
Abstract
β-Amino acids are structural motifs widely found in therapeutic natural products, novel biomimetic polymers and peptidomimetics. As a convergent method, the synthesis of stereoenriched β-amino amides through the asymmetric Mannich reaction requires specialized amide substrates or a metal catalyst for enolate formation. By a redesign of the Ugi reaction, a conceptually different solution to prepare chiral β-amino amides was established using ambiphilic ynamides as two-carbon synthons. The modulation of ynamides or oxygen nucleophiles concisely furnished three classes of β-amino amides with generally good efficiency as well as excellent chemo- and stereo-control. The utility is verified in the preparation of over 100 desired products that bear one or two contiguous carbon stereocentres, including those that directly incorporate drug molecules. This advance also provides a synthetic shortcut to other valuable structures. The amino amides could be elaborated into β-amino acids, anti-vicinal diamines, γ-amino alcohols and β-lactams or undergo transamidation with amino acids and amine-containing pharmaceuticals.

Similar content being viewed by others
Main
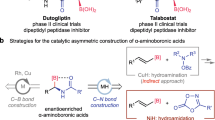
Work published by Seebach in 1996 showing that peptides composed of β-amino acids could form more stable helical secondary structures than their natural α-counterparts initiated numerous syntheses and structural studies on this class of peptidic oligomers1. With an additional methylene group between two functional termini, the self-assembly or distinctive conformations adopted by peptides with β-amino acid residues could modify and improve the properties of materials2,3,4. Special attention has been paid to the β-amino acid building blocks that are also common structural motifs in medicinally important natural products such as Taxol, andrimid and guineamide A (Fig. 1a)5,6,7,8. The related peptidomimetics could exhibit potent biological activity with enhanced metabolic stability and proteolytic resistance9,10. With possible substitution on C2 (β2-derivative), C3 (β3-derivative) or both positions (β2,3-derivative), β-amino acids present an adaptable platform to design chiral building blocks that could span a rather broad range of functional, regio- and stereo-chemical diversity.

a, Representative examples of β-amino acid-containing bioactive natural products. b, The classic Ugi reaction and our scheme to construct the β-amino amide scaffold. c, This work: CBA-catalysed four-component reaction for unified access to β3- and β2,3-amino amides and β3-acylamino amides. Compared to the Ugi reaction, this transformation is a more complex system and multiple competitive reaction sites are present. This entails a more stringent control of selectivity. The amino amide products could undergo versatile synthetic derivatizations to access other important chiral compounds. CBA, chiral Brønsted acid. Star, the substituents of the hydroxyl nucleophile, which could be carboxylic acid or water; triangle, substituent on the amine component; pentagon, substituent of aldehyde; circle, substituent of isocyanide. Rectangle and R, nitrogen substituents on ynamide; R', alkyne substituent. The filled symbol denotes the carbons originated from the alkyne on ynamide substrates.
While asymmetric Mannich reactions that unite imines and (latent) enolates through redox-neutral carbon–carbon bond formation are well-studied for the synthesis of β-amino carbonyl compounds, catalytic access to the carboxyl derivatives is hindered by the poor enolizability of substrates11,12,13,14,15,16. List’s group has recently published work on inventive methods for accessing free β-amino acids17 and the ester derivatives18 using their developed imidodiphosphorimidate catalysts. The former involves enantioselective addition of silyl ketene acetals to α-aminomethyl ethers as methylene imine equivalents, whereas the synthesis of esters employs electrophilic silyl nitronates derived from nitroalkanes. The reactivity constraint is even more remarkable as it uses simple amides as pronucleophiles19,20,21. In Yamashita’s successful endeavour, chiral enolates of simple amides were generated in situ by association of the potassium enolate with a chiral potassium Box salt22. Alternatively, Shibasaki and Kumagai employed tailored 7-azaindoline amides to impart high reactivity and stereoselectivity with the combined use of a Lewis-acidic chiral copper catalyst23,24,25,26,27.
To circumvent this inherent challenge in using a Mannich-type reaction for convergent access to β-amino amides without functional group manipulation, we envisaged that the asymmetric multicomponent reaction (AMCR) would constitute a promising arena for innovative reaction development. In particular, the atom- and step-efficiency of this chemistry would support diversity-oriented synthesis of these chiral motifs with vast therapeutic potential. As one representative MCR, the Ugi four-component reaction (Ugi-4CR) could implement one-pot assembly of α-peptide-like moieties from isocyanides, carboxylic acids, aldehydes and primary amines28. The original reaction has seen creative extensions, including the Ugi-3CR published by List that used water as the internal nucleophile in lieu of carboxylic acid29. More recently, our team addressed the decades-old stereochemical challenge in Ugi-4CRs to forge chiral α-amino amides using a chiral Brønsted acid (CBA) catalyst (Fig. 1b)30.
