
Abstract
Charge ordering creates a spontaneous array of differently charged ions and is associated with electronic phenomena such as superconductivity, colossal magnetoresistances (CMR), and multiferroicity. Charge orders are usually suppressed by chemical doping and site selective doping of a charge ordered array has not previously been demonstrated. Here we show that selective oxidation of one out of eight distinct Fe2+ sites occurs within the complex Fe2+/Fe3+ ordered structure of 2%-doped magnetite (Fe3O4), while the rest of the charge and orbitally ordered network remains intact. This ‘charge order within a charge order’ is attributed to the relative instability of the trimeron distortion surrounding the selected site. Our discovery suggests that similar complex charge ordered arrays could be used to provide surface sites for selective redox reactions, or for storing information by doping specific sites.
Similar content being viewed by others


Introduction
Charge ordering is associated with notable electronic phenomena such as superconductivity in copper1 and bismuth2 oxides, CMR in manganese oxide perovskites3 and multiferroicity in iron oxides4. Charge orders are usually suppressed by doping although in a few cases such as CMR manganese oxides, the entire array rearranges into a new periodicity5. The concept of charge ordering was first proposed to account for the Verwey transition at TV ≈ 125 K in magnetite6,7. The low-temperature order proved difficult to determine, but microcrystal X-ray diffraction has shown that the crystal structure changes from the cubic spinel type (space group \(Fd\bar 3m\)) to a complex monoclinic Cc superstructure below TV8,9. The Fe2+ sites within the Fe2+/Fe3+ charge-ordered array show a combination of orbital ordering and bonding to two Fe neighbours to form linear three-atom trimeron units, spin-polarised orbital molecules10. Orbital molecules associated with charge ordering have also been reported in other materials, such as CuIr2S411, AlV2O412,13, GaV2O414 and CaFe3O515. Complex charge orders may lead to spontaneous formation of structurally inequivalent cations of the same charge, for example, ordering of V4+/V5+ over eight inequivalent sites in α′-NaV2O516, and notably in the Cc magnetite superstructure where charges are localised over sixteen distinct octahedrally coordinated sites, eight for Fe2+ and eight for Fe3+. Inequivalent cations of the same charge should, in principle, have slightly different redox potentials, but site selective doping of a charge ordered array has not previously been demonstrated.
Previous work has shown that magnetite can be hole-doped over a small range, up to x ≈ 0.035, with suppression of TV and a change from sharp first order to broad second-order Verwey transitions reported around x = 0.01217. Equivalent effects were obtained for Fe-deficient samples Fe3(1-δ)O4 and Zn-doped materials Fe3-zZnzO4, where the hole-doping x = 3δ = z corresponds to the number of holes introduced per Fe3O4 unit. Here, we show that highly selective doping is observed in a lightly oxidised magnetite microcrystal, where one of eight distinct Fe2+ sites is preferentially oxidised, despite the disorder associated with chemical doping.
Results
Structural studies of doped magnetite microcrystals
We compare results for three magnetite microcrystals. Details of the syntheses and magnetic and structural investigations are shown in “Methods”. Sample #1 is the highly stoichiometric magnetite (x = 0) previously used to determine the Cc superstructure8, and a sharp Verwey transition at 123 K is observed in the magnetisation measurements shown in Fig. 1. Sample #2 is an Fe-deficient Fe3(1-δ)O4 material that also has a sharp transition at TV = 102 K, from which the estimated hole doping is x = 3δ = 0.0116. Zn2+ substitutes at tetrahedral cation sites that are not directly involved in the charge ordering, so a Fe3-zZnzO4 sample was used to access higher hole-doping levels. This sample #3 shows a broad Verwey transition at TV = 92 K from which doping x = z = 0.0228 is estimated.

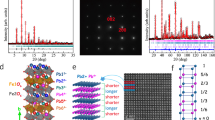
a Zero-field cooled magnetisations of the powder for sample #1 and the microcrystals #2 and #3, normalised to 140 K values. The fields used were 100 Oe (#1 and #2) and 500 Oe (#3). The Verwey transition for each sample is indicated by a vertical arrow. b Low-temperature splitting of the cubic-(440) powder X-ray diffraction peak for sample #3, demonstrating that a lattice distortion occurs at the Verwey transition between 85 and 90 K. c Thermal evolution of unit-cell parameters derived from fits to powder X-ray profiles for #3 showing the monoclinic splitting at low temperatures. Values for sample #1 at 90 K8, shown as stars, demonstrate that the magnitude of the monoclinic distortion is greatly reduced by doping.
