
Abstract
The partitioning behavior of platinum group elements in magmas is critical for their use as tracers of planetary accretion and in understanding magmatic sulfide deposits. Here we use laboratory experiments to determine sulfide liquid–silicate melt partition coefficients for platinum and palladium at 1.5 GPa, 1400 °C, and oxygen fugacity 1.5–2 log units above the fayalite–magnetite–quartz buffer. We find that the partitioning coefficients of these elements are 2.3 × 105 to 1.1 × 106 and are independent of the platinum and palladium concentration in the system. Combined with previous data obtained at oxygen fugacity below the fayalite–magnetite–quartz buffer, this indicates redox-controlled partitioning behavior whereby at oxidizing conditions platinum- and palladium-enrichments are achieved through their dissolution in sulfide liquids, while at reducing conditions the entrapment of platinum- and palladium-rich clusters in sulfide liquids is more critical. This redox-controlled partitioning behavior should be considered when studying crust–mantle differentiation and the formation of magmatic sulfide deposits.
Similar content being viewed by others


Introduction
Platinum-group elements (PGEs) are key tracers for planetary accretion and core–mantle–crust differentiation1,2,3,4,5 and the formation of magmatic sulfide deposits in Earth’s crust2,6,7. However, the valid use of PGEs as powerful tracers relies principally on the knowledge of PGE partitioning in the metallic melt–sulfide liquid–silicate melt systems. Therefore, sulfide liquid–silicate melt partition coefficients of PGEs (\({D}_{{PGE}}^{{Sul}/{Sil}}\)) were extensively determined8,9,10,11,12 during the 1990s; however, these experimental studies analyzed the samples using a bulk analytical technique, namely neutron activation analysis, and yielded largely discrepant \({D}_{{PGE}}^{{Sul}/{Sil}}\) values for each element (102–108), which could be caused by the presence of PGE-nuggets in the sulfide liquids and/or silicate melts, and/or an ineffective sulfide liquid–silicate melt separation13. In 2014, Mungall and Brenan2 published a high-quality dataset of \({D}_{{PGE}}^{{Sul}/{Sil}}\) (105–106) obtained at 1 bar and 1200 °C, using in-situ micro-analyses of laser ablation–inductively coupled plasma–mass spectrometry (LA–ICP–MS), which can effectively filter the contamination of PGE-nuggets in the silicate melts. These high \({D}_{{PGE}}^{{Sul}/{Sil}}\) values can readily explain the highly PGE-enriched magmatic sulfide deposits as the result of sulfide liquid segregation, such as the world-class Merensky reef and UG2 deposits of the Bushveld Complex of South Africa7,14. However, in the experiments of Mungall and Brenan2, ~0.5–12 wt.% of each Ru, Rh, Pt, and Pd was added in the sulfide liquids. Therefore, Zhang and Li15 performed an additional set of experiments to determine \({D}_{{Pt},{Pd}}^{S{ul}/{Sil}}\) as a function of the Pt and Pd concentrations in sulfide liquids (\({C}_{{Pt},{Pd}}^{{Sul}}\)) from ~100 µg/g to 2 wt% at 1 GPa and 1400 °C. They found that \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) increase positively from 2 × 103 to 3.5 × 105 with increasing \({C}_{{Pt},{Pd}}^{{Sul}}\), and their \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are systematically consistent with those of Mungall and Brenan2. Similar positive correlations between \({D}_{{Pt},{Pd},{Ru}}^{{Sul}/{Sil}}\) and \({C}_{{Pt},{Pd},{Ru}}^{{Sul}}\) were also recognized by Ballhaus and Sylvester16 using the data obtained from bulk analyses. They interpreted the dependence of \({D}_{{Pt},{Pd},{Ru}}^{{Sul}/{Sil}}\) on \({C}_{{Pt},{Pd},{Ru}}^{{Sul}}\) as resulting from the presence of Pt- and Pd-rich clusters in the sulfide liquids and/or non-Henrian law behavior15,16,17. These observations, together with the findings of insoluble PGE-nanonuggets in sulfide liquids18, suggest that the actual \({D}_{{PGE}}^{{Sul}/{Sil}}\) are below 104 in natural magmatic systems, and consequently, sulfide liquid–silicate melt partitioning may play a little role in the formation of some highly PGE-enriched sulfide deposits15,16,18.
We realize that the experiments of both Mungall and Brenan2 and Zhang and Li15, as well as the earlier experiments, were performed at oxygen fugacity (fO2) ranging from below FMQ − 2 to FMQ − 1 (FMQ = the fayalite–magnetite–quartz buffer), and thus far, \({D}_{{PGE}}^{{Sul}/{Sil}}\) have not ever been determined at oxidizing conditions (fO2 > FMQ). The determinations of \({D}_{{PGE}}^{{Sul}/{Sil}}\) at oxidizing conditions are fundamentally important, because most arc magmas are oxidized19 and oxidized magmas are also ultimately associated with the formation of magmatic sulfide deposits at various tectonic settings20,21,22,23,24. For example, the assimilation and presence of graphite determines the fO2 at <FMQ − 2 to FMQ − 1 for the Voisey’s Bay magmas, which host a world-class Ni–Cu sulfide deposit21,25. In contrast, the assimilation and presence of anhydrite determines the fO2 between FMQ + 1.5 and FMQ + 2 for the Kharaelakh intrusion, which is one of several sill-like, multiphase gabbroic intrusions that host world-class Ni–Cu–PGE sulfide deposits in the Noril’sk–Talnakh region23,26. Available studies13,27,28 show that the valence state of PGEs in silicate melts may change as a function of fO2. At fO2 > FMQ + 1.5, Pt dissolves mainly as Pt2+ oxide in silicate melts29,30, whereas at fO2 < FMQ − 1, Pt0 and/or Pt– may occur28,31. At fO2 > FMQ + 2, Pd dissolves mainly as Pd+ and Pd2+ in basaltic melts, whereas ~40% Pd occurs as Pd0 at fO2 of FMQ − 2 (ref. 27). In addition, PGEs and semimetals such as As and Sb can form clusters in sulfide liquids at reducing conditions, which can enhance the apparent dissolution of PGEs in sulfide liquids32, and thus the apparent \({D}_{{PGE}}^{{Sul}/{Sil}}\) (refs. 16, 18); however, semimetals may have a negligible role at oxidizing conditions due to their presence as cations33,34. Accordingly, \({D}_{{PGE}}^{{Sul}/{Sil}}\) could differ at oxidizing vs. reducing conditions but remains undetermined at oxidizing conditions.
