the possibility of gem-substituted undesired products16,17,18, and improving the efficiency of reactions to make the transformation synthetically useful.
Our group is quite interested in earth-abundant transition-metal-catalyzed reactions of alkenes and alkynes11,12,13,14,15,34,35,36,37, here, we report a cobalt-catalyzed chemo- and regioselective sequential hydrosilylation/hydrohydrazidation of terminal alkynes via ligand relay catalysis with hydrosilanes and diazo compounds to afford vicinal amino and silyl products bearing both N-H bond and Si-H bond which could be further derivatized (Fig. 2b).
Results
Reaction optimization
We began our investigation on sequential hydrosilylation/hydrohydrazidation of alkynes with selected initial reaction conditions, using ethynyl benzene 1a as a model substrate, diphenyl silane 2a as a silicon source, ethyl 2-diazo-2-phenylacetate 3a as a nitrogen source. Based on our previous studies on the cobalt-catalyzed sequential reaction of alkynes14, various cobalt complexes for alkyne hydrosilylation have been tested as a solo catalyst for the sequential reaction, however, reactions were messy and fewer desired products have been obtained. To achieve two reactions in one pot, a combined catalytic system using two different catalysts has been tested. One catalytic system is using Xantphos•CoBr2 and activator NaBHEt3 for the first hydrosilylation step38,39. The other catalytic system is using N-(2-(4,4-dimethyl-4,5-dihydrooxazol-2-yl)phenyl)-6-methylpicolinamide (OPPA) L1 and Co(OAc)2 for the second hydrohydrazidation step36. The combined catalytic system using PhSiH3 as a hydrogen donor in a solution of Et2O could promote the sequential hydrosilylation/hydrohydrazidation of alkynes to access 1-amino-2-silylalkane 4a in 12% yield (entry 1, Fig. 3). The use of different quinoline amine scaffolds improved the reactivity of reaction drastically (entries 2-6). The optimized ligand was N-oxazolinylphenyl 8-aminoquinoline (OPAQ) L5, delivering 4a in 81% yield. Altering several N-containing classical ligands such as OIP, PDI, PI, and Pybox, only trace amounts of 4a could be observed (entries 7-10). It is worth noting that this transformation in the absence of Co(OAc)2 could smoothly occur, delivering 4a in 84% yield, which presented a unique phenomenon of ligand relay (entry 11). With 3 equiv. of H2O as promotor, the reaction smoothly afforded 4a in 86% isolated yield (entry 12). The standard conditions are as shown in entry 12.

aReaction conditions: 1a (0.3 mmol), 2a (1.0 equiv.), 3a (1.0 equiv.), Xantphos•CoBr2 (1 mol%), NaBHEt3 (3 mol%), Co(OAc)2 (1 mol%), L1 (2 mol%) and PhSiH3 (1.2 equiv.) in Et2O (1.2 mL) at room temperature under nitrogen for 12 h. bYield was determined by 1H NMR using TMSPh as an internal standard. cIsolated yield was in parentheses. TMSPh = (Trimethylsilyl)benzene.
Substrate scope
With the optimized conditions in hand, substrate scope was explored as shown in Fig. 4. Substituents at ortho- and meta-positions on aryl rings can be well accommodated (4b-4h). Various functional groups, such as halo, ether, thiol, ether, free amine, Bpin, amide, acetal, and ester, could be tolerated (4i-4v). Substrates bearing fused rings and heterocycles, such as naphthyl, fluorenyl, indolyl, thiophene, benzothiophene, and pyridine, were also compatible in this system delivering corresponding products in 46-86% yields (4w-4ad). The alkyl substituted 1-amino-2-silylalkanes could hardly be observed in the process of aminosilylation of alkenes40 or ring-opening C(sp3)-Si cross-coupling of aziridines41,42,43, and it was challenging to obtain these compounds. Preliminary study found that hex-1-yne could be converted into 4ae in 32% yield (7/1 rr) under modified conditions. Aryl alkynes containing bioactive skeletons like estrone and geraniol could undergo the reaction to afford 4af and 4ag in 57-73% yields, which exhibited the underlying feasibility in late-stage modification of complicated molecules. Various hydrosilane, para-chloride and tert-butyl substituents diphenyl silanes and phenylsilane, could participate in this transformation smoothly to deliver 4ah and 4ai in 69-80% yields and 4aj bearing two silicon-hydrogen bonds in 55% yield. The diazo compounds with benzyl ester and dimethyl phosphate were also suitable for the system under standard conditions, delivering 4ak in 72% yield and 4al bearing three hetero atoms P, N, and Si in 61% yield.

