
Abstract
Platinum group elements are invaluable tracers for planetary accretion and differentiation and the formation of PGE sulfide deposits. Previous laboratory determinations of the sulfide liquid–basaltic melt partition coefficients of PGE (\({D}_{PGE}^{SL/SM}\)) yielded values of 102–109, and values of >105 have been accepted by the geochemical and cosmochemical society. Here we perform measurements of \({D}_{Pt,\,Pd}^{SL/SM}\) at 1 GPa and 1,400 °C, and find that \({D}_{Pt,\,Pd}^{SL/SM}\) increase respectively from 3,500 to 3.5 × 105 and 1,800 to 7 × 105, as the Pt and Pd concentration in the sulfide liquid increases from 60 to 21,000 ppm and 26 to 7,000 ppm, respectively, implying non-Henrian behavior of the Pt and Pd partitioning. The use of \({D}_{Pt,\,Pd}^{SL/SM}\) values of 2,000–6,000 well explains the Pt and Pd systematics of Earth’s mantle peridotites and mid-ocean ridge basalts. Our findings suggest that the behavior of PGE needs to be reevaluated when using them to trace planetary magmatic processes.
Similar content being viewed by others


Introduction
Platinum group elements (PGE: Os, Ir, Ru, Rh, Pt, and Pd) are powerful tracers for planetary accretion and core–mantle–crust differentiation1,2,3,4,5, as well as the formation of magmatic PGE sulfide deposits in Earth’s crust4,6,7. In planetary magmatic systems, PGE are primarily controlled by their partitioning into sulfide liquid. However, experimental determinations on the partition coefficients of PGE between sulfide liquid and silicate (basaltic) melt (\({D}_{PGE}^{SL/SM}\)) have yielded widely discrepant results, with \({D}_{PGE}^{SL/SM}\) values ranging from 102 to 109 (refs. 4,8,9,10,11,12,13). Studies in the 1990s determined \({D}_{PGE}^{SL/SM}\) by performing experiments at 1 bar10,11,12. Using a bulk analytical technique, i.e., neutron activation analysis, the obtained \({D}_{PGE}^{SL/SM}\) for a given element usually vary in the order of 102–106, which was late ascribed to inefficient separation of sulfide and silicate phases and/or the presence of PGE-sulfide nuggets in the silicate melt8. Experimental studies in the 2000s determined \({D}_{PGE}^{SL/SM}\) by measuring the solubility of PGE in sulfide liquid and silicate melt separately, which usually yielded \({D}_{PGE}^{SL/SM}\) values in the order of 105–109 (refs. 14,15,16). However, the disadvantage of this “indirect” method is that the formation of PGE-sulfide species in the silicate melt was not considered, which may result in artificially high \({D}_{PGE}^{SL/SM}\) (refs. 4,8,17). Most recent experimental studies determined \({D}_{PGE}^{SL/SM}\) using the in-situ micro-analytical technique of laser ablation–inductively coupled plasma–mass spectroscopy (LA–ICP–MS), such that the contribution of PGE-sulfide nuggets in the silicate melt can be identified and filtered4,18. The most precise \({D}_{PGE}^{SL/SM}\) values obtained at 1200 °C and 1 bar by Mungall and Brenan are larger than 105 (ref. 4), which have been widely accepted and used by the geochemical and cosmochemical society since their publication in 2014 (refs. 7,8,19,20). However, in ref. 4, a few wt.% of PGE was doped in the sulfide liquid to facilitate the analysis of PGE in the silicate melt. Whether \({D}_{PGE}^{SL/SM}\) obey Henry’s law when the sulfide liquid has such high PGE concentration has never been investigated. Using the bulk analytical technique, M.E Fleet, J.H Crocket and coauthors first recognized the dependence of \({D}_{PGE}^{SL/SM}\) on the PGE concentration in the sulfide liquid in the 1990s (refs. 11,12). However, this has not been further investigated since then, although LA–ICP–MS has been one of the most powerful in-situ micro-analytical techniques from the 2000s. Here we present experimental measurements of \({D}_{Pt,\,Pd}^{SL/SM}\) as a function of the Pt and Pd concentration in the sulfide liquid (\({C}_{Pt,\,Pd}^{SL}\)) using the in-situ analytical technique of LA–ICP–MS. We find that \({D}_{Pt,\,Pd}^{SL/SM}\) increase with increasing \({C}_{Pt,\,Pd}^{SL}\), and the use of \({D}_{Pt,\,Pd}^{SL/SM}\) values of < 104, which correspond to the \({C}_{Pt,\,Pd}^{SL}\) in Earth’s magmatic systems, can well explain the observed Pt and Pd systematics of Earth’s mantle peridotites and mid-ocean ridge basalts (MORB). These findings suggest that non-Henrian partitioning behavior of PGE should be considered when applying laboratory-determined \({D}_{PGE}^{SL/SM}\,\)in planetary magmatic systems.
Results and discussion
Pt and Pd partitioning between sulfide liquid and basaltic melt
Two sets of experiments were performed at 1 GPa and 1400 °C using a piston cylinder apparatus (Supplementary Data 1 and Methods). In Set-1 of 18 forward experiments, ~60–70 wt.% basalt, and ~30–40 wt.% sulfide (FeS) doped with 60–21,000 ppm Pt or 30–7000 ppm Pd, were loaded in graphite capsules. In Set-2 of four reversal experiments, ~200–7000 ppm Pd or 1000 ppm Pt was physically mixed with the basalt and then, ~60–70 wt.% basalt together with ~30–40 wt.% sulfide was loaded in graphite capsules. The formation of PGE nuggets in the silicate melt is related to the oxidation state of the starting silicate21. In order to reduce the formation of Pt or Pd nuggets in our basaltic melt, the starting basalt was prepared to contain only FeO without Fe2O3. The experimental durations ranged from 24 to 96 h. The experimental oxygen fugacity was close to the C–CO2 buffer22,23,24. All experiments produced coexisting basaltic melt and sulfide liquid. The basaltic melt was quenched into glass, and the sulfide liquid was qunched into large pyrrhotite crystals and minor Fe-Pt or Fe-Pd alloys, as shown in Fig. 1 and Supplementary Fig. 1. In the quenched sulfide liquid, the proportion of the Fe-Pt or Fe-Pd alloys increases with the mass of Pt or Pd doped in the experiments, and they occur alone the bouduary of pyrrhotite crystals. We interpret the Fe-Pt or Fe-Pd alloys as resulting from exsolution of sulfide liquid during quench, as interpreted previouly4. We found very small sulfide dots (< 0.1 µm) homogeneously distributed in the basaltic glass from all experiments, which we interpret as resulting from quench because of the significant drop in sulfide solubility (Fig. 1 and Supplementary Fig. 1). In a few experiments, we also found that Pt- or Pd-sulfide droplets (1–5 µm) dispersed as nuggets/inclusions in part of the basaltic glass, which we interpret as resulting from ineffective segregation from the silicate melt during the run (Supplementary Fig. 1e, f). The basaltic glasses that do not contain macroscopically visible sulfide nuggets (1–5 µm) are our analytical targets.