Our samples #2 and #3, respectively, lie in the first- and second-order Verwey transition regimes. It was originally thought that second-order samples did not show a structural transition at TV, but a subsequent study reported that a distortion of the cubic cell is present in the second-order regime, modelled with a rhombohedral \(R\bar 3m\) cell18. A recent Mössbauer and room temperature diffraction study suggested a change of lattice symmetry for Fe3-zZnzO4 between first- and second-order regimes19, but no corresponding change in phonon spectra was found by inelastic neutron scattering20. A lattice distortion is clearly seen in the low-temperature powder diffraction patterns of sample #3 (Fig. 1b), and our microcrystal structural analysis shows that the same Cc supercell as for the undoped material is present. Hence, there is no change of low-temperature structural symmetry on passing from first- to second-order Verwey regimes. Instead, this anomaly may be attributed to the site-selective doping discovered below.
The crystal structures of samples #2 and #3 below their Verwey transitions were determined from X-ray diffraction data using the same microcrystal method as for #18. Both structures show the same qualitative charge ordering arrangement as for the x = 0 material. In order to quantify any small changes in the charge localisation, three local symmetry mode parameters are used, as defined in “Methods”. Qrad measures the radial expansion of the FeO6 octahedra, equivalent to the average Fe–O distance, and hence is sensitive to the size difference between Fe2+ and Fe3+ cations. The tetragonal Jahn–Teller distortion due to the orbital order of high-spin 3d6 Fe2+ is given by QJT, as defined in ref. 6. (Fe3+ is orbitally non-degenerate and so is not subject to Jahn–Teller distortion.) The shortening of the distances from each Fe2+ to its two neighbours in each trimeron is quantified by the Qtri mode. Oxidation of an Fe2+ cation within the charge-ordered array is expected to lead to an decrease in Qrad as Fe3+ is smaller than Fe2+, an increase in QJT due to loss of Fe2+ Jahn–Teller compressions that have negative QJT values, and an increase in Qtri as shortened Fe–Fe distances are lost. Hence, −ΔQrad, ΔQJT and ΔQtri are all expected to increase with hole doping of Fe2+ sites. Doping trends in these parameters mostly evolve smoothly with x as shown in Fig. 2. To display the combined changes for the 8 Fe2+ sites, the differences −ΔQrad, ΔQJT and ΔQtri between values for undoped sample #1 and for the doped samples #2 and #3 are shown as a bar chart in Fig. 3. In total, 44 of the 48 Fe2+ ΔQ values are positive, demonstrating that the structure refinements are sensitive to the small changes induced by 1–2% hole doping. Most sites have values of the summed total distortion change ΔQtot = −ΔQrad + ΔQJT + ΔQtri in a common range between 0.01 and 0.12 Å, but it is striking that one site, B42 in crystal #3, has an outlying value of ΔQtot = 0.21 Å comprising large changes in all of the radial, Jahn–Teller and trimeron modes.

Plots from top to bottom show the Qrad radial distortion parameters for all 16 octahedral B sites, the QJT tetragonal Jahn–Teller distortions for the eight Fe2+ sites, and the Qtri trimeron shortening of Fe–Fe distances around the Fe2+ sites. B-site labels shown at the top are the same as used in previous studies8,9, with the eight Fe2+ sites in blue–green symbols to the left, and the eight Fe3+ sites with yellow–brown symbols to the right. Error bars are calculated from estimated standard deviations from the refinements.

Changes in radial (−ΔQrad), Jahn–Teller (ΔQJT) and trimeron (ΔQtri) modes for the eight distinct Fe2+ sites within the charge-ordered magnetite superstructure between the undoped sample #1 and samples #2 (left hand column for each site) or #3 (right hand columns) are shown. Negative values are shown below the horizontal axis.
