
Abstract
Developmental epileptic encephalopathies are devastating disorders characterized by intractable epileptic seizures and developmental delay. Here, we report an allelic series of germline recessive mutations in UGDH in 36 cases from 25 families presenting with epileptic encephalopathy with developmental delay and hypotonia. UGDH encodes an oxidoreductase that converts UDP-glucose to UDP-glucuronic acid, a key component of specific proteoglycans and glycolipids. Consistent with being loss-of-function alleles, we show using patients’ primary fibroblasts and biochemical assays, that these mutations either impair UGDH stability, oligomerization, or enzymatic activity. In vitro, patient-derived cerebral organoids are smaller with a reduced number of proliferating neuronal progenitors while mutant ugdh zebrafish do not phenocopy the human disease. Our study defines UGDH as a key player for the production of extracellular matrix components that are essential for human brain development. Based on the incidence of variants observed, UGDH mutations are likely to be a frequent cause of recessive epileptic encephalopathy.
Similar content being viewed by others
Introduction
Developmental epileptic encephalopathies are a clinically and genetically heterogeneous group of devastating disorders characterized by severe epileptic seizures that are accompanied by developmental delay or regression1. In several cases, a genetic etiology has been identified2. Germline mutations in these genes lead to different pathophysiological defects2, including ion channel dysfunction, synaptic impairment, transporter defects and metabolic abnormalities, such as deficiencies in glycosylation pathways3,4,5. However, the genetic cause of many epileptic encephalopathies remains unknown.
Defects of glycosylation are causing more than 100 rare human genetic disorders, most of these affecting the central and/or peripheral nervous systems. Patients typically show developmental delay or intellectual disability, seizures, neuropathy, and metabolic abnormalities in multiple organ systems3. Adding the correct sugar chains (glycans) to proteins and lipids significantly impacts their function. UGDH (MIM603370) codes for an enzyme that converts UDP-glucose (UDP-Glc) to UDP-glucuronic acid (UDP-GlcA) through the concomitant reduction of NAD+ into NADH6,7. UDP-GlcA is not only needed for detoxification via glucuronidation, but is also an obligate precursor for the synthesis of glycosaminoglycans (GAGs), and therefore an important component of proteoglycans of the extracellular matrix.
In this study, we establish UGDH as a gene responsible for autosomal recessive developmental epileptic encephalopathy in humans. We catalog a series of 30 patients from 25 families with biallelic germline UGDH variants. Using patients’ primary fibroblasts and biochemical assays, we demonstrate that these are loss-of-function alleles. While mutant ugdh zebrafish did not phenocopy the disease, we bring evidence that patient-derived cerebral organoids, which were smaller due to a reduced number of proliferating neuronal progenitors, can serve as an alternative disease-in-a-dish model for in vitro functional studies.
Results
Biallelic mutations in UGDH cause developmental epileptic encephalopathy
To identify the genetic cause of a developmental epileptic encephalopathy in a consanguineous Palestinian family with three affected siblings (Fig. 1a, F1), we performed exome sequencing on two affected siblings. No mutations in genes known to be associated with neurological disorders (either recessive or dominant) were found. As the consanguineous background and the pedigree suggested autosomal recessive inheritance, we focused on homozygous or compound heterozygous variants shared by the affected siblings. A rare homozygous variant c.131C > T in UDP-Glucose 6-Dehydrogenase (UGDH), which changes alanine into valine at position 44 of the UGDH protein, was the only segregating candidate variant. The UGDH p.A44V missense affects a highly conserved residue (Suppl. Fig. 1b and phyloP 100-way8 score 9.43), is extremely rare in public databases (not present in EVS65009, MAF of 0.0017% in ExAC10) and is a good candidate according to in silico prediction scores (CADD score11 of 33) (Suppl. Table 1). We then (i) screened the GENESIS12 database for additional patients with recessive UGDH variants, (ii) contacted the EuroEPINOMICS RES Consortium, and (iii) searched with the help of GeneMatcher13 for additional families with germline UGDH mutations. We uncovered 27 additional patients from 24 families carrying either compound heterozygous or homozygous UGDH variants (Fig. 1a and Suppl. Fig. 1a). All variants were absent or had an extremely low frequency (<0.01%) in the public databases ExAC/gnomAD10 and EVS6500 (Suppl. Table 2). Nineteen of the 20 identified missense variants are in highly conserved residues (Suppl. Fig. 1b and phyloP 100-way between 3.81 and 9.43). The A44V variant, identified in the Palestinian index family, was also found in two additional families from Puerto Rico (F11) and from Spain (F13) indicative of independent but recurrent mutation in this residue. In ExAC the A44V variant is observed in African (MAF 0.0096%) and European (Non-Finish) populations (MAF 0.0015%), however, it is not present in the Greater Middle East Variome.

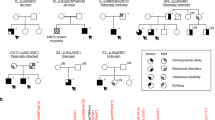
a Pedigrees of 19 families segregating autosomal recessive developmental epileptic encephalopathy. Countries of origin are specified above each pedigree. Filled black symbols, affected individuals. Crossed symbols, deceased individual. Mutations in UGDH protein are presented below pedigrees. Homozygous mutations are presented in bold (m in the pedigrees). Compound heterozygous mutations are presented according to the parental origin of the mutation with a maternal origin in the first row (m1 in the pedigrees), and a paternal, de novo or unknown origin in the second row (m2 in the pedigrees). Healthy siblings that could be sequenced are heterozygous [F6-II:2 (p.Arg65*), F11-II:2 (p.Ala44Val), and F18-II:3 (p.Arg317Gln)]. b Facial photographs of 14 affected individuals with mild craniofacial dysmorphisms, including short and flattened philtrum, protruding earlobes, ptosis blepharophimosis, and epicanthic folds. c Spectrum of MRI findings in exemplary patients showing no evidence for maldevelopment but displaying variable abnormalities ranging from abnormal myelination and/or cerebral or cerebellar atrophy, to normal findings. Patient F5-II:2 presented with a normal MRI, including normal myelination at 2 years of age. In contrast, MRI of patient F3-II:1 revealed some myelination of cerebellar peduncles at 5 months (arrow) and no progress of myelination on follow-up at 15 months, indicative of hypomyelination. In addition, repeated MRI revealed enlarged posterior ventricles over time (arrow heads). MRI of patient F6-II:1 at 7 days of age also proved normal, the circle indicates onset of myelination in the Posterior Limb of the Internal Capsule (PLIC) according to age. Patient F7-II:1 showed mild cerebellar atrophy at 4 years of age. Patient F9-II:1 showed slightly delayed myelination on axial T2 and cerebellar atrophy on coronal and sagittal T1 images (stars). Patient F14-II:1 showed a diffuse cerebral atrophy, ventriculomegaly, thin corpus callosum, vermian, and lobar cerebellar atrophy, with normal brainstem, hyperintensity of cerebellar cortex in T2-weighted images (white square). Patient F15-II:1 presented with normal MRI at 5 months, but with severe diffuse atrophy, bilateral symmetrical hyperintensities of thalami and globus pallidus (white square) at 8 months old. In all pictures, MRI pulse sequences (T1, T2, and Flair) and image orientation (S: sagittal, A: axial and C: coronal) are indicated in the upper left corner.
All 30 patients carrying biallelic mutations in UGDH presented with a common core phenotype consisting of marked developmental delay, epilepsy, mild dysmorphism, and motor disorder with axial hypotonia (Table 1 and Suppl. Data 1). Dysmorphic facial features such as short and flattened philtrum, outward protruding earlobes, ptosis, or blepharophimosis were mild but frequently present (Fig. 1b and Suppl. Data 1). Most patients have severe epilepsy ranging from neonatal onset developmental epileptic encephalopathy to infantile developmental epileptic encephalopathy (27 patients, 90%), of which 16 (53%) had infantile spasms (Table 1). Three patients have developmental encephalopathy, of which two had seizures in the setting of fever (F5-II:1 and F5-II:2). Only these two patients were seizure-free on sodium valproate. All other patients, except for one patient who seemed to benefit from ketogenic diet, did not respond to antiepileptic treatment. All patients had a severe motor disorder with axial hypotonia, while some patients presented with limb spasticity (43%), dystonia (17%), ataxia, chorea, and tremor, which were often present prior to onset of seizures. Twenty-four out of the 30 (80%) were noted to have swallowing difficulties and gastrostomy tubes were required for feeding in 12 infants. None but two patients (F5-II:1 and II:2) achieved sitting ability. A moderate to severe intellectual disability was observed in all patients. Three patients were deceased between 4 months and 6 years of age (Table 1 and Suppl. Data 1). Electroencephalography (EEG) was markedly abnormal with a burst suppression pattern in the neonatal period, hypsarrhythmia in affected children with infantile spasms, and focal and/or generalized spike-wave complexes in childhood (Suppl. Data 1). MRI revealed a spectrum of abnormalities with delayed myelination and enlarged ventricles probably due to cerebral and cerebellar atrophy in more severely affected patients without any signs of maldevelopment (Fig. 1c and Suppl. Data 1).
