
Abstract
Autosomal-dominant hypertension and brachydactyly syndrome (HTNB; Bilginturan syndrome) is known to cause stroke before age 50 when untreated. We report a novel PDE3A gene mutation in a mother and daughter affected with dominant brachydactyly of the hands and feet, a short stature, and hypertension. The hypertension was medically responsive to anti-hypertensive treatment. The 3-bp deletion in the PDE3A gene presented de novo in the mother. Here, we expand the list of PDE3A mutations identified in Bilginturan syndrome and emphasize the importance of standardized genetic testing of HTNB patients to improve diagnostics at an early age. We recommend extended phenotyping in patients with brachydactyly, a short stature or hypertension in clinical practice.
Similar content being viewed by others


Introduction
Hypertension and brachydactyly syndrome (HTNB; OMIM #112410) is characterized by severe salt-independent hypertension, a short stature, brachydactyly, and death from stroke before the age of 50 years when untreated. The syndrome was first described by Bilginturan et al. in 1973 in a Turkish family with brachydactyly, including shortening of the phalanges and metacarpals, and associated unexplained hypertension [1]. The pedigree, including six generations, showed an autosomal-dominant inheritance pattern with complete penetrance of brachydactyly and hypertension. Linkage analysis revealed a locus on chromosome 12p12.2-p11.2 with a LOD score of 9.29 [2].
In 2015, Maass and co-workers [3] performed whole-genome sequencing in the Turkish family and identified a heterozygous missense mutation in the PDE3A gene located on the short arm of chromosome 12. The identified autosomal-dominant inherited p.T445N mutation segregated with the disease and was not found in 200 Caucasian controls or in the publicly available 1000 Genomes Project and Exome Variant Server control databases (July 2013). Sequence analysis of five additional unrelated families with HTNB revealed five additional missense mutations in PDE3A. Functional characterization of the mutant PDE3A showed increased protein kinase A-mediated PDE3A protein phosphorylation in vitro. Hyperphosphorylation of PDE3A resulted in a gain of protein function with increased cAMP hydrolytic activity and enhanced cell proliferation [3]. The PDE3A mutations were suggested to lead to hypertension in the HTNB patients by increasing peripheral vascular resistance. Another PDE3A mutation was identified in a Japanese family with a clinical diagnosis of Bilginturan [4]. All identified mutations cluster in a highly conserved 5-amino acid domain in exon 4 of PDE3A.
Here, we describe a mother and daughter with a clinical diagnosis of HTNB who both carry a 3-bp deletion in PDE3A. We recommend standard genetic screening of suspected HTNB patients. Moreover, extended phenotyping is warranted for individuals with brachydactyly or hypertension, leading to early detection of HTNB. An early and accurate diagnosis provides treatment options that prevent stroke before the age of 50 in HTNB patients.
Methods
Patients
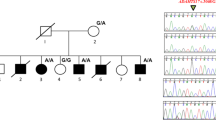
A mother and her daughter with HTNB were seen at the genetics outpatient clinic at the Department of Pediatrics of St. Elisabeth Hospital, Willemstad, Curaçao (Fig. 1). The family members consented to participate in this research and to publication of the study results. Peripheral blood samples were obtained from the two affected family members and their unaffected relatives. Genomic DNA was extracted from 5 ml of EDTA blood for array comparative genomic hybridization (CGH) and Sanger sequencing.

Pedigree of a family with Bilginturan syndrome, also known as the hypertension and brachydactyly syndrome (HTNB). Asterisks indicate that the DNA was analyzed for PDE3A gene variants by Sanger sequencing. A heterozygous PDE3A mutation (p.Thr445del) was found in the affected individuals (black symbols). ref reference allele, mut mutated allele
Genetic screening
Array CGH analysis was performed on the DNA of family members II.4 and III.2 (Fig. 1) using the Infinium CytoSNP-850K BeadChip (Illumina, San Diego, CA, USA). Additionally, exon 4 of the PDE3A gene (NM_000921.4; GRCh37), including the intron–exon boundaries, was screened for variants by Sanger sequencing. Forward 5′-GGTGATATTAAACCAATGTAATTTAG and reverse 5′-GGTTAGCCTTCCTTCTCCTG primers were applied. PCR conditions are available upon request.
Results
Clinical presentation: hypertension
A pregnant 24-year-old woman of Lebanese origin presented at the obstetric clinic with severe hypertension. She had been diagnosed with hypertension at age 15 and showed normalized blood pressures after treatment with anti-hypertensive medication (propranolol). She had one unaffected daughter (Fig. 1). A cesarean section was performed in the second pregnancy at 36+ weeks of gestation due to her history of hypertension. A female child was born in breech presentation with APGAR scores of 9 and 10 after 1 and 5 min, respectively. The premature child had a birth weight of 2870 grams (0 SD) and a head circumference of 34 cm (0 SD; normal for gestational age). Length at birth was 46 cm (−0.7 SD) and body surface area (BSA) was 0.18 m2. No dysmorphic features were seen at the time of birth. The child was admitted to the Department of Pediatrics a few hours after birth due to vomiting. Directly postpartum, hypertension with a systolic blood pressure of 90 mmHg (>95th percentile) was noted in the child (Table 1). Cardiac evaluation showed a systolic heart murmur grade II/VI, punctum maximum at the second left intercostal space.