Chemical doping of magnetite through introduction of Fe-vacancies in sample #2 or Zn-substitution in sample #3 occurs at high temperatures, so the defects are located randomly through the lattice with respect to the charge order that emerges below TV. Hence, a statistically uniform oxidation of Fe2+ sites that happen to be adjacent to a defect is expected, and this accounts for the distribution of similar ΔQtot values for most of the Fe2+ sites in Fig. 3. However, it is evident that site B42 is substantially more oxidised in crystal #3 (site labels in the charge-ordered structure are shown on Fig. 3 of ref. 9), and this reveals a remarkable site-selectivity between the spontaneously created inequivalent Fe2+ sites within the charge-ordered magnetite structure. It is notable that this effect is seen at such a small hole-doping level. The x = 0.0228 doping of sample #3 is only around one-fifth of the x = 1/8 = 0.125 level needed to fully oxidise one of the eight distinct Fe2+ sites in the charge-ordered array, and so corresponds to oxidation on average of only 0.7 of the 32 Fe2+ sites in the monoclinic unit cell. No selective doping of B42 or any other site is evident for crystal #2 at the lower doping of x = 0.0116. Hence, it is likely that the previously reported changeover from first- to second-order Verwey transitions near x = 0.012 is due to the localisation of holes at the B42 site above this limit, although studies of further samples would be needed to confirm this. Long-range ordering of trimerons likely persists throughout the existence range of doped magnetites (up to x ≈ 0.03517), and robustness of disordered local trimeron distortions to high temperatures above TV has also been reported recently21.
The low-temperature structure of magnetite was previously shown to consist of a network of corner-sharing trimerons, in which the extra t2g electron of the central Fe2+ donor is significantly delocalised onto two adjacent B-site acceptors8. Seven of the eight structurally distinct trimerons have Fe3+ cations as the two acceptors, i.e., they have charge configuration Fe3+–Fe2+–Fe3+. However, the B42 site is unique in having a Fe2+–Fe2+(B42)–Fe3+ configuration, where another Fe2+ acts as a terminal charge acceptor. The present discovery that B42 is more easily oxidised that other Fe2+ sites can thus be attributed to the anomalous nature of the B42 trimeron. Electron–electron repulsion between the localised electrons of the adjacent Fe2+’s in this trimeron lowers the potential for ionisation of the B42 site relative to other Fe2+ ions.
Discussion
The above results show that the complex spontaneous charge ordering in magnetite creates electron-localised Fe2+ sites of distinguishable redox activity. Although the charge-ordered superstructure of magnetite is very complex, accurate determination of the many variable structural parameters gives exquisite sensitivity to tiny changes in local charge states through the local radial, Jahn–Teller and trimeron-distortion modes. The discovered doping selectivity is remarkable, as it corresponds to a ‘charge order within a charge order’. It is also notable that the rest of the charge, orbital and trimeron network remains robust while the B42 sites are oxidised, at least up to the 2% doping in this study. This suggests that similar complex charge-ordered arrays could be used to provide surface sites for selective redox reactions, or for storing information by doping-specific sites. It is notable that the B42 trimerons lie parallel to the cubic-(100) plane of magnetite which can be grown as thin films, so it might be possible to access and directly measure the difference in ionisation potential between the B42 and other in-plane trimerons (Fig. 4b) using scanning nanoprobe microscopies.

a Trimeron distribution in the low-temperature magnetite Cc cell, with Fe2+/Fe3+ shown as blue/yellow spheres and trimeron ellipsoids in green. The selectively oxidised B42 site is labelled, and the unusual termination of its trimeron by another Fe2+ is marked by a square. b (100) surface of cubic magnetite showing trimerons in the surface layer. The B42 sites (blue and white ‘beachballs’) are expected to be oxidised in preference to other trimerons. Charge-ordered Fe2+/Fe3+ are shown as blue/yellow spheres, tetrahedral Fe/O sites are brown/red.
In summary, this work demonstrates that it is possible to selectively dope one site within the spontaneously created charge-ordered array of magnetite without disrupting the rest of the electronically ordered network. This opens new possibilities to create a ‘charge order within a charge order’, and such materials could be used to provide surface sites for selective redox reactions, or for storing information by doping-specific sites.
Methods
Sample preparation
Sample #1 is a highly stoichiometric magnetite Fe3(1-δ)O4 with 3δ = 0.0000 originally provided by Prof. J. Honig that has been used in previously studies8,9,21. Samples #2 and #3 were synthesised through a modified route from Honig17. Small microcrystals of Fe3(1-δ)O4 (sample #2) or of Fe3-zZnzO4 (sample #3) were obtained through firing Fe2O3 or an Fe2O3/ZnO mixture at 1580 °C under Ar flow. The stoichiometry of sample #2 Fe3(1-δ)O422 was adjusted by reannealing the crystals in a vertical furnace with a fixed CO2/H2 buffer gas flow, to control the oxygen partial pressure according to the thermodynamic equilibrium of magnetite17 and reference tables derived from the JANAF database23.