To adapt this venerable reaction for constructing β-amino amides, the identification of a suitable ambiphilic C2-synthon to substitute for the isocyanide as a C1-synthon in the Ugi reaction was necessary to accommodate this skeletal extension in target products. To this end, ynamides with Cα-electrophilicity and Cβ-nucleophilicity were envisaged to be a uniquely suitable linchpin that could bring together the imine and nucleophile components (Fig. 1b)31,32,33. The chemical reactivity of ynamides has offered numerous synthetic possibilities especially in cycloaddition, rearrangement and cycloisomerization reactions, but their engagement in catalytic asymmetric transformations has not been investigated as thoroughly34,35,36,37. Key advances have only emerged recently from the Ye group in which a chiral phosphoric acid (CPA) catalyst was shown to activate the alkyne and control the stereoselectivity for dearomatization of tethered (hetero)arenes38 and intramolecular hydroalkoxylation/Claisen rearrangement39 and cyclization reactions40. However, the reactivity of ynamides under the auspices of a CBA catalyst in an intermolecular setting has, to the best of our knowledge, not been reported. In our design, a CBA would be likely to activate a ynamide as an electrophilic keteniminium ion and then yield a hydrogen-bond-assisted ion pair to react with an oxygen nucleophile such as water or carboxylic acid. The generated enol amide then adds to the imine activated by the CBA via hydrogen bonding to furnish the β-amino amide. While strategically ideal, the incorporation of ynamides as a reacting component in an asymmetric four-component reaction that entails rigorous control of chemo-, enantio- and diastereo-selectivity could be compounded by their versatile reactivity. Compared to the classic Ugi reaction, unproductive pathways could outnumber the desired reaction due to this reactant switch. Markedly, the hydroacyloxylation of ynamides by the acid catalysts could lead to their deactivation41. The orchestration of this MCR sequence could be further disrupted by competitive amination of ynamides by amine42, hydration under acidic conditions or their susceptibility to undergo metathesis with aldehyde or imine species present in the reaction system43,44. These challenges notwithstanding, in this Article the development of such a CBA-catalysed AMCR of amines, aldehydes and ynamides with water or carboxylic acids to forge optically active β-amino amides is realized and described. This method presents an organocatalytic MCR for the streamlined construction of β3- and β2,3-amino amides and β3-acylamino amides in enantioenriched forms (Fig. 1c).
Results and discussion
Reaction optimization
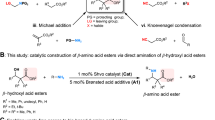
At the outset, the p-toluenesulfonyl (Ts)-protected terminal ynamide A1 was chosen as the model substrate, considering that this group would be a convenient handle for synthetic manipulation. The proof-of-concept experiment was performed with A1, 4-nitrobenzaldehyde B1 and 3,5-dibromoaniline C1 in dichloromethane (CH2Cl2) in the presence of (S)-C1 (10 mol%) at room temperature (r.t.) (Table 1, entry 1). Due to competitive addition of the phosphoric acid catalyst onto the ynamide to form an inactivated enol phosphate, the desired compound was not observed in this reaction system41. When the more acidic N-triflylphosphoramide derivative (S)-C2 was screened45, β3-amino amide 1 was formed in trace amount and 26% e.e. (Table 1, entry 2), which gave an initial hint at the feasibility of this reaction design. A solvent switch to methyl tert-butyl ether (MTBE) brought meaningful improvement of yield (36%) while 27% e.e. could be achieved for the target 1 (Table 1, entry 5). In investigating various axially chiral N-triflylphosphoramides, outstanding enantioinduction was consistently imposed by most N-triflylphosphoramides that bear substituents with an extended π system, regardless of the variations of other parameters (Table 1, entries 6–11). In particular, Me-SPINOL (4,4′-dimethyl-1,1-spirobiindane-7,7′-diol)-derived (R)-C8 promoted the model reaction with the best product yield and stereoselectivity control (Table 1, entry 11)46. An increased catalyst loading of 15 mol% slightly decreased the product yield and did not further improve the e.e. (Table 1, entry 12). Further optimization of reaction conditions by varying the catalyst loading and temperature did not further enhance the results (for details, see Supplementary Table 1). Therefore, following optimal conditions were used to survey the substrate generality: A1 (0.2 mmol) was treated with B1 (0.2 mmol) and C1 (0.1 mmol) in the presence of catalyst (R)-C8 (10 mol%) in MTBE (1.0 ml) at r.t. for 48 h. Using these conditions, desired product 1 was obtained in 80% isolated yield with 97% e.e. (Table 1, entry 11).
With viable conditions in hand for the transformation of terminal ynamides, the study was advanced with full exploration of substrate compatibility. As shown in Table 2, the CBA-catalysed AMCR was applicable for a range of aromatic aldehydes, aromatic amines and ynamides to afford β3-amino amides with excellent enantiocontrol for most cases (1–17). First, moderate to good yields (60–82%) were obtained for aromatic aldehydes with electron-withdrawing functionality such as meta and/or para substituents (1–6). For the aniline partner, trifluoromethyl (CF3) and halogen substituents could be diversely installed at the meta- and para-positions (7–10). From the performance of mono-substituted substrates, a positive influence of meta-substitution on the enantiocontrol could be discerned (11 versus 12). Compared to dibromosubstitution (7, 72%, 94% e.e.), the product yields for 4-bromoanilines (13) and 3-bromoanilines (14) decreased substantially to 28 and 30%, respectively. The effect on product e.e. was not as drastic where the 4-bromo analogue could achieve 86% e.e. while 96% e.e. was seen for the 3-bromo derivative. The use of electron-poor substituents at the meta- and/or para-positions on aldehydes and anilines was found to be important in facilitating the MCR. Benzaldehyde as well as the o-trifluoromethyl, p-methoxy and m-methoxy derivatives gave rise to only minute quantities of products. Similar outcomes were observed in the reactions of unsubstituted, p-methoxy, m-methoxy and o-trifluoromethyl anilines. The reaction details for these unsuccessful substrates can be found in Supplementary Fig. 3. On the other hand, the N-tosyl and N-methyl groups on the ynamides could also be varied, affording β3-amino amides 15–17 in good yields with 91–98% e.e.