a BSE image of run Z14, showing coexisting sulfide liquid and silicate melt in a graphite capsule. b Closer view of the quenched sulfide liquid with 5300 ppm Pt from run Z14, which includes large pyrrhotite crystals and exsolved Fe-Pt alloys at the boundary of pyrrhotite crystals. c Closer view of the quenched silicate melt in (a), showing homogeneously distributed small sulfide dots (< 0.1 µm) produced during quench. d BSE image of one reversal experiment (run Z28) for Pd partitioning, showing coexisting sulfide liquid and silicate melt. e Closer view of the quenched silicate melt in (d). f Closer view of the quenched sulfide liquid with 163 ppm Pd in (d).
Major and trace element compositions of the quenched basaltic melt and sulfide liquid were measured using electron probe microanalyzer (EPMA) and LA–ICP–MS with large beam sizes (Methods and Supplementary Data 2, 3). The time-resolved LA–ICP–MS signals of Pt and Pd are constant for most of the analyses, confirming the absence of Pt- or Pd-sulfide nuggets in the analyzed basaltic melt from both forward and reversal experiments (Supplementary Fig. 2). Any LA–ICP–MS analyses with Pt or Pd signals contaminated by sulfide nuggets in the silicate melt, as shown in Supplementary Fig. 2g, were discarded. The analytical results show that \({C}_{Pt,\,Pd}^{SL}\) equals 60–21,000 ppm and 26–7000 ppm (parts per million by weight), respectively, and the \({C}_{Pt,\,Pd}^{SL}\) values obtained using EPMA and LA–ICP–MS agree within 20% relative (Supplementary Fig. 3). \({C}_{Pt,\,Pd}^{SM}\) equals 10–160 ppb and 10–120 ppb (parts per billion by weight), respectively, which increases exponentially with increasing \({C}_{Pt,\,Pd}^{SL}\) (Fig. 2a, b and Supplementary Data 1). Nernst partition coefficients \({D}_{Pt,\,Pd}^{SL/SM}\), as calculated from the weight proportions of Pt and Pd in the sulfide liquid (\({C}_{Pt,\,Pd}^{SL}\)) and basaltic melt (\({C}_{Pt,\,Pd}^{SM}\)), range from 1800 to 7 × 105, which increase linearly with increasing the Pt and Pd concentration in the sulfide liquid (Fig. 2c, d and Supplementary Data 1). No correlation was observed between \({D}_{Pt,\,Pd}^{SL/SM}\), the FeO content in the silicate melt, or the atomic metal/S ratio in the sulfide liquid (Supplementary Figs. 4 and 5). At a given Pt and Pd concentration in the sulfide liquid, \({C}_{Pt,\,Pd}^{SL}\) and \({D}_{Pt,\,Pd}^{SL/SM}\) do not vary as a function of the experimental duration, and the forward and reversal experiments yielded consistent \({C}_{Pt,\,Pd}^{SL}\) and \({D}_{Pt,\,Pd}^{SL/SM}\) (Fig. 2). These observations suggest the approach of equilibrium partitioning of Pt and Pd. Previous \({C}_{Pt,\,Pd}^{SM}\) and \({D}_{Pt,\,Pd}^{SL/SM}\), as obtained by Mungall and Brenan using LA–ICP–MS (ref. 4), are systematically consistent with the present results (Fig. 2).

a, b The Pt and Pd concentration in the silicate melt increases exponentially with increasing the Pt and Pd concentration in the sulfide liquid. The dashed gray lines show the co-variation of the Pt and Pd concentration in the silicate melt and sulfide liquid if the partitioning of Pt and Pd between sulfide liquid and silicate melt obeys Henry’s law. c, d \({D}_{Pt,\,Pd}^{SL/SM}\) increase linearly with increasing the Pt and Pd concentration in the sulfide liquid. The literature data are taken from M&B 2014 (ref. 4), Fleet 1996 (ref. 10), Fleet 1999 (ref. 12), Stone 1990 (ref. 9), Bezmen 1994 (ref. 13), and Crocket 1997 (ref. 11). Note that the literature data of M&B 2014, obtained using in-situ micro-analytical technique of LA–ICP–MS, are systematically consistent with the present results. The other literature data (filled gray symbols) were obtained by neutron activation analysis of bulk samples, and an effective separation of the silicate and sulfide phases was questioned8. Source data are provided in Supplementary Data 1.
