
Abstract
Amines bearing γ-stereocenters are highly important structural motifs in many biologically active compounds. However, reported enantioselective syntheses of these molecules are indirect and often require multiple steps. Herein, we report a general asymmetric route for the one-pot synthesis of chiral γ-branched amines through the highly enantioselective isomerization of allylamines, followed by enamine exchange and subsequent chemoselective reduction. This protocol is suitable for establishing various tertiary stereocenters, including those containing dialkyl, diaryl, cyclic, trifluoromethyl, difluoromethyl, and silyl substituents, which allows for a rapid and modular synthesis of many chiral γ-branched amines. To demonstrate the synthetic utility, Terikalant and Tolterodine are synthesized using this method with high levels of enantioselectivity.
Similar content being viewed by others
Introduction
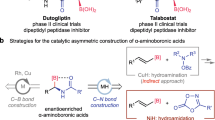
Aliphatic amines with adjacent stereocenters are prevalent in natural products and pharmaceuticals and are often key contributors to their potent biological activity1. In particular, enantiopure γ-branched amines represent an important subclass of bioactive amines, including many pharmaceutical agents (Fig. 1a). Despite the generality of this structure, the direct synthesis of chiral, γ-branched amines remains underdeveloped compared to the well-established methods for constructing α-branched and β-branched amines2,3,4,5, as well as distal stereocenters to other function groups such as ketones6, aldehydes7,8,9,10, and amides11.

Significance and syntheses of chiral γ-branched amines. a Biologically active molecules containing chiral γ-branched amine moiety. b Asymmetric hydrogenation or isomerization followed by reductive amination for the multistep synthesis. c Direct synthesis via a Cu–H catalyzed relay hydroamination reaction
Known catalytic approaches to install this subunit often require multistep synthetic sequences via chiral, β-branched carbonyl intermediates, which can hinder the rapid generation of compound libraries for high throughput screening in medicinal chemistry. For example, transition metal-catalyzed asymmetric hydrogenation of α,β-unsaturated acids or esters12,13,14 affords the enantiopure β-branched carbonyl intermediates, followed by a reductive amination to install the desired chiral γ-branched amines (Fig. 1b). However, varying substituents at the newly introduced stereocenters, such as aryl vs. alkyl, acyclic vs. cyclic, or carbon atom vs. heteroatom, often requires different metal/ligand scaffolds to achieve high enantioselectivity12,13,14. The redox neutral isomerization of allylic amines15,16 or alcohols17,18,19 provides a solution to this problem; however, current methods suffer from very limited substrate scope9,10,11,12,13. To the best of our knowledge, there is only one reported method for the direct synthesis of chiral γ-branched amines (Fig. 1c). Buchwald et al. have shown that 3,3-disubstituted allylic esters can undergo an enantioselective hydrocupration followed by β-alkoxide elimination and subsequent anti-Markovnikov hydroamination of the intermediate terminal olefin to afford γ-branched amines in one step20. Although this method demonstrates high enantioselectivity under a ligand-controlled hydrocupration of allylic esters, the preparation of electrophilic amines requires additional synthetic operations and limits the substrate scope to secondary alkyl amines20.
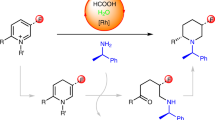
Our group has recently published a one-step synthesis of chiral, β-branched amides via Rh-catalyzed enantioselective isomerization of allylic amines, followed by enamine exchange, and subsequent oxidation21. The slow oxidation of the more sterically hindered diethyl enamine (i, when R = ethyl, Fig. 2a) compared to facile oxidation of enamine (ii) leads to exclusive formation of the desired amide product (iii). Based on this report, we proposed that chiral enamine intermediate (ii) could instead be reduced to afford the valuable enantiopure γ-branched amine (v).

Design of a tandem asymmetric isomerization—enamine exchange—reduction process. a The chemoselectivity is determined by the relative reduction rate of intermediates i and ii. When R = ethyl, exclusive formation of β-branched amide iii is observed in the presence of hydrogen acceptor. b One-pot synthesis of chiral γ-branched amine from allylic amine, exogenous amine nucleophile, and hydrogen donor
We report herein a nucleophilic amination of allylic amines with exogenous amine nucleophiles to afford chiral, γ-branched amines via a transfer hydrogenation (Fig. 2b). Both primary and secondary alkyl/aryl amines are effective nucleophiles, coupling with allylic diethylamine precursors to afford various γ-branched amine products with excellent enantioselectivities in a two-step one-pot manner.
