
Abstract
Metabolic diseases and their complications impose health and economic burdens worldwide. Evidence from past experimental studies and clinical trials suggests our body may have the ability to remember the past metabolic environment, such as hyperglycemia or hyperlipidemia, thus leading to chronic inflammatory disorders and other diseases even after the elimination of these metabolic environments. The long-term effects of that aberrant metabolism on the body have been summarized as metabolic memory and are found to assume a crucial role in states of health and disease. Multiple molecular mechanisms collectively participate in metabolic memory management, resulting in different cellular alterations as well as tissue and organ dysfunctions, culminating in disease progression and even affecting offspring. The elucidation and expansion of the concept of metabolic memory provides more comprehensive insight into pathogenic mechanisms underlying metabolic diseases and complications and promises to be a new target in disease detection and management. Here, we retrace the history of relevant research on metabolic memory and summarize its salient characteristics. We provide a detailed discussion of the mechanisms by which metabolic memory may be involved in disease development at molecular, cellular, and organ levels, with emphasis on the impact of epigenetic modulations. Finally, we present some of the pivotal findings arguing in favor of targeting metabolic memory to develop therapeutic strategies for metabolic diseases and provide the latest reflections on the consequences of metabolic memory as well as their implications for human health and diseases.
Similar content being viewed by others
Introduction
Metabolic diseases place a significant burden on global health systems.1 The prevalence of several metabolic diseases, encompassing diabetes, obesity as well as metabolism-associated fatty liver disease (MAFLD), has steadily increased over the past few decades. According to the Diabetes Atlas published in 2021, diabetes affects approximately 10.5% of adults globally, and the absolute amount of individuals suffering from diabetes is projected to rise in 2045 by 46%.2 Similarly, the prevalence of obesity and overweight exhibits a comparable growth pattern to that of diabetes.3 Since 1975, the prevalence of obesity increased almost twofold globally, with over 1.9 billion individuals categorized as overweight or obese by 2016.4 Approximately 25–30% of people worldwide are affected by MAFLD, which is the most common liver disease globally and has prevalence and incidence rates aligned with the escalating trends of obesity as well as type 2 diabetes mellitus (T2DM)5,6 Increasing incidences of these aberrant metabolism-related diseases and their consequential serious complications pose significant health challenges to human society. Consequently, it is imperative to investigate the pathological mechanisms involved in metabolic diseases as well as develop therapeutic interventions based on these scientific findings.
The initiation and evolution of metabolic diseases involve intricate mechanisms, requiring comprehensive therapeutic approaches for effective management.7,8,9 Recent studies have elucidated the persistent detrimental consequences that arise when cells are exposed to an abnormal metabolic environment. Even after the metabolic environment returns to a normal state, the cellular changes and characteristics of the abnormal metabolic state persist.10,11,12 These enduring cellular changes and characteristics represent the organism’s memory of an earlier metabolic state, exemplifying the phenomenon termed metabolic memory.
Conventional treatments for metabolic diseases are challenged by the existence of metabolic memory. For instance, glycemic control using hypoglycemic drugs was previously believed to be the primary approach to managing T2DM and its complications.13 However, it has been discovered that despite achieving great glycemic control, the organism continues to exhibit various inflammatory responses and complications associated with diabetes due to metabolic memory.14,15 This realization prompts further in-depth investigation into the molecular mechanisms underlying metabolic memory, aiming to develop corresponding therapeutic interventions that can effectively mitigate or eradicate the adverse effects associated with metabolic memory. This will ultimately enhance the treatment efficacy of various metabolic diseases.16 Accumulating evidence suggests that multiple intricate molecular and cellular mechanisms serve to establish and maintain metabolic memory, including epigenetic regulation, glycosylation end products, oxidative stress, etc. These interconnected mechanisms form a complex network that governs metabolic memory and can emerge as novel targets for both detection and intervention of metabolic diseases.17,18,19
Here, we seek to outline the research history and distinct features of metabolic memory. We also summarize the various molecular and cellular mechanisms that regulate metabolic memory. Furthermore, we emphasize the existence and profound impact of metabolic memory in numerous metabolic diseases and establish connections between these mechanisms and disease progression. Additionally, we specifically focus on the significant research advancements linking metabolic memory with cancer risk. Moreover, we explore the potential utility of the phenomenon of metabolic memory and its associated mechanisms as indicators and promising targets for the detection and therapeutic interventions of metabolic diseases.