aReaction conditions: 1 (0.3 mmol), 2 (1.0 equiv.), 3 (1.0 equiv.), Xantphos•CoBr2 (1 mol %), NaBHEt3 (3 mol %), L5 (2 mol %), PhSiH3 (1.2 equiv.), and H2O (3.0 equiv.) in Et2O (1.2 mL) under nitrogen at room temperature for 12 h, isolated yield of 4 with complete regioselectivity without additional indications. bStirred in THF, without H2O. cStirred for 24 h. dL11 (see Supplementary Information) was instead of L5, PhMeSiH2 was instead of PhSiH3, without H2O and stirred for 24 h (7/1 rr). eDPEphos•CoBr2 was instead of Xantphos•CoBr2, PhSiH3 was instead of Ph2SiH2.
To the best of our knowledge, the sequential hydrosilylation/hydrohydrazidation of alkynes for the synthesis of chiral vicinal N,Si compounds in an enantioselective manner has scarcely been reported. Thus, We preliminarily conducted asymmetric transformation of alkynes to afford chiral 1,2-N,Si compounds in moderate yields with good ee (Fig. 5). Under the optimized conditions, the standard substrates could be transformed to the chiral product in 52% yield with 86% ee. Both electron-withdrawing and electron-donating groups could be tolerated in the catalytic system, delivering the end-product in moderate yields with good ee. Alkynes bearing 2-naphthyl and benzothiophenyl underwent the reaction smoothly in 51% and 53% yields with 84% and 82% ee, respectively.

aReaction conditions: 1 (0.3 mmol), 2 (1.0 equiv.), 3 (1.0 equiv.), Xantphos•CoI2 (1 mol %), LiBHEt3 (3 mol %), L12 (2 mol %), PhSiH3 (1.2 equiv.), and H2O (3.0 equiv.) in EA to MeCN (1.2 mL) under nitrogen at 0 oC for 24 h, isolated yield of 4 with complete regioselectivity without additional indications. The details can be found in the Supplementary Information.
Gram-Scale reaction and synthetic applications
The gram-scale reaction could be smoothly conducted to access 4a in 82% yield (Fig. 6). The 1,2-N,Si compounds could be further derivatized via C-Si cleavage, Si-H transformation, and N-N cleavage. Firstly, these compounds could be easily transferred to alcohol 6 in excellent yield via Fleming-Tamao oxidation. Then, 1,2-N,Si compounds could be smoothly transformed to silanol 7, fluorosilane 8, and siloxane 9 via Si-H transformation in 67-84% yields. The product can also be converted into amide silane 10 in 48% yield via mild N-N bond cleavage and benzoyl group protection. These compounds containing both N-H bond and Si-H bond might be interesting and potentially useful, due to that, generally, N-H bond could provide a proton and Si-H bond could afford a hydride as a polar opposite reagent.