Having validated the applicability of terminal ynamides in the envisioned AMCR, the more challenging study to realize enantio- and diastereo-selective MCRs of internal ynamides was undertaken with A5 as the model substrate. This would give rise to β2,3-amino amides with two contiguous carbon stereocentres. Favourably, by applying (R)-C8 (15 mol%) and replacing MTBE with an ethyl acetate/diethyl ether (EtOAc/Et2O) mixture (1/1), β2,3-amino amide 18 could be obtained in 83% yield with >99% e.e. and >20:1 d.r. (for details, see Supplementary Table 2). Efforts then turned to verify the generality of this set of conditions in the assembly of enantioenriched β2,3-amino amides (Table 3). Generally, aromatic aldehydes with electron-withdrawing substituents gave the expected β2,3-amino amides in good yields with complete stereocontrol (18–23). The aldehyde with an electron-rich methoxy substituent and 2-naphthaldehyde with a more substantial structural variation delivered 24 and 25 in good yields with complete stereocontrol. However, 2-nitrobenzaldehyde could not provide any desired product (for details, see Supplementary Fig. 4). The compatibility of a panel of aromatic amines was next examined. When the meta-position was equipped with electron-withdrawing functionalities, anilines could be smoothly incorporated into amino amides 26–28 in serviceable yields and excellent enantioselectivities. Although noticeable erosions of stereoselectivities were observed for para-brominated aniline (29), excellent stereocontrol returned with improved yields when anilines bearing di- or tri-halogen substituents at the meta- and para-positions were utilized (30–33). In the presence of the electron-rich thiomethyl group, the aniline furnished 34 in moderate yield (72%) and poorer stereoselectivity (65% e.e., 2.3:1 d.r.). Similar to the aldehyde component, the ortho-substitution on aniline imposed a detrimental effect on the reaction. A representative substrate, 2-bromoaniline did not provide any desired product either (for details, see Supplementary Fig. 4). Subsequently, the tolerance of current chemistry towards different ynamides was assessed. A variety of groups that impart differentiated electronic properties could be effectively introduced on the terminal benzene ring (35–38 and 41–43). Ynamides connecting to the 3-thienyl (39) or 2-naphthyl (40) group were also applicable substrates. Generally, the reactions with 3,4-dibromoaniline as the amine component offered much higher yields but lower stereoselectivities than those with 3,5-dibromoaniline (35–40 versus 41–43). In addition, the estrone-based ynamide with more distinct structural features provided 44 in 93% yield with virtually perfect stereocontrol. We completed the scope survey by examining different protecting groups on ynamides. Expectedly, benzenesulfonyl and p-methoxybenzenesulfonyl (45 and 46) furnished the desired products in reasonable yields and remarkable stereocontrol. For these MCRs that involve water, the formation of side products accounts for the moderate product yields observed in some cases. The metathesis of a ynamide with an aldehyde would generate an α,β-unsaturated amide and a ynamide could also be hydrated to form an amide. The stereochemistry of products in this series was assigned based on the absolute configurations of 23 and 24 that have been confirmed by X-ray diffraction analysis (CCDC 2175112 and CCDC 2175113).
To further explore the capacity of this AMCR, the possibility of trapping the keteniumium intermediate with a carboxylate ion was hypothesized to produce β3-acylamino amides. To the best of our knowledge, this reaction mode has been elegantly verified in a racemic fashion47 but not been achieved enantioselectively. The reaction optimization commenced with benzoic acid D1 as the standard acid component (for details, see Supplementary Table 3). A complex reaction mixture with no target product 47 was observed when the reactants were mixed in CH2Cl2 at r.t. for 12 h. Based on Cui’s work that constructed β3-acylamino amides via MCR of ynamides47 and triazenyl alkenes48, we reasoned that the order of delivery of reactants might influence the reaction outcome. To generate the acyloxyenamide intermediate in readiness for electrophilic interception, A1 and D1 were first mixed in CH2Cl2 at r.t. before inclusion of B1, C1 and N-triflylphosphoramide (R)-C7. This led to formation of the postulated β3-acylamino amide product 47 in 35% yield with 96% e.e. (Supplementary Table 3, entry 6). Subsequent optimization by varying the solvent, temperature and catalyst loading led to the optimal conditions as follows: carboxylic acid D (0.12 mmol) was reacted with ynamide A (0.12 mmol) in CH2Cl2 (0.5 ml) for 6 h. Benzaldehyde B (0.12 mmol) and aniline C (0.1 mmol) were then added and the reaction mixture was stirred for 1 h followed by addition of (R)-C7 (10 mol%) and n-hexane (0.5 ml) at −40 °C. The reaction mixture was stirred for another 48 h at this temperature (Supplementary Table 3, entry 16).