The research history of metabolic memory
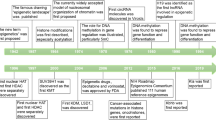
“Metabolic memory” originated from studies of diabetes and its complications (Fig. 1). In 1983, the Diabetes Control and Complications Trial (DCCT) was initiated to investigate “glucose hypothesis” of diabetic complications, which suggested that hyperglycemia critically affected long-term diabetic complications.20,21 There were 1441 individuals with type 1 diabetes mellitus (T1DM) attending the DCCT by 1989.22,23 Participants were randomized to either the intensive therapy group or the conventional therapy group. The intensive therapy participants received a minimum of 3 daily insulin injections or continuous insulin infusion using external pumps, aiming to achieve tight glycemic control comparable to non-diabetic people.24 The conventional therapy participants received 1 or 2 daily insulin injections with the goal of safely achieving asymptomatic glucose control.24 In 1993, after the mean follow-up period of 6.5 years, the study results demonstrated that intensive therapy provided a significant reduction in the development and evolution of diabetic retinopathy, nephropathy, and neuropathy versus conventional therapy. Notably, almost all beneficial effects were statistically attributed to the difference in mean glycosylated hemoglobin A1c (HbA1c) levels.25,26 These findings strongly support the glucose hypothesis by emphasizing blood glucose levels are the primary driving factor behind the development of diabetic complications.20,27

The research history of metabolic memory. The term “metabolic memory” originated from the study of the pathogenesis of long-term diabetic complications by the Diabetes Control and Complications Trial (DCCT) in 1983. In 1994, a long-term prospective, longitudinal, observational study conducted by Epidemiology of Diabetes Interventions and Complications (EDIC) found that the risk of complications in diabetic patients with regular glycemic control was higher in the conventional treatment group than in the early intensive control group. This phenomenon has been characterized as “metabolic memory” by the DCCT/EDIC. The following clinical trials, like UKPDS and Steno-2 trials, also revealed early intensive glycemic control might bring about prolonged benefits in diabetes care. Nowadays, the concept of metabolic memory and its implications have been expanded, especially in hyperglycemia, hyperlipidemia, hypoxia, and other metabolic disorders
After DCCT termination, a subsequent long-term observational follow-up study called the Epidemiology of Diabetes Interventions and Complications (EDIC) was initiated in 1994 and is still ongoing. The EDIC study involved 1394 surviving participants from the DCCT cohort.28 Individuals initially assigned to receive conventional treatment were subsequently exposed to intensive treatment, while every subject then returned to their respective healthcare provider to receive further treatment. During EDIC, HbA1c values in both original conventional and intensive treatment groups rapidly converged. Considering that mean HbA1c levels significantly influenced complication outcomes between intensive and conventional therapy groups over the period of DCCT, it would be reasonable to expect a similar trend in complication development between both treatment groups during EDIC. Surprisingly, though, the initial 4-year follow-up period of EDIC unveiled escalating disparities in complication rates between the two groups, with a notable reduction in retinopathy progression and nephropathy risk observed within the initially intensively treated group in comparison to the conventionally treated group.29 Results of the first 8-year follow-up further elucidated that earlier HbA1c levels largely influenced the long-term risk of complications and that the pathological alterations caused by hyperglycemia persisted after the hyperglycemic period, which was referred to as “metabolic memory”.14 Subsequent investigations and data analysis indicated that the biological impacts of metabolic memory peaked during the first decade and decayed thereafter.11 The findings of the DCCT/EDIC study and the discovery of metabolic memory highlight the great significance and prolonged benefits of early strict glycemic control, bringing about a dramatic change in diabetes management.
Comparable advantages have also been reported in the UK Prospective Diabetes Study (UKPDS) (referred to as the “legacy effect”) and the Steno-2 trial.30,31,32,33 During the period 1977–1991, 5102 newly diagnosed diabetic patients between 25 and 65 years old were recruited from 23 participating hospitals. Among them, 4209 eligible patients were randomly assigned for either the conventional or intensive glycemic control. The intensive therapy group exhibited a significant 25% risk reduction of microvascular lesions and a 16% risk reduction (P = 0.052) in myocardial infarction in the end.30 The UKPDS post-trial study, which followed patients with newly diagnosed T2DM on conventional and intensive therapy for up to 10 years, showed a significant reduction in endpoint events related to diabetes (including microvascular lesion, myocardial infarction, and death) in subjects receiving intensive therapy in comparison to conventional therapy. The findings imply that early intervention in blood glucose may make the most significant contribution to the prevention of T2DM complications.10 Furthermore, 160 participants with T2DM and microalbuminuria (with a mean age of 55) were randomized and allocated to undergo either conventional or intensive therapy in the Steno-2 trial, and they were followed for the mean 13.3-year duration.34 Despite the convergence of glycemic control after the end of the study, participants treated with intensive control experienced a decreased risk of cardiovascular incidents and cardiovascular and all-cause mortality.