Here we present experiments aimed at determining \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) at 1.5 GPa, 1400 °C, and fO2 between FMQ + 1.5 and FMQ + 2. We find that \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are as high as 105–106 and independent of \({C}_{{Pt},{Pd}}^{{Sul}}\). These findings, together with previous \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) obtained at reducing conditions2,15, reveal a redox-controlled sulfide liquid–basaltic melt partitioning of Pt and Pd, which places important constraints on the mechanisms for Pt- and Pd-enrichments in magmatic sulfide liquids formed at different redox conditions.
Results and discussion
Sulfide liquid–basaltic melt partitioning of Pt and Pd at oxidizing conditions
We loaded ~25 wt% sulfide (FeS), ~15 wt% anhydrite (CaSO4), and ~60 wt% silicate into olivine capsules, which were then placed into Re capsules. The sulfide and anhydrite powders were sandwiched by silicate powder, and the silicate has a major element composition like the mid-ocean ridge basalt (MORB) (Supplementary Table 1). Two sets of experiments were performed at 1.5 GPa and 1400 °C in a piston cylinder apparatus for 24–48 h (Supplementary Table 2). In Set-1 of six forward experiments, ~600–5000 µg/g Pt and ~500–10,000 µg/g Pd were doped in the sulfide; in Set-2 of three reversal experiments, ~2000–7000 µg/g Pt and Pd were mixed physically with the silicate powder. Also, in one experiment, we added a small piece of Pt metal in contact with the sulfide (run AN-06; Supplementary Table 2).
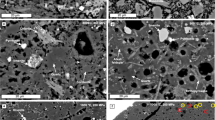
The experimentally produced assemblage of olivine, sulfide liquid, anhydrite, and basaltic melt (Fig. 1) would mimic the natural observations of anhydrite-assimilated and sulfide liquid-saturated mafic magmas23,26. The coexistence of sulfide liquid and anhydrite in basaltic melts determined the fO2 between FMQ + 1.5 and FMQ + 2, since only at this transitional oxidation state are both S2– and S6+ species important in basaltic melts23,35,36,37. Chemical compositions of the quenched samples were analyzed using electron probe microanalysis (EPMA) and LA–ICP–MS (Methods). Both basaltic melts and sulfide liquids are homogeneous in major elements (Supplementary Tables 3–4). The MgO and CaO contents of the silicate melts increase by up to 90% and 30%, respectively, due to the dissolution of olivine capsule and anhydrite (Supplementary Table 3). The basaltic melt NBO/T (the ratio of non-bridging oxygens to tetrahedrally coordinated cations) is ~1.5–1.9 (Supplementary Table 2), as calculated on an anhydrous basis and with the consideration of the number of Al3+ and Fe3+ that act as tetrahedral network-forming cations. Small sulfide dots (<0.3 μm) are homogeneously distributed in the quenched silicate melts (Fig. 1c), which we interpreted as resulting from the drop in sulfide solubility during quench. The sulfur contents in silicate melts are ~1.7–2.3 wt%, consistent with previous results obtained at similar conditions36,38. The sulfide liquids contain ~900–17,600 μg/g Pt, ~700–18,200 μg/g Pd, ~50–56 wt% Fe, ~40–42 wt% S, and ~1–2 wt% O (Supplementary Table 4). The \({C}_{{Pt},{Pd}}^{{Sul}}\) obtained using EPMA and LA–ICP–MS agrees within 20% relative (Supplementary Fig. 1). The sulfide liquid from run AN-06 contains ~17 wt% Pt, ~45 wt% Fe, ~36 wt% S, and ~1 wt% O (Supplementary Table 4). In addition, all sulfide liquids contain ~0.6–5.5 wt% Ni due to their chemical exchange with the olivine capsule walls (Supplementary Table 4).

a Reflected-light photomicrograph of run AN-03 (Supplementary Table 2) showing the coexistence of silicate melt, sulfide liquid, and anhydrite melt in an olivine-lined Re capsule. Note that the anhydrite melt globule was partially lost during polishing. b Back scattered electron image of the quenched anhydrite melt and sulfide liquid. In all experiments the round shape of the quenched anhydrite is consistent with its presence as immiscible melt at run conditions. EPMA analyses of the anhydrites show that they consisted almost entirely of CaO and SO3 (total >99 wt.%) and have presumably crystallized as anhydrite upon quenching. c Back scattered electron image of the homogenous distribution of small sulfide dots in silicate melts. These sulfide dots were produced during quenching the experiments, due to the drop of sulfide solubility, and they were included during LA–ICP–MS analysis to obtain the actual Pt and Pd concentrations in the silicate melts. d Back scattered electron image of the quenched sulfide liquid comprised monosulfide-solid-solution (MSS) and a phase with more S compared to MSS.