Non-Henrian behavior of \({D}_{Pt,\,Pd}^{SL/SM}\)
Both the non-linear increase of \({C}_{Pt,\,Pd}^{SM}\) and the linear increase of \({D}_{Pt,\,Pd}^{SL/SM}\) with increasing \({C}_{Pt,\,Pd}^{SL}\) (Fig. 2) indicate that the partitioning of Pt and Pd between sulfide liquid and basaltic melt follows a non-Henrian behavior. Assuming a chalcophile element M dissolves as M-oxide in the silicate melt, the partitioning of M between sulfide liquid and silicate melt can be described by the following exchange reaction:
where n denotes the valence state of M. The equilibrium constant (k) of Eq. (1) can be written as:
and
In Eqs. (2) and (3) \(\alpha\), \(x\), and \(\gamma\) denote activity, mole fraction, and activity coefficient, respectively, such as \({\alpha }_{M{S}_{n/2}}^{SL}\) denoting the activity of \(M{S}_{n/2}\) in the sulfide liquid. Rearranging Eq. (3) would yield:
Since \({D}_{M}^{SL/SM}\) equals \(C\cdot \frac{{x}_{M{S}_{n/2}}^{SL\,}}{{x}_{M{O}_{n/2}}^{SM\,}}\), where \(C\) is a constant, the following equation can be obtained:
All our experiments were performed at 1 GPa and 1400 °C with very similar silicate melt compositions (Supplementary Data 2); therefore, both \({k}_{{{{{{\rm{Eq}}}}}}.(1)}\) and \({({x}_{FeO}^{SM})}^{n/2}\cdot {({\gamma }_{FeO}^{SM})}^{n/2}\) are constant in our experiments. The sulfide liquid contains 60–21,000 ppm Pt or 26–7000 ppm Pd. We therefore assume that \({({x}_{FeS}^{SL})}^{n/2}\cdot {({\gamma }_{FeS}^{SL})}^{n/2}\) is also constant in our experiments. Accordingly, the observed increase of \({D}_{Pt,\,Pd}^{SL/SM}\) with increasing \({C}_{Pt,\,Pd}^{SL}\) can be ascribed to the decrease of the\(\,{\gamma }_{M{S}_{n/2}}^{SL\,}\) for Pt and Pd with increasing \({C}_{Pt,\,Pd}^{SL}\), or the increase of the \({\gamma }_{M{O}_{n/2}}^{SM\,}\)for Pt and Pd with increasing \({C}_{Pt,\,Pd}^{SM}\). However, when \({C}_{Pt,\,Pd}^{SL}\) increases from 60 to 6000 ppm and from 26 to 2000 ppm, respectively, \({C}_{Pt,\,Pd}^{SM}\) increases only by a factor of ~2 (Fig. 2a, b), but \({D}_{Pt,\,Pd}^{SL/SM}\) increase by a factor of ~30–100 (Fig. 2c, d). Therefore, the \({\gamma }_{M{O}_{n/2}}^{SM\,}\)for Pt and Pd may also be constant, and the increase of \({D}_{Pt,\,Pd}^{SL/SM}\) with increasing \({C}_{Pt,\,Pd}^{SL}\) most likely reflects that the\(\,{\gamma }_{M{S}_{n/2}}^{SL\,}\,\)for Pt and Pd decreases with increasing \({C}_{Pt,\,Pd}^{SL}\).
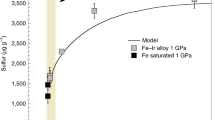
We take the advantage of available experimental results16 on coexisting Fe-Pt alloy and Fe-S-Pt sulfide liquid at 1200–1300 °C and 1 bar to thermodynamically understand the variation of the\(\,{\gamma }_{M{S}_{n/2}}^{SL\,}\,\)for Pt as a function of \({C}_{Pt}^{SL}\). Since the activity of Pt in the Fe-Pt alloy can be calculated thermodynamically16, we can calculate \(\frac{1}{k}\cdot {\gamma }_{Pt{S}_{n/2}}^{SL}\) (\(k\) is a constant as shown below) in the Fe-S-Pt sulfide liquid using the following approach. The partitioning of Pt between Fe-S-Pt sulfide liquid and Fe-Pt alloy can be written as:
where n is the valence state of Pt in the Fe-S-Pt sulfide liquid. The equilibrium constant of Eq. (6) can be written as:
and
which can be further arranged as:
In Eqs. (7)–(9)\(\,{\alpha }_{Pt}^{alloy}\,\)and \({\alpha }_{Pt{S}_{n/2}}^{SL}\) are the activity of Pt and \(Pt{S}_{n/2}\) in the Fe-Pt alloy and the Fe-S-Pt sulfide liquid, respectively;\(\,{\gamma }_{Pt{S}_{n/2}}^{SL}\)and \({x}_{Pt{S}_{n/2}}^{SL}\)are the activity coefficient and mole fraction of \(Pt{S}_{n/2}\) in the Fe-S-Pt sulfide liquid, respectively; and \(f\,{S}_{2}\) is sulfur fugacity. Since Pt is present mainly as Pt2+ in sulfide liquid at geologically relevant redox conditions16, Eq. (9) can be written as:
Using the experimental results obtained at the same temperatures and similar \(f{S}_{2}\) values in Fonseca et al.16, \(\frac{1}{{k}_{{{{{{\rm{Eq}}}}}}.(6)}}\cdot {\gamma }_{PtS}^{SL}\) was calculated as a function of the Pt concentration in the Fe-S-Pt sulfide liquid (Supplementary Data 4). Figure 3 shows that \(\frac{1}{{k}_{{{{{{\rm{Eq}}}}}}.(6)}}\cdot {\gamma }_{PtS}^{SL}\) decreases with increasing the Pt concentration in the Fe-S-Pt sulfide liquid. Therefore, \({\gamma }_{PtS}^{SL}\) must also decrease with increasing the Pt concentration in the Fe-S-Pt sulfide liquid at a given temperature, because \({k}_{{{{{{\rm{Eq}}}}}}.(6)}\) is a constant at a given temperature. These results thus support our inference that the\(\,{\gamma }_{M{S}_{n/2}}^{SL\,}\,\)for Pt and Pd decreases with increasing \({C}_{Pt,\,Pd}^{SL}\), and our explanations for the non-Henrian behavior of \({D}_{Pt,\,Pd}^{SL/SM}\). Here it should be noted that the dissolution of a fraction of Pt and/or Pd as sulfide species in the silicate melt4,17, which is not considered in Eqs. (1–5), does not change our explanations for the non-Henrian behavior of \({D}_{Pt,\,Pd}^{SL/SM}\).

At the same temperature and pressure and similar fS2, \(\frac{1}{k}\cdot {\gamma }_{PtS}^{SL}\) decreases with increasing the Pt concentration in the Fe-S-Pt sulfide liquid. Note that k is the equilibrium constant of Eq. (6) in the main text. The data used for these calculations were taken from Fonseca et al.16. Source data are provided in Supplementary Data 4.
Our experimental results and thermodynamic considerations conclusively demonstrate that \({D}_{Pt,\,Pd}^{SL/SM}\) follow a non-Henrian behavior. Therefore, the Pt and Pd concentration must be considered when \({D}_{Pt,\,Pd}^{SL/SM}\) are applied in a magmatic system, and the partitioning of Pt and Pd in the sulfide liquid is favored as the Pt and Pd concentration increases in the magma. The Pt and Pd concentration in sulfides of terrestrial mantle rocks and basalts is usually below 100 ppm25,26,27, so the \({D}_{Pt,\,Pd}^{SL/SM}\) values of < 104, rather than the values of >105 that have been widely accpeted7,8,20, should be used in terrestrial magmatic systems. We below show that the use of \({D}_{Pt,\,Pd}^{SL/SM}\) values of < 104 well explains the observed Pt and Pd systematics of Earth’s mantle peridotites and MORB.