UGDH mutations behave as hypomorphic alleles
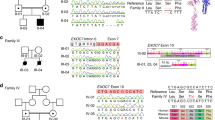
The UGDH oxidoreductase consists of three distinct domains14: the NAD-binding (N-terminal) and UDP-binding (C-terminal) domains, and an internal domain that bridges the two termini together14. The UGDH enzyme assembles into a disc-shaped double layer composed of a trimer of dimers6 (Suppl. Fig. 2a, b). This hexameric structure is a prerequisite for proper UGDH enzymatic function15. The 23 germline mutations presented in this study are distributed throughout the UGDH gene and its encoded protein (Fig. 2a). One of the variants in Family 12 mutates the first nucleotide of exon 8 (c.907 G > A; p.Val303Ile, Fig. 2a, b), which is predicted to affect the splice donor site16. Three different nonsense mutations were found in a compound heterozygous state with a missense mutation (Fig. 2a and Suppl. Table 2). All identified missense mutations are anticipated to be destabilizing according to DUET17 (Suppl. Table 2, ΔΔG). The missense mutations in residues Y14, I42 and A44, which are close to the NAD-binding site (Fig. 2c and Suppl. Fig. 2c) are expected to impair NAD+ reduction. Alteration of residues in the central domain such as I255, G271, M306, and R317 are expected to affect homo-dimerization18 (Fig. 2b and Suppl. Fig. 2d). The I116 residue (located in the NAD-binding domain), as well as the R393 and A410 residues (UDP-Glc binding domain) sit at the dimer-dimer interface19 (Fig. 2d), suggesting that these variants may prevent UGDH from assembling into a functional hexameric enzyme.

a UGDH genomic and protein domain structures. Type and positions of 22 germline UGDH mutations. 5′ and 3′ UTRs are shown in dark gray. NAD-binding (blue), central (pink), and UDP-binding (orange) domains are highlighted. Homozygous mutations are shown in bold. Compound heterozygous mutations that are in trans are linked by a line below the UGDH domain structure. b–d Three close-up views ribbon diagrams of the UGDH protein bound to UDP-Glc and NADH. b Interface between the central domains of subunits A and B. c NAD-binding site in NAD-binding domain of subunit A. Distances between NADH and mutated residues in patients are measured in Angström (Å). d Interface between the subunit A NAD-binding domain with the subunit C UDP-Glc-binding domain. In all the structures, residues carrying missense mutations in the patients are highlighted as 3D backbone. Residues Q110 and T325 known to interact together for dimer formation15; and residue V132, which is important for hexamerization15 are highlighted in black backbone. In all the structures, NAD-binding (blue), central (light/dark pink), and UDP-binding (orange) domains are shown. UDP-Glc (dark red) and NADH (midnight blue) are represented as colored carbon backbones. Adapted from PDB code 2Q3E6 using the Swiss-Pdb Viewer software67. For gels and graphs source data, please refer to the source data files 1 and 2.
To better understand the effect of the mutations on UGDH, we then derived and biobanked primary dermal fibroblasts from patients F3-II:1 (R393W/A410S), F4-II:1 (Y14C/S72P), F5-II:1 (A82T/A82T) and F6-II:1 (R65*/Y367C), and a non-affected parent F5-I:1 (WT/A82T). Endogenous UGDH messenger RNA (mRNA) levels were not significantly different in patients’ primary cells as compared to control fibroblasts (Fig. 3a, top panels). In contrast, we observed significant changes in endogenous UGDH protein levels for three of the four alleles studied. Fibroblasts with compound heterozygous R393W/A410S mutations displayed comparable UGDH levels relative to wild-type (WT) cells, while patients’ cells with R65*/Y367C, Y14C/S72P, or homozygous A82T mutations showed dramatically reduced endogenous UGDH levels (Fig. 3a, bottom panels). In contrast to the three nonsense mutations and the missense mutation potentially affecting splicing, which are likely to cause nonsense-mediated decay of the endogenous UGDH transcript, the missense mutations are most likely impacting the stability of the enzyme and/or its oxidoreductase activity. Consistently, we observed a significant decrease in the UGDH-catalyzed reduction of NAD+ to NADH in patients’ primary fibroblasts (R393W/A410S, Y14C/S72P, or homozygous A82T mutations) while the non-affected parent’s cells heterozygous for the A82T mutation showed intermediate level of NAD+ reduction (Fig. 3b, left panels). Patient’s cells with the homozygous A82T mutation also exhibited a reduction in the synthesis of hyaluronic acid (HA), which requires UDP-glucuronate, a product of UGDH enzymatic activity (Fig. 3b, right panel). When produced in bacteria, mutant UGDH had altered stability, kinetic, and biochemical properties as compared to WT-UGDH. Compared to the wild-type enzyme, mutant A44V and A82T UGDH (mutations found at the homozygous state in the patients from Families 1 and 5, respectively) were more susceptible to partial proteolysis by trypsin (Fig. 3c). The stability of UGDHA44V could be partially rescued upon incubation with substrate, product, or cofactor, while the UGDHA82T remained strongly sensitive to proteolysis regardless of the presence of any cofactor or substrate (Fig. 3c). A thermal stability study showed that the melting temperature of UGDHA44V was significantly reduced relative to WT and could only partially be rescued upon addition of substrate, product, reduced or oxidized cofactor, or any combination thereof (Fig. 3d). Notably, UGDHA82T was so intrinsically unstable that a melting temperature was unable to be ascertained. By gel filtration chromatography, we investigated the effect of the A44V and A82T mutations on UGDH oligomerization. When compared to UGDHWT, UGDH∆132 (an obligate hexamer15), and UGDHT325D (an obligate dimer15), we observed that UGDHA44V and UGDHA82T proteins were mainly eluted as dimer and monomer species, respectively, with virtually no stable hexameric population (Fig. 3e). This suggests that the A44V and A82T mutations may affect UGDH function by altering its capacity to form active hexamers. Finally, using equal amounts of recombinant enzyme, we determined that UGDHA44V and UGDHA82T were respectively 75 and 50% less efficient at reducing NAD+ to NADH as compared to UGDHWT (Fig. 3f). Similarly, comparison of the steady state Michaelis–Menten kinetic constants (summarized in Table 2) showed that UGDHA44V Vmax was only ~ 50% of the value of UGDHWT for both cofactor and substrate. In contrast, Km was not significantly different from UGDHWT for either cofactor or substrate, revealing that the mutation results in a reduced ability of the enzyme to catalyze the reaction, while still being able to associate with NAD+ and UDP-Glc. Taken together, our biochemical findings indicate that these missense mutations mainly impact the enzymatic function of UGDH by altering its quaternary structure and/or directly impairing its oxidoreductive activity.

a RT-qPCR (top), western blotting (bottom), and b enzymatic activity, assessed by measuring NADH production (left panel) and quantification of HA (right panel), for endogenous UGDH using patient-derived primary fibroblasts. a, b Control (WT/WT), unaffected mother F5-I:1 (WT/A82T) and 4 (in a) or 3 (in b) different patients’ fibroblasts (F5-II:1: A82T/A82T, F3-II:1: R393W/A410S, F4-II:1: Y14C/S72P, and F6-II:1: R65*/Y367C). a (top) Endogenous UGDH mRNA levels are normalized to β-ACTIN and GAPDH. Fold change relative to control (WT/WT) is plotted. a (bottom) Western blot analysis for endogenous UGDH protein using cellular extracts. GAPDH is used as a loading control. b (left) UGDH enzymatic activity measured as the conversion of NAD+ to NADH in whole-cell lysates. b (right) UGDH enzymatic activity measured as the HA production in conditioned media from primary fibroblast cultures. c Western blot analysis for UGDH sensitivity to limited proteolysis using purified WT and mutant (A44V and A82T) UGDH proteins in the absence or presence of its substrates and/or cofactors, as indicated. Results are representative of at least three experimental replicates. d Purified UGDH WT and A44V melting temperature (Tm) in the absence or presence of its substrates and/or cofactors, as indicated. Mean of three experiments ± S.D. is plotted for the Tm of each enzyme. e Representative traces at λ = 280 nm of purified WT and mutant UGDH proteins fractionated by size exclusion chromatography. WT, obligate dimer ∆13215, obligate hexamer T325D15, A44V and A82T UGDH are plotted in the graph. Dashed lines correspond to the known hexamer, dimer and monomer peak elution times. f Purified WT, A44V, and A82T UGDH enzymatic activity measured as the conversion of NAD + to NADH. Asterisks indicate p-values of p < 0.05(*), p < 0.01(**), and p < 0.001(***), NS: non-significant (p > 0.05) as determined by Student t-test. For gels and graphs source data, please refer to the source data files 1 and 2.