a Gram-scale reaction. b Synthetic applications. Reagents and conditions: (a) KF (4.0 equiv.), KHCO3 (4.0 equiv.), H2O2 (23.0 equiv.), MeOH/THF = 1/1, 65 oC, 12 h; (b) KHCO3 (1.0 equiv.), H2O2 (18.0 equiv.), MeOH/THF = 1/1, r.t., 12 h; (c) CuCl2 (4.0 equiv.), CuI (18 mol%), KF (2.4 equiv.), THF, r.t., 12 h; (d) Pd/C (10 wt%), MeOH/THF = 4/1, r.t., 12 h; (e) i) SmI2 (3.4 equiv.), MeOH/THF = 1/1, r.t., 12 h; ii) BzCl (3.0 equiv.), Et3N (4.0 equiv.), THF, r.t., 2 h. The details can be found in the Supplementary Information.
Mechanistic studies
To gain insight into the mechanism, two control experiments were conducted to illuminate possible mechanism (Fig. 7). With the existence of TEMPO, no desired product can be observed. The benzyl radical generated from MHAT process was trapped by TEMPO delivering the product in 9% yield, indicating that the reaction may go through radical process. The deuterium-labeling experiment was also conducted using PhSiD3 as a D source to afford 11 in 81% yield with 90% D in the adjacent methylene of silyl group (dr = 1/1), indicating that radical process may exist in this reaction. To further understand the ligand relay process, the absorption measurement was conducted (Fig. 8). Initially, we added NaBHEt3 to the solution of Xantphos•CoBr2 and it did not change the shape of absorption peaks. When adding OPAQ to the mixture, in the first 5 mins, there was no change in absorption peaks. A broad peak was observed at 468 nm in 30 mins (red line) which is cater to the absorption peak of OPAQ•CoBr2 activated by NaBHEt3 at 467 nm (blue line) indicating that it takes a period of time for OPAQ to coordinate with cobalt, the catalytic active species would emerge and keep active via ligand exchange process under N2 atmosphere while no peaks will emerge at 468 nm conducting the reaction under air (see Supplementary Information, Supplementary Figs. 14–15). Substrates will not influence the ligand exchange process (see Supplementary Information, Supplementary Figs. 14–17).

a Radical trapping experiment using TEMPO as radical trapping reagent. b Deuterium-labeling experiment using PhSiD3 as deuterated reagent.

The absorption spectra experiment is conducted under nitrogen using optic fiber spectrophotometer.
Several control experiments were also conducted. Firstly, the anti-Markovnikov hydrosilylation of alkynes could not successfully occur, delivering the α-adduct as the main product instead. Herein, Xantphos was essential for the first hydrosilylation step (Fig. 9a). Next, hydrohydrazidation of alkenyl silane failed in the absence of OPAQ, indicating that the relay process between Xantphos and OPAQ would promote the second step (Fig. 9b). From Fig. 9c we could know that OPAQ would slightly influence the anti-Markovnikov hydrosilylation step and 2% NMR yield of α-adduct was observed. In the end, Xantphos barely affected the hydrohydrazidation step, affording the 1-amino-2-silylalkanes in good yield (Fig. 9d).

a Investigation of hydrosilylation step using CoBr2 and L5. b Investigation of hydrohydrazidation step using Xantphos•CoBr2. c Investigation of hydrosilylation step using a combination of Xantphos, L5, and CoBr2. d Investigation of hydrosilylation step using a combination of Xantphos, L5, and CoBr2.
Based on the control experiments and previous studies38,39,44,45,46,47,48,49, we envisioned a possible mechanism shown in Fig. 10. Initially, Xantphos•CoBr2 was activated by NaBHEt3 to generate the cobalt hydride intermediate A which would coordinate with the alkyne. Then a following anti-Markovnikov type alkyne insertion into the cobalt hydride species gave the β-vinyl cobalt intermediate B which could undergo σ-bond metathesis with diphenyl silane to deliver the vinyl silane. The regenerated intermediate A would undergo a ligand exchange process to deliver OPAQ cobalt hydride species C. Selecting the appropriately exchangeable ligand to replace the former ligand is the key point in the ligand relay process. Here, the tridentate anionic N-ligand (OPAQ) presented a better coordination effect with cobalt ion than bidentate neutral P-ligand (Xantphos), ensuring the fluency of hydrohydrazidation of alkenyl silanes. The posterior reaction will not be influenced by the former metal catalyst which already loses the central metal ion. The newly emerging intermediate C could perform through metal hydride hydrogen atom transfer (MHAT)48 to afford a radical intermediate and cobalt intermediate D which could react with each other in the presence of diazo compound to form the intermediate E. The cobalt species E could then undergo alkyl group migration to deliver the azo enolate cobalt intermediate F, followed by σ-bond metathesis with PhSiH3 to give the silyl enol ester intermediate G and regenerate cobalt hydride intermediate C. In the presence of H2O, intermediate G would go through sequential hydrolysis and isomerization to deliver final product.