Results
Reaction discovery and optimization
To establish a method for the selective conversion of allylic amines to enantiopure γ-branched amines, we began our investigation by examining a variety of hydrogen donors in the reductive amination of geranyl diethylamine (1a) with morpholine (2b) under slightly modified conditions from our previous report21. Compared to the oxidative process, the reduction is more challenging as the hydrogenated starting material (Fig. 2a, iv) was often observed as the major byproduct in the amidation reaction21. Therefore, an appropriate selection of a hydrogen donor and starting material (R group) to allow for the rapid and chemoselective reduction of intermediate (ii) was the key challenge in our investigation. No conversion of 1a was observed in the presence of H2 donors, presumably due to protonation of the basic allylic nitrogen atom or coordination to the cationic catalyst, thereby impeding the initiation of the 1,3-hydride shift15,16. Sequential addition of the hydrogen donor after the isomerization/enamine exchange step led to higher conversion of starting material, with HCO2H showing superior reactivity and selectivity (Table 1, entries 1–4). Increasing the equivalency of amine nucleophile improved the ratio of 3a/3a’, but did not increase the yield of the desired product 3a (Table 1, entries 4–6). Different allylic amine precursors (1b–d) were then tested to compare both chemoselectivity and enantioselectivity (Table 1, entries 7–9). Elevated temperature was required to achieve high conversion for these substrates. Less sterically hindered dimethylamino substrate 1b afforded poor chemoselectivity and high enantioselectivity; however, bulkier allylic diisopropylamine 1c showed greater chemoselectivity but poor enantioselectivity. Secondary amine precursor 1d was less reactive and selective under these conditions.
Substrate scope
With these optimized conditions in hand, the amine nucleophile scope was investigated (Fig. 3). Secondary cyclic amines such as morpholine (3a), Boc-protected piperazine (3b), tetrahydroisoquinoline (3c), and 2-(piperazin-1-yl) pyrimidine (3d) all gave similarly excellent yields and enantiometric ratios. Without the addition of amine, 3e could be obtained in high yield and er. Surprisingly, more sterically hindered acyclic dialkyl amines 3f and 3g (compared to diethylamine) were effective nucleophiles in this reaction, indicating that the volatility of the resulting diethylamine byproduct is likely playing a larger role than steric hindrance in determining the chemoselectivity (vide infra). Enantiopure α-branched amine 2g afforded the desired product 3g and 3g’ with high er (>97:3) and dr (>20:1), demonstrating that the isomerization is not affected by the chirality of the nucleophile, but is instead controlled by the ligand. Importantly, no racemization of the chiral amine nucleophile occurred under the reaction conditions.

Scope of amine nucleophiles for the reductive amination of allylamine. a: General reaction conditions: 1a (0.24 mmol, 1.0 equiv, E/Z = 97.5:2.5, 40 °C for 1st step) or 1b (0.24 mmol, 1.0 equiv, E/Z > 99:1, 60 °C for 1st step) nucleophile 2 (1.2 equiv), hydrogen donor (3.0 equiv), THF (1.2 M). b: For 3a–3g, HCO2H used as H2 donor at 60 °C for 2nd step; For 3h–3n, NaBH4 (1.5 equiv) used as reductant at 0 °C to rt for 2nd step. c: 2c and 2d added together with HCO2H. d: No nucleophile added. e: (S)-BINAP used. f: 2k, 2l, and 2n added after isomerization. See supplementary methods for details. dr diastereomeric ratio
Under slightly modified conditions, primary aryl and alkyl amines were coupled with allylic diethylamine electrophiles to afford the chiral secondary amines, respectively. In these cases, NaBH4 proved to be a superior reductant than HCO2H. Both electron rich (3i) and poor (3j) anilines afforded the desired chiral amines with identically excellent enantiomeric ratios. In the presence of primary alkyl amines (with the exception of tBuNH2 2m), the isomerization of allylic diethylamine was completely prohibited; therefore, a sequential addition of nucleophile was required to reach high yields. Primary alkyl amines, α to 1°(3k), 2°(3l), and 3° (3m) carbons, all afforded desired products with moderate to good yields and excellent enantioselectivity. A nucleophile containing a tethered tertiary nitrogen atom (3n) was well tolerated.