Patient-derived cerebral organoids partially phenocopy human disease
Our attempts to model this developmental epileptic encephalopathy using the existing zebrafish hypomorphic loss-of-function ugdh (c.992 T > A; p.I331D) allele known as jekyll m15120,21,22 were unsuccessful (Suppl. Fig. 3). The behavioral activity of homozygous jekyll mutant larvae were recorded in presence or absence of the seizure-inducing drug pentylenetetrazol (PTZ)23. By quantitative PCR (qPCR), c-fos expression, which marks neural activity23,24, similarly increased in a dose-dependent manner upon PTZ treatment in all larvae regardless of genotypes (Suppl. Fig. 3a–d). Homozygous mutant larvae did not show signs of increased c-fos expression at basal state, suggesting that fish depleted of Ugdh activity do not exhibit spontaneous seizure and are equally responsive to PTZ treatment. As noted by reviewers, ugdh mutant fish do not have fully-inflated swim bladders, which may contribute to their reduced locomotor activity and demise before 14-dpf (Suppl. Fig. 3e). These in vivo experiments suggest that zygotic ugdh depletion in zebrafish does not satisfactorily model the human disease.
UGDH has been extensively studied in vertebrate model organisms where its complete knockout causes embryonic lethality around gastrulation25,26. To address its role in the context of central nervous system (CNS) development in humans, we attempted instead to model this disease in vitro by developing cerebral organoids27,28 from several patients with compound heterozygous R65*/Y367C, Y14C/S72P or homozygous A82T mutations, and from a non-affected parent (WT/A82T). After 10 weeks of differentiation, the volume of cerebral organoids from patients with biallelic UGDH mutations was on average 50% smaller and showed rougher edges than that of WT or carrier WT/A82T cerebral organoids (Fig. 4a, b and Suppl. Fig. 4a). Quantitative reverse transcription PCR (RT-qPCR) analysis revealed decreased levels of the early and intermediate neuronal progenitors markers PAX6 and TBR2, respectively, while the levels of neuronal marker TUJ1 were unchanged (Fig. 4c and Suppl. Fig. 4b). Immunofluorescence revealed similar amounts of peripheral neurons marked by TUJ1, and astrocytes marked by GFAP while ventricular zones marked by SOX2-positive neuronal progenitors were appreciably less proliferative. This was evidenced by reduced PCNA staining in mutant cerebral organoids relative to WT and WT/A82T sections (Fig. 4d and Suppl. Fig. 4c). These results argue that reduced UGDH activity is associated with impaired neuronal development in vitro, causing atrophy of patient-derived cerebral organoids. Even though our cerebral organoid data is congruent with our patients’ phenotype and biochemistry data, replicative studies with additional WT and complete UGDH knockout lines are warranted in light of the known variability in induced pluripotent stem cells (iPSCs’) response to differentiation protocols.

a Volumes (mean ± SD) and b representative images (scale bar = 1 mm) of cerebral organoids derived from iPSCs from WT (n = 18 organoids from the same batch), unaffected parent (UGDH WT/A82T, n = 15), and patients (UGDH A82T/A82T (n = 10), Y14C/S72P (n = 7), and R65*/Y367C (n = 6) after 10 weeks of differentiation. Lower right panel: close-up views of the edges of indicated cerebral organoids. Scale bar = 500 μm. c RT-qPCR for neuronal differentiation markers (PAX6, TBR2, and TUJ1) in WT (n = 4 cerebral organoids), unaffected parent (WT/A82T, n = 3), and patients (A82T/A82T, Y14C/S72P, and R65*/Y367C, n = 3 each) cerebral organoids. Levels of expression are normalized to GAPDH. Mean ± SD fold change relative to WT is plotted. d Representative images of consecutive sections of cerebral organoids derived from iPSCs from WT (N = 5 cerebral organoids, n = 40 ventricle-like zones), unaffected parent (WT/A82T, N = 4, n = 15), and patients (A82T/A82T N = 3, n = 40, Y14C/S72P N = 4, n = 18, and R65*/Y367C N = 2, n = 9) stained with H&E, and immunostained with markers TUJ1/PCNA/DAPI, SOX2/DAPI, and GFAP/DAPI. Scale bar = 100 μm. a, c Asterisks indicate p-values of p < 0.05(*), p < 0.001(***), NS: non-significant (p > 0.05) as determined by ANOVA test with Bonferroni correction. a–c Cerebral organoids represented here are all from batch 2 and derived from iPSCs clone 1 for each genotype, see Suppl. Fig. 4 for more information. For graphs source data, please refer to the source data file 2.
To understand whether mutations in UGDH directly affect neuronal function, we also differentiated WT, non-affected parent (WT/A82T), and patient (Y14C/S72P) iPSCs into neuro-precursor cells (NPCs), which were subsequently matured into neurons over a period of 21 days. Using a multi-electrode array (MEA) system, and in contrast to neurons mutant for CAMK2A29, no significant differences between controls and mutant UGDH neurons were recorded for either the total number of spontaneous spikes or the mean firing rate (Suppl. Fig. 4d). Altogether, these in vitro experiments suggest that while UGDH is not required for proper function of isolated neurons in culture, its absence significantly affects neuronal differentiation in cerebral organoids, which may provide a powerful platform to study the pathogenesis for the new disease in vitro.
Discussion
In this study, we described disease-causing mutations in UGDH in humans. These 23 coding variants represent an allelic series of germline mutations, which when inherited recessively are responsible for epileptic encephalopathy with variable degrees of developmental delay. We propose to name this novel Mendelian disease Jamuar Syndrome, a member of the early infantile epileptic encephalopathies (EIEE). The genetic, biochemical, cellular and developmental findings reveal that these UGDH germline mutations behave as loss-of-function alleles. This was confirmed in vitro using patient-derived cerebral organoids, which showed marked underdevelopment. In zebrafish, we found that hypomorphic ugdhI331D/I331D mutant larvae did not show signs of increased seizures at baseline or after PTZ treatment. The brain-specific UGDH phenotype in humans may come as a surprise since in Drosophila, zebrafish, and mouse, complete knockout of Ugdh cause early and lethal gastrulation defects by hindering FGF signaling25,30,31,32. One potential explanation for this incongruity is that other proteoglycans not reliant on UGDH activity for the synthesis of UDP-GlcA or UDP-Xylose (UDP-Xyl) may be solicited and help bypass the need for UGDH during human gastrulation. Alternatively, a complete knockout of UGDH in humans may not be viable as we did not identify any homozygous or compound heterozygous truncating mutations. A search in ExAC for homozygous variants resulted in mostly synonymous or non-coding variants. Only two homozygous missense variants were detected, but no homozygous truncating mutations were seen (Suppl. Table 3). This suggests that the severity of the epileptic encephalopathy may correlate with the amount of residual UGDH activity, the extent of which may be sufficient to allow gastrulation to take place during early human embryonic stages but may be limiting for neuronal development thereafter.
As UDP-GlcA is the major product of the UGDH enzyme, it is possible that reduced levels of UDP-GlcA may trigger a cascade of secondary pathogenic events resulting in neurodevelopmental delay and encephalopathy. In support of this, is the recent demonstration that a homozygous loss-of-function mutation in the upstream enzyme UGP2 is also responsible for a severe form of developmental epileptic encephalopathy in humans33. UDP-GlcA is not only needed for detoxification via glucuronidation, but is also a key component of glycosaminoglycans (GAGs). UGDH deficiency might parralel other neurological diseases with defects in GAG synthesis, modification, and degradation. For example, EXTL3 and CHSY1 mutations, which affect heparan sulfate and chondroitin sulfate synthesis, respectively, cause developmental delay and intellectual disabilities34,35. Defects in heparan sulfate modification caused by NDST1 mutations are responsible for intellectual disability associated with epilepsy36. Moreover, mucopolysaccaridoses, diseases caused by defects in GAG degradation, affect cognitive development37. In addition, proteoglycans containing GlcA derived from UDP-GlcA are major components of the extracellular matrix (ECM) and key players in neuronal development and plasticity38, particularly in areas important for neuronal migration39. In human, various psychiatric and intellectual disorders are caused by mutations in genes involved in ECM homeostasis and may be driven by neuronal migration defects38. The central role of UDP-GlcA may open a window for early therapeutic interventions. In plants and lower animals, including zebrafish, UDP-GlcA can be synthesized by two alternative pathways. Apart from UGDH, UDP-GlcA can be generated via the myo-inositol oxygenation pathway from glucuronic acid by glucuronokinase and UDP-glucuronic acid pyrophosphorylase40. If a similar route exists in humans, supplementation of glucuronate may help to enhance this alternative pathway and increase levels of UDP-GlcA levels and its essential metabolites. To this date, however, the existence of human homologs of glucuronokinase and UDP-glucuronic acid pyrophosphorylase remains to be proven.
Conservative estimates of disease frequency resulting from germline UGDH mutations projects a prevalence of 1:14,000,000 to 1:2,000,000 (Suppl. Note 1). Considering that developmental epileptic encephalopathies are most commonly caused by de novo dominant mutations, this estimated prevalence seems relatively frequent.
Methods
Ethical approval
Written informed consent was obtained from the parents of the underage patients for diagnostic procedures and next-generation sequencing, as well as for the publication of identifying facial images in Fig. 1b. The study has been approved by the local Institutional Review Board of the Medical Faculty of the University of Tübingen, Germany (vote 180/2010BO1).