With this established working process, the scope and limitation of this reaction were evaluated by systematic examination of different components (Table 4). Interestingly, this reaction was characterized by ample scope with respect to the acid component. Benzoic acids bearing a CF3, halogen, alkyl or phenyl substituent at the para-position furnished the chiral β3-acylamino amides 47–54 in 52–83% yields with >99% e.e. in most cases. The outcome was upheld with ortho- and meta-substitutions as well as with 2-naphthoic acid (55–57). The use of aromatic acids was not mandatory: a linear alkyl carboxylic acid was converted into 58 in moderate yield with absolute enantiocontrol and 3-phenylpropiolic acid (59) and 2-octynoic acid (60) were transformed into the corresponding products with excellent results. The generality of this strategy was reinforced in the direct engagement of anti-Duchenne muscular dystrophy (DMD) drug ataluren and anti-acne drug adapalene in AMCRs with A1, B1 and C1. Both drug molecules were enantioselectively incorporated into β3-acylamino amides (61 and 62) with reasonable yields, signifying utility in direct diversification of pharmaceuticals and bioactive molecules. In line with previous observations, it could be gleaned from experimental data of 63–75 that higher yields could arise from the electron-poor aldehydes compared to the electron-rich (66) or electron-neutral analogues (71, 72 and 75). Thiophene-3-carboxaldehyde also competently provided 76 in a good yield with high enantiopurity. Although imines formed from aliphatic aldehydes showed insufficient reactivity in a previous study47, the present catalytic system efficiently facilitated the transformation of these aldehydes after slight adjustment of the reaction conditions (for details, see Supplementary Table 4). This result bears significance as aliphatic groups are native substituents of the α-amino acid subunit found in biologically relevant compounds, especially for the severe acute respiratory syndrome coronavirus 2 main protease (SARS-CoV-2 Mpro) inhibitors49. 3,3-Dimethylbutyraldehyde delivered the desired product 77 in 81% yield and moderate enantiopurity. The enantiomeric purity was facilely upgraded to 98% e.e. after one recrystallization with CH2Cl2/isopropanol. When tri-substituted anilines were employed, amino amides (78–83) derived from aliphatic aldehydes could be furnished in good yields with 80–90% e.e. Aldehydes that bear an α-substituent including 3,3-dimethylbutanal and pivaldehyde did not form any target product, possibly due to steric influence (for details, see Supplementary Fig. 5). We next conducted the focused screening of anilines, verifying the compatibility of derivatives with meta- and/or para-positions occupied by mono- (84–89 and 95), di- (90–93) or tri-substitution (94). CF3, cyano (CN), nitro (NO2) and halogen functionalities endowed suitable substrate compatibility to achieve excellent enantiocontrol and generally good efficiencies. Compared to the meta-bromo-substituted substrate, inferior product yields were achieved for the chloro substituent and when an additional methyl substituent was introduced (85 versus 86 and 90). In this method, while the yield dropped substantially when the N-methyl group on the ynamide was replaced by N-ethyl (98, 40% yield), modification of the sulfonyl group (96 and 97) was well tolerated. In these reactions that formed β3-acylamino amides, the hydrolysis of the iminium intermediate before Mumm rearrangement could be observed. The side product was found to be optically active and possessed the same e.e. as the β3-acylamino amide product. The compatibility of other nucleophiles such as ethanol, benzyl alcohol, 2-nitrophenol, 2-hydroxypyridine and an aromatic sulfonic acid was examined as well (for details, see Supplementary Fig. 6). None of them were found to undergo this MCR. The absolute configuration of 47 was determined by X-ray diffraction analysis and the stereochemistry of other products from this series was assigned by analogy (CCDC 2175114).
To verify the preparative utility of this catalytic AMCR, the gram-scale syntheses of 1 (1.0 g), 30 (1.0 g) and 47 (1.2 g) were performed (Fig. 2a). The good efficiencies and remarkable selectivities paved the way for large-scale production. The synthetic manipulations of β-amino amides provide a shortcut to other appealing compounds (Fig. 2b–d). Hydrolysis of 1 yielded amino acid 99 that could be further diversified to chiral anti-vicinal diamine derivative 100 upon treatment with diphenyl azidophosphate (DPPA) in toluene at 80 °C (Fig. 2b). Similarly, amino amide 30 could be hydrolysed to amino acid 101, followed by conversion into the anti-vicinal diamine 102 when subjected to DPPA (Fig. 2c). The chiral diamines are valuable building blocks in the synthesis of medicinal agents, together with chiral ligands as well as the catalysts50,51. The enantiopurities were perfectly retained while the absolute configurations of 100 (CCDC 2175115) and the minor product (102b, CCDC 2175116) generated alongside 102a were determined by X-ray diffraction analysis. Amide 30 could be reduced by LiAlH4 to deliver γ-amino alcohol 103 in >99% yield without affecting the stereocentre. In view of the broad representation of the β-lactam ring within a range of drugs and bioactive molecules52,53, the generation of β-lactam 104 from 30 was realized in 86% yield by using caesium carbonate (Cs2CO3) to trigger intramolecular cyclization (Fig. 2d). Likewise, amino amides 44 and 42 provided β-lactams 105 and 106 in moderate yields and excellent enantiopurities. Encouraged by the facile aminolysis of 30 by 1-octylamine (to give 107), the late-stage modification of drug molecules was pursued by treating β-amino amides 30 and 1 with homoveratrylamine (to give 108) and amlodipine (to give 110), respectively. β/α-Dipeptide 109 was also assembled by ready attachment of protected α-amino acid l-Tyr(tBu)-OtBu to the obtained β-amino amide 30. This reactivity might have important implications in designing biomimetic polymers with distinctive folding patterns and biological properties that surpass the natural analogues.

