
Abstract
In recent years, several post-interventional analyses of large-scale randomized controlled clinical trials have given us a new concept regarding the risk management of hypertension and cardiovascular diseases. The beneficial effects of intensive treatments were extended even after the interventions ended. This phenomenon is known as “metabolic memory” or “legacy effect”, and we recognized its clinical significance. A certain level of evidence in human and animal studies employing organ transplantation techniques has indicated that this type of “memory” resides in each organ and could be transferrable, erasable, and rewritable, which is similar to neuronal and immune “memory”. In this review, we define this memory as “organ memory” and summarize the current picture and future direction of this concept. “Organ memory” can be observed in many clinical settings, including in the control of hypertension, diabetes mellitus, and dyslipidemia. Several intensive treatments were demonstrated to have the potential to rewrite “organ memory”, leading to the curability of targeted diseases. “Organ memory” is the engraved phenotype of altered organ responsiveness acquired by a time-dependent accumulation of organ stress responses. Not only is the epigenetic change of key genes involved in the formation of “organ memory” but the alteration of multiple factors, including low molecular weight energy metabolites, immune mediators, and tissue structures, is involved as well. These factors intercommunicate during every stress response and carry out incessant remodeling in a certain direction in a spiral fashion through positive feedback mechanisms. Future studies should be directed toward the identification of the core unit of “organ memory” and its manipulation.
Similar content being viewed by others
“Memory phenomenon” and “organ memory”
In several randomized controlled clinical trials (RCTs), the “memory phenomenon” or “legacy effect” has been proposed by clinical evidence to show that, even after the cessation of the clinical trial, the superiority of one treatment over the other persists. This proposal is now highlighted for its medical significance. The existence of “memory” acquisition by transient interventions has been experienced in many clinical contexts of non-communicable diseases (NCDs) such as lifestyle-related diseases (diabetes mellitus, hypertension, and so on). As described later, we have come to recognize that this kind of “memory” is supposed to reside in each organ, so we designate it “organ memory”.
The actual entity of “organ memory”, however, has barely been examined. “Organ memory” is the engraved phenotype of altered organ responsiveness acquired by time-dependent accumulation of organ stress responses and is expected to have a great potential to offer a new insight into medical science. In this review, we delineate the concept of “organ memory” from multiple aspects including clinical settings, mechanistic insight, and molecular basis.
“Memory phenomenon” in clinical settings
In clinical settings, the “memory phenomenon” can cause both beneficial and deleterious effects. Previous insufficient risk control was revealed to have persistent effects on the occurrence of lifestyle-related diseases, such as hypertension and diabetes, or the progression of their vascular complications, even though the present control was satisfactory. Past history of obesity, hypertension, hyperglycemia, and smoking is recognized to continuously generate a harmful effect on the occurrence of cardiovascular events. Although it is assured that smoking cessation is beneficial for risk reduction, most large-scale cohort studies have indicated that ex-smokers still have a residual risk of vascular diseases compared with never smokers [1, 2]. As another example, we previously reported that past obesity could influence the progression of diabetic nephropathy even if the current body weight is within the normal range [3].
In contrast, persistent beneficial effects of the transient intervention for preventing cardiovascular events and its related mortality were observed even after completion of the intervention in various large-scale clinical trials. In regard to the RCTs on intensive glucose lowering in type 2 diabetes, a 10-year post-trial follow-up of UKPDS (United Kingdom Prospective Diabetes Study) revealed that a relative reduction of cardiovascular risks in the intensive treatment group lasted long after the intervention ended, although superior glucose lowering by the intensive treatment was lost soon after the trial ended [4]. The DCCT (Diabetes Control and Complications Trial)/EDIC (Epidemiology of Diabetes Interventions and Complications) study demonstrated that intensive therapy in type 1 diabetes significantly slowed the progression of diabetic retinopathy and nephropathy during 6.5 years of the intervention period (DCCT) [5]. The reduction resulting from intensive therapy persisted for at least 4 years of the post-trial follow-up period (EDIC) [6], despite the fact that glycemic control between the intensive and standard group was similar during the post-trial period. For blood pressure control, the ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation)-ON showed a beneficial post-trial “memory phenomenon” with respect to reductions in the risk of death from cardiovascular causes [7]. In a lipid-lowering study, WOSCOPS (the West of Scotland Coronary Prevention Study) demonstrated that cardiovascular events and related deaths were reduced even during the subsequent 10-year post-trial follow-up period after the 5-year treatment of hypercholesterolemia by pravastatin [8]. The 4 S (the Scandinavian Simvastatin Survival Study) study also showed a “memory phenomenon” by a transient treatment of hypercholesterolemia by simvastatin [9].