Exome sequencing
To unravel the molecular cause of the disease exome sequencing were performed at different genetic institutes using next-generation sequencing techniques according to local standard protocols. Variants were confirmed via Sanger sequencing using standard methods and chemicals (primer sequences are available on request).
Family 1: Exome sequencing for two affected siblings was performed on a HiSeq2500 System (Illumina, CA) after enrichment with SureSelectXT Human All Exon V5 (Agilent, Santa Clara CA). FASTQ files were imported into GENESIS (http://thegenesisprojectfoundation.org/)12 for further analysis using a pipeline build on BWA41, Picard, and FreeBayes. Variants were filtered for changes that segregated in an autosomal recessive fashion and passed the following filter criteria: (i) frequency in public databases (ExAC10 minor allele frequency (MAF) <0.1%), (ii) present in <5 families within GENESIS ( ∼ 4,300 exomes), (iii) conserved (PhyloP 100-way score >2 or PhastCons (100 vertebrate genomes) >0.75), (iv) CADD score >12, and (v) sufficient quality scores (Genotype Quality >75). In addition, variants had to be present in exomes from both siblings. This resulted in a list of seven variants (Suppl. Table 1), out of which only the homozygous missense variant c.131 C > T in the UGDH gene segregated with the third affected sibling.
Families 2, 9, 10: Using genomic DNA from the proband and parents, the exonic regions and flanking splice junctions of the genome were captured using the Agilent SureSelect Human All Exon V4 (50 Mb) or the Clinical Research Exome kit (Agilent Technologies, Santa Clara, CA). Massively parallel (NextGen) sequencing was done on an Illumina system with 100 bp or greater paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool. Additional sequencing technology and variant interpretation protocol has been previously described42. The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/). After variant stratification based on population frequencies within an internal database and ExAC10, inheritance, in silico predictors such as Provean, Mutation Taster and CADD, GeneDX reported only the UGDH variants to be the best potentially pathogenic candidates and connected to this project via GeneMatcher entries.
Family 3: Samples of the oldest sibling and both parents were sequenced in context of the EUROCORES project EuroEPINOMICS-RES, for which the technical details have been reported before43. Briefly, the trio underwent exome sequencing at the Wellcome Trust Sanger Institute (Hinxton/Cambridge, UK). Capturing of the exome was performed using the SureSelect Human All Exon 50 Mb exome kit (Agilent). The enriched exome libraries were then sequenced on a HiSeq2000 platform (Illumina) as 75 bp paired-end reads. BWA was used to align the sequenced reads to the reference genome (hg19). De novo analysis of these data did not reveal any variants. As the younger sib later developed a similar disorder, exome sequencing was also performed locally on the second sibling: for library preparation, genomic DNA was sheared to the average size 150 bp (Covaris) and the genome libraries prepared using KAPA HTP Lib Prep Kit Illumina 96 rxns (07138008001). Exome capturing was performed using the SeqCap EZ Human Exome v3.0 capture system and the sample was sequenced on the NextSeq500 platform using NextSeq500 High-output V2 kit. Mapping to the human reference genome (Hg19) and variant calling were performed with the CLC Genomics Workbench. Subsequent annotation and filtering were executed with GenomeComb44 (http://genomecomb.sourceforge.net/). Exome sequencing results of the trio and the second sibling were merged and reannotated and the family was reanalyzed as a quartet. Variants were filtered based on following quality parameters: coverage >7, quality >50 and not located in homopolymers >8 or tandem repeats. Only variants present with a frequency <0.5% in control population databases ExAC10 and Exome Variant Server, seen <3 times in the local exome sequencing database, and with predicted impact on the encoded protein (missense, nonsense, frameshift, deletions, insertions and (essential) splice site) were retained for further analysis. Remaining variants were filtered under an autosomal recessive (homozygous or compound heterozygous) and x-linked hypothesis. In addition, heterozygous variants called in both siblings and absent in the parents were selected under a parental mosaics for which the ExAC10 filter was set at <2 calls. This analysis revealed only this one compound heterozygous UGDH variants.
Family 4: Parent-proband trio exomes were prepared using the SureSelect Target Enrichment System (Agilent, Santa Clara CA) and sequenced on a HiSeq2000 System (Illumina, CA). Data processing, bioinformatics pipeline (for alignment, variant calling, annotation, and genetic model filtering), and analyses were previously described45. The compound heterozygous rare missense alterations in the UGDH gene, c.214T > C and c.41A > G, were interpreted as the only candidate genetic etiology.
Family 5: The exome library was prepared on an ION OneTouch System and sequenced on an Ion Proton instrument (Life Technologies, Carlsbad, CA, USA) using one ION PI chip. Sequence reads were aligned to the human GRCh37/hg19. Variants were filtered for common SNPs using the NCBI’s “common and no known medical impacts” database (ClinVar), ExAC10, as well as an in-house database of 406 sequenced samples. Additional filters were applied to retain proband’s exonic variants that were homozygous while heterozygous in both parents. Out of 5 homozygous variants, only one missense variant c.244G > A in the UGDH gene was found to segregate with the disease.
Family 6: Exome sequencing was performed on a NextSeq 500 System (Illumina, CA USA), with a 2 × 150 bp high-output sequencing kit after enrichment with Seq Cap EZ MedExome kit (Roche, Basel Switzerland). Sequence alignment, variant calling, and variant annotation was performed by Genosplice Technology (Paris France) with BWA 0.7.12, picard-tools-1.121, GenomeAnalysisTK-2014.3-17-g0583018 and SNPEff-4.2 with additional annotations from ClinVar and HGMD. The compound heterozygous UGDH variants were selected to be the most promising candidates and were thus submitted to GeneMatcher.
Families 7, 8, 16–25: Exome sequencing was performed essentially as described before46 for families 7, 8, and 16–25. Target regions were enriched using the Agilent SureSelectXT Human All Exon 50 Mb Kit. Whole-exome sequencing was performed on the Illumina HiSeq platform (BGI, Copenhagen, Denmark) followed by data processing with BWA (read alignment,) and GATK (variant calling) software packages. Variants were annotated using an in-house developed pipeline. Prioritization of variants was done by an in-house designed “variant interface” and manual curation. As four families with similar phenotype shared the homozygous p.R317Q as best candidate, a GeneMatcher entry was made and the in-house database was systematically screened for other potentially pathogenic UGDH variants. This allowed the identification of families 18 to 25.
Family 11: The sequencing was performed at Claritas Genomics (Cambridge, USA). Extracted genomic DNA was amplified using the AmpliSeq system and sequenced using an IonTorrent Proton Instrument. Alignment and variant calling of the nuclear DNA was done on Proton data using Torrent Suite 4.4 Software. Nuclear variants were filtered for quality using a custom filtering tool. In addition, extracted genomic and mitochondrial DNA was also run on an Agilent Clinical Research Exome capture sequence and then sequenced using an Illumina NextSeq instrument. Alignment and variant calling on NextSeq data was performed by an implementation of GATK Best Practices Pipeline. Genomic DNA results from the two NGS runs on the proband were combined and annotated by a custom bioinformatics pipeline. Besides a heterozygous SLC6A5 missense variant inherited from the unaffected father, the compound heterozygous UGDH variants were the only candidates reported that had a minor allele frequency (MAF) of < or = 0.01% that passed the laboratory’s quality metrics and were not de novo, X-linked or had biallelic variants.
Family 12: Isolated genomic DNA from peripheral blood leukocytes of proband and parents was captured with the Agilent Sure Select Clinical Research Exome (CRE) kit (v2). Sequencing was carried out with 150 bp paired-end reads on the Illumina HiSeq 4000. Reads alignments to the GRCh37/UCSC hg19 build were achieved using BWA (BWA-MEM v0.7.13). Variants were called using GATK (v3.7 (reference: http://www.broadinstitute.org/gatk/). Annotated Variants were filtered and prioritized using the Bench lab NGS v5.0.2 platform (Agilent technologies). The full exome analysis revealed the compound heterozygous variants in UGDH. The family was linked to this cohort via GeneMatcher.
Family 13: The exonic regions and flanking splice junctions of the genome were captured using proprietary GeneDx tools. Sequencing was performed on an Illumina system with 100 bp or greater paired-end reads. Reads were aligned to the human genome build GRCh37/UCSC hg19. A custom-developed analysis tool (Xome Analyzer) was used to call sequence variants. Sanger sequencing was used to confirm all potentially pathogenic variants identified in available family members. Additional variants not included in this report are available upon request.
Family 14: The patient F14-II:1 was enrolled in the ongoing “Undiagnosed Patients Program” at the Ospedale Pediatrico Bambino Gesù, Rome. Targeted enrichment (SureSelect All Exon V.4, Agilent) used genomic DNA extracted from circulating leukocytes for the affected subject and both parents, and parallel sequencing was performed using an Illumina HiSeq2000 platform, obtaining about 70 million reads. The data analysis was performed using an in-house implemented pipeline, which mainly take advantage of the Genome Analysis Toolkit (GATK V.3.7)47 framework, as previously reported48,49). The functional annotation of variants was achieved using SnpEff and dbNSFP (V.3.0)50,51,52. The functional impact of variants was analyzed by Combined Annotation Dependent Depletion (CADD) V.1.3, M-CAP V.1.0, and InterVar V.0.1.6 algorithms11,53,54. Two compound heterozygous private missense variants in the UGDH gene, c.347T > C and c.1328G > A, were interpreted as the only candidate genetic etiology.