Our LA–ICP–MS detection limits (DLs) of the Pt and Pd in silicate melts are ~2–7 ng/g and ~1–6 ng/g, respectively (Supplementary Table 3). The time-resolved LA–ICP–MS signals of the Pt and Pd in silicate melts are constant and covary with those of the other major and trace elements, indicating negligible contamination from Pt- or Pd-bearing nuggets (Fig. 2). The measured Pt and Pd concentrations in silicate melts (\({C}_{{Pt},{Pd}}^{{Sil}}\)) are 3–158 ng/g and 7–44 ng/g, respectively, except for that the \({C}_{{Pt}}^{{Sil}}\) of runs AN-03, AN-04, AN-05, and ANr-02, and the \({C}_{{Pd}}^{{Sil}}\) of runs AN-05, AN-06, and ANr-02, are below the DLs (Supplementary Table 2; Supplementary Fig. 2). The total mass of Pt or Pd in sulfide liquid and silicate melt of each individual sample is generally consistent with the Pt or Pd mass doped in the starting material (Supplementary Table 2). Figure 3a, b show that \({C}_{{Pt},{Pd}}^{{Sil}}\) increase linearly with increasing \({C}_{{Pt},{Pd}}^{{Sul}}\). Figure 3a, b also show that at given \({C}_{{Pt},{Pd}}^{{Sul}}\) our \({C}_{{Pt},{Pd}}^{{Sil}}\) obtained at oxidizing conditions are lower than those obtained at reducing conditions15, and all of the \({C}_{{Pt},{Pd}}^{{Sil}}\) obtained at reducing conditions2,15 together show a non-linear dependence on \({C}_{{Pt},{Pd}}^{{Sul}}\).

a–f The signals of Pt and Pd are constant, without showing a considerable contribution of Pt- or Pd-rich nuggets.

a, b The Pt and Pd concentrations in silicate melts increase linearly with increasing the Pt and Pd concentrations in sulfide liquids, in contrast with the data of Mungall and Brenan2 and Zhang and Li15. The symbols with downward arrows are plotted with Pt or Pd detection limits in silicate melts, and the true values are below the detection limits. c, d \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) remain nearly constant, in contrast with the data of Mungall and Brenan2 and Zhang and Li15, which show a linear increase with increasing the Pt and Pd concentrations in sulfide liquids. The symbols with upward arrows are \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) calculated using the Pt or Pd detection limits in silicate melts, and the true \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) values should be larger than the values plotted. Error bars are 1σ standard deviation. Only data obtained by LA–ICP–MS analysis were plotted.
The \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\), as calculated from \({C}_{{Pt},{Pd}}^{{Sul}}\)/\({C}_{{Pt},{Pd}}^{{Sil}}\), are 0.7–1.3 × 106 and 2.3–4.5 × 105, respectively. Figure 3c, d show that our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) obtained at oxidizing conditions are comparable to the high endmembers of previous data obtained at reducing conditions by Mungall and Brenan2 and Zhang and Li15; however, our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are one more order of magnitude larger than previous data when \({C}_{{Pt},{Pd}}^{{Sul}}\) are below 1 wt%. All of the \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) data obtained at reducing conditions2,15 together show a positive dependence on \({C}_{{Pt},{Pd}}^{{Sul}}\); however, our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are independent of \({C}_{{Pt},{Pd}}^{{Sul}}\). Our larger \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) appear to be caused by the lower \({C}_{{Pt},{Pd}}^{{Sil}}\) (Fig. 3a, b). In addition, we do not find any correlation between \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\), the atomic metal/S ratios of sulfide liquids, or the FeOtot contents of basaltic melts (Supplementary Fig. 3).
The approach of equilibrium partitioning of Pt and Pd in this study is indicated by the following evidence. First, our samples have homogeneous major element and Pt and Pd concentrations (\({C}_{{Pt},{Pd}}^{{Sil}}\) and \({C}_{{Pt},{Pd}}^{{Sul}}\)), independent of the run duration from 24–48 h (Supplementary Table 2; Supplementary Fig. 4). Second, our forward and reversal experiments yielded systematically consistent \({C}_{{Pt},{Pd}}^{{Sil}}\) and \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) (Fig. 3).
Redox-controlled dissolution and partitioning of Pt and Pd
Figure 3a, b show that the dissolution behavior of Pt and Pd differs in reduced vs. oxidized silicate melt, which should have caused the different partitioning behavior of Pt and Pd at different redox conditions (Fig. 3c, d). Previous studies28 show that Pt solubility in silicate melt at a given temperature and melt composition decreases with decreasing fO2 down to ~FMQ + 1, but then slightly increases with further decreasing fO2 (see also Supplementary Fig. 5), which indicates the change of Pt valence state from Pt2+ predominance29,30 at fO2 > FMQ + 1 to Pt0 and/or Pt− predominance28,31 at fO2 < FMQ − 1. Similarly, Pd occurs mainly as Pd+ and Pd2+ in silicate melt at fO2 > FMQ + 1, but ~40% Pd occurs as Pd0 at fO2 of FMQ − 2 (ref. 27). Therefore, the observed lower \({C}_{{Pt},{Pd}}^{{Sil}}\) at oxidizing conditions than at reducing conditions at given \({C}_{{Pt},{Pd}}^{{Sul}}\) (Fig. 3a, b) could be partially ascribed to the presence of Pt0/Pt− and Pd0 that can enhance the dissolution of Pt and Pd in the reduced silicate melts. Previous studies2,39,40 also show that the presence of S2− can enhance the dissolution of Pt and Pd in silicate melt through the formation of Pt- and Pd-sulfide species, but it remains unknown whether the effect of S2– is the same or different at different redox conditions when Pt and Pd have different valence states. If S2– has a stronger effect at reducing conditions than at oxidizing conditions, then the observed larger \({C}_{{Pt},{Pd}}^{{Sil}}\) at reducing conditions can also be partially explained.