Magnetisation
Magnetic measurements were recorded with a Quantum Design SQUID magnetometer after zero-field cooling in fields of 100 Oe (samples #1 and #2) or 500 Oe (sample #3) during warming. Microcrystals were fixed in Eicosane within the gelatine capsule. Hole-doping levels x = 0.0116 and 0.0228 of samples #2 and #3, respectively, were estimated from their measured Verwey transition temperatures using the TV-x doping correlation plot in ref. 17.
X-ray diffraction
Microcrystal X-ray diffraction data were collected on the diffractometer ID11 at ESRF (France). The wavelengths were all close to λ = 0.16 Å. The microcrystals (40–60 μm) were suspended in paratone oil in a litholoop. Cooling in a magnetic field from ambient temperature to 80–90 K (as described in ref. 6) was used to align the magnetic easy axis of the crystal with the field, thus helping to create a unique monoclinic c-axis below the Verwey transition. Temperature was controlled with a nitrogen cryostream. Data sets were recorded at 90 K (samples #1 and #2) or 80 K (sample #3), with varying omega-step sizes, exposure times, beam fluxes and at different phi settings, for high redundancy and to observe both low angle fundamental diffraction peaks and high-angle weak superstructure reflections.
Data reduction and integration for sample #2 were performed using the same procedure as for #1 in ref. 8, via Bruker SMART and SAINT software24. For crystal #3, data were indexed and integrated using CrysalisPro25 using an increased integration box (×2), and a filter for reflections above 50,000 counts to ensure detector linearity. Integrated results for each collection run were then processed using Sadabs26, applying a spherical absorption correction. The data were finally merged for refinement using Xprep27 to a maximum resolution of 0.28 Å. Structure refinements were carried out using SHELXL software28 and took both monoclinic and orthorhombic twin domains into account (excluding inversion twinning). Samples #2 and #3 were found to show substantial monoclinic twinning. All 168 of the variable atomic coordinates were refined independently, subject to the origin constraints of space group Cc.
High-resolution powder diffraction patterns for sample #3 were recorded at the ID22 beamline of ESRF from 55 to 325 K using a helium cryostat with λ = 0.354224 Å. Rietveld refinements were performed using Fullprof software29, and the monoclinic unit-cell parameters at 80 K were used as the values for the single-crystal refinement.
Distortion modes
Local distortion mode parameters are used to quantify the magnitude and changes of local distortions of the low-temperature magnetite structure with doping. Distortions of FeO6 octahedra have been previously described by local modes Q in the Oh point group8.
The radial expansion (breathing) mode Qrad is calculated from Eq. (1).
where di are the six Fe–O distances in the octahedron and <d> is the global average octahedral Fe–O distance. This breathing mode describes changes in the cation size, and so is sensitive to charge ordering of large Fe2+ against small Fe3+.
The orbital ordering is characterised by the tetragonal Jahn–Teller mode QJT, calculated as described in Supplementary Information to ref. 8.
A trimeron mode Qtri sensitive to Fe–Fe distances is similarly defined for each Fe2+ in Eq. (2).
This is summed over the two Fe–Fe distances BBi within the trimeron, where <BB> is the average of all nearest-neighbour B–B distances within the structure.
Data availability
Data that support the findings of this study have been deposited at https://doi.org/10.7488/ds/2783.
References
Keimer, B., Kivelson, S. A., Norman, M. R., Uchida, S. & Zaanen, J. From quantum matter to high-temperature superconductivity in copper oxides. Nature 518, 179–186 (2015).
Sleight, A. W. Bismuthates: BaBiO3 and related superconducting phases. Phys. C. 514, 152–165 (2015).
Dagotto, E. Open questions in CMR manganites, relevance of clustered states and analogies with other compounds including the cuprates. N. J. Phys. 7, 67 (2005).
Van Den Brink, J. & Khomskii, D. I. Multiferroicity due to charge ordering. J. Phys. Condens. Matter 20, 434217 (2008).
Loudon, J. C. et al. Weak charge-lattice coupling requires reinterpretation of stripes of charge order in La1−xCaxMnO3. Phys. Rev. Lett. 94, 097202 (2005).
Verwey, E. J. W. Electronic conduction of magnetite (Fe3O4) and its transition at low temperatures. Nature 144, 327 (1939).
Walz, F. The Verwey transition—a topical review. J. Phys.: Condens. Matter 14, R285–R340 (2002).