Two-dimensional echocardiography was performed two days after birth and showed asymmetric thickened ventricular hypertrophy of the left ventricular posterior wall with a normal shortening fraction. The left end-diastolic diameter was 16 mm, end-systolic septum diameter was 11 mm, end-diastolic interventricular septum diameter was 3.1 mm (Z-score = 0.3), and end-diastolic left ventricular posterior wall diameter was 4.5 mm (Z-score = 2.5; ≥2 SD)[5]. A left ventricular outflow tract obstruction was not present on the Doppler echocardiographic examination. No mitral valve insufficiency nor aortic stenosis or coarctation was noted. No other cardiac abnormalities were present. These echocardiography findings led to a diagnosis of mild (asymmetric) hypertrophic cardiomyopathy most likely caused by hypertension.
In the search for the causes of hypertension, an ultrasound examination of the kidneys and adrenals was performed. No abnormalities were shown. Doppler ultrasound of the renal arteries excluded renal artery stenosis and renal vein thrombosis. Plasma electrolyte levels, creatinine, thyroid function, and urinalysis were all normal. The neonate was discharged from the hospital on anti-hypertensive medication (propranolol 1 mg/kg/day) (Table 1). During outpatient follow-up, other causes for hypertension were investigated. Analysis of catecholamines in a 24-h urine collection initially showed a relatively high HVA/5-HIAA ratio. However, magnetic resonance imaging (MRI) of the abdomen and a metaiodobenzylguanidine (MIBG) uptake analysis showed no suspicion of a pheochromocytoma. No cardiac cause was found for the hypertension in the first years of childhood. Fortunately, treatment with anti-hypertensive medications—metoprolol and lisinopril—normalized the child’s blood pressure at age 10 (Table 1).
Clinical presentation: short stature and brachydactyly
At the age of six, patient III.2 (Fig. 1) presented with complaints of headache and concomitant vomiting. On neurological examination it was noted that both the mother and daughter had a short stature (−2 SD and <−2.5 SD, respectively; Fig. 2) and brachydactyly of the hands and feet (Fig. 3a, b). X-rays of the hands and feet at age seven years revealed a uniform and symmetric brachydactyly involving all phalanges, metacarpal, and metatarsal bones (Fig. 3c). Furthermore, coned-shaped epiphyses were seen in the proximal phalanges of the hands and feet with premature fusion of the epiphyses with the metaphyses in the metacarpal bones and middle phalanges of the hands and the middle and distal phalanges of the feet.

Growth curve indicating height related to age for patient III.2 Dots represent height measurements for patient III.2. Curves represent reference growth curves for children. Up to an age of 2 years, the growth of patient III.2 was equal to the −2 SD curve. From age 2 years until age 10 years, the growth moved below the −2.5 SD curve

Imaging of the hands and feet. a Pictures of the hands and feet of patient II.4 showing brachydactyly. b Pictures of the hands and feet of patient III.2 showing brachydactyly. c X-rays were taken of the hands and feet from patient III.2 at age seven. Brachydactyly was observed involving all phalangeal, metacarpal, and metatarsal bones. Coned-shaped epiphyses were seen in the proximal phalanges of the hands and feet. A premature fusion of the epiphyses with the metaphyses was noted in the metacarpal bones and middle phalanges of the hand and the middle and distal phalanges of the feet, indicating premature closure of the growth plates
MRI of the brain and abdomen show no abnormalities
MRI of the brain was performed at the age of seven years due to chronic complaints of fatigue and headache in patient III.2, but no abnormalities were seen. To investigate possible causes for the persistent hypertension (e.g., an obstruction of the renal artery), MRI of the abdomen was performed at age eight and repeated at age nine. The kidneys showed normal sizes and normal pyelocaliceal systems. No obstructions of the renal arteries were identified.