Family 15: genomic DNA from of the proband and parents were enriched for exonic sequences with the SureSelect Human All Exon 50 Mb V5 Kit (Agilent Technologies, Santa Clara, California, USA). The HiSeq2500 (Illumina, San Diego, California, USA) was used to generate 125-bp paired-end runs of sequences. Reads were aligned and variant called with DNAnexus (Palo Alto, California, USA) using the reference human genome assembly hg19 (GRCh37). A mean coverage of 104x was achieved for the proband. Data analysis was preformed using an in-house bioinformatics pipeline. The compound heterozygous UGDH variants were selected to be the most promising candidates and were thus submitted to GeneMatcher.
Brain magnetic resonance imaging
Magnetic resonance images (MRI) have been recorded on 1.5 or 3 Tesla scanners at the different clinical sites. Sagittal, transversal, and coronal images of the brain have been acquired with standard sequences, including T1, T2, and Flair images.
Cell culture
Primary dermal fibroblast cultures were established from skin biopsies obtained from individuals F3-II:1, F4-II:1, F5-I:1, and F5-II:1 according to standard procedures55. In brief, primary fibroblasts were derived from biopsy samples and cultured in Dulbecco's modified Eagle medium (DMEM; HyClone, SH30243.01) supplemented with 10% fetal bovine serum (Biological Industries) and 2 mM l-glutamine (Biological Industries). Written informed consent of healthy probands and parents of UGDH patients were received prior to biopsy according to the ethical approvals of the local Institutional Review Boards (IRB).
Reverse transcription (RT-PCR) and quantitative PCR
Total RNAs were extracted using the RNeasy Mini Kit (Qiagen). RNA (1 µg) was reverse transcribed using the Iscript™ complementary DNA (cDNA) Synthesis Kit (Bio-Rad). Quantitative real-time PCRs were performed using Power SYBR green master mix (Applied Biosystems) on the 7900HT Fast real-time PCR system (Applied Biosystems). qPCR primer sequences are as follows: UGDH (between exons 6 and 7) 5′CTTGCCCAGAGAATAAGCAG3′ and 5′CAAATTCAGAACATCCTTTTGGA3′; β-ACTIN 5′ATGTTTGAGACCTTCACACC3′ and 5′AGGTAGTCAGTCAGGTCCCGGCC3′; GAPDH 5′TGAACCACCAACTGCTTAGC3′ and 5′GGCATGGACTGTGGTCATGAG3′.
Protein isolation and analysis
Cells were lysed using ice-cold RIPA buffer (250 mM Tris, pH: 7.5; 150 mM NaCl; 1% NP-40; 0.5% Na deoxycholate; protease inhibitors P2714 [Sigma-Aldrich, USA]). The total protein concentration of cell lysates was determined using the BCA Protein assay Kit (Thermo Fisher Scientific, USA). Sixty micrograms of total proteins were reduced in Laemeli loading buffer, denatured at 95 °C for 10 min, separated by 4–20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Invitrogen, Germany) electrophoresis and transferred onto Immun-Blot® Low Fluorescence PVDF Membranes (BIORAD). Protein detection was performed using anti-UGDH (1:500, Sigma-Aldrich, USA, HPA036657) and anti-GAPDH (1:2000, Santa Cruz Biotechnology Inc., USA, SC 47724) antibodies. Secondary antibodies conjugated to peroxidase (1:4000, Santa Cruz Biotechnology Inc., USA) were used and blots were developed using an enhanced chemiluminescence system, Pierce™ ECL Plus (Thermo Scientific), followed by detection on autoradiographic films.
HA quantification
HA content was compared in the culture-conditioned media from fibroblasts expressing WT or mutant UGDH using a competitive binding assay as previously described19. Fibroblasts were grown in 10 cm plates, 1 mL of conditioned media was aspirated from technical replicates, and cells were counted. HA concentration was interpolated from a standard curve, normalized to cell number, and plotted as mean ± SD. Statistical significance was assessed by Student’s t-test with at least three technical triplicates.
Specific activity measurement of UGDH
Fibroblasts expressing WT or mutant UGDH were assayed for UGDH-specific activity essentially as previously described56. Fibroblasts were cultured in 15 cm plates, washed three times with cold 1x PBS, and centrifuged at 1500 rpm for 5 min. Cells were resuspended in twice the pellet volume of Lysis Buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail). Samples were transferred to tubes with an equal volume of acid washed glass beads (Sigma) and lysed in the Bullet Blender 24 (Next Advance) at speed 8 for 3 min. The resulting supernatant was centrifuged at 13,000 rpm for 15 min to obtain final lysates. Enzymatic activity of the lysates (50 µg) was assayed with 1 mM UDP-glucose and 1 mM NAD+ in the presence or absence of 1 mM UDP-xylose, a UGDH-specific inhibitor, and monitored for changes in NADH, A340. Reaction rates for samples containing UDP-Xyl were subtracted from samples without UDP-Xyl to obtain UGDH-specific activity reported as [NADH] in nmol min−1 mg−1 lysate as described above. Each fibroblast cell line analyzed contained three or more technical replicates for reactions with and without UDP-Xyl plotted as mean ± SD. Statistical significance was assessed by one-way ANOVA (Prism).
Generation and purification of UGDH point mutants
Point mutants of human UGDH were generated from the codon optimized E. coli expression construct, WT-UGDH pET28a, using polymerase chain reaction mutagenesis with appropriate primers as previously described19,57. Sequences were verified by Eurofins MWG Operon (Huntsville, AL). The UGDH mutant constructs were expressed in E. coli strain BL21(DE3) grown in 2xYT medium containing 50 mg L−1 kanamycin at 37 °C. At an OD600 of 0.6-0.8, protein expression was induced with the addition of IPTG at a final concentration of 0.5 mM, and cultures were incubated at 18 °C overnight. Cells were harvested by centrifugation and lysed by sonication. All UGDH point mutants were expressed in the soluble fraction, and enzymes were purified by affinity chromatography using a HisTrap FF column (GE Healthcare) according to the manufacturer’s protocol. The average protein yields were: ~20 mg L−1 for UGDH WT and T325D, ~1.5 mg L−1 for UGDH A82T, and ~6 mg/L for UGDH A44V. Purified protein was dialyzed against 20 mM Tris-HCl pH 7.4 containing 1 mM dithiothreitol (DTT), concentrated, flash frozen in liquid nitrogen, and stored at –80 °C.
Analytical gel filtration
Purified recombinant UGDH WT and all point mutants were analyzed by size exclusion chromatography as previously described57. All samples were centrifuged prior to loading. Each apoprotein sample was injected into a 250 µL loop and separated by FPLC in 1x PBS containing 1 mM DTT at a flow rate of 0.5 mL min−1 on a Superdex 300 10/200 GL gel filtration column (GE Healthcare). Elution was monitored by A280 and plotted to compare alterations in oligomeric state.
Trypsin susceptibility assay
Purified recombinant WT-UGDH and all point mutants were assessed by limited trypsin proteolysis as previously described19. UGDH WT, A44V (10 µg), and A82T (14.2 µg) were digested with 10 ng trypsin in 1x PBS pH 7.4 for 2.5 h at room temperature in the absence or presence of 1 mM UDP-glucose, 1 mM UDP-glucuronate, 5 mM NAD+, 5 mM NADH, or combinations that yielded abortive and productive ternary complexes. Samples were analyzed by western blot probed for UGDH as previously described58.
Thermal stability measurement
Recombinant UGDH WT and A44V protein were assessed for thermal stability as previously described19 with minor alterations. All samples of UGDH WT and A44V (~15 µg) were incubated in 1x PBS and Sypro Orange dye (Invitrogen; 1:500 dilution) in the absence or presence of 1 mM UDP-glucose, 1 mM UDP-glucuronate, 5 mM NAD+, 5 mM NADH, or in combinations that yielded abortive and productive ternary complexes. Samples were handled at room temperature and transferred to an iCycler MyiQ thermocycler (Bio-Rad) for incremental thermal denaturation. Tm was plotted as the mean ± SD for seven replicates. Statistical significance was assessed by two-way ANOVA (Prism).