Our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are independent of \({C}_{{Pt},{Pd}}^{{Sul}}\), indicating that \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) follow Henry’s law. Positive correlations between \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) and \({C}_{{Pt},{Pd}}^{{Sul}}\), recognized by refs. 15, 16, were interpreted as resulting from non-Henrian law behavior and/or the presence of PGE-nuggets in sulfide liquids. Our results rule out non-Henrian law behavior as a main cause for such positive correlations. This is consistent with the limited variation of the activity coefficient of PtS in sulfide liquids, by a factor of less than7, as \({C}_{{Pt}}^{{Sul}}\) varies from ~100 to 21,000 μg/g (ref. 15). Accordingly, the presence of PGE-nuggets may be the most plausible explanation for the positive correlations between \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) and \({C}_{{Pt},{Pd}}^{{Sul}}\), and the about three orders of magnitude variation in \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) (Fig. 3c, d). Available observations on both synthetic and natural samples show the easy formation of PGE-rich clusters in sulfide liquids and monosulfide-solid-solution17,18,32,41. This holds particularly true for Pt and Pd at reducing conditions, which with semimetals, such as As and Sb in the form of anions, can form clusters and stick to sulfide liquids due to the low surface energy32,34. Our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) are independent of \({C}_{{Pt},{Pd}}^{{Sul}}\), which suggests the absence of considerable Pt- and Pd-rich nuggets in our sulfide liquids at oxidizing conditions. This is highly possible because our sulfide liquids contain only 5–20 µg/g of combined semimetals As, Bi, Sb, Se, and Te (Supplementary Table 4), and most of these semimetals may occur as cations at presently oxidizing conditions33,34.
Implications for Pt- and Pd-enrichments in magmatic sulfide liquids
Our new \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) obtained at oxidizing conditions, together with previous \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) obtained at reducing conditions2,15, suggest an important role for redox conditions in governing the partitioning of Pt and Pd between sulfide liquid and silicate melt. The redox-controlled \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) could have new implications for understanding the behavior of Pt and Pd in magmas, and the Pt- and Pd-enrichments in magmatic sulfide liquids formed at different redox conditions (Fig. 4). The high \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) (105–106) at oxidizing conditions imply that the sulfide liquids segregated from oxidized magmas could be enriched in Pt and Pd through chemical dissolution of Pt and Pd in sulfide liquids (Fig. 4a). This mechanism may explain the Pt-and Pd-enrichments in sulfide deposits hosted by oxidized magmas with fO2 between FMQ + 1 and FMQ + 2, such as the Jinchuan, Stillwater, and Noril’sk–Talnakh sulfide deposits in intraplate tectonic setting21,23, and the Mascot sulfide deposit in convergent margin tectonic setting24. However, the positive correlations between \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) and \({C}_{{Pt},{Pd}}^{{Sul}}\) at reducing conditions, caused by the presence of PGE-rich clusters in sulfide liquids, indicate that the sulfide liquids segregated from reduced magmas cannot acquire significant Pt and Pd through chemical dissolution. This is because the actual \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) could be significantly below 104 in natural magmatic systems where \({C}_{{Pt},{Pd}}^{{Sul}}\) are usually below 100 µg/g (Fig. 3c, d; see also refs. 15, 16, 18). In this case, Pt- and Pd-rich clusters or platinum-group minerals (PGMs), which exsolve or crystallize from reduced magmas but entrapped in sulfide liquids formed later, could make a more important contribution to the Pt- and Pd-enrichments in sulfide liquids (Fig. 4b). This mechanism could be applied to the Pt- and Pd-enrichments in sulfide deposits hosted by reduced magmas, such as the Merensky reef sulfide deposits hosted by magmas16 with the presence of graphite and corresponding fO2 of ~FMQ − 2.

a The sulfide liquids saturated from oxidized magmas (fO2 > FMQ + 1) would be enriched in Pt and Pd due to the high \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) (=105–106). Note that the assimilation of anhydrite-bearing sediments would lead to oxidation of mantle-derived magmas, as indicated by the presence of anhydrite. b The sulfide liquids saturated from reduced magmas (fO2 < FMQ–1) could be enriched in Pt and Pd due to the entrapments of Pt- and Pd-semimetal clusters in sulfide liquids, which formed earlier than sulfide liquids. Note that the assimilation of organic matter-bearing sediments would lead to reduction of mantle-derived magmas, as indicated by the presence of graphite.
We tentatively tested the above-proposed two different mechanisms for Pt- and Pd- enrichments in sulfide liquids, through the comparison of Pt and Pd distribution in the Merensky reef and Noril’sk 1 sulfide deposits, as these two deposits represent two endmembers that are hosted by magmas of very different redox conditions. In the Merensky reef deposit hosted by magmas16 with fO2 of ~FMQ − 2, 95% Pt is stored in PGMs, and 5% Pt in sulfide minerals. While, in the Noril’sk 1 deposit hosted by magmas26 with fO2 of FMQ + 1.5 to FMQ + 2, 40% Pt is stored in PGMs, and 60% Pt in sulfide minerals6,42. The Pd stored in sulfide minerals is also higher in the Noril’sk 1 deposit than in the Merensky reef deposit (~95% vs. 80%)6,42. We understand that PGMs in magmatic sulfide deposits may form during subsolidus transformation of sulfides, and the formation of PGMs also depends on the availability of semimetals43,44. However, both laboratory experiments and natural observations demonstrate that the coexistence of sulfide minerals and PGMs in some cases may represent high-temperature magmatic signatures17,18,32,41,45, and PGMs can crystallize directly from high-temperature magmas46,47,48. We therefore believe that the observed different distributions of Pt and Pd in the Merensky reef and Noril’sk 1 sulfide deposits at least qualitatively support the mechanisms we proposed for Pt- and Pd-enrichments in sulfide liquids formed at different redox conditions.