Model the behavior of Pt and Pd during terrestrial magmatic process
The newly determined \({D}_{Pt,\,Pd}^{SL/SM}\) as a function of \({C}_{Pt,\,Pd}^{SL}\) allow us to reconsider the behavior of Pt and Pd during partial melting of Earth’s mantle and MORB differentiation. A large dataset of PGE in Earth’s mantle peridotites has been established by previous studies28,29, and considerable variations of the PGE content in Earth’s mantle peridotites, as shown in Fig. 4, were explained as resulting from petrogenetic processes such as partial melting and the percolation of mantle fluids/melts28,30. To model the Pd content and Pt/Pd ratio in Earth’s mantle peridotites (Fig. 4 and Supplementary Data 5), a near-fractional melting model31 was used. Whole rock Al2O3 content was taken as a melt depletion indicator, and the melt extraction model parameters for Al2O3 were taken from ref. 32. During paritial melting, all sulfide was assumed to be sulfide liquid33. In addition, the following conditions were used: (i) the starting silicate composition is Earth’s primitive mantle34,35; (ii) the S concentration at sulfide saturation is 1000 ppm in the generated basaltic melt36; (iii) Earth’s primitive mantle contains 150–300 ppm S as sulfide (refs. 37,38); (σ) the \({D}_{Pt,\,Pd}^{SL/SM}\) during partial melting of Earth’s primitive mantle are 4000 and 2000, respectively; (τ) Pd is completely incompatible in silicate and oxide minerals5; (υ) the partition coefficients of Pt between mantle minerals and silicate melt are 0.009 for olivine, 0.8 for clinopyroxene, 2.2 for orthopyroxene, and 0.22 for spinel39,40,41,42. Figure 4 shows the modeled Pd content and Pd/Pt ratio in Earth’s mantle peridotites, which will cover the observed values, particularly if the variations of the S, Pt, and Pd content in Earth’s primitive mantle are considered. Accordingly, partial melting of Earth’s primitive mantle with \({D}_{Pt,\,Pd}^{SL/SM}\) values not exceeding 104 can largely explain the observed Pt and Pd systematics of Earth’s mantle peridotites.

a, b Our model calculations were performed by partial melting of Earth’s primitive mantle (PM), with Al2O3 content (wt.%) in the residual mantle as a proxy for the degree of partial melting. The used \({D}_{Pt,\,Pd}^{SL/SM}\) values are 4000 and 2000, respectively. The black ellipses refer to the Pd content and Pd/Pt ratio of the PM (refs. 34, 35). Mantle peridotite Pt, Pd, and Al2O3 data were taken from refs. 28, 29 and references therein. This figure illustrates that the Pd content and Pd/Pt ratio in Earth’s mantle peridotites can be largely explained by PM partial melting with \({D}_{Pt,\,Pd}^{SL/SM}\) values of 4000 and 2000, respectively. Source data are provided in Supplementary Data 5.
We have also modeled the behavior of Pd and Pt during MORB differentiation (Fig. 5 and Supplementary Data 6). The Cu/Pt and Cu/Pd ratios are used as effective tools to identify the timing of sulfide liquid saturation during magmatic differentiation; the Cu/Pt and Cu/Pd ratios in magmas will increase if sulfide liquid saturation occurs26,39,43. To model the evolution trends of Cu/Pt and Cu/Pd during MORB differentiation, we used a fractional crystallization model and assumed that MORB crystallizes isobarically at 0.1 GPa. The relative mass fractions of crystallized minerals (olivine, clinopyroxene, and plagioclase), and the major element composition of the basaltic melt, during MORB differentiation were determined using the Petrolog3 program44. We used the SCSS model of Smythe et al.45 to calculate the S concentration at sulfide saturation in the basaltic melt and the mass of sulfide precipitated, with an assumption that S is removed as sulfide liquid which contains 46 wt.% Fe, 10 wt.% Ni, 7 wt.% Cu, 2 wt.% O, and 35 wt.% S (ref. 26). Three different groups of Cu, Pt, and Pd content (Cu = 110 ppm, Pt = 3.8 ppb, and Pd = 6.5 ppb; Cu = 110 ppm, Pt = 1.2 ppb, and Pd = 2.1 ppb; Cu = 110 ppm, Pt = 0.25 ppb, and Pd = 0.40 ppb) were used for the parental MORB to differentiate, considering that the parental MORB could have very different Pt and Pd contents5,43. The used \({D}_{Pt,\,Pd}^{SL/SM}\) values ranged from 2000 to 6000, and the partition coefficients of Pt between minerals and silicate melt are 0.009 for olivine, 0.8 for clinopyroxene, and 0.3 for plagioclase39,46. The partition coefficients of Cu between sulfide liquid and basaltic melt were fixed at 800 (refs. 23,24,47). The partition coefficients of Cu between minerals and basaltic melt are 0.05 for olivine, 0.049 for clinopyroxene, and 0.115 for plagioclase48,49. Figure 5 shows that during MORB differentiation, the Cu/Pt and Cu/Pd ratios increase with decreasing the MgO content (wt.%) in the silicate melt, indicating the saturation of sulfide liquid during MORB differentiation26,50,51. Figure 5 also shows that the modeled Cu/Pt and Cu/Pd ratios, using \({D}_{Pt,\,Pd}^{SL/SM}\) values of 2000–6000, well cover the observed Cu/Pt and Cu/Pd ratios in the differentiated MORB. Therefore, the observed Pt and Pd systematics of MORB can be explained by the use of \({D}_{Pt,\,Pd}^{SL/SM}\) values of 2000–6000 for MORB differentiation.

a, b The \({D}_{Pt,\,Pd}^{SL/SM}\) values of 2000–6000 were applied in the modeled differentiation trends, and the observed Cu/Pt and Cu/Pd ratios in MORB can be largely explained. Note that the solid and dashed differentiation lines were modeled using different initial Pt and Pd contents (see main text for details). The MORB Cu, Pt, and Pd data were taken from refs. 5, 43. SKR Southern Kolbeinsey Ridge, EPR East Pacific Rise; SWIR (A, B): Southwest Indian Ridge Zone A and B; SWIR(C): Southwest Indian Ridge Zone C, HD: HREE-depleted MORBs. Source data are provided in Supplementary Data 6.