Saturating enzymatic activity and kinetic characterization
Enzyme activity of recombinant UGDH WT and all point mutants was characterized as described previously57 with minor alterations. Enzymatic activity was calculated by NADH turnover using the NADH extinction coefficient of 6220 M−1 cm−1. UGDH A82T activity was converted to [NADH] in nmol min−1 mg−1 UGDH and subsequently normalized to the fractional purity of UGDH in the sample preparation. Samples were run in triplicate and statistical significance was determined using Student’s t-test. Michaelis rate constants, Km and Vmax, were determined for UGDH WT and A44V as previously described using a 96-well plate assay to measure the change in NADH (A340) with respect to both the substrate, UDP-glucose, and cofactor, NAD+.
iPSCs reprogramming
WT, WT/A82T, and A82T/A82T fibroblasts were reprogrammed using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, A16517) in accordance with the manufacturer’s instructions. Briefly, fibroblasts were transduced and plated after 7 days onto Matrigel Basement Membrane Matrix (Corning, 354234) in mTeSR1 medium (STEMCELL Technologies, 85850). iPSC colonies were picked between days 17–28 and maintained in Matrigel Basement Membrane Matrix and mTeSR1 for expansion. R393W/A410S, R65*/Y367C, Y14C/S72P fibroblasts were reprogrammed using the ReproRNA™-OKSGM kit (Stemcell Technologies, 05930) in accordance with the manufacturer’s instructions. Briefly, fibroblasts were plated onto Matrigel Basement Membrane Matrix (Corning, 354234) and transfected with ReproRNA™-OKSGM cocktail. Puromycin selection was carried out 1 day after transfection. iPSC colonies were picked between 20 and 28 days after transfection and maintained in Matrigel Basement Membrane Matrix and mTeSR1 for expansion. Between 1 and 3 clones per genotype were maintained for further experiments.
Neuronal and cerebral organoid differentiation
Neuronal and cerebral organoid differentiation was performed as previously described27. Briefly, on day 0 of organoid culture, iPSCs were dissociated by accutase (STEMCELL Technologies, 07920) treatment to generate single cells. In total, 9000 cells were then plated per well of an ultra-low-binding 96-well plate (Corning) in MEDI medium [Knockout SR 20% (Thermo Fisher scientific, 10828-028), l-glutamine 2 mM (Thermo Fisher scientific, 200 mM, 25030-081), Non-essential amino-acid (NEAA) 1 × (Thermo Fisher scientific, 100× , 11140-050), sodium pyruvate 1× (Thermo Fisher scientific, 100 mM, 11360-070), and β-mercaptoethanol 1/1000 in neurobasal medium (Thermo Fisher scientific, A2477-501)] complemented with 10 µM Rho-associated protein kinase (ROCK) inhibitor (STEMCELL technology, Y-27632) and 0.05% polyvinyl alcohol. Embryoid bodies were fed with this medium every other day for 6 days. Then, MEDI medium was replaced with NIM-I medium [N2 1× (Thermo Fisher scientific, 100× , 17502-048), l-glutamine 2 mM, NEAA 1× , and heparin 1 µg mL−1 (Sigma-Aldrich, H3149) in neurobasal medium] every other day for 6 additional days for the neuroepithelial tissues induction. On day 11, tissues exhibiting neural ectoderm were transferred to droplets of cold Matrigel (BD Biosciences) on a sheet of Parafilm with dimples in a low-adhesion 6-well plate (Corning). These droplets were allowed to gel at 37 °C and were subsequently removed from the Parafilm and grown in differentiation medium Dif-M [N2 0.5 × , B27 without vitamin A 1× (Thermo Fisher scientific, 50 × , 12587-010), l-glutamine 2 mM, NEAA 1× , and β-mercaptoethanol 1/1000 in neurobasal medium] for 4 days. On day 15, the medium was replaced by Dif-M II medium [N2 0.5× , B27 1× (Thermo Fisher scientific, 50× , 17504-001), l-glutamine 2 mM, NEAA 1× , and β-mercaptoethanol 1/1000 in neurobasal medium], and the plates are placed on a horizontal shaker rotating at 85 rpm. Cerebral organoids were grown up to 10 weeks with 75% of the medium changed weekly.
Histology and immunofluorescence of organoids
Cerebral organoids were fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, then washed in PBS for 10 min and dehydrated by incubations in Ethanol (70%, 95% then 100%) for 1 h at 4 °C followed by two times 1 h incubation with Xylene 100% at room temperature. The cerebral organoids were then embedded in Paraplast Plus (Leica, 39602004) and sectioned at 30 µm. Tissue sections were stained with Haematoxylin and Eosin (H&E) or used for immunostaining. For immunofluorescence, antigen retrieval was performed by using the Antigen Retriever buffer (Citrate Buffer pH 6.0, Sigma, C 9999, 10× ). Sections were then blocked and permeabilized in blocking buffer (0.5% Triton X-100 and 1% BSA in PBS) for 20 min. Sections were then incubated with primary antibodies in blocking buffer at the following dilutions: SOX2 (mouse, R&D systems, MAB2018, 1:200), TUJ1 (mouse, Biolegend MMS-435P, 1:3000), GFAP (Rabbit, Dako Z0334, 1/2500), PCNA (Rabbit, abcam ab18197, 1 µg mL−1). For visualization, an antibody anti-mouse immunoglobulin G (anti–mouse IgG) Alexa Fluor 594 conjugate (Invitrogen, Molecular Probes) and an anti-rabbit IgG Alexa Fluor 488 conjugate (Invitrogen, Molecular Probes) was applied. DNA was stained by DAPI (1/500) and sections were mounted in ProLong Diamond Antifade mountant (Thermo Fisher Scientific, P36965). Images were collected by using an Olympus FV3000 RS with a 20x objective.
RT and RT-qPCR
Total RNA of individual cerebral organoids was extracted using the RNeasy Mini Kit (Qiagen, 74104). Total RNA (0.5 µg) was reverse transcribed using the Iscript™ cDNA Synthesis Kit (Bio-Rad, 1708891). Real-time quantitative PCRs were performed using the Power SYBR green master mix (Applied Biosystems, 4309155) on the 7900HT Fast real-time PCR system (Applied Biosystems). qPCR primers were as previously described:59 PAX6 5′CCGTGTGCCTCAACCGTA3′ and 5′CACGGTTTACTGGGTCTGG3′; TBR2 5′AAATGGGTGACCTGTGGCAAAGC3′ and 5′CTCCTGTCTCATCCAGTGGGAA3′; TUJ1 5′TCAGCGTCTACTACAACGAGGC3′ and 5′GCCTGAAGAGATGTCCAAAGGC3′.
Neural induction
Neural induction was carried out by dual SMAD inhibition60. iPSC were first dissociated into single cells and seeded in low attachment 96-well u-bottom plate at a density of 10,000 cells/well in Neural Induction Medium [DMEM/F12 (Thermo Fisher Scientific, 10565-018) supplemented with B27 (Thermo Fisher Scientific, 17504044), N2 (Thermo Fisher Scientific, 17505048), 0.2 mM NEAA (Thermo Fisher Scientific, 11140-050), 100 nM LDN 193189 (STEMCELL Technologies, 72148), 10 µM SB431542 (STEMCELL Technologies, 72234) and 10 µM Y-27632 (STEMCELL Technologies, 72304)]. After 6 days, cells were attached onto matrigel coated-plate in Neural Expansion Medium [DMEM/F12 (Thermo Fisher Scientific, 10565-018) supplemented with B27 (Thermo Fisher Scientific, 17504044), N2 (Thermo Fisher Scientific, 17505048), 0.2 mM NEAA (Thermo Fisher Scientific, 11140-050) and 20 ng mL−1 βFGF (Stemgent, 03-0002)]. After 3–6 days, rosette structures were manually cut out and expanded as suspension culture in neural expansion medium.
Neuronal differentiation
iPSCs-derived Neuro-Precursor Cells (NPCs) were differentiated into neurons for 21 days using a previously published protocol61 Briefly, NPCs were plated at a density of 50,000 cells cm−2 in a poly-l-ornithine and laminin-coated plates, cultured in N2B27 medium supplemented BDNF (20 ng mL−1), GDNF (20 ng mL−1), cAMP (N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate; Sigma; 0.3 mM) and ascorbic acid (0.2 mM).
Multi-electrode Array (MEA) recordings
Neurons on day 21 were dissociated and replated on 0.1 polyethylenimine (Sigma)-coated 48-well MEA plates (Axion Biosystems) in BrainPhys media supplemented with BDNF, GDNF, cAMP, and ascorbic acid as previously described62. Spontaneous neuronal activity was observed and recorded at 37o C for 5 min every 2–3 days using the Maestro MEA System (Axion Biosystem).
Zebrafish strains and maintenance
All zebrafish husbandry procedures were performed in compliance with the Singapore National Advisory Committee on Laboratory Animal Research Guidelines from the Institutional Animal Care and Use Committee (IACUC, IACUC number 161172). The jek ugdhm151 mutant line63 was used for this study and was maintained in the AB background. All embryos were raised in egg water (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4).
Genomic DNA extraction
Genomic DNA from larvae used for the locomotion assay was extracted by larval fin clipping as previously described64. Briefly, microscopic caudal fin slices of 3-dpf larvae were sectioned under a stereomicroscope. The fins were then digested with proteinase K (Invitrogen, 25530049) overnight at 65 °C, and the supernatant was used as a source of genomic DNA for genotyping. In 7-dpf larvae, heads were used for c-fos qPCR analysis, while the rest of the bodies was used for genotyping.