In conclusion, our experiment work reveals that the sulfide liquid–basaltic melt partitioning of Pt and Pd is redox-controlled, and the redox-controlled \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) imply different mechanisms for the Pt- and Pd-enrichments in magmatic sulfide liquids formed at different redox conditions. The redox-controlled \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) should be considered in any future models for the crust–mantle differentiation in general, and the formation of magmatic sulfide deposits in particular. Finally, we suggest that the sulfide liquid–basaltic melt partitioning of PGEs could be more complex than we previously thought, and inter-laboratory experiments should be performed at various redox conditions to determine \({D}_{{PGE}}^{{Sul}/{Sil}}\) with µg/g level PGE concentrations in the systems, so as to have a better understanding of the behavior of PGEs in magmas of different tectonic settings.
Methods
Starting materials
Starting materials loaded in each sample capsule included synthetic silicate, sulfide (FeS), and reagent–grade CaSO4 (anhydrite). The synthetic silicate was prepared from analytical grade oxides SiO2, Al2O3, FeO, MgO, MnO, TiO2, and Cr2O3, and carbonates Na2CO3, CaCO3, and K2CO3. To minimize absorbed water contents, SiO2, TiO2, Al2O3, Cr2O3, and MgO powders were each heated at 1000 °C overnight, MnO2 at 400 °C and CaCO3 at 200 °C for 5 h, and Na2CO3 and K2CO3 at 110 °C for 5 h. After drying, the weighed oxides and carbonates were mixed and ground in ethanol in an agate mortar for 2 h, and then dried at room temperature. The dried mixture was heated in a muffle furnace at 1000 °C overnight to decarbonate. The FeO powder was finally added to the decarbonated mixture. The final mixture was then ground again in ethanol in an agate mortar for 1 h and dried at room temperature. In Set-1 of the forward experiments, ~600–5000 µg/g Pt and ~500–10,000 µg/g Pd were doped in the sulfide powder. In Set-2 of the reversal experiments, ~2000–7000 µg/g Pt and Pd were mixed physically with the silicate powder in ethanol in an agate mortar for 2 h, which was then dried at room temperature. All dried silicate and sulfide materials were stored in a vacuumed oven at 110 °C for > 24 h before loading into sample capsules for experiments.
High-pressure experiments
All experiments were conducted in an end-loaded solid-media piston cylinder apparatus (Max Voggenreiter LPC250), using 3/4-inch diameter talc–Pyrex assemblies with graphite heaters. A friction correction of 18% was applied to the nominal pressure based on calibration of the quartz–coesite and kyanite–sillimanite transitions. The total pressure uncertainty was less than 0.1 GPa. The experimental temperatures were monitored by C-type (W95Re05–W74Re26) thermocouples with an uncertainty of ~10 °C, and were controlled to ±2 °C. The experiments were pressurized at room temperature to the target pressures, and then the temperatures were raised at 100 °C/min to the target temperatures. All experiments were quenched to below 100 °C within 10–20 s by turning off electrical power to the graphite heaters. The recovered samples were mounted in epoxy resin and polished for EPMA and LA–ICP–MS analyses.
Electron probe microanalysis (EPMA)
Major element compositions of the quenched silicate melts and sulfide liquids were measured with a JEOL–JXA–8230 electron microprobe, using wavelength-dispersive mode and a ZAF matrix correction. For the measurements of major elements in the quenched silicate melts, 15 kV accelerating voltage, 10 nA beam current, and a defocused beam of 20 μm were used for both the standardizations and samples. Both natural and synthetic standards were used to calibrate the instrument, and the used standards were albite (Na), plagioclase (Si), almandine (Al), magnetite (Fe), olivine (Mg), Cr-diopside (Ca), orthoclase (K), rhodonite (Mn), and rutile (Ti). The peak counting time was 20 s for Si, Al, Fe, Mg, Ca, and Ti, 10 s for K and Na, and 40 s for Mn. The sulfur contents in the quenched silicate melts were analyzed with 15 kV/70 nA, 20 μm beam diameter, and 180 s peak counting time using a barite standard. The quenched anhydrite melts were analyzed using the same conditions as used for analyzing the silicate melts. The sulfide liquids were analyzed with 20 kV/20 nA and 20–30 μm beam diameter. Iron and sulfur were calibrated on a synthetic pyrrhotite with a well-known Fe: S ratio. Ni, Cu, Pt, Pd, and Re were calibrated on pure metals, and O was calibrated on magnetite. The peak counting time were 20 s for Fe and S, and 40 s for Ni, Cu, Pt, Pd, Re and O.