Implications for the geochemical behavior of PGE in planetary magmas
The modeling results shown in Figs. 4 and 5 demonstrate that the use of \({D}_{Pt,\,Pd}^{SL/SM}\) values of 2000–6000 well explains the observed Pd and Pt systematics of Earth’s mantle peridotites and MORB, with \({D}_{Pt,\,Pd}^{SL/SM}\) values of >105 not required. The use of \({D}_{PGE}^{SL/SM}\) values of >105 readily explains the extreme enrichments of PGE in a volumetrically small sulfide fraction and the formation of world-class PGE deposits, such as the Stillwater and Bushveld Complex-hosted reef deposits6,7,52. However, our findings, based on both experimental results (Fig. 2) and modeling results (Figs. 4 and 5), suggest that the application of \({D}_{Pt,\,Pd}^{SL/SM}\) values of >105 in terrestrial magmas is inappropriate, and sulfide immiscibility alone cannot explain the observed high Pt and Pd abundance in the Stillwater and Bushveld Complex-hosted reef deposits, which have PGE enrichment factors of 105–106 (refs. 6,7,19). The other mechanisms, such as direct crystallization of PGE-bearing minerals from magmas53,54, and/or secondary enrichment of PGE in immiscible sulfide liquid55, may be necessary conditions for the formation of world-class magmatic PGE sulfide deposits.
Our experimental and modeling results also have significant implications for planetary accretion. The \({D}_{Pt,\,Pd}^{SL/SM}\) values of < 104 when \({C}_{Pt,\,Pd}^{SL}\) < 100 ppm suggest that the capacity of segregating sulfide liquid (Hadean matte) in extracting Pt and Pd from Earth’s solidifying magma ocean is not as strong as previously thought3,56. In this context, the models proposed for Earth’s accretion and core-formation based on \({D}_{Pt,\,Pd}^{SL/SM}\) values of >105 (refs. 3,56) need to be revised to explain the depleted but near-chondritic relative PGE abundance in the bulk silicate Earth. The non-Henrian behavior of \({D}_{Pt,\,Pd}^{SL/SM}\) may also suggest that the partitioning of PGE between metallic and silicate melts does not obey Henry’s law either. A dependence of the partition coefficients of Pd between metallic and silicate melts (160–3 × 105) on the Pd concentration in the metallic melt (3000–89,000 ppm) has indeed been observed by Wheeler et al.57. Consequently, the distribution of PGE between planetary core and mantle may be controlled by not only temperature and pressure1,2,3 but also the PGE concentration in the system. We speculate that it may be possible that the depleted but near-chondritic relative PGE abundance in planetary silicate mantles58 can be explained by core-formation alone, i.e., a late veneer not being required, if all PGE follow a non-Henrian partitioning behavior between metallic and silicate melts. This speculation deserves future investigations.
Our study conclusively demonstrates that the partitioning of PGE between sulfide liquid and silicate melt is more complex than previously thought. Future laboratory studies must be performed to fully understand the partitioning behavior of PGE between metallic melt, sulfide liquid, and silicate melt, with PGE concentrations close to the natural systems. This will be critically important for tracing many planetary processes, such as the formation of magmatic PGE sulfide deposits in Earth’s crust, the origin of the near-chondritic relative PGE abundance in planetary silicate mantles, and the accretion and differentiation of terrestrial planets.
Methods
Starting materials
Starting materials loaded in each sample capsule included ~60–70 wt.% of a synthetic silicate and ~30–40 wt.% sulfide (FeS). The synthetic silicate with major element compositions similar to those of average MORB were prepared from analytical grade oxides and carbonates. To minimize absorbed water, SiO2, TiO2, Al2O3, Cr2O3 and MgO powder were each heated overnight at 1000 °C, MnO2 at 400 °C, CaCO3 at 200 °C, and Na2CO3 and K2CO3 at 110 °C. After drying, all the oxides and carbonates were mixed and ground in acetone, then dried at room temperature overnight. Well-mixed oxide and carbonate powder was sintered in a high purity alumina crucible at 1000 °C overnight to decarbonate. The decarbonated powder was finally mixed with FeO powder and ground in acetone, so as to limit the introduction of ferric iron into subsequent partitioning experiment, which was proved to be an efficient way to prevent the formation of PGE nuggets in silicate melt21. In the forward experiments, ~20–20,000 ppm Pt or Pd was doped in the sulfide powder, which was then sintered in a graphite capsule at 1100 °C and 1.5 GPa for 10 h. In the reversal experiments, ~200–7000 ppm Pd or 1000 ppm Pt was physically mixed with the silicate powder in acetone in an agate mortar, which was then dried at room temperature. All dried silicate and sulfide materials were stored in a vacuumed oven at 110 °C for > 24 h before loading into graphite capsules for high-pressure experiments.
High temperature and pressure experiments
All experiments were conducted at 1.0 GPa and 1400 °C in an end-loaded solid media piston cylinder apparatus, using 3/4-inch diameter talc-Pyrex assemblies with graphite heaters. Pressure was calibrated against the quartz-coesite and kyanite-sillimanite transitions, and a friction correction of 18% was applied to the nominal pressure. The total pressure uncertainty is less than 0.1 GPa. The experimental temperatures were monitored by C-type (W95Re05-W74Re26) thermocouples, and temperature was controlled to ±2 °C and was accurate to ±10 °C. For each experiment, in order to reduce the porosity of the graphite capsules and thus prevent leakage of sulfide liquid, the sample was first heated to 850 °C and held for 2–4 h and then raised to the target temperature. The experimental durations ranged from 24 to 96 h. After quenching by turning off electric power to the graphite heaters, the recovered capsules were sectioned longitudinally into two halves, mounted in epoxy, and polished for EPMA and LA–ICP–MS analyses.