A survey of 3,3-disubstituted allylic amine electrophiles revealed that a wide variety of tertiary stereocenters can be installed under these reductive amination conditions (Fig. 4). Several 3,3-aryl,alkyl allylic diethylamines (5a–c) were tested and all afforded products with good yields and enantioselectivities. An ortho substituent on the aryl ring (5c) has no effect on the enantioselectivity of the isomerization, and the standard reaction conditions were amenable to aryl bromides, with no proteodebromination byproducts observed. The use of β,β-dialkyl allylic diethylamine (5d–f) was successful, enabling the highly enantioselective synthesis of γ-dialkyl amines, even with minimally differentiated substituents (5d, nPent vs. nBu). When more challenging 3,3-diaryl allylic diethylamines (5g–i) were subjected to the reaction conditions, amine products bearing γ-diaryl stereocenters, a common moiety in pharmaceutical agents, can be formed with excellent enantioselectivity22,23,24,25. Substrates bearing electron-rich (5h) and electron-poor (5i) aryl substituents afforded good yields and excellent enantiomeric ratios. This method can be used to set stereocenters containing sterically and electronically similar phenyl and para-tolyl groups with excellent selectivity (5 g, 96.5:3.5 er). Chiral γ-cyclic amines containing five-membered, six-membered, and seven-membered rings (5j-l) can be obtained as well with high enantioselectivity under identical conditions26,27.

Scope of allylamine. a: General reaction conditions: allylic diethylamine 4 (0.24 mmol, 1.0 equiv, E/Z > 99:1 unless otherwise noted), nucleophile 2 (1.2 equiv), HCO2H (3.0 equiv), THF (1.2 M). b: Substrate E/Z = 96.7:3.3. c: Substrate Z/E > 99:1. d: 1,4-dioxane used. e: Substrate Z/E = 95.6:4.4. f: Toluene used. See supplementary methods for details. The absolute configuration of product is determined by alkene configuration
Due to the superior reactivity and broad substrate tolerance of this catalyst, we sought to further develop this method for the construction of highly valuable stereocenters containing CF3, CF2H, and SiR3 substituents (Fig. 4). In order to effect suitable conversion, modification of the reaction solvent and increased temperatures were required. This may be attributed to the difficult isomerization of the more hindered allylic amines. Under these new conditions, difficult to synthesize enantiopure γ-trifluromethylated (5m–o) and difluoromethylated (5p) amines can be accessed with moderate to good yields and excellent enantioselectivities28,29. It is worth noting that the (Z)-CF3 allylic amine (5n) was slightly more reactive under these conditions compared to the (E)-isomer (5m), as higher conversion was observed for 5n. Phenyldimethylsilyl substituted allylic diethylamines (5q) afforded good yields and enantioselectivities under these conditions as well. It is noteworthy that the chiral silyl group can be installed, as this can be converted to a range of functionalities30.
Synthetic application
This methodology was applied in the enantioselective syntheses of biologically active Terikalant and Tolterodine as illustrated in Fig. 5. Substrate 4k and nucleophile 2o were prepared according to literature procedures. The presence of 2o proved to inhibit the isomerization of allylic amine 4k. Therefore, the addition of nucleophile along with formic acid after the isomerization step was found effective, giving 75% yield as well as excellent er (96.7:3.3) for Terikalant (Fig. 5a), a significant improvement over the current synthesis utilizing chiral resolution31. A highly enantioselective synthesis of (R)-Tolterodine was then demonstrated in Fig. 5b32,33,34. The (E)-vinyl bromide 6, prepared from trans-cinnamyl chloride35, was coupled with aryl boronic acid 7 to afford the diastereopure (E)-allylic amine 8 in 91% yield. A sequential addition of catalyst, hydrogen donor, and strong acid afforded the desired (R)-Tolterodine in 88% overall yield and 96.0:4.0 er. Although diisopropylamine was not a sufficient nucleophile to perform the enamine exchange with the diethyl enamine, the isomerization of allylic diisopropylamine 8 also proceeds in a highly enantioselective fashion. It is worth noting that the reaction was carried out on the 1.0 mmol scale with half the catalyst loading compared to the aminations performed on the smaller scale. Compared to state-of-the-art Tolterodine synthesis, which requires the ortho-hydroxyl substituent to direct the asymmetric hydrogenation34, our method allows for a modular and rapid synthesis of Tolterodine derivatives, including those without the ortho-hydroxyl functionality (5g–i).