LA–ICP–MS analysis
Major and trace elements of the quenched silicate melts and sulfide liquids were analyzed by laser–ablation ICP–MS, using an Agilent 7900 Quadrupole ICP–MS coupled to a Photon Machines Analyte HE 193-nm ArF Excimer Laser Ablation system. Argon was used as the make-up gas (0.87 L/min) and mixed with the He carrier gas (0.9 L/min) via a T-connector before entering the ICP. The quenched silicate melts were analyzed with a laser beam size of 90–110 μm at 8 Hz with energy of ~4 J/cm2 for 40 s after measuring the gas blank for 20 s, and the sulfide liquids were analyzed with a laser beam size of 40 μm at 7 Hz with energy of ~2 J/cm2. The isotopes 23Na, 24Mg, 27Al, 29Si, 39K, 44Ca, 47Ti, 55Mn, 57Fe, 60Ni, and 63Cu were measured with a dwell time of 5 ms, and 75As, 77Se, 118Sn, 121Sb, 125Te, and 209Bi were measured with a dwell time of 10 ms; whereas, the 185Re, 105Pd, 106Pd, and 195Pt were measured with a dwell time of 80 ms. NIST SRM 610 glass was used as external standard for all analyses, which contains 3.15 μg/g Pt and 1.05 μg/g Pd, whereas Si and Fe determined by electron microprobe were used as internal standard for the silicate melt and sulfide, respectively. Ten to twenty spots were measured for the silicate melt per sample, and five to twelve spots were measured for the sulfide liquid. Repeated analyses of the MASS-1 sulfide standard yielded an agreement between our LA–ICP–MS measurements and the certified Pt concentration within 10–22 % relative. In addition, the Pt and Pd concentrations in sulfide liquid, measured by LA–ICP–MS, agree with those measured by EPMA within 20% relative (Supplementary Fig. 1).
The detection limits (DLs) for measuring the Pt and Pd concentrations in the silicate melts were calculated as three times the standard deviation of three replicate measurements of a sample that contains zero analyte15,49. Our obtained DLs for Pt and Pd in the silicate melts were between 2–7 ng/g, and between 1–6 ng/g, respectively. Pt concentration in silicate melts (\({C}_{{Pt}}^{{Sil}}\)) was generally below the DLs when the Pt concentration in sulfide liquids was below ~1900 μg/g, whereas Pd concentration in silicate melts (\({C}_{{Pd}}^{{Sil}}\)) was generally below the DLs when the Pd concentration in sulfide liquids was below ~700 μg/g (Fig. 2 and Supplementary Fig. 2; Supplementary Table 2). In the runs with \({C}_{{Pt},{Pd}}^{{Sil}}\) above the DLs, the Pt and Pd concentrations in silicate melts were 3–158 ng/g and 7–44 ng/g, respectively. Most samples have \({C}_{{Pt},{Pd}}^{{Sil}}\)/DLave ratios in the range of 3–33 (DLave = average of DL values for a given sample); three samples show relatively low \({C}_{{Pt}}^{{Sil}}\)/DLave values (1–2; Supplementary Table 3). We do not believe that isobaric interferences from 179Hf16O, 155Gd40Ar, 65Cu40Ar, and 66Zn40Ar considerably affect our measured Pt and Pd concentrations in silicate melts, because our synthetical silicate is nominally free of Hf, Gd, Cu, and Zn. The Pt and Pd concentrations in silicate melts of several experiments are below DLs, as mentioned above, which indicates that any potential effects of isobaric interferences would be smaller than our DLs of Pt and Pd. In addition, any potential contributions of isobaric interferences to our measured Pt and Pd concentrations in silicate melts would reinforce our conclusions in the main text, that the \({D}_{{Pt},{Pd}}^{{Sul}/{Sil}}\) obtained at oxidizing conditions are higher than those obtained at reducing conditions.
Estimation of sample oxygen and sulfur fugacity
Our experimental oxygen fugacity (fO2) was mainly controlled by the starting materials. The fO2 for coexisting of sulfide and anhydrite in basaltic melt is generally larger than FMQ + 1.5 but lower than FMQ + 2 (refs. 35,36,37). Therefore, the fO2 of our experiments should be in a rather narrow range of fO2, FMQ + 1.5 to FMQ + 2. Our estimated S2–/Stot ratios in silicate melts, with the S2– contents in silicate melts calculated using the SCSS model of ref. 50. are also consistent with an fO2 range of FMQ + 1.5 to FMQ + 2. Our experimental sulfur fugacity (fS2) can be estimated from the P–T, fO2, and silicate melt composition using the following equation2:
where T is temperature in K, P is pressure in kbar, ΔFMQ denotes logfO2 relative to the FMQ buffer, and \({X}_{{{Fe}}_{2}{O}_{3}}\) is the total iron oxide content expressed as ferric oxide in wt%. The calculated logfS2 in our experiments ranged from 3.1 to 3.6 at fO2 of FMQ + 1.5, whereas the calculated logfS2 ranged from −1.0 to −0.6 at fO2 of FMQ − 2 for the experiments of Zhang and Li15. Consistent with the observations in previous studies17,51, the sample fS2 correlates positively with the fO2 in fO2-buffered but fS2-unbuffered experiments (Supplementary Fig. 6).
Data availability
All data supporting the findings of this study are available within the paper and Supplementary Information. All of these data are archived in Zenodo (https://doi.org/10.5072/zenodo.4242752).
References
Rehkämper, M. et al. Ir, Ru, Pt, and Pd in basalts and komatiites: new constraints for the geochemical behavior of the platinum-group elements in the mantle. Geochim. et Cosmochim. Acta 63, 3915–3934 (1999).
Mungall, J. E. & Brenan, J. M. Partitioning of platinum-group elements and Au between sulfide liquid and basalt and the origins of mantle-crust fractionation of the chalcophile elements. Geochim. et Cosmochim. Acta 125, 265–289 (2014).
Barnes, S. J., Mungall, J. E. & Maier, W. D. Platinum group elements in mantle melts and mantle samples. Lithos 232, 395–417 (2015).
Day, J. M. D., Brandon, A. D. & Walker, R. J. Highly siderophile elements in earth, mars, the moon, and asteroids. Rev. Mineralo. Geochem. 81, 161–238 (2016).
Rubie, D. C. et al. Highly siderophile elements were stripped from Earth’s mantle by iron sulfide segregation. Science 353, 1141–1144 (2016).