Genetic analysis shows a de novo PDE3A gene mutation
The combination of autosomal-dominant inherited brachydactyly, short stature and unexplained hypertension suggested the clinical diagnosis of Bilginturan syndrome. At the first genetic consultation, the underlying genetic cause of HTNB was still unknown. Therefore, an array CGH was performed as a first genetic diagnostic approach to identify copy number variations but showed no abnormalities. When PDE3A mutations were reported to cause HTNB [3], we performed Sanger sequencing of exon 4 of the PDE3A gene (NM_000921.4; GRCh37) and revealed a heterozygous 3-bp in-frame deletion (c.1333_1335del; p.Thr445del) (Fig. 4). Segregation analysis of the family showed that the variant was de novo in the affected mother (II.4) and was inherited by the affected daughter (III.2). The variant has not previously been described in the following publicly available databases, which were all accessed in April 2017: 1000 Genomes Project, http://www.internationalgenome.org [6]; ExAC, http://exac.broadinstitute.org/[7]; gnomAD, http://gnomad.broadinstitute.org [7] GoNL, http://www.nlgenome.nl [8]; and Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, http://evs.gs.washington.edu/EVS/.

Molecular genetic analysis of the PDE3A gene in a family with hypertension and brachydactyly syndrome (HTNB). A 3-bp deletion in the PDE3A gene (NM_000921.4; GRCh37), c.1333–1335delACC; p.T445del, was identified in the two affected family members (II.4 and III.2). a Exon–intron structure of the PDE3A gene. b Chromatograms showing the PDE3A mutation identified in patient III.2 compared to a control individual
Comparison of clinical features of HTNB patients
We compared the phenotypes of our patients to the clinical features of HTNB patients who were described previously (Table 2) [1, 4, 9]. A large Turkish HTNB family was described in 1973 by Bilginturan et al. [1], including 31 affected family members in six generations (family A in Table 2). Hypertension and brachydactyly were present in all patients in this family. Moreover, all affected family members showed a short stature compared to the unaffected family members and to unrelated people from the same geographic area. Apart from that, the patients responded to anti-hypertensive treatment [10]. Comparison with four other HTNB families (families B, C, and D in Table 2), including our patients described in the current study (family E in Table 2), revealed striking similarities. All patients showed moderate to severe hypertension and all patients responded well to anti-hypertensive treatment. Brachydactyly of the hands and feet was present in all patients. A short stature was present in most patients except for the affected members of a Hispanic family described by Chitayat et al. [9], who were significantly shorter than their unaffected relatives but were not significantly shorter than unrelated people from the same area. Another important similarity is the autosomal-dominant inheritance pattern with complete penetrance in all described families with PDE3A mutations. Differences include the occurrence of neurovascular anomalies and dysmorphic features, such as a stocky build and round faces, which were reported only in the Turkish family [1]. Different cardiovascular features were present in the Canadian, Hispanic, Japanese, and Lebanese families, which were probably secondary effects of the consistent hypertension.
Discussion
In the current study, we identified a PDE3A gene mutation in a mother and daughter with Bilginturan syndrome, also known as hypertension and brachydactyly syndrome (HTNB). The mutation was de novo in the affected mother and was inherited by the affected daughter. Herein, we extend the spectrum of PDE3A mutations identified in HTNB patients. A comparison of clinical features observed in previously reported HTNB patients revealed that essential hypertension, brachydactyly, and a short stature were present in (almost) all patients and are thus key symptoms of Bilginturan syndrome.
The PDE3A gene encodes phosphodiesterase 3A, which is known to degrade the small intracellular signaling molecules cyclic AMP (cAMP) and cyclic GMP (cGMP) to AMP and GMP, respectively, in the cardiovascular system and developing limbs [11, 12]. Maass et al. [3] identified six missense mutations in PDE3A in six unrelated HTNB families of diverse ethnic backgrounds. Similar to the 3-bp deletion identified in this study, the previously identified mutations (p.T445N, p.T445A, p.T445S, p.A447T, p.G449V, and p.A447V) cluster in amino acids 445–449 of PDE3A, which underlines the importance of this highly conserved domain for protein function [13]. Functional analyses showed that the p.T445N variant caused PDE3A protein hyperphosphorylation and thereby increased PDE3A activity, leading to less hydrolysis of cAMP and thus increased cAMP signaling. In addition, vascular smooth muscle cells with PDE3A mutations show increases in both mitotic activity and peripheral vascular constriction. Activating mutations in amino acids 445–449 of PDE3A contribute to an increase in peripheral vascular resistance, which probably causes hypertension in HTNB patients. Moreover, PDE3A is known to be involved in osteogenesis and limb development [3, 12]. The exact pathogenesis of the brachydactyly and short stature in HTNB requires in vivo studies on wild-type and mutant PDE3A function. Since (1) the 3-bp deletion described here is located in the same five-amino acid domain of PDE3A, (2) the variant is completely absent from public variant databases, and (3) the mutation fully segregates with the disease in this family, we can conclude that c.1333_1335del; p.Thr445del affects PDE3A protein function, resulting in HTNB.