In some cases, the beneficial effects of the intensive treatment first became evident during the post-trial follow-up period. In the DCCT/EDIC study, the reduction in major cardiovascular events, which consisted of nonfatal myocardial infarction, nonfatal stroke, and cardiovascular death, was not shown during the intervention period. The significant reduction associated with the intensive treatment became evident during the post-trial period [10]. The ACCORD-BP (Action to Control Cardiovascular Risk in Diabetes blood pressure trial) examined the effect of intensive blood pressure control in high-risk patients with type 2 diabetes. A significant reduction in major cardiovascular events was not observed during the 5 years of the intervention period [11]. The reduction caused by the early intensive treatment became evident in a subgroup analysis of the ACCORD Follow-On trial (ACCORDION) in which ACCORD participants were subsequently followed for 4 years [12].
In addition to the reduction in cardiovascular events, the “memory phenomenon” after a transient intervention has been observed in the onset of hypertension. The TROPHY (the Trial of Preventing Hypertension) study demonstrated that the risk of the progression from the prehypertension stage to hypertension was reduced by a 2-year transient inhibition of the renin–angiotensin system (RAS) by candesartan, and this reduction was observed even after the trial period was finished [13]. We have shown this kind of “memory phenomenon” in hypertension-prone animal models [14,15,16], which we termed as “angiotensin block memory”. In contrast, the activation of RAS in youth has been shown to induce hypertension, even after the cessation of RAS activation [17].
In humans, salt intake in the neonatal stage could determine late-stage blood pressure, which is also considered a “memory phenomena”. A randomized trial was conducted and demonstrated that sodium restriction in newborn infants influenced blood pressure in a 15-year post-trial follow-up [18]. We recently reported that a persistent hypertensive phenotype was acquired by transient high salt intake in two experimental models of hypertension (spontaneously hypertensive rat stroke prone, SHR-SP, and the Dahl salt sensitive hypertensive rat) and proposed this phenotype as “salt memory” [19].
A systematic review of the “memory phenomenon”
To better understand the characteristics of the “memory phenomenon” in clinical settings, we performed a systematic review of RCTs of cardiovascular diseases that investigated the “memory phenomenon”, namely, the post-trial effects of the transient intervention, as we have reported recently [20]. In that systematic review, “memory phenomenon” was identified as positive when the cardiovascular outcome was significantly superior in the intervention group even at the end of the post-trial observational follow-up period. At that point, the significant reduction of a risk factor (blood glucose, blood pressure, or lipid level) was lost owing to stopping the intervention. Usually, the effect of an intervention to reduce cardiovascular risk factors is lost soon after the cessation of the intervention. In some cases, however, the effect of a transient intervention on the control of risk factors significantly persisted, even during the post-trial observational period, with the significant suppression of the outcome. We defined the prolonged effect of the intervention as “carry-over effect” and distinguished it from “memory phenomenon”.
Twenty-one articles were found to describe the “memory phenomenon” as positive, after careful examination of the full-text of the 907 articles retrieved from the initial screening. Only three articles were judged as describing the “memory phenomenon” as negative. Five articles showed “carry-over effects” of the intervention (Table 1). The systematic review revealed that each intervention type, that is, glucose lowering, blood pressure lowering, or LDL-cholesterol lowering, possessed unique characteristics of the “memory phenomenon” (details are described in our report [20]). Most of the observational periods of these studies lasted for > 10 years. Therefore, “memory phenomenon” was suggested to last for a long time and is thought to have a considerable effect on the clinical course of NCDs [21,22,23].
“Memory” resides in organs
Based upon the clinical evidence described above, we should reach the consensus that “memory phenomenon” truly exists. Our next question is “where these memories are”. A certain series of organ transplantation studies would provide the possible answer.
One study reported that a kidney transplantation from a donor with a history of hypertension conveyed increased blood pressure to the recipient [24]. An opposite phenomenon was observed after transplanting a kidney from a donor with normal blood pressure; the recipient’s hypertension subsided [25]. In a cohort study of heart transplantation, it was demonstrated that recipient outcomes could be predicted by the donor’s characteristics, among which, donor diabetes was associated with recipient mortality [26]. In pancreas transplantations, donor obesity can influence both recipient and allograft survival [27].