Barnes, S.-J. & Ripley, E. M. Highly siderophile and strongly chalcophile elements in magmatic ore deposits. Rev. Mineral. Geochem 81, 725–774 (2016).
Mungall, J. E. & Naldrett, A. J. Ore deposits of the platinum-group elements. Elements 4, 253–258 (2008).
Fleet, M. E., Crocket, J. H. & Stone, W. E. Partitioning of platinum-group elements (Os, Ir, Ru, Pt, Pd) and gold between sulfide liquid and basalt melt. Geochim. et Cosmochim.Acta 60, 2397–2412 (1996).
Stone, W. E., Crocket, J. H. & Fleet, M. E. Partitioning of palladium, iridium, platinum, and gold between sulfide liquid and basalt melt at 1200 °C - ScienceDirect. Geochim. et Cosmochim. Acta 54, 2341–2344 (1990).
Crocket, J. H., Fleet, M. E. & S, W. E. Implications of composition for experimental partitioning of platinum-group elements and gold between sulfide liquid and basalt melt: the significance of nickel content. Geochim. et Cosmochim. Acta 61, 4139–4149 (1997).
Fleet, M. E., Crocket, J. H., Liu, M. & Stone, W. E. Laboratory partitioning of platinum-group elements (PGE) and gold with application to magmatic sulfide–PGE deposits. Lithos 47, 127–142 (1999).
Bezmen, N. I., Asif, M., Brügmann, G., Romanenko, I. M. & Naldrett, A. J. Distribution of pd, rh, ru, jr, os, and au between sulfide and silicate metals. Geochim. et Cosmochim. Acta 58, 1251–1260 (1994).
Brenan, J. M., Bennett, N. R. & Zajacz, Z. Experimental results on fractionation of the Highly Siderophile Elements (HSE) at variable pressures and temperatures during planetary and magmatic differentiation. Rev. Mineral. Geochem. 81, 1–87 (2015).
Naldrett, A. J., Wilson, A., Kinnaird, J. & Chunnett, G. PGE tenor and metal ratios within and below the merensky reef, bushveld complex: implications for its genesis. J. Petro. 50, 625–659 (2009).
Zhang, M. & Li, Y. Breaking of Henry’s law for sulfide liquid-basaltic melt partitioning of Pt and Pd. Nat. Commun. 12, 5994 (2021).
Ballhaus, C. & Sylvester, P. Noble metal enrichment processes in the Merensky Reef, Bushveld Complex. J. Petrol. 41, 545–561 (2000).
Liu, Z. & Li, Y. Experimental constraints on the behavior of Pt and Re in oxidized arc magmas. Earth Planet. Sci. Lett. 603, https://doi.org/10.1016/j.epsl.2022.117986 (2023).
Anenburg, M. & Mavrogenes, J. A. Noble metal nanonugget insolubility in geological sulfide liquids. Geology https://doi.org/10.1130/g47579.1 (2020).
Frost, D. J. & McCammon, C. A. The redox state of earth’s mantle. Annu. Rev. Earth Planet. Sci. 36, 389–420 (2008).
Naldrett, A. J. in Magmatic Ni-Cu and PGE Deposits: Geology, Geochemistry, and Genesis Vol. 17 (eds Chusi Li & Edward M. Ripley) 1–50 (Society of Economic Geologists, 2011).
Barnes, S. J. et al. Sulfide-Olivine Fe-Ni exchange and the origin of anomalously Ni Rich magmatic sulfides. Econ. Geol. 108, 1971–1982 (2013).
Cao, Y., Wang, C. Y. & Wei, B. Magma oxygen fugacity of mafic-ultramafic intrusions in convergent margin settings: insights for the role of magma oxidation states on magmatic Ni-Cu sulfide mineralization. Am. Mineral. 105, 1841–1856 (2020).
Li, C., Ripley, E. M. & Naldrett, A. J. A new genetic model for the giant ni-cu-pge sulfide deposits associated with the Siberian flood basalts. Econ. Geol. 104, 291–301 (2009).
Manor, M. J., Scoates, J. S., Nixon, G. T. & Ames, D. E. The Giant Mascot Ni-Cu-PGE Deposit, British Columbia: mineralized conduits in a convergent margin tectonic setting. Econ. Geol. 111, 57–87 (2016).
Brenan, J. M. & Li, C. Constraints on oxygen fugacity during sulfide segregation in the Voisey’s Bay Intrusion, Labrador, Canada. Econ. Geol. 95, 901–915 (2000).
Li, C., Ripley, E. M., Naldrett, A. J., Schmitt, A. K. & Moore, C. H. J. G. Magmatic anhydrite-sulfide assemblages in the plumbing system of the Siberian Traps. Geology 37, 259–262 (2009).
Zhang, X., Maciag, B. J., Brenan, J. M. & Mungall, J. E. Experimental study of the partitioning of some platinum group elements (Pd and Ir) between orthopyroxene and silicate melt. Geochim. et Cosmochim. Acta 339, 127–138 (2022).
Bennett, N. R., Brenan, J. M. & Koga, K. T. The solubility of platinum in silicate melt under reducing conditions: Results from experiments without metal inclusions. Geochim. et Cosmochim. Acta 133, 422–442 (2014).
Borisov, A. & Palme, H. Experimental determination of the solubility of platinum in silicate melts. Geochim. et Cosmochim. Acta 61, 4349–4357 (1997).
Ertel, W., O’Neill, H. S. C., Sylvester, P. J. & Dingwell, D. B. Solubilities of Pt and Rh in a haplobasaltic silicate melt at 1300 °C. Geochim. Cosmochim. Acta 63, 2439–2449 (1999).