Several recent research studies have expanded the concept of metabolic memory with its implications in various pathologic states. Similar to hyperglycemia, abnormal fat and cholesterol levels contribute to prolonged cellular alterations and tissue damage.35,36 For instance, a set of works by Crisóstomo et al. discovered the correlation between the early high-fat diet and irreversible alterations in testicular lipid content and metabolism. These changes appear to be related to lasting impairments in sperm quality in the future, and switching to a regular diet cannot reinstate the quality of sperm.37,38 This phenomenon, known as “inherited metabolic memory” caused by exposure to an elevated-fat diet, alters fatty acid metabolism in the testes with harmful effects on sperm that can last for up to two generations. “Inherited metabolic memory” is reported to be associated with sperm small non-coding RNAs (sncRNAs) content.12,39,40 The high-fat diet alters the accessibility of mice liver chromatin, with a substantial proportion of loci remaining altered after the diet returns to normal. These long-lasting chromatin accessibility changes were discovered to be correlated to specific transcription factors as well as histone modifications, indicating that long-term risk of metabolic diseases may be impacted by persistent epigenetic modifications induced by high-fat diets.41 A recent study of the medaka fish also illustrated that early nutritional conditions may consistently influence the animal’s metabolism. The study found that the medaka fish fed with high-fat food during early life developed hepatic steatosis with substantial hepatocyte gene expression alternations. Prolonged normal feeding reversed a majority of epigenetic modulations induced by the previous high-fat diet, whereas some loci around genes associated with hepatofibrosis and hepatocarcinogenesis still showed non-reversible changes.42
Long-term pathological alterations are likewise mediated by abnormal metabolic reprogramming in hypoxia. It was found that fibroblasts in hypoxic environments are also capable of generating metabolic memory. Hypoxia-induced pulmonary hypertension causes fibroblasts to undergo metabolic reprogramming, shifting the metabolic paradigm toward aerobic glycolysis, accompanied by increased free nicotinamide adenine dinucleotide (NADH) and NADH/ nicotinamide adenine dinucleotide (NAD) ratios. Increased free NADH further activates C-terminal binding protein 1 (CtBP1), driving the proliferation and pro-inflammatory phenotype of fibroblasts in turn.43 Significantly, the same metabolic reprogramming event, along with enduring inflammation and fibrosis, was observed when these fibroblasts returned to normoxic culture conditions. Hypoxia also evokes the generation of metabolic memory in cardiac fibroblasts via inducing alterations of expression of DNA methyltransferase (DNMT) enzymes and develops a long-lasting pro-fibrotic milieu.44,45 Recent studies on hypoxia-mediated cell metabolic reprogramming in the tumor microenvironment have provided novel perspectives regarding cancer pathogenesis. Hypoxia has been recognized to be an important cancer hallmark and is positively associated with cancer progression, metastasis, and therapeutic resistance.46 Hypoxia-inducible factor-1α (HIF-1α) mediates the adaptive response of tumors to hypoxia and was found to be highly overexpressed in the majority of solid tumors and their metastases.47,48,49 It has been demonstrated that hypoxia can upregulate transcriptional activity and stability of HIF-1α expression through a range of epigenetic modifications and influences the expression of numerous epigenetic modulators in a manner dependent on HIF-1α.50,51 The persistence of transcriptional reprogramming induced by the hypoxic tumor microenvironment leads to upregulation of the glycolytic program and increased lipolysis, driving cancer cell proliferation, migration, and immune escape.52,53,54,55
In addition, a number of recent research studies suggest that high levels of uric acid could influence the immune response through persistent epigenetic modifications, resulting in an altered functional state of immune cells that persists after removing the initial stimulus. The methylation level of the C-C motif chemokine ligand 2 (CCL2) promoter is dramatically reduced in Chinese Han male gout patients.56 A recent DNA methylation sequencing of gout patients and healthy individuals showed differential DNA methylation of numerous genes in signaling pathways linked to innate and adaptive immunity as well as osteoclastogenesis, including interleukin 17 (IL-17), signal transducer and activator of transcription 2 (STAT2), interferon regulatory factor 1 (IRF1), and myocyte-specific enhancer factor 2 C (MEF2C), etc.57 Peripheral biological mononuclear cells (PBMC) from gout patients and PMBC from healthy subjects treated with uric acid produce enhanced levels of pro-inflammatory cytokines stimulated by toll-like receptor (TLR) agonist compared to controls and maintain a high response potential at stimulation intervals.58 Treatment with histone methyltransferase inhibitors reversed the persistent effects of urate. Another study reported that romidepsin, a histone deacetylase (HDAC) 1/2 inhibitor, reduced pro-inflammatory cytokine production in PBMC stimulated with monosodium urate (MSU) crystals.59 Long-term effects of uric acid-mediated epigenetic changes on hyperuricemic complications and targets for intervention require further studies.
In summary, metabolic memory, as a concept initially proposed in studies on diabetes and its complications, described possible adverse effects of short-term abnormalities in glucose metabolism on long-term health. Recent research on hyperglycemic memory revealed that hyperglycemia may lead to persistent complication progression even after glycemic control, suggesting the importance of early and strict control of hyperglycemia. The field of metabolic memory has also been expanded by many recent studies to encompass a number of metabolic activities except glucose metabolism, including lipid metabolism, oxygen metabolism, uric acid metabolism, and others, all of which may have far-reaching effects on the host through underlying mechanisms. In the review, we define metabolic memory as the ability of an individual to retain memory of the damage caused by aberrant metabolism that persists after normalization of metabolism rather than a separate description of long-term adverse effects or toxicity of glucose.
The characteristics of metabolic memory
Metabolic memory refers to the distinct phenomenon in which detrimental impacts of a transient abnormal metabolic state on the body remain after normalized metabolism. Several basic experiments have previously provided valuable insights into some important characteristics of metabolic memory.