In terms of animal studies, similar transferrable memories were reported. Transplantation of kidney allografts from hypertension-prone rats resulted in a rise in blood pressure in the normotensive recipient rats. In contrast, kidney allografts from hypertension-resistant rats resulted in a sharp decrease in blood pressure in the hypertensive recipient rats [28, 29]. In addition, we recently identified “salt memory” in hypertensive rats, and this memory was also transferrable [19]. We demonstrated that transplanting the kidneys of hypertensive rats after transient salt loading to normotensive animals resulted in the development of hypertension in the recipient (Fig. 1). The transplantation of normotensive rat kidneys to hypertensive rats with “salt memory” normalized their blood pressure, and vice versa. These results indicated that the kidney contains “memory” as a single organ. Thus, the memory “resides” in the organ. Interestingly, in addition to these substantial organs, gut microbiota could also act as a bearer of “metabolic memory”. It has been known that treatment with antibiotics in the neonatal period could cause a transient change in the microbiome and induce obesity later in adult life [30], and this “metabolic memory” was transferrable when the microbiota from early-life treatment with antibiotics was transplanted into germ-free recipients [31]. Based on these findings, we assume that clinically observed memory is stored in organs. Therefore, this kind of memory can be defined as “organ memory.”
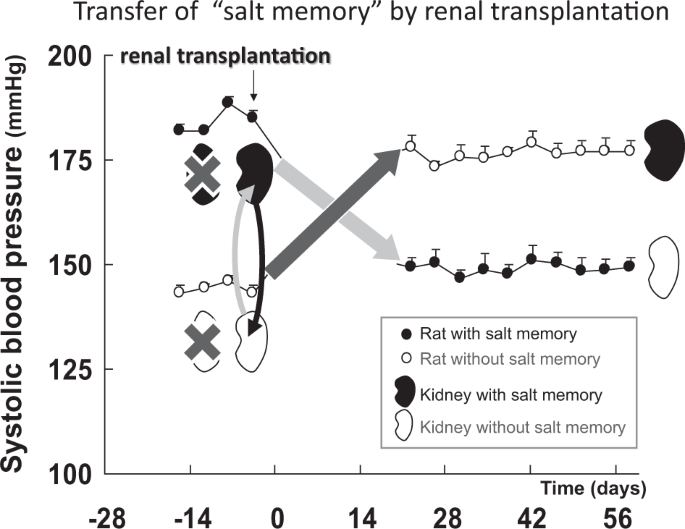
“Memory” resides in organs–transfer of “salt memory” by renal transplantation. After transplantation of the kidney of a rat with salt memory that had received transient salt loading (a black kidney), the blood pressure in normotensive rats without salt memory (○) increased. After transplantation of the kidney of a rat without salt memory (a white kidney), the blood pressure (●) in the rat with salt memory was normalized (quoted from reference 19)
The nature of “organ memory”—the “updating” process
The observation of “organ memory” in clinical settings and animal studies leads us to believe that “organ memory” participates in an incessant “updating” process provoked by external stimuli loaded on the organ. Memory is not merely an engraved mark that lacks elasticity and is unidirectional but originally contains multi-faceted phenomena, including “learning”, “imprinting”, “forgetting”, “recalling”, “erasing”, and “rewriting”. In the field of neuroscience, recent behavioral studies as well as molecular mechanistic studies have revealed that a series of “updating” processes is indispensable in maintaining neuronal memory [32, 33]. In other words, biological phenomena that exhibit the updating process can be referred to as “memory”. The “updating” of neuronal memory is completed through three processes, namely, 1. “recalling” of the memory, 2. “erasing” of the original memory, and 3. “rewriting” it with a new version of the memory.
Organs cannot possess “consciousness”, so it might not be appropriate to consider whether a “recalling” process exists in organs. However, the properties of organs are incessantly altered by external stimuli loaded on organs as if it is “updating”. Thus, “organ memory” should be distinctive from an existing concept of “organ remodeling” or “irreversible deviation”, because it is continuously “updating” through dynamic adaptive process owing to the repetitive external stimuli loaded on organs. For example, when repetitive exogenous noxious stimuli cause DNA damage in the cells of organs, they not only induce the DNA repair process each time but also influence the epigenetic status of DNA, which contributes to the long-term alteration of gene activation in organs (described in detail later). Importantly, epigenetic status is variously modulated, depending upon the kinds, severity, duration, and intervals of the stimuli. The precise relation of DNA repair and epigenetic modulation is explained in a recent review, and this process is analogous to neuronal memory “updating” [34].