Synthetic application of rhodium-catalyzed asymmetric reductive amination of allylamines. a Enantioselective synthesis of Terikalant from allylic diethylamine 4k. b Enantioselective and modular synthesis of (R)-Tolterodine from vinyl bromide 6. See supplementary methods for details
Control experiments and mechanistic discussion
To gain insight into the overall selectivity of this tandem process, a series of control reactions were carried out under optimized conditions (Fig. 6a–d). The selectivity of the enamine exchange step was first investigated. In general, less sterically hindered amine nucleophiles (compared to diethylamine) led to higher selectivity of desired product enamine 9 over the diethylenamine 10a (Fig. 6a). For sterically similar dibutylamine and dibenzylamine, 9 was found to be the major product, presumably due to a combination of the relative amine volatilities, stoichiometry of the reaction, and enamine stability. When equimolar amounts of nucleophile and substrate were subjected to the reaction conditions, similar product distributions were observed regardless of the permutation of allylic amine versus nucleophile (Fig. 6b). This implies that the exchanging product distribution is controlled by a thermodynamic equilibrium under standard reaction conditions. When the nucleophiles and hydrogen donor were added simultaneously into the reaction after the isomerization step (Fig. 6c), the observed selectivities are similar to those shown in Fig. 6a, indicating that the exchange step is faster than the reduction. Finally, various secondary amine nucleophiles were studied under standard conditions (Fig. 6d). Higher selectivities were observed compared to those in Fig. 6a, implying that the reduction of desired enamine intermediates is faster than the diethylenamine 10a. Therefore, the chemoselectivity of this two-step one-pot reaction comes from both steps, favoring the desired product. A proposed mechanism is shown in Fig. 6e: the basic nitrogen atom of the allylic amine substrate coordinates to the cationic rhodium to form A, followed by β-hydride elimination and re-insertion of in situ generated conjugated iminium B to afford the chiral enamine C. A thermally controlled enamine exchange leads to D, which then undergoes subsequent transfer hydrogenation upon addition of formic acid. A rhodium-mediated transfer hydrogenation mechanism is proposed, as lower conversion was observed in the absence of metal catalyst when investigating the reduction of pre-made enamine (see Supplementary Figure 5). An in situ formed rhodium formate species F can undergo decarboxylation to generate Rh hydride species G36,37. Subsequent iminium E inserts into Rh–H G to give the desired chiral γ-branched amine and regenerate rhodium formate F.

Control experiments and proposed catalytic cycles. a Selectivity of the enamine exchange step. b Thermodynamic equilibrium for the enamine exchange. c Selectivity of the transfer hydrogenation step (simultaneous addition of amine and hydrogen donor). d Chemoselectivity for various secondary amine nucleophiles under standard conditions. e Proposed catalytic cycles: enantioselective isomerization and transfer hydrogenation, X = BAr4F
Discussion
We have developed conditions for a highly enantioselective, modular synthesis of chiral γ-branched amines. This method enables a rapid assembly of various stereocenters as well as amine functionalities via a tandem isomerization–enamine exchange transfer hydrogenation process. Stereocenters bearing diaryl, cyclic, fluoroalkyl, and silyl substituents are established using same catalyst under similar conditions.
Methods
Direct asymmetric synthesis of γ-branched amines
[Rh(COD)Cl]2 (2.0 mg, 1.5 mol %), (R)-BINAP (4.5 mg, 3.0 mol %), NaBAr4F (6.4 mg, 3.0 mol %), and THF (0.2 mL) were added to a oven-dried 4 mL vial equipped with a stir bar in the glove box under nitrogen atmosphere. To the vial was added sequentially allylic diethylamine (1, 0.24 mmol, 1.0 equiv), and secondary amine (2, 0.29 mmol, 1.2 equiv). The resulting solution was allowed to stir for 22 h at 40 °C (unless otherwise noted). After 22 h, formic acid (0.36 mmol, 3.0 equiv) was added into reaction vial via syringe and the reaction was allowed to stir for another 2 h at 60 °C (unless otherwise noted). The crude reaction was quenched by the addition of DCM, concentrated in vacuo and then purified by basic alumina chromatography to afford the desired product 3.
Data availability
All data is available from the corresponding authors upon reasonable request.
References
Nugent, T. C. Chiral Amine Synthesis: Methods, Developments and Applications (John Wiley & Sons, Hoboken, New Jersey, 2010).
Hoffmann, S., Nicoletti, M. & List, B. Catalytic asymmetric reductive amination of aldehydes via dynamic kinetic resolution. J. Am. Chem. Soc. 128, 13074–13075 (2006).