Persistence
The first distinct hallmark is that metabolic memory promotes persistent harmful effects, including inflammatory changes, premature cell senescence, apoptosis, etc. Vascular smooth muscle cells (VSMCs) in diabetes models show remarkably enhanced expression of pro-inflammatory-related genes as well as associated inflammatory molecules.60,61 Interestingly, even after glucose normalization, VSMCs derived from diabetic mice continue to show elevated oxidative stress levels and enhanced inflammatory signaling pathway activation. This indicates that metabolic memory confers a pro-inflammatory phenotype on VSMCs, contributing to increased vascular dysfunction and atherosclerosis that occur in patients with diabetes. Furthermore, studies of human endothelial cells raised in high glucose concentration environments have observed prolonged and sustained upregulation of fibronectin gene expression, even when transferred to a medium with normal glucose. This is considered to be correlated to diabetic complications progression.62 Further research successfully replicated this metabolic memory phenomenon in animal models with diabetic nephropathy or retinopathy.63,64,65 These studies well implicated the association between metabolic memory and persistently aberrant expression of anti-oxidant and inflammatory genes.66,67,68,69
The abnormal metabolic microenvironments are well known to accelerate the senescence process in multiple cell types by causing mitochondrial dysfunction, increasing the generation of advanced glycation end products (AGEs) and reactive oxygen species.70,71,72,73 Prematurely senescent cells show heightened metabolic activity that enhances the release of proinflammatory cytokines, chemokines, as well as growth factors, collectively termed senescence-associated secretory phenotype (SASP). This leads to further development of inflammatory damage and establishes a harmful positive feedback loop in diabetes.74,75 Furthermore, studies have shown that the activity of secreting pro-inflammatory factors in senescent critical immune cells like macrophages remains upregulated after transient exposure to high glucose concentration, suggesting that metabolic memory promotes sustained cellular senescence and release of SASP factors.76,77,78 Metabolic memory also allows pro-apoptotic activation to persist despite termination of hyperglycemia.79 A series of apoptosis-associated genes, such as the tumor necrosis factor (TNF) receptor and ligand and the B-cell lymphoma-2 (Bcl-2) family, remained elevated in retinal cells after re-establishment of good glycemia management of diabetic rats. Finally, and notably, the duration of these adverse consequences mediated by metabolic memory varies depending on the source, extent, and duration of the stimulus. Most experimental results vary from study to study depending on the investigator’s protocol design. With respect to current in vivo and in vitro studies of glucose and lipid stimuli, the duration of metabolic memory after stimulus elimination is at least as long as the duration of the previously received stimulus.62,66,80
Progressivity
The second hallmark of metabolic memory is the long-term adverse effects on metabolic complications, which depend on early control; subsequent metabolic control does not prevent progressive complications. One of the earliest associated studies published in 1987 compared and analyzed the incidence of retinopathy in dogs with poor glycemic control, good glycemic control, and good glycemic control after a period of poor glycemic control.81 The results demonstrated that the incidence of retinopathy in dogs with good glycemic control after poor glycemic control was similar to that in the poor glycemic control group and higher than that in the good control group. Moreover, by the completion of the trial, the severity of retinopathy in the last group was greater compared to the end of their period of poor glycemic control, indicating that subsequent control failed to prevent the continued diabetic complications progression caused by early hyperglycemia. Lack of intensive management in the early stage of diabetes can lead to prolonged, irreversible inflammatory responses and oxidative stress in tissues like kidneys and retina. The Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) and Veterans Affairs Diabetes Trial (VADT), comprising individuals with uncontrolled glycemia of long duration, found no significant benefit of intensive glycemic management on major cardiovascular disease incidents.82,83 These results suggest that the longer the duration of hyperglycemia, the less impact intensive glycemia control has on diabetic complications.84 Consequently, diabetic complications progression may perpetuate even when removed from the hyperglycemic environment.
Maternal high-fat dietary interventions also affect tissue immunity as well as metabolic homeostasis in offspring in a timing-dependent intervention.85 Compared with offspring from experimental groups in which maternal mice were switched from a high-fat diet to a normal-fat diet 9 weeks before pregnancy, offspring from experimental groups switched 1 or 5 weeks before pregnancy showed earlier and more severe glucose intolerance, hepatocyte degeneration and adipose tissue inflammation.86,87 Enhanced adipogenic genes and hyperactivation of inflammatory signaling pathways were found in these offspring, accompanied by reduced expression of insulin receptor substrates and blunted insulin signaling.88 Thus, the early state of metabolic control is critical for the long-term memory impacts conducted by metabolic disorders, and traditional interventions may not be effective in alleviating progressive complications. The reversibility of adverse effects depends on the timing of intervention and therapeutic measures.
Epigenetic modifications regulation
Finally, recent studies have highlighted the close association between sustained detrimental influences of metabolic memory and epigenetic regulation. A series of studies by Kowluru et al. demonstrated that transient or prior hyperglycemia led to various persistent epigenetic modulations, including DNA methylation, histone methylation, and histone acetylation. These modulations lead to sustained activation of pro-inflammatory pathways as well as oxidative stress.68,80,89,90 Consequently, these epigenetic modifications contribute to the enduring adverse effects of early abnormal metabolic conditions on cellular functions, perpetuating the pro-inflammatory and pro-destructive metabolic memory and driving diabetic complications progression after subsequent normoglycemia. As is similar to the long-term effects mediated by hyperglycemia, the short-term of a high-fat diet leads to disruption of the expression of key markers correlated with the regulation of cholesterol and lipid metabolism, with long-term adverse effects mediated by persistent epigenetic modifications.91,92 As previously described, epigenetic modifications in the liver were found in the early-life high-fat diet mice and would be maintained to varying degrees after resumption of a normal food diet.41 In addition, high-fat nutritional status in early life triggers irreversible epigenetic changes at specific gene locus in medaka, which are primarily histone modifications of the acetylation of lysine 27 on histone 3 (H3K27ac) and the methylation of lysine 9 on histone 3 (H3K9me3).42 ATAC-seq analyses identified multiple genes associated with hepatic fibrosis and hepatocellular carcinoma to show sustained gene signaling changes, including Ephrin type-A receptor 5 (epha5), raft linking protein (raftlin), and HERV-H LTR-associating protein 2 (hhla2b1), which may lead to an increased propensity for liver inflammation, fibrosis, and carcinoma. Maternal high-fat diets lead to persistent alterations in hepatic DNA methylation and histone modifications in offspring fetuses, increasing susceptibility to metabolic syndrome and steatohepatitis.93,94 To investigate the regulation mechanisms of epigenetic changes in metabolic memory maintenance, various animal models, including atherosclerosis, diabetic nephropathy, diabetic retinopathy, and other diseases, have been established. These disease models yield valuable insights into the intricate relationship between epigenetic modification and the persistence of metabolic memory.