Genotyping of zebrafish
Genotyping was performed via nested PCR to amplify the exon 7 of ugdh, and restriction fragment length polymorphism as described previously63. The first PCR was performed with the following primers, 5′-TGTATGTGCAGGTGATTGACA-3′ and 5′-TGTAGGTCACAGGTTTTTGACA-3′. PCR products were cleaned-up by Exonuclease I (New England Biolabs, M0293L) and FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher, EF0651) treatment, and were then used as templates for a second PCR with the following primers: 5′-GACATGAATGAATATCAGAGAAAGAG-3′ and 5′-AGGAGAAACCCAACAACGC-3′. These PCR products were digested with the MluI enzyme (New England Biolabs, R3198L) that only cuts when the mutation is present.
c-fos qPCR experiments
7-dpf larvae were first incubated with increasing concentrations (up to 15 mM) of PTZ (Sigma, P6500) for 45 min and were then decapitated. The bodies were used for genotyping and 20 larvae heads of the same genotype were pooled together for total RNA extraction using the RNeasy Mini Kit (Qiagen, 74104). Total RNA (0.5 µg) was reverse transcribed using the Iscript™ cDNA Synthesis Kit (Bio-Rad, 1708891). Real-time quantitative PCRs were performed using the Power SYBR green master mix (Applied Biosystems, 4309155) on the 7900HT Fast real-time PCR system (Applied Biosystems). qPCR primers were as follows: c-fos23 5′-AACTGTCACGGCGATCTCTT-3′ and 5′-GCAGGCATGTATGGTTCAGA-3′; gapdh 5′-GTGGAGTCTACTGGTGTCTT-3′ and 5′-GTGCAGGAGGCATTGCTTAC-3′.
Twenty-four-well locomotion and convulsion test assay
A Basler Ace (acA1300-200um; 1280 × 1024) camera was used to acquire videos of larval zebrafish at 50 fps. A custom hardware setup was designed to acquire full frame videos of 24-well flat bottom plates using a 25 mm lens attachment to the camera placed 65 cm above the plate. The 24-well plates were backlit using a white light LED lightbox that delivered uniform lighting across the entire field. 7-dpf larval fish were placed individually in each well in 500 µl of egg water and acclimated to the setup for 10 min. Then, 500 µl of either egg water (negative control) or PTZ 30 mM (15 mM final concentration, Sigma, P6500) was added to the well. Three videos of 2 min each were acquired for a duration of 10 min for each condition tested. Fish locomotion was tracked online during video recording on a custom written software in LabView (www.critta.org). Analysis of locomotion was automated and performed blind offline after each experiment using custom written Python scripts (available on Github: https://github.com/mechunderlyingbehavior/24-Well-Larval-Locomotion.git). In this assay, low and high speed were defined as average speeds of 0 to 8 mm/s and >8 mm s−1, respectively, based on a recent publication measuring convulsive-induced speed changes in zebrafish larvae65.
Data availability
The data that support the findings of this study are available within the paper and its supplementary information files. All identified variants have been deposited in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) under the name “UGDH001”. Whole dataset from Family F3 was already published66 and deposited in the European Genome-phenome Archive, accession numbers EGAS00001000190, EGAS00001000386, and EGAS00001000048. Whole datasets from Families F5 and F15 are available upon request from the corresponding authors. Consent restrictions preclude deposition of sequencing data of the other families, however, specific information (e.g., secondary variants etc., but not full datasets) can be obtained upon request from the corresponding authors. Lists of primers and antibodies are in Supplementary Tables 4 and 5, respectively. The source data underlying Fig. 3a–f, Fig. 4a, and c, Suppl. Fig. 3a–d, Suppl. Fig. 4a, b, and d are provided as a Source Data file 1 (for gels) and 2 (for graphs).
Code availability
The custom written Python scripts used to track fish locomotion is available following this link: https://github.com/mechunderlyingbehavior/24-Well-Larval-Locomotion.git.
References
Scheffer, I. E. et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 512–521 (2017).
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A. & Scheffer, I. E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316 (2016).
Freeze, H. H., Eklund, E. A., Ng, B. G. & Patterson, M. C. Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125 (2015).
Morava, E. et al. Defining the phenotype in congenital disorder of glycosylation due to ALG1 mutations. Pediatrics 1034–1039 (2012). https://doi.org/10.1542/peds.2011-2711
Wu, X. et al. Deficiency of UDP-GlcNac:dolichol phosphate N-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum. Mutat. 22, 144–150 (2003).
Egger, S., Chaikuad, A., Kavanagh, K. L., Oppermann, U. & Nidetzky, B. Structure and mechanism of human UDP-glucose 6-dehydrogenase. J. Biol. Chem. 286, 23877–23887 (2011).
Egger, S. et al. Structural and kinetic evidence that catalytic reaction of human UPD-glucose 6-dehydrogenase involves covalent thiohemiacetal and thioester enzyme intermediates. J. Biol. Chem. 287, 2119–2129 (2012).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
EVS. Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP) (EVS, 2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Gonzalez, M. et al. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum. Mutat. 36, 950–956 (2015).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Rajakannan, V. et al. Structural basis of cooperativity in human UDP-glucose dehydrogenase. PLoS ONE 6, e25226 (2011).
Sommer, B. J., Barycki, J. J. & Simpson, M. A. Characterization of human UDP-glucose dehydrogenase: CYS-276 is required for the second of two successive oxidations. J. Biol. Chem. 279, 23590–23596 (2004).
Desmet, F. O. et al. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucl. Acids Res. 37, 1–14 (2009).
Pires, D. E. V., Ascher, D. B. & Blundell, T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucl. Acids Res. 42, 314–319 (2014).
Egger, S., Chaikuad, A., Kavanagh, K. L., Oppermann, U. & Nidetzky, B. UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochem. Soc. Trans. 38, 1378–1385 (2010).
Hyde, A. S., Thelen, A. M., Barycki, J. J. & Simpson, M. A. UDP-glucose dehydrogenase activity and optimal downstream cellular function require dynamic reorganization at the dimer-dimer subunit interfaces. J. Biol. Chem. 288, 35049–35057 (2013).
Neuhauss, S. C. et al. Mutations affecting craniofacial development in zebrafish. Development 123, 357–367 (1997).
Stainier, D. Y. et al. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 123, 285–292 (1996).
Driever, W. et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46 (1996).
Baraban, S. C., Taylor, M. R., Castro, P. A. & Baier, H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 131, 759–768 (2005).
Zhang, J. et al. C-Fos regulates neuronal excitability and survival. Nat. Genet. 30, 416–420 (2002).
García-García, M. J. & Anderson, K. V. Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell 114, 727–737 (2003).
Choksi, S. P. et al. Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development 141, 3410–3419 (2014).
Lancaster, M. A. & Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014).
Torisawa, T. et al. Katanin p80, NuMA and cytoplasmic dynein cooperate to control microtubule dynamics. Sci. Rep. 7, 1–16 (2017).
Chia, P. H. et al. A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability. Elife 7, 1–19 (2018).
Hacker, U., Lin, X. & Perrimon, N. The Drosophila sugarless gene modulates Wingless signaling and encodes an enzyme involved in polysaccharide biosynthesis. Development 124, 3565–3573 (1997).
Lin, X., Buff, E. M., Perrimon, N. & Michelson, A. M. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development 126, 3715–3723 (1999).
Superina, S., Borovina, A. & Ciruna, B. Analysis of maternal-zygotic ugdh mutants reveals divergent roles for HSPGs in vertebrate embryogenesis and provides new insight into the initiation of left-right asymmetry. Dev. Biol. 387, 154–166 (2014).
Perenthaler, E. et al. Loss of UGP2 in brain leads to a severe epileptic encephalopathy, emphasizing that bi- allelic isoform specific start-loss mutations of essential genes can cause genetic diseases. bioRxiv (2019). https://doi.org/10.1101/799841.
Volpi, S. et al. EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J. Exp. Med. jem.20161525 (2017). https://doi.org/10.1084/jem.20161525.
Tian, J. et al. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am. J. Hum. Genet. 87, 768–778 (2010).
Reuter, M. S. et al. NDST1 missense mutations in autosomal recessive intellectual disability. Am. J. Med. Genet. Part A 164, 2753–2763 (2014).
Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 50, 4–12 (2011).
Soleman, S., Filippov, M. A., Dityatev, A. & Fawcett, J. W. Targeting the neural extracellular matrix in neurological disorders. Neuroscience 253, 194–213 (2013).
Nishimura, K., Ishii, M., Kuraoka, M., Kamimura, K. & Maeda, N. Opposing functions of chondroitin sulfate and heparan sulfate during early neuronal polarization. Neuroscience 169, 1535–1547 (2010).
Gangl, R., Behmüller, R. & Tenhaken, R. Molecular cloning of a novel glucuronokinase/putative pyrophosphorylase from zebrafish acting in an UDP-glucuronic acid salvage pathway. PLoS ONE 9, e89690 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Tanaka, A. J. et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am. J. Hum. Genet. 97, 457–464 (2015).
Suls, A. et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with dravet syndrome. Am. J. Hum. Genet. 93, 967–975 (2013).
Reumers, J. et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 30, 61–68 (2011).
Farwell, K. D. et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586 (2015).
Neveling, K. et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 34, 1721–1726 (2013).