Proposed reaction pathway starts from Xantphos•Co-H species to catalyze the hydrosilylation step, followed by the key ligand relay process to generate the OPAQ•Co-H to catalyze the hydrohydrazidation step.
Discussion
In summary, we reported a cobalt-catalyzed sequential hydrosilylation/hydrohydrazidation of readily available terminal alkynes with hydrosilanes and diazo compounds to deliver 1,2-N,Si compounds with good functional group tolerance. These products bearing both N-H bond and Si-H bond can be easily transformed to hydrazino alcohol, diversified silicon-substituted hydrazino silanes, and amide silane. Two mechanistically distinctive transformations including cobalt-catalyzed alkyne insertion for alkyne hydrosilylation and the HAT process for hydrohydrazidation of alkenyl silane could be conducted using 1 mol% of cobalt as a solo metal in one pot. The asymmetric reaction could also be carried out to afford chiral products with up to 86% ee. A phenomenon of ligand relay is found to deliver cobalt from diphosphine ligand to NNN-tridentate ligand which is also illustrated by absorption measurement. Ligand relay could offer an opportunity as a potentially powerful strategy for metal-catalyzed sequential reactions using a solo metal. Further studies on ligand relay guided sequential reactions and the synthesis of organosilicon compounds will be continuously explored in our laboratory.
Methods
Materials
For NMR spectra of compounds in this manuscript, see Supplementary Information. For gram-scale reaction, see Supplementary Figure 1. For synthesis of amide silane, see Supplementary Fig. 2. For radical trapping experiment, isotopic labeling experiment, time course study, control experiment and absorption measurement, see Supplementary Figs. 3–17 and Table 1.
Standard conditions A for the synthesis of 1,2-N,Si compounds
To a 25 mL Schlenk flask equipped with a magnetic stirrer and a flanging rubber plug was dried with flame under vacuum. When cooled to ambient temperature (10–25 oC), it was vacuumed and flushed with N2. This degassed procedure was repeated for three times. To the flame-dried Schlenk flask Xantphos•CoBr2 complex (0.0030 mmol, 1 mol%), 1.2 mL (0.25 M) of Et2O and diphenylsilane (0.30 mmol, 1.0 equiv.) (or other silanes) were added by dropwise sequentially. After that, NaBHEt3 (9 μL, 1.0 M in THF, 0.0090 mmol) and alkyne (0.30 mmol, 1.0 equiv.) were added to the mixture sequentially and stirred for 5 s, and then L5 (0.0060 mmol, 2 mol%), phenylsilane (0.36 mmol, 1.2 equiv.), phenyldiazoacetate (0.30 mmol, 1.0 equiv.) (or other aryldiazoacetates), H2O (0.90 mmol, 3.0 equiv.) were added by dropwise sequentially. Pinholes were sealed with silicone grease and the flanging rubber plug was wrapped with sealing film tightly. The mixture was stirred at ambient temperature for 12 h, and then quenched by 5 mL of petroleum ether (PE) and stirred for 5 min until catalyst precipitated. The resulting solution was filtered through a pad of silica gel and the Schlenk flask and silica gel were washed by PE/EtOAc (5/1) (15 mL× 3) (or other suitable solvent). The combined filtrates were concentrated in vacuo. NMR yield was monitored by 1H NMR analysis using TMSPh as internal standard. The crude mixture was purified by short flash column chromatography to give the corresponding product.