In summary, metabolic memory refers to the phenomenon where adverse impacts of transiently abnormal metabolic conditions persist even after metabolic normalization. It has three distinct hallmarks (Fig. 2). Firstly, metabolic memory promotes persistent harmful effects including inflammatory changes, premature cell senescence, apoptosis, etc. Secondly, the long-term adverse effects of metabolic complications depend on early control, while subsequent metabolic control does not prevent progressive complications. Thirdly, the establishment of metabolic memory is highly correlated to epigenetic regulation, resulting in the enduring adverse effects and progression of metabolic complications, which may be transmitted to offspring.

The characteristics of metabolic memory. Metabolic memory has three distinct hallmarks. Firstly, the long-term adverse effects on diabetic complications depend on early glycemic control, as subsequent glycemic control does not prevent progression. Secondly, metabolic memory promotes inflammatory changes, premature cell senescence, and ongoing apoptosis, perpetuating the harmful effects even after hyperglycemia is resolved. Thirdly, the establishment of metabolic memory is highly associated with epigenetic modifications, contributing to the enduring adverse effects and progression of diabetic complications. The figure was created with the assistance of Servier Medical Art (https://smart.servier.com/)
Molecular mechanisms of metabolic memory
Mounting evidence has emerged indicating a close association between epigenetic modifications and metabolic memory in recent years. Epigenetic alterations have been detected in diverse target cells when exposed to disrupted metabolic circumstances, and these changes persist after metabolism levels return to normal, suggesting that epigenetic modifications may serve as the underlying molecular mechanism for metabolic memory.95,96,97,98 Chromosomes consist of DNA–protein complexes known as chromatin, which are composed of nucleosomes as their subunits.99 Each nucleosome consists of a complex of octameric histone, composed of dimers of core histones H2A, H2B, H3, and H4, intricately enveloped by 147 base pairs of DNA. Specific gene expression is regulated by epigenetic modifications without altering the original DNA sequence.100 These different modifications occur at distinct levels of nucleic acids and histones, encompassing DNA methylation, modifications to histones, as well as non-coding RNAs (ncRNAs), working synergistically to govern gene function and expression.101 In general, altered metabolic circumstances lead to the initiation of cellular metabolic reprogramming, resulting in changes in metabolites that subsequently impact epigenome-modifying enzymes that use intermediate metabolites as substrates.102 These regulatory processes, mediated by epigenetic changes, enable cells to respond rapidly to ever-changing environmental stimuli and acquire long-term responsiveness even when the initial stimuli are removed (Fig. 3).103,104

An overview of the interplay between epigenetic modifications and metabolic reprogramming during metabolic memory. The molecular mechanisms of metabolic memory mainly include epigenetic modifications and metabolic reprogramming. Accumulation of metabolic intermediates induces epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNAs (ncRNAs). DNA methylation and histone modifications take place at the level of chromatin, while ncRNAs modulate gene expression mainly at post-translational level. Epigenetic modifications could induce persistent expression of metabolic disease-related genes and pro-inflammatory genes, which interact and work together
DNA methylation
DNA methylation is a prevalent biochemical process that involves the addition of methyl groups to DNA molecules. In mammals, the process of DNA methylation occurs primarily at dinucleotides consisting of cytosine-phosphate-guanine (CpG), leading to the formation of 5-methylcytosines, although non-CpG methylation may also occur.105 The CpG-rich regions are predominantly situated in regulatory domains and play an instrumental role in gene transcription. DNA methylation in promoter regions typically suppresses gene expression by hindering the interaction between transcription factors or by enlisting chromatin inactivation complexes in conjunction with methyl-CpG binding domain proteins, leading to the suppression of transcription. On the other hand, methylation occurring in the gene’s body could impact both transcription elongation and alternative splicing.106 The process of DNA methylation is significantly influenced by the crucial involvement of DNMTs. DNMT1 is responsible for recognizing and methylating CpG islands located on the newly formed DNA strand, thereby transmitting epigenetic information across cell generations to maintain methylation. Meanwhile, the initiation of de novo DNA methylation is attributed to DNMT3a and DNMT3b.107,108 The process of DNA methylation is a dynamic modification that can be reversed by inhibiting the activity of DNMT1 and activating DNA demethylases, such as the ten-eleven translocation (TET) methylcytosine dioxygenases, which actively eradicate the methyl group from 5-methylcytosines.