Electron microprobe analyses of major elements
Major elements compositions of the quenchend sulfide liquid and silicate melt were measured with a JEOL JXA-8230 microprobe. The analyses were performed in wavelength-dispersive mode, and a PAP matrix correction was applied to the raw data. For the measurements of major elements in the silicate melt, a defocused beam of 20 µm, 15 kV accelerating voltage, and 10 nA beam current were used for both the standardizations and sample measurements. Natural and synthetic standards were used to calibrate the instrument, and the used standards were andradite (Si), MnTiO3 (Ti), spinel (Al), metal Fe (Fe), MnTiO3 (Mn), forsterite (Mg), wollastonite (Ca), albite (Na), orthoclase (K), Gallium phosphite (P), and metal Ni (Ni). The peak counting time was 20 s except for Na and K, which were measured for 10 s. The quenched sulfide liquid was analyzed with 20 kV acceleration voltage and 20 nA beam current. Fe and S were calibrated on a synthetic pyrrhotite with a well-known Fe:S ratio, Ni, Co, Cu, Pd and Pt were calibrated on pure metals, and O was calibrated on magnetite. A defocused beam of 30 µm diameter was used for all standardizations and sample measurements, as this was sufficient to average small-scale quench-phase inhomogeneity. Sulfur in the quenched silicate melts was analyzed with 50 nA beam current and 60 s peak counting time using the synthetic pyrrhotite standard.
LA–ICP–MS analyses of Pt and Pd
Major and trace elements of the quenched silicate melt and sulfide liquid were measured using a laser–ablation ICP–MS. These analyses were carried out on an Agilent 7900 Quadrupole ICP-MS coupled to a Photon Machines Analyte HE 193-nm ArF Excimer Laser Ablation system. The quenched silicate melt was analyzed with 10 Hz, 80 mJ, and laser beam sizes of 90–110 μm, whereas the quenched sulfide liquid was analyzed with 7 Hz, 70 mJ, and laser beam sizes of 50–60 μm. A typical time-resolved analysis involved ~20 s of background acquisition, followed by laser ablation for 40 s. The sample chamber was flushed with He at a rate of 0.4 L/min, to which 5 ml/min H2 was added on the way to the ICP–MS. The isotopes 23Na, 24Mg, 27Al, 29Si, 32P, 39K, 42Ca, 49Ti, 52Cr, 55Mn, 57Fe, 59Co, 62Ni, 65Cu, 75As, 121Sb, and 209Bi were measured with a dwell time of 10 ms. To precisely measure 105Pd and 195Pt in the silicate melt, 105Pd and 195Pt were measured with a dwell time of 50 ms. NIST SRM 610 glass was used as external standard for all analyses, which contains 3.15 ppm Pt and 1.05 ppm Pd59, whereas Si and Fe determined by electron microprobe were used as the internal standard for the silicate melt and sulfide, respectively. Mass-1 sulfide standard was used to check the accuracy of the LA-ICP-MS analyses. Repeated analyses of the Mass-1 sulfide standard after analyzing each ten sample spots yielded Pt and Pd concentrations consistent with the certified values within 6–17% relative, which demonstrates the validity of using NIST SRM 610 glass as the standard for measuring the Pt and Pd concentration in the quenched sulfide liquid.
As done in previous studies60,61, the detection limits (DL) for measuring the Pt and Pd concentration (\({C}_{Pt,\,Pd}^{SM}\)) in the silicate melt were calculated as three times the standard deviation of three replicate measurements of a sample that contains zero analyte62. Each analytical point has a own DL because the laser beam size and the interval of signal used for integration may change, althouth a dwell time of 50 ms was always used. The calculated minimum to maximum DLmin-max values for the analyses of each sample were given in Supplementary Data 1, which are 3 (min)–7 ppb (max) for Pt and 3 (min)–8 ppb (max) for Pd. Our measured \({C}_{Pt,\,Pd}^{SM}\) are significantly higher than DLmin-max. Most samples have \({C}_{Pt,\,Pd}^{SM}\)/DLave ratios in the range of 3–39 (DLave = average of DL values for a given sample; Supplementary Data 1). Only five of twenty-two samples show relatively low \({C}_{Pt,\,Pd}^{SM}\)/ DLave ratios (2.6–2.9); however, these \({C}_{Pt,\,Pd}^{SM}\) values are still well around the quantification limits. The low \({C}_{Pt,\,Pd}^{SM}\) (down to ~4 ppb) determined by previous study was confirmed to be reliable under similar analytical conditions4,21. In addition, the measured \({C}_{Pt,\,Pd}^{SM}\) values of most of our samples have one sigma standard deviation (σ) < 30% relative (Supplementary Data 1). All of these demonstrate the high sensitivity of our LA–ICP–MS analyses of Pt and Pd in the silicate melt and the homogeneous distribition of Pt and Pd in the silicate melt.
References
Righter, K., Humayun, M. & Danielson, L. Partitioning of palladium at high pressures and temperatures during core formation. Nat. Geosci. 1, 321–323 (2008).
Mann, U., Frost, D. J., Rubie, D. C., Becker, H. & Audétat, A. Partitioning of Ru, Rh, Pd, Re, Ir and Pt between liquid metal and silicate at high pressures and high temperatures - Implications for the origin of highly siderophile element concentrations in the Earth’s mantle. Geochim. Cosmochim. Acta 84, 593–613 (2012).
Rubie, D. C. et al. Highly siderophile elements were stripped from Earth’s mantle by iron sulfide segregation. Science 353, 1141–1144 (2016).
Mungall, J. E. & Brenan, J. M. Partitioning of platinum-group elements and Au between sulfide liquid and basalt and the origins of mantle-crust fractionation of the chalcophile elements. Geochim. et. Cosmochim. Acta 125, 265–289 (2014).
Rehkämper, M. et al. Ir, Ru, Pt, and Pd in basalts and komatiites: new constraints for the geochemical behavior of the platinum-group elements in the mantle. Geochim. Cosmochim. Acta 63, 3915–3934 (1999).
Mungall, J. E. & Naldrett, A. J. Ore Deposits of the Platinum-Group Elements. Elements 4, 253–258 (2008).
Barnes, S.-J. & Ripley, E. M. Highly Siderophile and Strongly Chalcophile Elements in Magmatic Ore Deposits. Rev. Mineral. Geochem. 81, 725–774 (2015).
Brenan, J. M., Bennett, N. R. & Zajacz, Z. Experimental Results on Fractionation of the Highly Siderophile Elements (HSE) at Variable Pressures and Temperatures during Planetary and Magmatic Differentiation. Rev. Mineral. Geochem. 81, 1–87 (2015).