Lu, Z., Wilsily, A. & Fu, G. C. Stereoconvergent amine-directed alkyl-alkyl Suzuki reactions of unactivated secondary alkyl chlorides. J. Am. Chem. Soc. 133, 8154–8157 (2011).
Zhu, S. & Buchwald, S. L. Enantioselective CuH-catalyzed anti-markovnikov hydroamination of 1,1-disubstituted alkenes. J. Am. Chem. Soc. 136, 15913–15916 (2014).
Czekelius, C. & Carreira, E. M. Catalytic enantioselective conjugate reduction of β,β -disubstituted nitroalkenes. Angew. Chem. Int. Ed. 42, 4793–4795 (2003).
Werner, E. W., Mei, T. S., Burckle, A. J. & Sigman, M. S. Enantioselective heck arylations of acyclic alkenyl alcohols using a redox-relay strategy. Science 338, 1455–1458 (2012).
Mei, T. S., Werner, E. W., Burckle, A. J. & Sigman, M. S. Enantioselective redox-relay oxidative heck arylations of acyclic alkenyl alcohols using boronic acids. J. Am. Chem. Soc. 135, 6830–6833 (2013).
Mei, T. S., Patel, H. H. & Sigman, M. S. Enantioselective construction of remote quaternary stereocentres. Nature 508, 340–344 (2014).
Patel, H. H. & Sigman, M. S. Palladium-catalyzed enantioselective heck alkenylation of acyclic alkenols using a redox-relay strategy. J. Am. Chem. Soc. 137, 3462–3465 (2015).
Zhang, C., Santiago, C. B., Kou, L. & Sigman, M. S. Alkenyl carbonyl derivatives in enantioselective redox relay heck reactions: accessing α,β-unsaturated systems. J. Am. Chem. Soc. 137, 7290–7293 (2015).
Wang, Z. X., Bai, X. Y., Yao, H. C. & Li, B. J. Synthesis of amides with remote stereocenters by catalytic asymmetric γ-alkynylation of α,β-unsaturated amides. J. Am. Chem. Soc. 138, 14872–14875 (2016).
Verendel, J. J., Pàmies, O., Diéguez, M. & Andersson, P. G. Asymmetric hydrogenation of olefins using chiral crabtree-type catalysts: Scope and limitations. Chem. Rev. 114, 2130–2169 (2014).
Khumsubdee, S. & Burgess, K. Comparison of asymmetric hydrogenations of unsaturated carboxylic acids and esters. ACS Catal. 3, 237–249 (2013).
Li, J.-Q. et al. Asymmetric hydrogenation of allylic alcohols using Ir–N,P-complexes. ACS Catal. 6, 8342–8349 (2016).
Tani, K. et al. Highly enantioselective isomerization of prochiral allylamines catalyzed by chiral diphosphine rhodium(I) complexes. preparation of optically active enamines. J. Am. Chem. Soc. 106, 5208–5217 (1984).
Inoue, S. et al Mechanism of the asymmetric isomerization of allylamines to enamines catalyzed by 2,2’-Bis(diphenylphosphino)-1,1’-binaphthyl-rhodium complexes. J. Am. Chem. Soc. 112, 4897–4905 (1990).
Tanaka, K., Qiao, S., Tobisu, M., Lo, M. M. C. & Fu, G. C. Enantioselective isomerization of allylic alcohols catalyzed by a rhodium/phosphaferrocene complex [18]. J. Am. Chem. Soc. 122, 9870–9871 (2000).
Mantilli, L., Gérard, D., Torche, S., Besnard, C. & Mazet, C. Iridium-catalyzed asymmetric isomerization of primary allylic alcohols. Angew. Chem. Int. Ed. 48, 5143–5147 (2009).
Arai, N., Sato, K., Azuma, K. & Ohkuma, T. Enantioselective isomerization of primary allylic alcohols into chiral aldehydes with the tol-binap/dbapen/ ruthenium(II) catalyst. Angew. Chem. Int. Ed. 52, 7500–7504 (2013).
Zhu, S., Niljianskul, N. & Buchwald, S. L. A direct approach to amines with remote stereocentres by enantioselective CuH-catalysed reductive relay hydroamination. Nat. Chem. 8, 144–150 (2016).
Wu, Z., Laffoon, S. D., Nguyen, T. T., McAlpin, J. D. & Hull, K. L. Rhodium-catalyzed asymmetric synthesis of β-branched amides. Angew. Chem. Int. Ed. 56, 1371–1375 (2017).