a, Gram-scale preparation of compounds 1, 30 and 47. b, Representative amino amide product 1 was shown to undergo hydrolysis to amino acid 99, which could be subjected to DPPA for intramolecular cyclization towards chiral anti-vicinal diamine derivative 100. c, Another model product 30 was subjected to hydrolysis to amino acid 101 or reduction to amino alcohol 103. In the presence of DPPA, intramolecular cyclization proceeded for β-amino acid 101 to form chiral anti-vicinal diamine 102. d, Treatment with caesium carbonate triggers lactamization of several model amino amide products (30, 44 and 42) into β-lactams 104, 105 and 106, respectively. In addition, the amide group of the products could be replaced through transamidation of 1 or 30 with alkyl amine (to give 107), amino acid l-Tyr(tBu)-OtBu (to give 109) and amine drugs such as homoveratrylamine (to give 108) and amlodipine (to give 110). These post-synthetic modifications occurred with excellent retention of optical purities. TEA, triethylamine. Triangle, substituent on the amine component; pentagon, substituent on aldehyde; star, substituent on hydroxyl nucleophile, which could be carboxylic acid or water. The filled symbol denotes the carbons originated from the alkyne on ynamide substrates.
Mechanistic investigations
To elucidate the mechanistic details, we probed the intermediacy of amides that were isolated in reactions involving H2O as the internal nucleophile. The use of standard reaction conditions did not yield 1 and 18, suggesting that product formation did not occur via the hydration of ynamides (Fig. 3a). Studies of the nonlinear effect for the three model reactions (1, 18 and 47) were also performed, where a linear relationship between the enantiopurities of product and catalyst was invariably determined. These studies indicated that only one catalyst has participated in the enantiodetermining step54,55 (Fig. 3b).

a, Control experiments. The amides derived from representative terminal and internal ynamides were subjected to standard reaction conditions. The failure to form target products (1 and 18) implies that the hydration of ynamides is not involved in product formation. b, Nonlinear effect studies. As the e.e. values of product and catalyst are linearly correlated in the formation of amino amides 1, 18 and 47, a single chiral catalyst is likely to be involved in the stereodetermining transition state. c, Proposed reaction mechanism. In the catalytic cycle on the left, the CBA activates the ynamide substrate in the form of a chiral ion pair that comprises the keteminium ion and CBA anion (Int-1). The acid-catalysed addition of water onto the keteminium ion (Int-2) yields an enol amide that undergoes anti- and stereo-selective addition with the imine (generated from condensation of the aldehyde and amine) via a bifunctional activation mode (Int-3). This releases the β3- or β2,3-amino amide and catalyst for turnover. The catalytic cycle on the right depicts the pathway to form β3-acylamino amides from terminal ynamides. The initial formation of the ketiminium ion is succeeded by addition of the carboxylic acid to give the acyloxyenamide. The catalyst then activates the imine intermediate for stereoselective addition with the acyloxyenamide (Int-2′). A rapid catalyst-mediated Mumm rearrangement (Int-3′) furnishes the β3-acylamino amide product and regenerates the catalyst. Triangle, substituent on the amine component; pentagon, substituent on aldehyde; R, terminal alkyne substituent on ynamides; R', aliphatic or aromatic substituent of carboxylic acid component.
Guided by these experimental insights as well as previous reports47,56,57,58, a plausible mechanistic pathway for the established AMCR was postulated (Fig. 3c). The reaction proceeds with the ynamide being catalytically activated by the formation of a keteniminium ion (Int-1) that associates with the CBA anion as an ion pair. The nucleophilic interception is promoted by the acid catalyst that concurrently orients the H2O molecule via hydrogen bonding (Int-2). The catalyst then organizes the generated enol amide and imine via a bifunctional catalytic mode (Int-3). This leads to stereoselective nucleophilic addition that occurs with anti-selectivity to yield a β2,3-amino amide and releases the catalyst for further turnover. On the other hand, the order of addition of reactants influences the pathway of reaction that employs carboxylic acids as external nucleophiles: the union of a carboxylic acid with a ynamide first gives rise to an acyloxyenamide. The imine intermediate could be activated by the CBA as the hydrogen bonded species (Int-1′), followed by the stereoselective nucleophilic addition of an acyloxyenamide to form Int-3′ through Int-2′ via a mono-activation mode59. This is followed by a rapid Mumm rearrangement promoted by the chiral N-triflylphosphoramide to release the β3-acylamino amide and regenerate the catalyst.
Conclusion
Grounded in the use of ynamides as ambiphilic two-carbon synthons, a redesign of the asymmetric Ugi reaction was realized for efficient synthesis of β-amino amides with excellent yields and diastereo- and enantio-selectivities. The utility of this strategy was demonstrated in the preparation of three classes of analogues, covering more than 100 β-amino amides that bear one or two well-defined carbon stereocentre(s). In addition to atom- and step-efficiency, the protocol demonstrates good scalability and could operate on drug molecules in native form. The suitability of amide products to synthesize the related amino acids, amino alcohols, β-lactams and chiral anti-vicinal diamines or undergo direct transamidation with amine-containing drugs and amino acids was verified. As a strategic step forward for MCRs, this approach bodes well for adoption in natural product synthesis as well as the development of therapeutic agents and peptidic oligomers.