It is essential to diagnose HTNB at early age, since early anti-hypertensive treatment can prevent hypertension-related organ damage. However, early diagnosis of HTNB can be difficult since patients may be asymptomatic or the symptoms may become more apparent at a later age. Since hypertension was detected in our patients (II.4 and III.2) at age 15 and directly postpartum, respectively, we plead for early recognition of the syndrome with immediate blood pressure monitoring after birth in suspected HTNB patients. In 2010, Toka et al. suggested clinical criteria for the HTNB diagnosis [15]. Affected children were determined to have a significantly lower birth weight on average (2600 g) than unaffected babies (3205 g) in the same family. Furthermore, the short stature and brachydactyly became clinically recognizable in children aged 10 to 17 years. Almost all affected children can be classified as hypertensive, even at age 3. No strokes occurred in patients treated with anti-hypertensive medication [15]. Thus, the conclusive diagnosis of HTNB at a young age benefits the patients tremendously through effective anti-hypertensive treatment and prevention of stroke before age 50.
Here, we emphasize that pediatricians, cardiologists, nephrologists, neurologists, and general practitioners who see these patients in their clinics should look beyond the clinical features of hypertension and realize that the combination of brachydactyly with a short stature suggests an early recognizable syndrome. Blood pressure measurements should be performed in individuals with brachydactyly, whereas hands, feet, and height should be examined in patients with hypertension. Thus, a physical examination combined with immediate PDE3A gene screening forms a solid and standardized approach for the diagnosis of HTNB. Therefore, PDE3A mutation analysis has been implemented in our DNA diagnostics laboratory. This approach not only results in the best clinical care for HTNB patients but also prevents unnecessary expensive additional diagnostics.
References
Bilginturan N, Zileli S, Karacadag S, Pirnar T. Hereditary brachydactyly associated with hypertension. J Med Genet. 1973;10:253–9.
Schuster H, Wienker TE, Bahring S, Bilginturan N, Toka HR, Neitzel H et al. Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat Genet. 1996;13:98–100.
Maass PG, Aydin A, Luft FC, Schachterle C, Weise A, Stricker S et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet. 2015;47:647–53.
Boda H, Uchida H, Takaiso N, Ouchi Y, Fujita N, Kuno A et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet. 2016;61:701–3.
Pettersen MD, Du W, Skeens ME, Humes RA. Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: an echocardiographic study. J Am Soc Echocardiogr. 2008;21:922–34.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Exome aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–25.
Chitayat D, Grix A, Balfe JW, Abramowicz JS, Garza J, Fong CT et al. Brachydactyly-short stature-hypertension (Bilginturan) syndrome: report on two families. Am J Med Genet. 1997;73:279–85.
Schuster H, Toka O, Toka HR, Busjahn A, Oztekin O, Wienker TF et al. A cross-over medication trial for patients with autosomal-dominant hypertension with brachydactyly. Kidney Int. 1998;53:167–72.
Begum N, Hockman S, Manganiello VC. Phosphodiesterase 3A (PDE3A) deletion suppresses proliferation of cultured murine vascular smooth muscle cells (VSMCs) via inhibition of mitogen-activated protein kinase (MAPK) signaling and alterations in critical cell cycle regulatory proteins. J Biol Chem. 2011;286:26238–49.
Wakabayashi S, Tsutsumimoto T, Kawasaki S, Kinoshita T, Horiuchi H, Takaoka K. Involvement of phosphodiesterase isozymes in osteoblastic differentiation. J Bone Miner Res. 2002;17:249–56.
Houslay M. Hypertension linked to PDE3A activation. Nat Genet. 2015;47:562–3.
Schuster H, Wienker TF, Toka HR, Bahring S, Jeschke E, Toka O et al. Autosomal dominant hypertension and brachydactyly in a Turkish kindred resembles essential hypertension. Hypertension. 1996;28:1085–92.
Toka O, Maass PG, Aydin A, Toka H, Hubner N, Ruschendorf F et al. Childhood hypertension in autosomal-dominant hypertension with brachydactyly. Hypertension. 2010;56:988–94.
Naraghi R, Schuster H, Toka HR, Bahring S, Toka O, Oztekin O et al. Neurovascular compression at the ventrolateral medulla in autosomal dominant hypertension and brachydactyly. Stroke. 1997;28:1749–54.
Toka HR, Bahring S, Chitayat D, Melby JC, Whitehead R, Jeschke E et al. Families with autosomal dominant brachydactyly type E, short stature, and severe hypertension. Ann Intern Med. 1998;129:204–8.
Acknowledgements
We thank the family who participated in this research. The author KYR was financed by the European Community’s Seventh Framework Programme (FP7/2009) under Grant Agreement 305608; EURenOmics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Renkema, K.Y., Westermann, J.M., Nievelstein, R.A.J. et al. PDE3A gene screening improves diagnostics for patients with Bilginturan syndrome (hypertension and brachydactyly syndrome). Hypertens Res 41, 981–988 (2018). https://doi.org/10.1038/s41440-018-0094-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-018-0094-5