Lee, J. C. H., McDonald, R. & Hall, D. G. Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem. 3, 894–899 (2011).
Li, Y., Dong, K., Wang, Z. & Ding, K. Rhodium(I)-catalyzed enantioselective hydrogenation of substituted acrylic acids with sterically similar β,β-diaryls. Angew. Chem. Int. Ed. 52, 6748–6752 (2013).
Yan, Q., Kong, D., Zhao, W., Zi, G. & Hou, G. Enantioselective hydrogenation of β,β-disubstituted unsaturated carboxylic acids under base-free conditions. J. Org. Chem. 81, 2070–2077 (2016).
Tolstoy, P. et al. Iridium-catalyzed asymmetric hydrogenation yielding chiral diarylmethines with weakly coordinating or noncoordinating substituents. J. Am. Chem. Soc. 131, 8855–8860 (2009).
Guo, S., Yang, P. & Zhou, J. Nickel-catalyzed asymmetric transfer hydrogenation of conjugated olefins.Chem. Commun. 51, 12115–12117 (2015).
Guo, S. & Zhou, J. N,N-dimethylformamide as hydride source in nickel-catalyzed asymmetric hydrogenation of α,β-unsaturated esters. Org. Lett. 18, 5344–5347 (2016).
Dong, K., Li, Y., Wang, Z. & Ding, K. Catalytic asymmetric hydrogenation of α-CF3- or β-CF3-Substituted acrylic acids using Rhodium(I) complexes with a combination of chiral and achiral ligands. Angew. Chem. Int. Ed. 52, 14191–14195 (2013).
Chen, J., Yuan, P., Wang, L. & Huang, Y. Enantioselective β-protonation of enals via a shuttling strategy. J. Am. Chem. Soc. 139, 7045–7051 (2017).
Colvin, E. W. Recent synthetic applications of organosilicon reagents. Chem. Org. Silicon Compd. 2, 1667–1685 (1998).
Barreau, M.; Hardy, J.-C.; Martin, J.-P.; Renault, C. Benzopyran derivatives, their preparation and pharmaceutical compositions containing them. US patent 4,994,470 (1990).
Hedberg, C. & Andersson, P. Catalytic asymmetric total synthesis of the muscarinic receptor antagonist (R)-tolterodine. Adv. Synth. Catal. 347, 662–666 (2005).
Roesner, S. & Aggarwal, V. K. Enantioselective synthesis of (R)-tolterodine using lithiation/borylation–protodeboronation methodology. Can. J. Chem. 90, 965–974 (2012).
Wang, X. et al. Highly enantioselective hydrogenation of styrenes directed by 2′-hydroxyl groups. Org. Lett. 13, 1881–1883 (2011).
Limberger, J., Claudino, T. S. & Monteiro, A. L. Stereoselective synthesis of (E)-3,3-diaryl and (E)-3-aryl-3-aryloxy allylamines and allylalcohols from trans-cinnamyl chloride and alcohol. RSC Adv. 4, 45558–45565 (2014).
Fornika, R., Dinjus, E., Görls, H. & Leitner, W. Structure and reactivity of dimeric rhodium(I) formate complexes: X-ray crystal structure analysis of [{(cod)Rh(μ−κ2O,O′ HCO2)}2] and phosphane-induced hydride transfer to give an η3-cyclooctenyl complex. J. Organomet. Chem. 511, 145–155 (1996).
Leitner, W., Brown, J. M., Leitner, W., Brunner, H. & Leitner, W. Mechanistic aspects of the rhodium-catalyzed enantioselective transfer hydrogenation of α,β-unsaturated carboxylic acids using formic acid/triethylamine (5:2) as the hydrogen source. J. Am. Chem. Soc. 115, 152–159 (1993).
Acknowledgements
We would like to acknowledge the University of Illinois, NSF CAREER (0332333), and Sloan Research Fellowship for their generous support. S.D.L. is grateful to the NSF GRFP (DGE-1144245). Finally, we would like to thank the SCS NMR and Mass Spectrometry Facilities for their assistance.
Author information
Authors and Affiliations
Contributions
Z.W. and K.L.H. conceived and designed the project. Z.W. and S.D.L. performed the experiments. Z.W. wrote the manuscript with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Z., Laffoon, S.D. & Hull, K.L. Asymmetric synthesis of γ-branched amines via rhodium-catalyzed reductive amination. Nat Commun 9, 1185 (2018). https://doi.org/10.1038/s41467-018-03535-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-03535-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