Senn, M. S., Wright, J. P. & Attfield, J. P. Charge order and three-site distortions in the Verwey structure of magnetite. Nature 481, 173–176 (2012).
Senn, M. S., Loa, I., Wright, J. P. & Attfield, J. P. Electronic orders in the Verwey structure of magnetite. Phys. Rev. B 85, 125119 (2012).
Attfield, J. P. Orbital molecules in electronic materials. APL Mater. 3, 041510 (2015).
Radaelli, P. G. et al. Formation of isomorphic Ir3+ and Ir4+ octamers and spin dimerization in the spinel CuIr2S4. Nature 416, 155–158 (2002).
Horibe, Y. et al. Spontaneous formation of vanadium “molecules” in a geometrically frustrated crystal: AlV2O4. Phys. Rev. Lett. 96, 086406 (2006).
Browne, A. J., Kimber, S. A. J. & Attfield, J. P. Persistent three- and four-atom orbital molecules in the spinel AlV2O4. Phys. Rev. Mater. 1, 052003(R) (2017).
Browne, A. J., Lithgow, C., Kimber, S. A. & Attfield, J. P. Orbital molecules in the new spinel GaV2O4. Inorg. Chem. 57, 2815–2822 (2018).
Hong, K. H., Arevalo-Lopez, A. M., Cumby, J., Ritter, C. & Attfield, J. P. Long range electronic phase separation in CaFe3O5. Nat. Commun. 9, 2975 (2018).
Sawa, H. et al. Low-temperature structure of the quarter-filled ladder compound α‘-NaV2O5. J. Phys. Soc. Jpn. 71, 385–388 (2002).
Honig, J. M. Analysis of the Verwey transition in magnetite. J. Alloy. Compd. 229, 24–39 (1995).
Kakol, Z. et al. The effect of doping on global lattice properties of magnetite Fe3-xMexO4 (Me=Zn, Ti and Al). J. Solid State Chem. 192, 120–126 (2012).
Chlan, V. et al. Effect of low doping on the Verwey transition in magnetite single crystals: Mössbauer spectroscopy and x-ray diffraction. Phys. Rev. B 98, 125138 (2018).
Kolodziej, T. et al. Nuclear inelastic scattering studies of lattice dynamics in magnetite with a first- and second-order Verwey transition. Phys. Rev. B 85, 104301 (2012).
Perversi, G. et al. Co-emergence of magnetic order and structural fluctuations in magnetite. Nat. Comm. 10, 2857 (2019).
Wriedt, H. A. The Fe-O (iron-oxygen) system. J. Phase Equilib. 12, 170–200 (1991).
Deines, P., Nafziger, R. H., Ulmer, G. C. & Woermann, E. Temperature-oxygen fugacity tables for selected gas mixtures in the system C-H-O at one atmosphere total pressure. Bull. Earth Miner. Sci. 88, 1–129 (1974).
Bruker, SMART and SAINT (Bruker AXS Inc., Madison, Wisconsin, 2012).
Agilent, CrysAlis PRO (Agilent Technologies Ltd, Yarnton, Oxfordshire, 2014).
Sheldrick, G. M. Sadabs (University of Göttingen, Germany, 1996).
XPREP 2014. X-ray Data Preparation and Reciprocal Space Exploration Program (Bruker AXS Inc., Madison, Wisconsin, 2014).
Sheldrick, G. M. A short history of SHELX. Acta Cryst. A 64, 112–122 (2008).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B 192, 55–69 (1993).
Acknowledgements
We thank Prof. J. Honig for providing the stoichiometric magnetite sample, Dr. Ian B. Butler for assistance with the gas mixing furnace, and Dr. Catherine Dejoie for help with data collection at ID22. Financial support from ERC and EPSRC is acknowledged. Access to ESRF was provided by STFC.
Author information
Authors and Affiliations
Contributions
E.P., J.C., G.P. and J.P.A. designed the concept for this study. E.P., J.C., G.P. and J.P.W. performed the experimental work and data analysis. E.P. and J.P.A. wrote the paper with inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Sian Dutton, Andrzej Oleś and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pachoud, E., Cumby, J., Perversi, G. et al. Site-selective doping of ordered charge states in magnetite. Nat Commun 11, 1671 (2020). https://doi.org/10.1038/s41467-020-15504-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-15504-5
This article is cited by
-
Slow oxidation of magnetite nanoparticles elucidates the limits of the Verwey transition
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