McKenna, S. The genome analysis toolkit. Proc. Int. Conf. Intellect. Capital, Knowl. Manag. Organ. Learn. 254–260 (2009). https://doi.org/10.1101/gr.107524.110.20
Flex, E. et al. Biallelic mutations in TBCD, encoding the tubulin folding cofactor D, perturb microtubule dynamics and cause early-onset encephalopathy. Am. J. Hum. Genet. 99, 962–973 (2016).
Bauer, C. K. et al. Mutations in KCNK4 that affect gating cause a recognizable neurodevelopmental syndrome. Am. J. Hum. Genet. 103, 621–630 (2018).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin). 5, 29–30 (2013).
Liu, X. et al. “dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations.” Hum. Mutat. 34, E2393–402 (2013).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581–1586 (2016).
Li, Q. & Wang, K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280 (2017).
Bonifert, T. et al. Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 137, 2164–2177 (2014).
Spicer, A. P. Molecular cloning and characterization of the human and mouse UDP-glucose dehydrogenase genes. J. Biol. Chem. 273, 25117–25124 (1998).
Grady, G. et al. Inhibiting hexamer disassembly of human UDP-glucose dehydrogenase by photoactivated amino acid cross-linking. Biochemistry 55, 3157–3164 (2016).
Zimmer, B. M. et al. Loss of exogenous androgen dependence by prostate tumor cells is associated with elevated glucuronidation potential. Horm. Cancer 7, 260–271 (2016).
Yakoub, A. M. & Sadek, M. Development and characterization of human cerebral organoids: an optimized protocol cell transplantation (2018). https://doi.org/10.1177/0963689717752946.
Chambers, S. M. et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 (2009).
Xu, X. et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 8, 619–633 (2017).
Xu, X., Radulescu, C. I., Utami, K. H. & Pouladi, M. A. Obtaining multi-electrode array recordings from human induced pluripotent stem cell–derived neurons. Bio-Protoc. 7, e2609 (2017).
Walsh, E. C. & Stainier, D. Y. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Sci. (N. Y., NY) 293, 1670–1673 (2001).
Wilkinson, R. N., Elworthy, S., Ingham, P. W. & van Eeden, F. J. M. A method for high-throughput PCR-based genotyping of larval zebrafish tail biopsies. Biotechniques 55, 314–316 (2013).
Pena, I. A. et al. Pyridoxine-dependent epilepsy in zebra fish caused by Aldh7a1 deficiency. 207, 1501–1518 (2017).
Appenzeller, S. et al. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370 (2014).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (1997).
Acknowledgements
We are grateful to the families of affected individuals whose cooperation made this study possible. We would like to thank Dr. Mary-Claire King from the University of Washington (Seattle, USA) and Dr. Ephrat Levy-Lahad (Medical Genetics Institute Shaare Zedek Medical Center, Israel) who independently investigated Family 1 and completed the segregation of the UGDH mutations in available family members. This study has been supported by the German Research Foundation (DFG, grant SCHO754/5-2 to L.S. and H.M.M.), by the European Union through funding for the NEUROMICS network (F5-2012-305121 to L.S.), by A Strategic Positioning Fund on Genetic Orphan Diseases (GODAFIT) and an Industry Alignment Fund on Singapore Childhood Undiagnosed Diseases Program (SUREKids) from the Biomedical Research Council, A*STAR, Singapore. T.M.P. was supported by the Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases. M.T. was supported by the Fondazione Bambino Gesù (Vite Coraggiose) whose study was in the frame of the ongoing “Undiagnosed Patients Program”, Ospedale Pediatrico Bambino Gesù, Rome. B.C. is supported by operating grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada. S.W. is supported by the Eurocores program EuroEPINOMICS, the Fund for Scientific Research Flanders (FWO), the International Coordination Action (ICA) grant G0E8614N, and the University of Antwerp (research fund). H.S. is a Ph.D fellow of the Fund for Scientific Research Flanders (1125416 N). D.J.L. received financial support obtained from the Netherlands Organization for Scientific Research (ZONMW VIDI grant 91713359 to D.J.L.). S.S.J. is supported by National Medical Research Council, Singapore (NMRC/CISSP/0003/2016). B.R. is a fellow of the Branco Weiss Foundation, an A*STAR Investigator, a Young EMBO Investigator, a Senior NRF and AAA fellow. K.H.U was supported by the FRAXA Foundation. Furthermore, we would like to thank the EuroEPINOMICS-RES NLES Working Group for their support by screening their database and matching Family 3. T.B. is supported by the Netherlands Organisation for Scientific Research (ZonMW Veni, grant 91617021) and by an NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation.
Author information
Authors and Affiliations
Contributions
B.R., C.B.-L., E.S.-R., H.H., L.S. designed the study. B.R., C.B.-L., H.H., L.S. co-wrote the paper. B.R., E.S.-R., H.H., L.S. supervised the study. D.J.L., E.S.-R., J.J.B., M.A.S. contributed to writing the paper. S.S.J. (F5) recognized this disease as a new clinical entity with the F5. H.H. coordinated the clinical data and phenotyping. A.R. analyzed dysmorphic features, and reviewed of all available pictures. I.K.-M. interpreted the MRI data. F.S.A. (F11, F20) and M.M.A. (F23) coordinated the local clinical study. H.V. (F3) and S.W. (F3) designed and supervised local genetic study. A.A. (F11, F20), M.A. (F11, F20), R.H.G. (F13), M.J.M. (F2), H.V. (F3), D.H. (F6), D.A.K. (F7, F8, F16-F19), T.M.P. (F9), W.G.M. (F9), and H.H. made clinical diagnosis. H.M.M. (F1), N.H. (F1), R.K. (F1), W.D. (F1), M.C.W. (F10), M.V.A. (F10), L.H.R. (F11), T.B. (F12), R.H.G. (F13), T.G. (F15), A.A.H.M. (F16-F19), E.A.F. (F16-F19), L.A.J. (F16-F19), S.M.A. (F16-F19), A.A.A. (F16-F19), A.M.T. (F21), M.O.H (F21), H.V. (F3), S.W. (F3),S.D. (F4), S.N. (F4), I.H. (F4, F9), E.T. (F5), J.L. (F5), T.T. (F5), M.A.A.P.W. (F7), R.O. (F8), D.M. (F14) M.D.S. (F23), M.M.A. (F23), and H.M.D. (F24, F25) collected the clinical data. H.V. (F3), S.W. (F3) designed and supervised local genetic study. M.C.W. (F10), M.V.A. (F10), E.W.K. (F13), L.S.R. (F13), M.G.-A. (F2), M.J.M. (F2), and H.M.D. (F24, F25) provided genetic evaluation. A.Y.J.N. (F5), D.A.K(F7, F8, F16-F19) collected DNA samples. H.H. (F1), P.B. (F1), A.B. (F2, F9, F10), A.T. (F2, F9, F10), H.S. (F3), S.W. (F3), S.Ta. (F4), A.Y.J.N. (F5), B.V. (F5), C.B. (F5), S.To. (F5), B.K. (F6), D.A.K. (F7, F8, F16-F19), R.P. (F7, F8, F16-F19), T.M.P. (F9), S.B. (F14),, H.M.-S. (F15), S.M. (F22), and R.S. performed WES sequencing and analyses. H.H. (F1), S.G. (F1), F.S.A. (F11, F20), C.B. (F5), M.T. (F14), and S.M. (F22) identified the gene. B.R., L.S. did matchmaking with GeneMatcher, and R.S. with Genesis. M.A.S. designed biochemical and cell culture studies. J.J.B. designed structural and biochemical studies. D.J.L. provided glycosylation expertize. C.B.-L., D.J.L., G.G., S.B.P. performed biochemical studies. E.L. (F6), C.B.-L., G.G. performed cell culture experiments. T.T., G.N., and M.Y.E. performed reprogramming experiments. K.H.U. and M.A.P. designed and performed neuronal cell culture and multi-electrode array. C.B.-L. performed cerebral organoids differentiation, staining and analysis. B.C. shared the jekyll m151 zebrafish line. A.C.-C., A.S.M., J.C.S. designed hardware and software, and A.S.M., H.L. designed automated scripts for zebrafish behavior studies. B.C., M.Y. performed functional genetic studies, and C.B.-L. performed behavior and PTZ experiments in zebrafish.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: P.B. is employed by and receives a salary from Centogene AG (Exome sequencing is among the commercially available tests). S.Ta. is employed by and receive a salary from Ambry Genetics (exome sequencing is a commercially available test). A.B. and A.T. are employees of GeneDx (exome sequencing is a commercially available test). S.S.J. is co-founder of Global Gene Corporation Pte Ltd., however, there are no competing financial interests. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hengel, H., Bosso-Lefèvre, C., Grady, G. et al. Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat Commun 11, 595 (2020). https://doi.org/10.1038/s41467-020-14360-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14360-7
This article is cited by
-
UDP-glucuronate metabolism controls RIPK1-driven liver damage in nonalcoholic steatohepatitis
Nature Communications (2023)
-
Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort
European Journal of Human Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