Data availability
The authors declare that the data Supplementary the findings of this study are available within the paper and its Supplementary Information file. The experimental procedures and characterization of all new compounds are provided in the Supplementary Information. Source data are provided with this paper.
References
Ciriminna, R. et al. The sol-gel route to advanced silica-based materials and recent applications. Chem. Rev. 113, 6592–6620 (2013).
Ojima, I. In The Chemistry of Organic Silicon Compounds; (eds. Patai, S., Rappoport, Z.) (Wiley: Chichester, 1989).
Denmark, S. E. & Sweis, R. F. Design and implementation of new, silicon-based, cross-coupling reactions: importance of silicon-oxygen bonds. Acc. Chem. Res. 35, 835–846 (2002).
Min, G. K., Hernández, D. & Skrydstrup, T. Efficient routes to carbon-silicon bond formation for the synthesis of silicon-containing peptides and azasilaheterocycles. Acc. Chem. Res. 46, 457–470 (2013).
Franz, A. K. & Wilson, S. O. Organosilicon molecules with medicinal applications. J. Med. Chem. 56, 388–405 (2013).
Meanwell, N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 54, 2529–2591 (2011).
Barraza, S. J. & Denmark, S. E. Synthesis, reactivity, functionalization, and ADMET properties of silicon-containing nitrogen heterocycles. J. Am. Chem. Soc. 140, 6668–6684 (2018).
Ramesh, R. & Reddy, D. S. Quest for novel chemical entities through incorporation of silicon in drug scaffolds. J. Med. Chem. 61, 3779–3798 (2018).
Zeng, X. Recent advances in catalytic sequential reactions involving hydroelement addition to carbon-carbon multiple bonds. Chem. Rev. 113, 6864–6900 (2013).
Cheng, Z., Guo, J. & Lu, Z. Recent advances in metal-catalysed asymmetric sequential double hydrofunctionalization of alkynes. Chem. Commun. 56, 2229–2239 (2020).
Guo, J., Wang, H., Xing, S., Hong, X. & Lu, Z. Cobalt-catalyzed asymmetric synthesis of gem-Bis(silyl)alkanes by double hydrosilylation of aliphatic terminal alkynes. Chem 5, 881–895 (2019).
Cheng, Z. et al. Highly regioselective sequential 1,1-dihydrosilylation of terminal aliphatic alkynes with primary silanes. Chin. J. Chem. 37, 457–461 (2019).
Hu, M.-Y., Lian, J., Sun, W., Qiao, T.-Z. & Zhu, S.-F. Iron-catalyzed dihydrosilylation of alkynes: efficient access to geminal bis(silanes). J. Am. Chem. Soc. 141, 4579–4583 (2019).
Guo, J., Shen, X. & Lu, Z. Regio- and enantioselective cobalt-catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Angew. Chem. Int. Ed. 56, 615–618 (2017).
Chen, J., Shen, X. & Lu, Z. Cobalt-catalyzed Markovnikov selective sequential hydrogenation/hydrohydrazidation of aliphatic terminal alkynes. J. Am. Chem. Soc. 142, 14455–14460 (2020).
Niljianskul, N., Zhu, S. & Buchwald, S. L. Enantioselective synthesis of alpha-aminosilanes by copper-catalyzed hydroamination of vinylsilanes. Angew. Chem., Int. Ed. 54, 1638–1641 (2015).
Nishino, S., Hirano, K. & Miura, M. Cu-catalyzed reductive gem-difunctionalization of terminal alkynes via hydrosilylation/hydroamination cascade: concise synthesis of α-aminosilanes. Chem. Eur. J. 26, 8725–8728 (2020).