The manipulation of “organ memory”
It is clinically important to investigate whether “organ memory” can be “updated” by our behaviors. Possible manipulation of “organ memory” has been demonstrated in recent reports on cardiovascular diseases. Salt memory for hypertension has been erased by combining treatment with a calcium channel blocker and an angiotensin receptor blocker [19]. Established hypertensive nephrosclerosis and arteriosclerosis were also reversed by transient intensive RAS inhibition [35, 36]. The transient blockade of RAS reversed established glomerulosclerosis caused by Adriamycin administration [37]. In recent parabiosis (the joining of two animals) experiments using an older mouse and a younger mouse, rejuvenation occurred in the older mouse [38, 39]. That is, aging phenotypes (cardiomegaly, muscle fibrosis, and cognitive decline) observed in older mice were reported to recover as if reverting to a young state. Therefore, the modulation of humoral factors has the potential to update “organ memory”. In the clinical field, it has been reported that glomerular expansion was reduced and advanced stage diabetic nephropathy regressed owing to the transplantation of a healthy pancreas into a patient with type 1 diabetes [40]. We have also demonstrated that the alteration of the clinical course of hypertension was partly achievable by inhibiting RAS transiently (STAR CAST study) [41]. These results indicate that “organ memory” can be “updated”. The manipulation of “organ memory” is a worthwhile challenge that could be a new medical approach for NCDs.
Mechanistic insight into “organ memory”—the spiral model
As described above, even if the same stress is loaded on an organ, the organ response to the stress would differ in extent, speed, or direction from the previous response. Depending on the stress burden, the organ undergoes continuous remodeling of its structure and function to adapt to the stimulation. Therefore, after being subjected to a transient stress, the components of the organ shift to a different state/dimension, which arouses a different stress response. This adaptive time-dependent change in the organ stress responsiveness is “organ memory”. Then, the next question is “What is the mechanistic basis for “organ memory”?
It is becoming clear that many biological systems are governed by non-linear processes that are regulated by positive feedback mechanisms. These processes may switch between discrete states based on a functional relationship with the memory of input stimuli. The concept of “hysteresis” has been used to describe such memory-dependent non-linear processes in material sciences and has recently been applied to biological processes [42]. Cell fate commitment during oocyte maturation is a known example of such process that is governed by “hysteresis”. In oocytes, mitogen-activated protein kinase and cell-division cycle protein kinase Cdc2 are organized into positive feedback loops. Such time-dependent positive feedback mechanisms could produce an actively maintained “memory” and could explain the irreversibility of maturation [43]. In these contexts, we propose that “organ memory” is incessantly updated by transient stress loaded on organs, which results in the production of different organ responses in a spiral fashion (“the spiral model”, Fig. 2), driven by positive feedback mechanisms. We hypothesize that “organ memory” has a nature that is governed by “hysteresis” and repeated stimuli would designate the organ to a certain different state, which may lead to the occurrence and progression of diseases.

The spiral model of “organ memory”. Spatio-temporal changes in organ responsiveness. After experiencing a transient stress, the organ components shift to a different state or dimension, which arouses different reactions, even if the organ experiences the same stress again. We propose that “organ memory” is generated in a spiral fashion. P, organ parenchymal cells. S, stromal cells. C, circulating cells (immune cells). M, intercellular mediators (hormones, cytokines)
Repeated stimuli can continuously change the status of an organ through the signal networks governed by positive feedback loops. The multiple factors in the organ would consist of such networks that generate “organ memory” in a spiral fashion. Within the organ, the intricate cellular interactions of organ parenchymal cells, interstitial cells, and circulating cells that are delivered through blood and lymph vessels are postulated to participate in the generation of “organ memory.” Intercellular mediators, including hormones, cytokines, micro RNA, exosomes, and so on, also play significant roles in cellular communications that are crucial for the generation of “organ memory”. Alteration in tissue structure, such as the modification of the extracellular matrix through the generation of Amadori products due to hyperglycemia in diabetes mellitus, should affect “organ memory” [44]. Intracellular events, including the remodeling of organelle dynamics and metabolic processes and epigenetic modifications of gene expression, should play essential roles in the generation of “organ memory”.
Once “organ memory” is established in an organ, “organ memory” in each organ can influence one another and affect “organ memory” in a different organ. We assume that organ failure that occurs in NCDs may develop in such inter-organ communication of “organ memory”, as demonstrated in Fig. 3.