Several experimental and clinical studies have presented convincing findings supporting the correlation between DNA methylation and the persistence of metabolic memory. Previous investigations have demonstrated that environment factors and dietary choices may affect epigenetic modifications, there contributing to individuals’ vulnerability to metabolic diseases.109,110 Analysis of DNA methylation in blood/DNA samples obtained from participants in the DCCT/EDIC study has revealed a significant correlation between long-term preceding glycemic history and DNA methylation changes. A total of 186 CpGs were identified as being associated with the average level of HbA1c in DCCT.111 Importantly, the majority of these HbA1c-associated CpGs exhibited significant enrichment within enhancers or transcription-related regions of blood cells and hematopoietic stem cells, particularly at CCAAT/enhancer binding protein (C/EBP) binding sites. C/EBPs exert a vital role in governing hematopoiesis and the differentiation of blood cells and are associated with oxidative stress and inflammation in both blood cells and target cells involved in diabetic complications.112 Changes in the DNA methylation patterns at CpG sites associated with HbA1c have an impact on hematopoietic cells and other target cells, promoting immune response and inflammation and ultimately contributing to disease development. These results highlight the significance of DNA methylation at specific CpG sites in the progression of complications associated with diabetes. Notably, the persistence of DNA methylation differences at HbA1c-associated CpGs further strengthens the link between DNA methylation and metabolic memory.113
Various reports have documented different methylation levels of genes associated with diabetes. Differential methylation levels were found between individuals with T1DM and healthy subjects at four CpG loci near the insulin gene encoding pre-insulin. Specifically, CpG-19, 135, and 234 exhibited hypomethylation, whereas CpG-180 displayed hypermethylation, all of which were associated with an elevated susceptibility to the development of T1DM.114 Similar results have been documented in research investigating specific complications. The global levels of DNA methylation exhibited a significant increase in individuals with T2DM who presented albuminuria, as compared to those without albuminuria. Moreover, a positive association was observed between the severity of albuminuria and the identified increased levels.115 Furthermore, genome-wide analysis of DNA methylation in DNA derived from peripheral blood cells of individuals diagnosed with T1DM, both with or without diabetic nephropathy, unveiled the discovery of 19 CpG loci linked to the susceptibility to diabetic kidney disease.116 A higher methylation level of T1DM patients was observed at a specific CpG island positioned upstream of the transcriptional start site of the unc-13 homolog B (UNC13B) gene, which was previously been associated with the development of diabetic nephropathy.116 In addition, high glucose also induces activation and overexpression of DNMT1 and promotes apoptosis and oxidative stress through DNMT1-mediated methylation of peroxisome proliferator-activated receptor α (PPARα), leading to exacerbation of diabetic retinopathy.117 Elevated expression of DNMT1 in histiocytes was found in diabetic mice, as well as in peripheral immune cells of diabetic patients. This upregulation is associated with the activation of multiple inflammatory pathways.118,119
More importantly, it has been discovered that altered methylation levels mediated by early metabolic abnormalities are not reversed as metabolism returns to normal. In the retinas of diabetic rats induced by streptozotocin, there was an increase in methylation levels within the promoter region of polymerase gamma (POLG1), which is responsible for encoding the catalytic subunit of mitochondrial DNA replicase.89 The hypermethylation was observed even after glucose levels were restored to their normal range. Similar results were noted in the retinal endothelial cells that were exposed early to high glucose.89 On the contrary, a decrease in global DNA methylation was noted in fibroblasts from diabetic foot ulcers when compared to fibroblasts from non-diabetic feet.120 This DNA methylation pattern remained consistent across multiple cell cultivation sessions under normoglycemic conditions. Moreover, studies conducted on diabetic zebrafish and rats induced by STZ also demonstrated a chronic hyperglycemia-induced overall DNA hypomethylation that perpetuated under normoglycemic conditions.121,122
Short-term high-fat diets have demonstrated the ability to induce long-term modifications in DNA methylation. Kim et al. found that a regular diet administered for 9 weeks after a high-fat diet of the same duration was able to reverse the non-alcoholic fatty liver disease phenotype, but elevated serum triglyceride levels and changes in gut microbiome composition persisted.123 Analysis of the changes in microbiome composition revealed a persistent enrichment of Odoribacter, which is known to produce butyrate with histone deacetylation inhibitor effects.124,125 Further genome-wide DNA methylation studies have revealed persistent alterations in methylation patterns at loci associated with lipid and cholesterol metabolism, such as hypomethylation of the apolipoprotein A4 (Apoa4) gene, which is considered to contribute to elevated triglyceride transport from liver to serum. Offspring exposed to a maternal high-fat diet are more susceptible to hepatic steatosis and inflammatory responses, with sustained changes in DNA methylation levels of key genes relevant to tissue development, metabolism, and cellular adhesion and communication. These genes include fibroblast growth factor 21 (Fgf21), peroxisome proliferator-activated receptor γ coactivator 1-beta (Ppargc1β), and von Willebrand factor (VWF), among others.94
Taken collectively, these findings propose that aberrant metabolic stimuli can trigger alterations in promoter methylation and exert a persistent impact on the expression levels of genes associated with oxidative stress, mitochondrial dysfunction, and apoptosis. This intricate process may contribute significantly to the establishment of metabolic memory.126
Histone modifications
Histone modifications are a range of post-translational modifications that occur on specific residues of the N-terminal amino acids of histones. These modifications include but are not limited to acetylation, methylation, phosphorylation, ubiquitination, and others.127 These modifications can regulate the interplay between histones and DNA as well as other nuclear proteins, thereby either inhibiting or activating gene transcription. The impact on gene expression is contingent upon the type and degree of modification, along with the specific location of the altered amino acid residues.128 Different levels and types of histone modifications collaboratively facilitate epigenetic regulation to affect cellular metabolism by controlling the expression of relevant genes through intricate and diverse mechanisms.