Stone, W. & Crocket, J. & Fleet Partitioning of palladium, iridium, platinum, and gold between sulfide liquid and basalt melt at 1200 °C. Geochim. Cosmochim. Acta 54, 2341–2344 (1990).
Fleet, M., Crocket, J. & Stone, W. J. Partitioning of platinum-group elements (Os, Ir, Ru, Pt, Pd) and gold between sulfide liquid and basalt melt. Geochim. Cosmochim. Acta 60, 2397–2412 (1996).
Crocket, J. & Fleet, M. Implications of composition for experimental partitioning of platinum-group elements and gold between sulfide liquid and basalt melt: the significance of nickel content. Geochim. Cosmochim. Acta 61, 4139–4149 (1997).
Fleet, M., Crocket, J., Liu, M. & Stone, W. Laboratory partitioning of platinum-group elements (PGE) and gold with application to magmatic sulfide–PGE deposits. Lithos 47, 127–142 (1999).
Bezmen, N., Asif, M., Brügmann, G., Romanenko, I. & Naldrett, A. Distribution of Pd, Rh, Ru, Jr, Os, and Au between sulfide and silicate metals. Geochim. Cosmochim. Acta 58, 1251–1260 (1994).
Andrews, D. R. & Brenan, J. M. J. The solubility of ruthenium in sulfide liquid: implications for platinum group mineral stability and sulfide melt–silicate melt partitioning. Chem. Geol. 192, 163–181 (2002).
Fonseca, R. O. C., Mallmann, G., O’Neill, H. S. C., Campbell, I. H. & Laurenz, V. Solubility of Os and Ir in sulfide melt: Implications for Re/Os fractionation during mantle melting. Earth Planet. Sci. Lett. 311, 339–350 (2011).
Fonseca, R. O. C., Campbell, I. H., O’Neill, H. S. C. & Allen, C. M. Solubility of Pt in sulphide mattes: Implications for the genesis of PGE-rich horizons in layered intrusions. Geochim. Cosmochim. Acta 73, 5764–5777 (2009).
Laurenz, V. et al. The solubility of palladium and ruthenium in picritic melts: 2. Eff. Sulfur. Geochim. Cosmochim. Acta 108, 172–183 (2013).
Brenan, J. M. Re–Os fractionation by sulfide melt–silicate melt partitioning: a new spin. Chem. Geol. 248, 140–165 (2008).
Barnes, S. J., Holwell, D. A. & Le Vaillant, M. Magmatic Sulfide Ore Deposits. Elements 13, 89–95 (2017).
Day, J. M. D. Geochemical constraints on residual metal and sulfide in the sources of lunar mare basalts. Am. Mineral. 103, 1734–1740 (2018).
Médard, E., Schmidt, M. W., Wälle, M., Keller, N. S. & Günther, D. Platinum partitioning between metal and silicate melts: Core formation, late veneer and the nanonuggets issue. Geochim. Cosmochim. Acta 162, 183–201 (2015).
Médard, E., McCammon, C. A., Barr, J. A. & Grove, T. L. Oxygen fugacity, temperature reproducibility, and H2O contents of nominally anhydrous piston-cylinder experiments using graphite capsules. Am. Mineralogist 93, 1838–1844 (2008).
Li, Y. & Audétat, A. Partitioning of V, Mn, Co, Ni, Cu, Zn, As, Mo, Ag, Sn, Sb, W, Au, Pb, and Bi between sulfide phases and hydrous basanite melt at upper mantle conditions. Earth Planet. Sci. Lett. 355-356, 327–340 (2012).
Kiseeva, E. S. & Wood, B. J. A simple model for chalcophile element partitioning between sulphide and silicate liquids with geochemical applications. Earth Planet. Sci. Lett. 383, 68–81 (2013).
Alard, O., Griffin, W. L., Lorand, J. P., Jackson, S. E. & O’Reilly, S. Y. J. N. Non-chondritic distribution of the highly siderophile elements in mantle sulphides. Nature 407, 891–894 (2000).
Patten, C., Barnes, S.-J., Mathez, E. A. & Jenner, F. E. Partition coefficients of chalcophile elements between sulfide and silicate melts and the early crystallization history of sulfide liquid: LA-ICP-MS analysis of MORB sulfide droplets. Chem. Geol. 358, 170–188 (2013).
Peach, C., Mathez, E. & Keays, R. J. Sulfide melt-silicate melt distribution coefficients for noble metals and other chalcophile elements as deduced from MORB: Implications for partial melting. Geochim. Cosmochim. Acta 54, 3379–3389 (1990).
Lorand, J.-P., Luguet, A. & Alard, O. Platinum-group element systematics and petrogenetic processing of the continental upper mantle: a review. Lithos 164-167, 2–21 (2013).
Barnes, S. J., Mungall, J. E. & Maier, W. D. Platinum group elements in mantle melts and mantle samples. Lithos 232, 395–417 (2015).
Lorand, J.-P. & Luguet, A. Chalcophile and Siderophile Elements in Mantle Rocks: trace Elements Controlled By Trace Minerals. Rev. Mineral. Geochem. 81, 441–488 (2015).
Lee, C.-T. A. et al. Copper systematics in arc magmas and implications for crust-mantle differentiation. Science 336, 64–68 (2012).
Walter, M. J. J. Melt extraction and compositional variability in mantle lithosphere. TrGeo 2, 568 (2003).
Zhang, Z. & Hirschmann, M. M. Experimental constraints on mantle sulfide melting up to 8 GPa. Am. Mineralogist 101, 181–192 (2016).
Becker, H. et al. Highly siderophile element composition of the Earth’s primitive upper mantle: Constraints from new data on peridotite massifs and xenoliths. Geochim. Cosmochim. Acta 70, 4528–4550 (2006).
Fischer-Gödde, M., Becker, H. & Wombacher, F. Rhodium, gold and other highly siderophile elements in orogenic peridotites and peridotite xenoliths. Chem. Geol. 280, 365–383 (2011).
Mavrogenes, J. A. & O’Neill, H. S. C. The relative effects of pressure, temperature and oxygen fugacity on the solubility of sulfide in mafic magmas. Geochim. Cosmochim. Acta 63, 1173–1180 (1999).
Salters, V. J. & Stracke, A. Composition of the depleted mantle. Geochem. Geophys. Geosyst. 5, 5 (2004).
McDonough, W. F. & Sun, S.-S. The composition of the Earth. Chem. Geol. 120, 223–253 (1995).