Methods
Procedure for the enantioselective syntheses of 1–17
To a dry Schlenk tube (10 ml) were added an amine (0.1 mmol), an aldehyde (0.2 mmol), a ynamide (0.2 mmol), (R)-C8 (10 mol%) and MTBE (1.0 ml). Then the mixture was stirred at r.t. for 48 h. The reaction mixture was purified directly by preparative thin-layer chromatography (TLC) (petroleum ether/EtOAc = 5:1) to give the pure product.
Procedure for the enantioselective syntheses of 18–46
To a dry Schlenk tube (10 ml) were added an amine (0.1 mmol), an aldehyde (0.15 mmol), a ynamide (0.15 mmol), (R)-C8 (15 mol%) and EtOAc/Et2O (0.5 ml/0.5 ml). Then the mixture was stirred at r.t. for 48 h. The reaction mixture was purified directly by preparative TLC (petroleum ether/EtOAc = 5:1) to give the pure product.
Procedure for the enantioselective syntheses of 47–76 and 84–98
To a dry Schlenk tube (10 ml) were added an acid (0.12 mmol), a ynamide (0.12 mmol) and dry CH2Cl2 (0.5 ml). The resulting solution was stirred until the ynamide was completely consumed (for carboxylic acids with poor solubility, it was appropriately heated to 60 °C until the ynamide was completely consumed). An amine (0.1 mmol) and an aldehyde (0.12 mmol) were then added and the mixture was stirred for about 1 h followed by addition of (R)-C7 (10 mol%) and n-hexane (0.5 ml) at −40 °C. The reaction mixture was stirred for another 48 h at this temperature. The mixture was purified directly by preparative TLC (petroleum ether/CH2Cl2 = 1:3) to give the pure product.
Procedure for the enantioselective syntheses of 77–83
To a dry Schlenk tube (10 ml) were added benzoic acid (0.12 mmol), a ynamide (0.12 mmol) and dry CH2Cl2/toluene (1.0 ml, v/v = 1:4). The resulting solution was stirred for 6 h at r.t. until the ynamide was completely consumed. An amine (0.1 mmol), an aldehyde (0.12 mmol) and (S)-C9 (10 mol%) were then added at −30 °C. The reaction mixture was stirred for another 48 h at this temperature. The mixture was purified directly by preparative TLC (petroleum ether/CH2Cl2 = 1:3) to give the pure product.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2175112 (23), CCDC 2175113 (24), CCDC 2175114 (47), CCDC 2175115 (100) and CCDC 2175116 (102b). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. The data supporting the findings of this work are provided in the Supplementary Information including experimental procedures and characterization of new compounds.
References
Seebach, D. et al. β-Peptides: synthesis by Arndt-Eistert homologation with concomitant peptide coupling. Structure determination by NMR and CD spectroscopy and by X-ray crystallography. Helical secondary structure of a β-hexapeptide in solution and its stability towards pepsin. Helv. Chim. Acta 79, 913–941 (1996).
Szefczyk, M. Peptide foldamer-based self-assembled nanostructures containing cyclic beta-amino acids. Nanoscale 13, 11325–11333 (2021).
Kulkarni, K., Habila, N., Borgo, M. P. D. & Aguilar, M.-I. Novel materials from the supramolecular self-assembly of short helical β3-peptide foldamers. Front. Chem. 7, 70 (2019).
Cheng, R. P., Gellman, S. H. & DeGrado, W. F. β-Peptides: from structure to function. Chem. Rev. 101, 3219–3232 (2001).
Kudo, F., Miyanaga, A. & Eguchi, T. Biosynthesis of natural products containing β-amino acids. Nat. Prod. Rep. 31, 1056–1073 (2014).
Wani, M. C., Taylor, H. L., Wall, M. E., Coggon, P. & McPhail, A. T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 93, 2325–2327 (1971).
Fredenhagen, A. et al. Andrimid, a new peptide antibiotic produced by an intracellular bacterial symbiont isolated from a brown planthopper. J. Am. Chem. Soc. 109, 4409–4411 (1987).
Tan, L. T., Sitachitta, N. & Gerwick, W. H. The guineamides, novel cyclic depsipeptides from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscule. J. Nat. Prod. 66, 764–771 (2003).
Aguilar, M.-I. et al. β-Amino acid-containing hybrid peptides—new opportunities in peptidomimetics. Org. Biomol. Chem. 5, 2884–2890 (2007).
Seebach, D. & Gardiner, J. β-Peptidic peptidomimetics. Acc. Chem. Res. 41, 1366–1375 (2008).
Córdova, A. The direct catalytic asymmetric Mannich reaction. Acc. Chem. Res. 37, 102–112 (2004).
Marques, M. M. B. Catalytic enantioselective cross-Mannich reaction of aldehydes. Angew. Chem. Int. Ed. 45, 348–352 (2006).
Ting, A. & Schaus, S. E. Organocatalytic asymmetric Mannich reactions: new methodology, catalyst design, and synthetic applications. Eur. J. Org. Chem. 2007, 5797–5815 (2007).
Verkade, J. M. M., van Hemert, L. J. C., Quaedflieg, P. J. L. M. & Rutjes, F. P. J. T. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 37, 29–41 (2008).
Karimi, B., Enders, D. & Jafari, E. Recent advances in metal-catalyzed asymmetric Mannich reactions. Synthesis 45, 2769–2812 (2013).