Acetylation represents a highly dynamic process mediated by histone acetyltransferases (HATs), such as p300 and CREB-binding protein, along with HDACs encompassing HDAC1–11 and sirtuins. These enzymes are pivotal for orchestrating chromatin remodeling events.99 On one hand, the acetylation of lysine residues in the histone tails results in the reduction of their positive charge, thereby decreasing the binding affinity between histones and negatively charged DNA. This impedes the interplay between DNA and histones, facilitating chromatin opening and promoting gene transcription. What’s more, acetylation can contribute to the recruitment of specific transcription factors and cofactors that further augment expression levels of genes.99 Histone acetylation at gene promoters (e.g., H3K9ac, H3K14ac, and H3K56ac) is generally linked to the activation of transcription, while the elimination of acetyl groups is associated with histone condensation and gene repression.129 Hyperglycemia promotes the activation of HATs, leading to the acetylation of lysine residues on histones located in the promoter regions of proinflammatory genes, thereby amplifying the expression of inflammatory factors.107,130 For instance, retinal capillary endothelial cells cultured in hyperglycemic conditions exhibited a reduction in both expression and functionality of Class III HDAC sirtuin 1 (SIRT1). This decrease persisted even following the resolution of hyperglycemia, implying a correlation between metabolic memory and histone acetylation.131 High glucose levels lead to the inhibition of SIRT1, resulting in increased acetylation of target genes, including forkhead box o1 (Foxo1) and nuclear factor kappa B (NF-κB) subunit p65, ultimately leading to heightened oxidative stress and inflammatory responses.132 Additionally, it was observed that the expression of HDAC3 and HDAC4 is enhanced under high glucose conditions, exacerbating inflammation and fibrosis.133,134 Besides, histone acetylation on DNMT proteins could potentially contribute to the regulation of DNA methylation.135 Specifically, H3K9 acetylation of the DNMT1 promoter can activate DNMT1 by downregulating SIRT1.136
The impact of histone methylation on gene transcription encompasses various aspects, as it can either facilitate or inhibit gene transcription based on the modified amino acid residues and level of methylation. Transcriptional activation is linked to four methylation sites on histones: H3K4me1/2/3, H3K36me2/3, H3K48me3, and H3K79me3. Conversely, H3K9me3, H3K27me3, and H4K20me3 are associated with transcriptional repression.99,137 Histone methyltransferases (HMTs) selectively transfer methyl groups from S-adenosyl-l-methionine to lysine or arginine residues in a highly specific manner, which can be reversed by histone demethylases (HDMs).138 Multiple studies on diabetic rat models have revealed the reduced levels of H3K9me3 and H3K27me3, mediating the release of transcriptional repression at the promotor/enhancer regions of genes associated with fibrosis and inflammation. The upregulation of these genes, including Il-6, monocyte chemotactic protein-1(Mcp-1), collagen type 1 alpha 1(Col1a1), and plasminogen activator inhibitor-1(Pai-1), ultimately lead to enhanced inflammation and disease progression.97,139,140,141 Conversely, markers associated with transcriptional activation, such as H3K4me1, were found to be upregulated at the promotor site of p65 in a high glucose environment and persisted after normalization of glucose levels.66,67 Mice subjected to an 8-week high-fat diet followed by an 8-week normal diet showed persistent lipid accumulation and elevated triglyceride levels. Formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq) analysis indicated persistent changes in the accessibility of chromatin for transcription factors, such as hepatocyte nuclear factor 4alpha (HNF4α), which were correlated with increased specific repressive histone modifications. Enrichment of H3K9me2 was found at sites with reduced chromatin accessibility.41 Similar to histone acetylation, histone methylation also modulates DNA methylation by affecting DNMT proteins.142 The methylation of H3K4 initiates de novo DNA methylation, leading to the activation of transcription. Conversely, transcription repression occurs as a result of the interaction between DNMT and H3K4 at the promoter level. Moreover, the recruitment of ubiquitin-like containing PHD and RING finger domains 1 (UHRF1) proteins by H3K9 with DNA methylation enhances the binding affinity of DNMTs to DNA, thereby promoting transcriptional repression.143,144
Histone phosphorylation can also influence histone–DNA interactions by altering the change of histones. The process of histone phosphorylation primarily occurs on serine, threonine, and tyrosine residues. A recent investigation unveiled an elevation in H3Ser10 phosphorylation within glomerular endothelial cells derived from diabetic patients, which mediates the amplified vascular cell adhesion protein 1 (VCAM-1).145 VCAM-1 promotes leukocyte adhesion and migration on the endothelium, thereby correlating with the advancement of diabetic nephropathy.146 Histone ubiquitination is typically observed at specific lysine residues located in the C-terminal tail of both histone H2A and histone H2B.147 The presence of high glucose levels induces H2A ubiquitination while reducing H2B ubiquitination, which activates the transforming growth factor-beta (TGF-β) pathway and accelerates disease progression.148 In the past few years, there has been a growing interest in studying histone lactylation as it plays a significant role in influencing gene transcription and metabolic regulation.149 This emerging field calls for further exploration into the association between histone lactylation and metabolic disorders, which holds significant potential for advancing our understanding in this area.