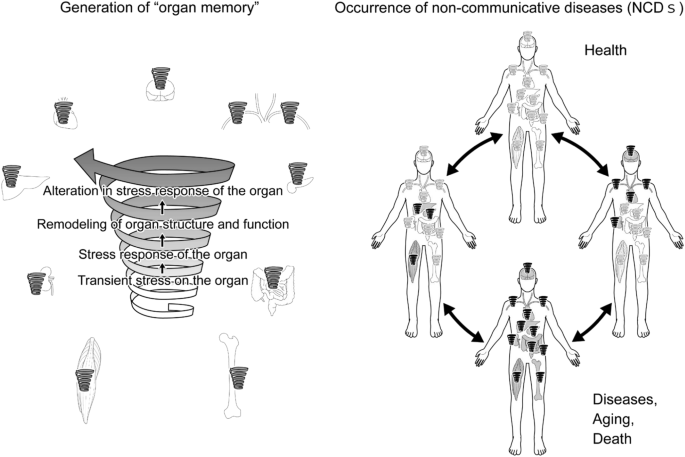
Schematic representations of the occurrence of non-communicable diseases (NCDs) through the generation of “organ memory”. Details are described in the manuscript
“Organ memory” and epigenetics
One of the significant elements responsible for “organ memory” is the epigenetic regulation of gene expression (Fig. 4). In the process of neuronal memory formation, epigenetic mechanisms have been proven to play an important role [45]. In the case of pro-inflammatory activation of cardiac fibroblasts, epigenetic changes in histone acetylation have been suggested to memorize a stimulation by angiotensin II [46]. For “angiotensin block memory”, which we originally reported as an organ memory, actual epigenetic changes occurred as a key step in the formation of memory in cases of human glomerulopathy, including diabetic nephropathy [47]. We demonstrated that the expression of the transcription factor, Kruppel-like Factor 4 (KLF4) was reduced in podocytes in patients with glomerular diseases. The reduced expression of KLF4 in podocytes leads to an increased methylation level of the nephrin promoter and decreased nephrin expression, resulting in proteinuria. Concerning the mechanism of the epigenetic regulation of the nephrin promoter, overexpression of KLF4 reduced the binding of DNA methyltransferase 1 to the nephrin promoter [48]. We further reported that angiotensin II, a crucial mediator of kidney diseases, suppressed KLF4 expression to modulate the long-term epigenetic status of the nephrin gene [37].
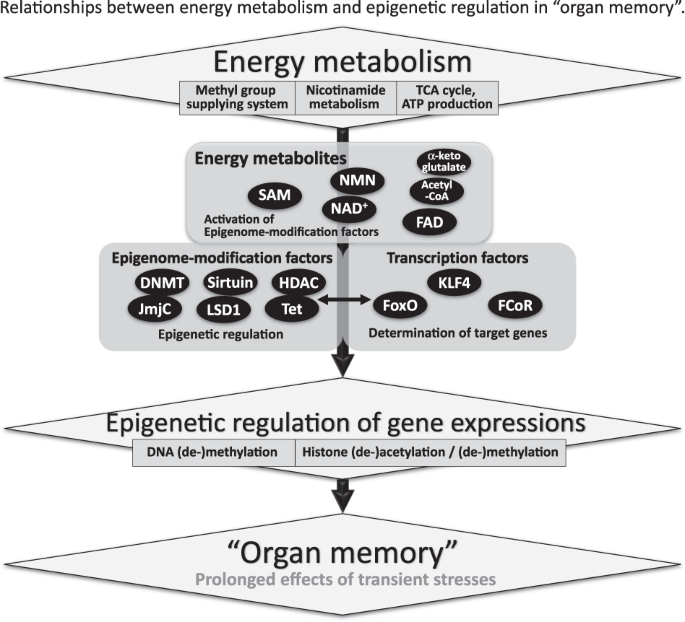
Relationships between energy metabolism and epigenetic regulation in “organ memory”. Energy metabolites, epigenome-modification factors, and transcription factors are essential for the generation of “organ memory”. Epigenetic control of gene expression could be responsible for the generation of “organ memory”. Transcription factors play a critical role in gene-specific epigenetic regulation. Low molecular weight energy metabolites, such as acetyl-CoA, nicotinamide adenine dinucleotide (NAD), and S-adenosylmethionine (SAM), are essential for the epigenetic regulation of gene expression. The activity of epigenetic modification factors is regulated by low molecular weight energy metabolites. DNMT: DNA methyltransferase, HDAC: histone-deacetylase, JmjC: jumonji C, LSD1: lysine specific demethylase l, Tet: ten–eleven translocation, KLF4: Kr û ppel-like factor 4, FoxO: forkhead box O, FCoR: FoxO1-corepressor
In these ways, both epigenome-modification enzymes, such as histone deacetylases and DNA methyltransferases, and transcription factors seem to be essential for the epigenetic regulation of gene expression, which is involved in the generation of “organ memory” (Fig. 4) [49]. Forkhead box O1 (FoxO1) is a transcription factor that can directly bind to the nicotinamide adenine dinucleotide (NAD)-dependent histone-deacetylase, Sirt1 [50]. Interactions between Sirt1, FoxO1, and the FoxO1-corepressor, which we identified and that represses activity of FoxO1 [51], are another example of the complex of transcription factors and epigenome-modification factors that would be involved in the generation of “organ memory”.