Wenzel, A. G. & Jacobsen, E. N. Asymmetric catalytic Mannich reactions catalyzed by urea derivatives: enantioselective synthesis of β-aryl-β-amino acids. J. Am. Chem. Soc. 124, 12964–12965 (2002).
Zhu, C., Mandrelli, F., Zhou, H., Maji, R. & List, B. Catalytic asymmetric synthesis of unprotected β2‑amino acids. J. Am. Chem. Soc. 143, 3312–3317 (2021).
Das, S. et al. Harnessing the ambiphilicity of silyl nitronates in a catalytic asymmetric approach to aliphatic β3-amino acids. Nat. Catal. 4, 1043–1049 (2021).
Saito, S. & Kobayashi, S. Highly anti-selective catalytic aldol reactions of amides with aldehydes. J. Am. Chem. Soc. 128, 8704–8705 (2006).
Saito, S., Tsubogo, T. & Kobayashi, S. Direct-type catalytic Mannich reactions of amides with imines. Chem. Commun. 1236–1237 (2007).
Suzuki, H., Sato, I., Yamashita, Y. & Kobayashi, S. Catalytic asymmetric direct-type 1,4-addition reactions of simple amides. J. Am. Chem. Soc. 137, 4336–4339 (2015).
Yamashita, Y., Noguchi, A., Fushimi, S., Hatanaka, M. & Kobayashi, S. Chiral metal salts as ligands for catalytic asymmetric Mannich reactions with simple amides. J. Am. Chem. Soc. 143, 5598–5604 (2021).
Yin, L., Brewitz, L., Kumagai, N. & Shibasaki, M. Catalytic generation of α-CF3 enolate: direct catalytic asymmetric Mannich-type reaction of α-CF3 amide. J. Am. Chem. Soc. 136, 17958–17961 (2014).
Brewitz, L. et al. Direct catalytic asymmetric Mannich-type reaction of α- and β-fluorinated amides. J. Am. Chem. Soc. 137, 15929–15939 (2015).
Sun, Z., Weidner, K., Kumagai, N. & Shibasaki, M. Direct catalytic asymmetric Mannich-type reaction of α-N3 amide. Chem. Eur. J. 21, 17574–17577 (2015).
Arteaga, F. A. et al. Direct catalytic asymmetric Mannich-type reaction of alkylamides. Org. Lett. 18, 2391–2394 (2016).
Sun, B., Balaji, P. V., Kumagai, N. & Shibasaki, M. α-Halo amides as competent latent enolates: direct catalytic asymmetric Mannich-type reaction. J. Am. Chem. Soc. 139, 8295–8301 (2017).
Ugi, I., Meyr, R., Fetzer, U. & Steinbrückner, C. Versuche mit Isonitrilen. Angew. Chem. 71, 386 (1959).
Pan, S. C. & List, B. Catalytic three-component Ugi reaction. Angew. Chem. Int. Ed. 47, 3622–3625 (2008).
Zhang, J. et al. Asymmetric phosphoric acid-catalyzed four-component Ugi reaction. Science 361, eaas8707 (2018).
Hu, L. et al. Ynamides as racemization-free coupling reagents for amide and peptide synthesis. J. Am. Chem. Soc. 138, 13135–13138 (2016).
Wang, X.-N. et al. Ynamides in ring forming transformations. Acc. Chem. Res. 47, 560–578 (2014).
Pan, F., Shu, C. & Ye, L.-W. Recent progress towards gold-catalyzed synthesis of N-containing tricyclic compounds based on ynamides. Org. Biomol. Chem. 14, 9456–9465 (2016).
Lynch, C. C., Sripada, A. & Wolf, C. Asymmetric synthesis with ynamides: unique reaction control, chemical diversity and applications. Chem. Soc. Rev. 49, 8543–8583 (2020).
Hong, F.-L. & Ye, L.-W. Transition metal-catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis. Acc. Chem. Res. 53, 2003–2019 (2020).
Zhou, B. et al. Stereoselective synthesis of medium lactams enabled by metal-free hydroalkoxylation/stereospecific [1,3]-rearrangement. Nat. Commun. 10, 3234 (2019).
Chen, Y.-B., Qian, P.-C. & Ye, L.-W. Brønsted acid-mediated reactions of ynamides. Chem. Soc. Rev. 49, 8897–8909 (2020).
Zhang, Y.-Q. et al. Asymmetric dearomatization catalysed by chiral Brønsted acids via activation of ynamides. Nat. Chem. 13, 1093–1100 (2021).
Chen, P.-F., Zhou, B., Wu, P., Wang, B. & Ye, L.-W. Brønsted acid catalyzed dearomatization by intramolecular hydroalkoxylation/Claisen rearrangement: diastereo- and enantioselective synthesis of spirolactams. Angew. Chem. Int. Ed. 60, 27164–27170 (2021).
Wang, Z.-S. et al. Synthesis of axially chiral N-arylindoles via atroposelective cyclization of ynamides catalyzed by chiral Brønsted acids. Angew. Chem. Int. Ed. 134, e202201436 (2022).
An, D., Zhang, W., Pan, B. & Zhao, Y. Metal-free hydrophosphoryloxylation of ynamides: rapid access to enol phosphates. Eur. J. Org. Chem. 2021, 314–317 (2021).