Gao, D.-W. et al. Cascade CuH-catalysed conversion of alkynes into enantioenriched 1,1-disubstitued products. Nat. Catal. 3, 23–29 (2020).
Jin, S., Liu, K., Wang, S. & Song, Q. Enatioselective cobalt-catalyzed cascade hydrosilylation and hydroboration of alkynes to access enantioenriched 1,1-silylboryl alkanes. J. Am. Chem. Soc. 143, 13124–13134 (2021).
You, Y. & Ge, S. Cobalt-catalyzed one-pot asymmetric difunctionalization of alkynes to accesss chiral gem-(Borylsilyl)alkanes. Angew. Chem. Int. Ed. 60, 1–6 (2021).
Organotransition Metal Chemistry: From Bonding to Catalysis (ed. Hartwig, J.) (University Science Books: Sausalito, CA, 2010).
The Organometallic Chemistry of the Transition Metals, Sixth Edition (ed. Crabtree, R. H.) (John Wiley: Hoboken, NJ. 2014).
Duursma, A. et al. First examples of improved catalytic asymmetric C–C bond formation using the monodentate ligand combination approach. Org. Lett. 5, 3111–3113 (2003).
Reetz, M. T., Sell, T., Meiswinkel, A. & Mehler, G. A new principle in combinatorial asymmetric transition-metal catalysis: mixtures of chiral monodentate P ligands. Angew. Chem. Int. Ed. 42, 790–793 (2003).
Reetz, M. T. Combinatorial transition-metal catalysis: mixing monodentate ligands to control enantio-, diastereo-, and regioselectivity. Angew. Chem. Int. Ed. 47, 2556–2588 (2008).
Pignataro, L. et al. Combination of a binaphthol-derived phosphite and a C1-symmetric phosphinamine generates heteroleptic catalysts in Rh- and Pd-mediated reactions. Chem. Commun. 24, 3539–3541 (2009).
Teichert, J. F. & Feringa, B. L. Phosphoramidites: privileged ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 49, 2486–2528 (2010).
Wieland, J. & Breit, B. A combinatorial approach to the identification of self-assembled ligands for rhodium-catalysed asymmetric hydrogenation. Nat. Chem. 2, 832–837 (2010).
Fors, B. P. & Buchwald, S. L. A multiligand based Pd catalyst for C-N cross-coupling reactions. J. Am. Chem. Soc. 132, 15914–15917 (2010).
Gao, S., Liu, Y. & Ma, S. CuCl-catalyzed aerobic oxidation of 2,3-allenols to 1,2-allenic ketones with 1:1 combination of phenanthroline and bipyridine as ligands. Beilstein J. Org. Chem. 7, 396–403 (2011).
Sheng, J., Ni, H. Q., Liu, G., Li, Y. & Wang, X. S. Combinatorial nickel-catalyzed monofluoroalkylation of aryl boronic acids with unactivated fluoroalkyl iodides. Org. Lett. 19, 4480–4483 (2017).
Liu, W. et al. Iron-catalyzed intramolecular amination of aliphatic C–H bonds of sulfamate esters with high reactivity and chemoselectivity. Org. Lett. 21, 2673–2678 (2019).
Li, W. et al. Design of Ru(II)-NHC-diamine precatalysts directed by ligand cooperation: applications and mechanistic investigations for asymmetric hydrogenation. J. Am. Chem. Soc. 142, 7100–7107 (2020).
Chen, J., Guo, J. & Lu, Z. Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis. Chin. J. Chem. 36, 1075–1109 (2018).
Chen, J., Shen, X. & Lu, Z. Cobalt-catalyzed Markovnikov-type selective hydroboration of terminal alkynes. Angew. Chem. Int. Ed. 60, 690–694 (2021).
Shen, X. et al. Ligand-promoted cobalt-catalyzed radical hydroamination of alkenes. Nat. Commun. 11, 783 (2020).