Epigenetics and cellular metabolism
We also focused on the fact that “energy metabolites” with low molecular weights, such as NAD, flavin adenine dinucleotide, acetyl-CoA, and S-adenosylmethionine (SAM), are essential for epigenetic regulation (Fig. 4) [52]. These energy metabolites have been shown to regulate the activity of epigenome-modification factors, including sirtuins, histone deacetylases, DNMTs, and jumonji C. Recent reports have demonstrated that epigenetic modulation through these small molecule metabolites is involved in the progression of various diseases [53].
Several reports have demonstrated that dysregulation of nicotinamide metabolism causes widespread changes in the epigenetic regulation of gene expression. We reported that the activity of the histone-deacetylase Sirt1 in podocytes in patients with diabetes was reduced owing to an impairment in the transportation of the NAD precursor nicotinamide mononucleotide from the renal proximal tubules to the podocytes. Consequently, the expression of claudin-1 in podocytes was epigenetically upregulated, which led to impaired glomerular barrier function and albuminuria [54]. Nicotinamide N-methyltransferase (NNMT) is an enzyme that methylates nicotinamide using SAM as a methyl donor. Overexpression of NNMT has been shown to consume a large amount of SAM and impair cellular methylation potential. Conversely, NNMT inhibition increases cellular SAM levels, promotes methylation of histone H3K4 and, thereby, augments gene expression. The genetic depletion of NNMT in white adipose tissue and the liver has been reported to protect against diet-induced obesity in mice by upregulating the expression of genes related to energy expenditure through histone H3K4 methylation [55]. In cancer cells, overexpression of NNMT has been shown to contribute to tumorigenesis by decreasing histone methylation, which alters the expression of protumorigenic genes [56]. In these ways, cellular metabolism can epigenetically modulate the expression of genes that are related to the progression of diseases.
Transient changes in organ cellular metabolism can produce prolonged effects on the organ through the epigenetic regulation of gene expression. Transient hyperglycemia has been shown to induce long-lasting epigenetic changes in the promoter of the nuclear factor κB p65 subunit gene in cultured aortic endothelial cells, which resulted in the promotion of monocyte recruitment [57]. After completion of the DCCT/EDIC study [5, 6], the extent of H3K9 acetylation/methylation in monocytes of the participants was revealed to relate to diabetic vascular complications (“glucose memory”) [58]. The elucidation of these interactions between cellular metabolism and epigenetics would contribute to the further understanding of “organ memory”.
Future direction of “organ memory” research
In the present review, we proposed that “organ memory” is another concept of memory, in addition to two types of “memory” that are recognized in brain and immune sciences. Future research on “organ memory” could be expanded in many directions, including the following approaches that would be effective in suppressing or regressing the chronic progression of NCDs, including hypertension and its organ complications.
-
1.
Mathematical modeling of the processes of “organ memory” generation to predict the directions of organ responsiveness to stress that could occur in the future.
-
2.
Identification of the core unit of “organ memory” and its manipulation.
-
3.
Engineering technologies to modify “organ memory”, that is, to “produce organ memory” in vitro and to “transplant organ memory” in vivo.
These kinds of research approaches regarding “organ memory” could result in new treatments for NCDs and generate a new dimension of medical science.
References
Colditz GA, Bonita R, Stampfer MJ, Willett WC, Rosner B, Speizer FE, et al. Cigarette smoking and risk of stroke in middle-aged women. N Engl J Med. 1988;318:937–41.