Park, J.-W., Campbell, I. H. & Arculus, R. J. Platinum-alloy and sulfur saturation in an arc-related basalt to rhyolite suite: evidence from the Pual Ridge lavas, the Eastern Manus Basin. Geochim. Cosmochim. Acta 101, 76–95 (2013).
Brenan, J. M., McDonough, W. F. & Dalpé, C. Experimental constraints on the partitioning of rhenium and some platinum-group elements between olivine and silicate melt. Earth Planet. Sci. Lett. 212, 135–150 (2003).
Brenan, J. M., Finnigan, C. F., McDonough, W. F. & Homolova, V. Experimental constraints on the partitioning of Ru, Rh, Ir, Pt and Pd between chromite and silicate melt: The importance of ferric iron. Chem. Geol. 302-303, 16–32 (2012).
Ely, J. C. & Neal, C. R. Using platinum-group elements to investigate the origin of the Ontong Java Plateau, SW Pacific. Chem. Geol. 196, 235–257 (2003).
Yang, A. Y. et al. Chalcophile elemental compositions of MORBs from the ultraslow-spreading Southwest Indian Ridge and controls of lithospheric structure on S-saturated differentiation. Chem. Geol. 382, 1–13 (2014).
Danyushevsky, L. V. & Plechov, P. Petrolog3: Integrated software for modeling crystallization processes. Geochem. Geophys. Geosyst. 12, 7 (2011).
Smythe, D. J., Wood, B. J. & Kiseeva, E. S. The S content of silicate melts at sulfide saturation: new experiments and a model incorporating the effects of sulfide composition. Am. Mineral. 102, 795–803 (2017).
Chazey, W. J. III & Neal, C. R. Platinum-group element constraints on source composition and magma evolution of the Kerguelen Plateau using basalts from ODP Leg 183. Geochim. Cosmochim. Acta 69, 4685–4701 (2005).
Kiseeva, E. S. & Wood, B. J. The effects of composition and temperature on chalcophile and lithophile element partitioning into magmatic sulphides. Earth Planet. Sci. Lett. 424, 280–294 (2015).
Liu, X., Xiong, X., Audétat, A. & Li, Y. Partitioning of Cu between mafic minerals, Fe–Ti oxides and intermediate to felsic melts. Geochim. Cosmochim. Acta 151, 86–102 (2015).
Liu, X. et al. Partitioning of copper between olivine, orthopyroxene, clinopyroxene, spinel, garnet and silicate melts at upper mantle conditions. Geochim. Cosmochim. Acta 125, 1–22 (2014).
Jenner, F. E., O’Neill, H. S. C., Arculus, R. J. & Mavrogenes, J. A. The Magnetite Crisis in the Evolution of Arc-related Magmas and the Initial Concentration of Au, Ag and Cu. J. Pet. 51, 2445–2464 (2010).
Li, Y. Comparative geochemistry of rhenium in oxidized arc magmas and MORB and rhenium partitioning during magmatic differentiation. Chem. Geol. 386, 101–114 (2014).
Mudd, G. M. Key trends in the resource sustainability of platinum group elements. Ore Geol. Rev. 46, 106–117 (2012).
Kamenetsky, V. S. & Zelenski, M. J. G. Origin of noble-metal nuggets in sulfide-saturated arc magmas: a case study of olivine-hosted sulfide melt inclusions from the Tolbachik volcano (Kamchatka, Russia). Geology 48, 620–624 (2020).
Anenburg, M. & Mavrogenes, J. A. Experimental observations on noble metal nanonuggets and Fe-Ti oxides, and the transport of platinum group elements in silicate melts. Geochim. Cosmochim. Acta 192, 258–278 (2016).
Anenburg, M. & Mavrogenes, J. A. Noble metal nanonugget insolubility in geological sulfide liquids. Geology 48, 939–943 (2020).
Laurenz, V., Rubie, D. C., Frost, D. J. & Vogel, A. K. The importance of sulfur for the behavior of highly-siderophile elements during Earth’s differentiation. Geochim. Cosmochim. Acta 194, 123–138 (2016).
Wheeler, K. T., Walker, D. & McDonough, W. F. Pd and Ag metal-silicate partitioning applied to Earth differentiation and core-mantle exchange. Meteorit. Planet. Sci. 46, 199–217 (2011).
Dale, C. W. et al. Late accretion on the earliest planetesimals revealed by the highly siderophile elements. Science 336, 72–75 (2012).
Sylvester, P. J. & Eggins, S. M. J. Analysis of Re, Au, Pd, Pt and Rh in NIST glass certified reference materials and natural basalt glasses by laser ablation ICP‐MS. Geostand. Newsl. 21, 215–229 (1997).
Jégo, S., Pichavant, M. & Mavrogenes, J. A. Controls on gold solubility in arc magmas: an experimental study at 1000 °C and 4 kbar. Geochimica et. Cosmochimica Acta 74, 2165–2189 (2010).
Jégo, S. & Pichavant, M. Gold solubility in arc magmas: experimental determination of the effect of sulfur at 1000 °C and 0.4 GPa. Geochim. Cosmochim. Acta 84, 560–592 (2012).
Longerich, H. P., Jackson, S. E. & Günther, D. Inter-laboratory note. Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation. J. Anal. At. Spectrom. 11, 899–904 (1996).
Acknowledgements
We thank Fangyue Wang and Andreas Audétat for helpful discussion on LA–ICP–MS analyses. This work is supported by National Key R&D Program of China (2018YFA0702600) and the National Natural Science Foundation of China (42021002) to Y.L.
Author information
Authors and Affiliations
Contributions
M.D.Z. performed the experiments and analyses as part of his Ph.D. dissertation. Y.L. conceived and supervised the study and interpreted the data. M.D.Z. and Y.L. co-wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks xxxx for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, M., Li, Y. Breaking of Henry’s law for sulfide liquid–basaltic melt partitioning of Pt and Pd. Nat Commun 12, 5994 (2021). https://doi.org/10.1038/s41467-021-26311-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26311-x
This article is cited by
-
Redox control of the partitioning of platinum and palladium into magmatic sulfide liquids
Communications Earth & Environment (2024)
-
Sulfide saturation in reduced magmas during generation of the Gangdese juvenile lower crust: Implications for porphyry Cu–Au mineralization in the Gangdese belt, Tibet
Mineralium Deposita (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