Médard, E., Schmidt, M. W., Wälle, M., Keller, N. S. & Günther, D. Platinum partitioning between metal and silicate melts: core formation, late veneer and the nanonuggets issue. Geochim. et Cosmochim. Acta 162, 183–201 (2015).
Helmy, H. M. et al. Noble metal nanoclusters and nanoparticles precede mineral formation in magmatic sulphide melts. Nat. Commun. 4, 2405 (2013).
Liang, Q.-L. et al. Thermodynamic conditions control the valences state of semimetals thus affecting the behavior of PGE in magmatic sulfide liquids. Geochim. et Cosmochim. Acta 321, 1–15 (2022).
Helmy, H. M. & Bragagni, A. Platinum-group elements fractionation by selective complexing, the Os, Ir, Ru, Rh-arsenide-sulfide systems above 1020 °C. Geochim. et Cosmochim. Acta 216, 169–183 (2017).
Li, Y., Audétat, A., Liu, Z. & Wang, F. Chalcophile element partitioning between Cu-rich sulfide phases and silicate melt and implications for the formation of Earth’s continental crust. Geochim. et Cosmochim. Acta 302, 61–82 (2021).
Jugo, P. J., Luth, R. & Richards, J. An experimental study of the sulfur content in basaltic melts saturated with immiscible sulfide or sulfate liquids at 1300 C and 1.0 GPa. J. Petrol. 46, 783–798 (2005).
Jugo, P. J., Wilke, M. & Botcharnikov, R. E. Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: implications for S speciation and S content as function of oxygen fugacity. Geochim. et Cosmochim. Acta 74, 5926–5938 (2010).
Feng, L. & Li, Y. Comparative partitioning of Re and Mo between sulfide phases and silicate melt and implications for the behavior of Re during magmatic processes. Earth Planet. Sci. Lett. 517, 14–25 (2019).
Laurenz, V. et al. The solubility of palladium and ruthenium in picritic melts: 2. The effect of sulfur. Geochim. et Cosmochim. Acta 108, 172–183 (2013).
Wang, W. & Li, Y. Platinum solubility in silicate melts: the effects of sulfur (S2–), temperature, and melt composition. Geochim. et Cosmochim. Acta 361, 113–132 (2023).
Zelenski, M. & Kamenetsky, V. S. Origin of noble-metal nuggets in sulfide-saturated arc magmas: a case study of olivine-hosted sulfide melt inclusions from the Tolbachik volcano (Kamchatka, Russia). Geology 48, 620–624 (2020).
Dare, S. A. S., Barnes, S.-J. & Prichard, H. M. The distribution of platinum group elements (PGE) and other chalcophile elements among sulfides from the Creighton Ni–Cu–PGE sulfide deposit, Sudbury, Canada, and the origin of palladium in pentlandite. Mineralium Deposita 45, 765–793 (2010).
Mansur, E. T. & Barnes, S.-J. The role of Te, As, Bi, Sn and Sb during the formation of platinum-group-element reef deposits: examples from the Bushveld and Stillwater Complexes. Geochim. et Cosmochim. Acta 272, 235–258 (2020).
Mansur, E. T., Barnes, S.-J. & Duran, C. J. An overview of chalcophile element contents of pyrrhotite, pentlandite, chalcopyrite, and pyrite from magmatic Ni-Cu-PGE sulfide deposits. Mineralium Deposita 56, 179–204 (2021).
Helmy, H. M. et al. How Pt and Pd are hosted in magmatic sulfides, substitutions and/or inclusions?. Contrib. Mineral. Petrol. 178, https://doi.org/10.1007/s00410-023-02018-8 (2023).
Maier, W. D. et al. Petrogenesis of the ~2·77 Ga Monts de Cristal Complex, Gabon: evidence for direct precipitation of Pt-arsenides from Basaltic Magma. J. Petrol. 56, 1285–1308 (2015).
Kamenetsky, V. S. et al. Crystallization of platinum-group minerals from silicate melts: evidence from Cr-spinel–hosted inclusions in volcanic rocks. Geology 43, 903–906 (2015).
González-Jiménez, J. M. et al. Magmatic platinum nanoparticles in metasomatic silicate glasses and sulfides from Patagonian mantle xenoliths. Contrib. Mineral. Petrol. 174, https://doi.org/10.1007/s00410-019-1583-5 (2019).
Longerich, H. P., Jackson, S. E. & Günther, D. Inter-laboratory note. Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation. J. Anal. Atomic Spectr. 11, 899–904 (1996).
Smythe, D. J., Wood, B. J. & Kiseeva, E. S. The S content of silicate melts at sulfide saturation: New experiments and a model incorporating the effects of sulfide composition. Am. Mineral. 102, 795–803 (2017).
Xue, S. & Li, Y. Pyrrhotite–silicate melt partitioning of rhenium and the deep rhenium cycle in subduction zones. Geology 50, 232–237 (2021).
Wang, W. & Li, Y. Supplementary Data for “Redox control of the partitioning of platinum and palladium into magmatic sulfide liquids”. Zenodo, https://doi.org/10.5072/zenodo.42427 (2024).
Acknowledgements
This study received support from the National Science Fund for Distinguished Young Scholars (Grant No. 42225302) to Y.L.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
W.W. performed the experiments and analyzed the samples as part of her Ph.D. dissertation. Y.L. conceived, designed, and supervised the study. Y.L. and W.W. interpreted the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth and Environment thanks Shitou Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Renbiao Tao and Joe Aslin. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, W., Li, Y. Redox control of the partitioning of platinum and palladium into magmatic sulfide liquids. Commun Earth Environ 5, 190 (2024). https://doi.org/10.1038/s43247-024-01366-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-024-01366-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