Rosenberg L, Kaufman DW, Helmrich SP, Shapiro S. The risk of myocardial infarction after quitting smoking in men under 55 years of age. N Engl J Med. 1985;313:1511–4.
Meguro S, Kabeya Y, Tanaka K, Kawai T, Tomita M, Katsuki T, et al. Past obesity as well as present body weight status is a risk factor for diabetic nephropathy. Int J Endocrinol. 2013;2013:590569.
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–86.
The Diabetes Control and Complications Trial Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381–9.
Zoungas S, Chalmers J, Neal B, Billot L, Li Q, Hirakawa Y, et al. ADVANCE-ON Collaborative Group. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N Engl J Med. 2014;371:1392–406.
Ford I, Murray H, Packard CJ, Shepherd J, Macfarlane PW, Cobbe SM. Long-term follow-up of the west of scotland coronary prevention study. N Engl J Med. 2007;357:1477–86.
Strandberg TE, Pyorala K, Cook TJ, Wilhelmsen L, Faergeman O, Thorgeirsson G. et al. Mortality and incidence of cancer during 10-year follow-up of the scandinavian simvastatin survival study (4S). Lancet. 2004;364:771–7.
Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ. et al. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–53.
Cushman WC, Evans GW, Byington RP, Goff DC,Jr, Grimm RH,Jr, Cutler JA. et al. The ACCORD Study Group Effects of intensive Blood-pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362:1575–85.
Buckley LF, Dixon DL, Wohlford GF 4th, Wijesinghe DS, Baker WL, Van Tassell BW. Effect of intensive blood pressure control in patients with type 2 diabetes mellitus over 9 years of follow-up: a subgroup analysis of high-risk ACCORDION trial participants. Diabetes Obes Metab. 2018;20:1499–502.
Julius S, Nesbitt SD, Egan BM, Weber MA, Michelson EL, Kaciroti N. et al. Trial of Preventing Hypertension (TROPHY) Study Investigators Feasibility of treating prehypertension with an angiotensin-receptor blocker. N Engl J Med. 2006;354:1685–97.
Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F, Lever AF. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension. 1990;16:603–14.
Wu JN, Berecek KH. Prevention of genetic hypertension by early treatment of spontaneously hypertensive rats with the angiotensin converting enzyme inhibitor captopril. Hypertension. 1993;22:139–46.
Nakaya H, Sasamura H, Hayashi M, Saruta T. Temporary treatment of prepubescent rats with angiotensin inhibitors suppresses the development of hypertensive nephrosclerosis. J Am Soc Nephrol. 2001;12:659–66.
Togashi N, Maeda T, Yoshida H, Koyama M, Tanaka M, Furuhashi M, et al. Angiotensin ii receptor activation in youth triggers persistent insulin resistance and hypertension--a legacy effect? Hypertens Res. 2012;35:334–40.
Geleijnse JM, Hofman A, Witteman JC, Hazebroek AA, Valkenburg HA, Grobbee DE. Long-term effects of neonatal sodium restriction on blood pressure. Hypertension. 1997;29:913–7.
Oguchi H, Sasamura H, Shinoda K, Morita S, Kono H, Nakagawa K, et al. Renal arteriolar injury by salt intake contributes to salt memory for the development of hypertension. Hypertension. 2014;64:784–91.
Itoh H, Kurihara I, Miyashita K, Tanaka M. Clinical significance of “cardiometabolic memory”: a systematic review of randomized controlled trials. Hypertens Res. 2017;40:526–34.
Holman RR, Paul SK, Bethel MA, Neil HA, Matthews DR. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N Engl J Med. 2008;359:1565–76.
Bosch J, Lonn E, Pogue J, Arnold JM, Dagenais GR, Yusuf S. HOPE/HOPE-TOO Study Investigators Long-term effects of ramipril on cardiovascular events and on diabetes: results of the hope study extension. Circulation. 2005;112:1339–46.
Buch P, Rasmussen S, Abildstrom SZ, Køber L, Carlsen J, Torp-Pedersen C. TRACE investigators The long-term impact of the angiotensin-converting enzyme inhibitor trandolapril on mortality and hospital admissions in patients with left ventricular dysfunction after a myocardial infarction: follow-up to 12 years. Eur Heart J. 2005;26:145–52.
Strandgaard S, Hansen U. Hypertension in renal allograft recipients may be conveyed by cadaveric kidneys from donors with subarachnoid haemorrhage. BMJ. 1986;292:1041–4.
Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, et al. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309:1009–15.