Cheng, Z. et al. Regio-contollable cobalt-catalyzed sequential hydrisilylation/hydroborylation of arylacetylenes. Angew. Chem. Int. Ed. 60, 22454–22460 (2021).
Cheng, B., Lu, P., Zhang, H., Cheng, X. & Lu, Z. Highly enantioselective cobalt-catalyzed hydrosilylation of alkenes. J. Am. Chem. Soc. 139, 9439–9442 (2017).
Wu, C., Teo, W. J. & Ge, S. Cobalt-catalyzed (E)-selective anti-Markovnikov hydrosilylation of terminal alkynes. ACS Catal. 8, 5896–5900 (2018).
Yang, Y. et al. Iron-catalyzed intermolecular 1,2-difunctionalization of styrenes and conjugated alkenes with silanes and nucleophiles. Angew. Chem. Int. Ed. 56, 7916–7919 (2017).
Fleming, I., Frackenpohl, J. & Ila, H. Cleavage of sulfonamides with phenyldimethylsilyllithium. J. Chem. Soc., Perkin Trans. 1, 1229–1236 (1998).
Takeda, Y., Shibuta, K., Aoki, S., Tohnai, N. & Minakata, S. Catalyst-controlled regiodivergent ring-opening C(sp3)-Si bond-forming reactions of 2-arylaziridines with silylborane enabled by synergistic palladium/copper dual catalysis. Chem. Sci. 10, 8642–8647 (2019).
Yi, H. & Oestreich, M. Regiodivergent and stereospecific aziridine opening by copper-catalyzed addition of silicon Grignard reagents. Chem. Eur. J. 25, 6505–6507 (2019).
Li, W. et al. New electrophilic addition of α-diazoesters with ketones for enantioselective C–N bond formation. J. Am. Chem. Soc. 133, 15268–15271 (2011).
Zheng, J., Qi, J. & Cui, S. Fe-catalyzed olefin hydroamination with diazo compounds for hydrazone synthesis. Org. Lett. 18, 128–131 (2016).
Waser, J., Gaspar, B., Nambu, H. & Carreira, E. M. Hydrazines and azides via the metal-catalyzed hydrohydrazination and hydroazidation of olefins. J. Am. Chem. Soc. 128, 11693–11712 (2006).
Gui, J. et al. Practical olefin hydroamination with nitroarenes. Science 348, 886–891 (2015).
Green, S. A. et al. The high chemofidelity of metal-catalyzed hydrogen atom transfer. Acc. Chem. Res. 51, 2628–2640 (2018).
Gu, Q.-S., Li, Z.-L. & Liu, X.-Y. Copper(I)-catalyzed asymmetric reactions involving radicals. Acc. Chem. Res. 53, 170–181 (2020).
Acknowledgements
Financial supports were provided by National Key R&D Program of China (2021YFF0701603 and 2021YFA1500200), NSFC (21922107 and 21772171), and Zhejiang Provincial Natural Science Foundation of China (LR19B020001), Center of Chemistry for Frontier Technologies.
Author information
Authors and Affiliations
Contributions
Z.L. proposed this project. Y.S., J.G. and X.S. provided initial results. Y.S. performed the experiments. Z.L. and Y.S. prepared the manuscript. Y.S. prepared the Supplementary Information.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liqun Jin and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Y., Guo, J., Shen, X. et al. Ligand relay catalysis for cobalt-catalyzed sequential hydrosilylation and hydrohydrazidation of terminal alkynes. Nat Commun 13, 650 (2022). https://doi.org/10.1038/s41467-022-28285-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28285-w
This article is cited by
-
Dual ligands relay-promoted transformation of unstrained ketones to polyfluoroarenes and nitriles
Science China Chemistry (2023)
-
Catalytic asymmetric silicon-carbon bond-forming transformations based on Si-H functionalization
Science China Chemistry (2023)
-
Cobalt-catalyzed branched selective hydroallylation of terminal alkynes
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