Wang, Y. et al. Nonmetal-catalyzed hydroamination of ynamides with amines. Org. Chem. Front. 8, 6244–6251 (2021).
Ao, C. et al. Zinc-catalyzed alkyne–carbonyl metathesis of ynamides with isatins: stereoselective access to fully substituted alkenes. J. Org. Chem. 84, 15331–15342 (2019).
Shindoh, N., Takemoto, Y. & Takasu, K. Atropisomerism of α,β-unsaturated amidines: stereoselective synthesis by catalytic cascade reaction and optical resolution. Chem. Eur. J. 15, 7026–7030 (2009).
Nakashima, D. & Yamamoto, H. Design of chiral N-trifyl phosphoramide as a strong chiral Brønsted acid and its application to asymmetric Diels–Alder reaction. J. Am. Chem. Soc. 128, 9626–9627 (2006).
Li, S., Zhang, J.-W., Li, X.-L., Cheng, D.-J. & Tan, B. Phosphoric acid-catalyzed asymmetric synthesis of SPINOL derivatives. J. Am. Chem. Soc. 138, 16561–16566 (2016).
Huang, B., Zeng, L., Shen, Y. & Cui, S. One-pot multicomponent synthesis of β-amino amides. Angew. Chem. Int. Ed. 56, 4565–4568 (2017).
Wang, C., Lai, Z., Xie, H. & Cui, S. Triazenyl alkynes as versatile building blocks in multicomponent reactions: diastereoselective synthesis of β-amino amides. Angew. Chem. Int. Ed. 60, 5147–5151 (2021).
Gao, K. et al. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 64, 16922–16955 (2021).
Zhou, Z. et al. Enantioselective ring-closing C–H amination of urea derivatives. Chem 6, 2024–2034 (2020).
Swan, S. P. & Mohanty, S. Imidazolidinones and imidazolidine-2,4-diones as antiviral agents. ChemMedChem 14, 291–302 (2019).
Sammes, P. G. Recent chemistry of the β-lactam antibiotics. Chem. Rev. 76, 113–155 (1976).
da Silveira Pinto, L. S., Alves Vasconcelos, T. R., Gomes, C. R. B. & de Souza, M. V. N. A brief review on the development of novel potentially active azetidin-2-ones against cancer. Curr. Org. Chem. 24, 473–486 (2020).
Chen, M. & Sun, J. Catalytic asymmetric N-alkylation of indoles and carbazoles through 1,6-conjugate addition of aza-para-quinone methides. Angew. Chem. Int. Ed. 56, 4583–4587 (2017).
Ma, D., Miao, C.-B. & Sun, J. Catalytic enantioselective House–Meinwald rearrangement: efficient construction of all-carbon quaternary stereocenters. J. Am. Chem. Soc. 141, 13783–13787 (2019).
Zhu, B.-H. et al. Catalytic hydrative cyclization of aldehyde-ynamides with water for synthesis of medium-sized lactams. Sci. China Chem. 64, 1985–1989 (2021).
Pinto, A. et al. Hydrative aminoxylation of ynamides: one reaction, two mechanisms. Chem. Eur. J. 24, 2515–2519 (2018).
Zhu, B.-H. et al. Regio- and stereoselective synthesis of diverse 3,4-dihydro-2-quinolones through catalytic hydrative cyclization of imine- and carbonyl-ynamides with water. ACS Catal. 11, 1706–1713 (2021).
Parmar, D., Sugiono, E., Raja, S. & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 114, 9047–9153 (2014).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (No. 22231004 to B.T., No. 21825105 to B.T. and No. 22271135 to S.-H.X.), the China Postdoctoral Science Foundation (No. 2021M691416 to J.W.), the National Key R&D Program of China (No. 2021YFF0701604 to B.T.), Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002 to B.T.), Guangdong Innovative Program (2019BT02Y335 to B.T.) and the Shenzhen Science and Technology Program (JCYJ20210324120205016 to B.T., KQTD20210811090112004 to B.T. and JCYJ20210324105005015 to S.-H.X.). We appreciate the assistance of the SUSTech Core Research Facilities. Dedicated to Professor Guofu Zhong on the occasion of his 60th birthday.
Author information
Authors and Affiliations
Contributions
B.T. and S.-H.X. conceived and directed the project. J.W. designed and performed the experiments. J.Z., S.-H.X. and J.K.C. helped with the collection of some new compounds and data analysis. B.T., J.W., S.-H.X. and J.K.C. wrote the paper with input from J.Z. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–9, Tables 1–9, experimental procedures, synthetic procedures and characterization data.
Supplementary Data 1
Crystallographic data for compound 23; CCDC reference 2175112.
Supplementary Data 2
Crystallographic data for compound 24; CCDC reference 2175113.
Supplementary Data 3
Crystallographic data for compound 47; CCDC reference 2175114.
Supplementary Data 4
Crystallographic data for compound 100; CCDC reference 2175115.
Supplementary Data 5
Crystallographic data for compound 102b; CCDC reference 2175116.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, J., Zhang, J., Cheng, J.K. et al. Modular enantioselective access to β-amino amides by Brønsted acid-catalysed multicomponent reactions. Nat. Chem. 15, 647–657 (2023). https://doi.org/10.1038/s41557-023-01179-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-023-01179-0
This article is cited by
-
Amino amide assembly
Nature Chemistry (2023)