Khush KK, Menza R, Nguyen J, Zaroff JG, Goldstein BA. Donor predictors of allograft use and recipient outcomes after heart transplantation. Circ Heart Fail. 2013;6:300–9.
Fridell JA, Mangus RS, Taber TE, Goble ML, Milgrom ML, Good J, et al. Growth of a nation part i: impact of organ donor obesity on whole-organ pancreas transplantation. Clin Transplant. 2011;25:E225–32.
Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med. 1974;47:435–48.
Rapp JP, Knudsen KD, Iwai J, Dahl LK. Genetic control of blood pressure and corticosteroid production in rats. Circ Res. 1973;1:139–49.
Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–6.
Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–21.
Lee JL. Reconsolidation: maintaining memory relevance. Trends Neurosci. 2009;32:413–20.
Almeida-Correa S, Amaral OB. Memory labilization in reconsolidation and extinction--evidence for a common plasticity system? J Physiol Paris. 2014;108:292–306.
Hayashi K, Hishikawa A, Itoh H. DNA damage and epigenetic changes in kidney diseases - focused on transcription factors in podocytes. Curr Hypertens Rev. 2016;12:105–11.
Hayashi K, Sasamura H, Ishiguro K, Sakamaki Y, Azegami T, Itoh H. Regression of glomerulosclerosis in response to transient treatment with angiotensin ii blockers is attenuated by blockade of matrix metalloproteinase-2. Kidney Int. 2010;78:69–78.
Ishiguro K, Hayashi K, Sasamura H, Sakamaki Y, Itoh H. “Pulse” treatment with high-dose angiotensin blocker reverses renal arteriolar hypertrophy and regresses hypertension. Hypertension. 2009;53:83–9.
Hayashi K, Sasamura H, Nakamura M, Sakamaki Y, Azegami T, Oguchi H, et al. Renin-angiotensin blockade resets podocyte epigenome through kruppel-like factor 4 and attenuates proteinuria. Kidney Int. 2015;88:745–53.
Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–4.
Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring systemic gdf11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–52.
Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75.
Sasamura H, Nakaya H, Julius S, Tomotsugu N, Sato Y, Takahashi F. et al. STAR CAST investigators Feasibility of regression of hypertension using contemporary antihypertensive agents. Am J Hypertens. 2013;26:1381–8.
Angeli D, Ferrell JE Jr, Sontag ED. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc Natl Acad Sci USA. 2004;101:1822–7.
Xiong W, Ferrell JE. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426:460–5.
Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–21.
Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–18.
Somanna NK, Valente AJ, Krenz M, McDonald KS, Higashi Y, Noda M, et al. Histone deacetyltransferase inhibitors Trichostatin A and Mocetinostat differentially regulate MMP9, IL-18 and RECK expression, and attenuate Angiotensin II-induced cardiac fibroblast migration and proliferation. Hypertens Res. 2016;39:709–16.
Hayashi K, Itoh H. Transcription factors and epigenetic modulation: its therapeutic implication in chronic kidney disease. Arch Immunol Ther Exp (Warsz). 2015;63:193–6.
Hayashi K, Sasamura H, Nakamura M, Azegami T, Oguchi H, Sakamaki Y, et al. Klf4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J Clin Invest. 2014;124:2523–37.
Hervouet E, Vallette FM, Cartron PF. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics. 2009;4:487–99.
Accili D, Arden KC. Foxos at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–6.
Nakae J, Cao Y, Hakuno F, Takemori H, Kawano Y, Sekioka R, et al. Novel repressor regulates insulin sensitivity through interaction with foxo1. EMBO J. 2012;31:2275–95.
Su X, Wellen KE, Rabinowitz JD. Metabolic control of methylation and acetylation. Curr Opin Chem Biol. 2016;30:52–60.
Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–8.
Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19:1496–504.
Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, et al. Nicotinamide n-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–62.
Ulanovskaya OA, Zuhl AM, Cravatt BF. Nnmt promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013;9:300–6.
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–17.
Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X. et al. DCCT/EDIC Research Group Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014;63:1748–62.
Acknowledgements
We express sincere gratitude to Dr. Masami Tanaka (Keio University) for helpful contributions to this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Itoh, H., Kurihara, I. & Miyashita, K. Organ memory: a key principle for understanding the pathophysiology of hypertension and other non-communicable diseases. Hypertens Res 41, 771–779 (2018). https://doi.org/10.1038/s41440-018-0081-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-018-0081-x
This article is cited by
-
Pre-emptive medicine for hypertension and its prospects
Hypertension Research (2019)