Non-coding RNAs
Non-coding RNAs (ncRNAs) are a type of RNA molecule that does not possess the ability to produce proteins. Instead, they play a part in regulating gene expression at both the post-transcriptional and translational stages.150 ncRNAs can be primarily classified into two categories: structural RNAs, including rRNAs and tRNAs, and regulatory RNAs. The regulatory RNA group consists of sncRNAs, which have a length below 200 nucleotides (nt), and long ncRNAs (lncRNAs), which possess a length above 200 nt.151 The sncRNAs subgroup further comprises microRNAs (miRNAs), small interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), and other sncRNAs. The lncRNAs can be categorized based on their origin into intergenic lncRNA, bidirectional lncRNA, sense/antisense lncRNA, intronic lncRNA, and enhancer lncRNA.152,153 Recent evidence has provided new evidence indicating that the control of gene expression and modulation of growth factors and inflammatory factors associated with metabolic diseases can be influenced by both miRNAs and lncRNAs.
miRNAs are a group of endogenously encoded non-coding RNAs that are typically characterized by their short length, usually ranging from 17 to 25 bp. They operate by specifically binding to the mRNA of genes that encode proteins through base pairing, ultimately causing either degradation or inhibition after transcription.154 A single miRNA possesses the capacity to regulate numerous target genes implicated in diverse pathogenic pathways in metabolic memory.155,156,157 Conversely, it is also possible for a specific target gene to be modulated simultaneously by multiple miRNAs.158 The dysregulated expression of various miRNAs is responsible for the development of metabolic complications by impacting crucial pathological mechanisms, including angiogenesis, apoptosis, inflammation, and oxidative stress.159,160 For instance, in a hyperglycemic environment, there is an upregulation of miRNA-21 (miR-21) expression, which activates TGF-β and NF-κB signaling pathways, leading to inflammatory responses and apoptosis.161 Therefore, these important miRNAs exhibit potential as diagnostic and prognostic biomarkers, along with being prospective therapeutic targets for metabolic diseases and their associated complications in the future.162
LncRNAs, which are transcribed by RNA polymerase II or III and resemble protein-coding mRNAs, belong to a group of transcripts that cannot be translated.163,164 They are typically observed at minimal levels under normal circumstances but play crucial roles in regulating vital cellular physiological activities, including cell proliferation, differentiation, and senescence.165,166 However, their aberrant expression is strongly associated with the advancement of specific diseases.167,168,169 Recent investigations have revealed that lncRNAs actively participate in the regulation of several metabolic disorders through their influences on epigenetic modifications, transcriptional regulation, and post-transcriptional modulation.170 For instance, the lncRNA maternally expressed gene 3 (MEG3) has been reported to be downregulated in the retina of mice with STZ-induced diabetes, which exacerbates retinal microvascular dysfunction by activating phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway.171 Further investigation showed that the decrease in MEG3 expression is controlled by DNMT1-mediated methylation occurring at the promoter region of MEG3, thereby expediting the progression of endothelial-mesenchymal transition (endMT) in individuals with diabetic retinopathy.172
Cellular mechanisms in metabolic memory
Numerous studies have presented evidence supporting the involvement of various cellular mechanisms in metabolic memory. These mechanisms, encompassing oxidative stress, non-enzymatic glycation of proteins, and low-grade inflammation, operate in a cascade and mutually reinforce each other, contributing to the persistence and progression of the deleterious effects of aberrant metabolism on the organism. This ultimately leads to abnormalities in cellular structure and function as well as organ pathology. Furthermore, environmental changes triggered by inflammation and oxidative stress further induce aberrant intracellular epigenetic modifications. These modifications subsequently boost the activation of genes associated with inflammation and programmed cell death, establishing a positive feedback loop that collectively sustains metabolic memory.173
Oxidative stress
The initial findings by Brownlee et al. elucidated the generation of reactive oxygen species (ROS) is excessively enhanced, serving as a distinguishing feature in hyperglycemia-related reactions to various pathological states.174 Under hyperglycemic conditions, the electron transport chain in the tricarboxylic acid (TCA) cycle of metabolism experiences an increase in electron donors like NADH and flavin adenine dinucleotide (FADH2), resulting in an elevation of the voltage gradient across the mitochondrial membrane. The elevated voltage gradient promotes excessive generation of superoxide (O2−) and reactive oxygen species (ROS).175,176 Meanwhile, elevated blood sugar levels increase the production of diglycerides (DAG), triggering protein kinase C (PKC) activation and increasing NADPH oxidase activity. Consequently, this results in an augmented generation of O2−. ROS, such as peroxynitrite (ONOO−), readily penetrate cell membranes, disrupting a variety of intracellular structures and contributing to nuclear and mitochondria DNA destruction and damage.177,178,179 It should be noted that mitochondrial DNA is highly sensitive to oxidative stress, causing both structural and functional impairments within the mitochondria. This damage further triggers cumulative ROS, perpetuating a detrimental cycle.180 Sustained overproduction of ROS can explain hyperglycemic metabolic memory even after normalization of glycemic level.181 Notably, despite the short half-life of excess free ROS, their presence endures after the normalization of blood glucose, contributing to the memory phenomenon and cell-damaging effects.63,65,182 These detrimental effects of ROS can be antagonized by broad-spectrum antioxidants acting at the mitochondrial level.19,183 In addition, it has been observed that an excessive amount of ROS can drive a range of pathological cellular mechanisms, including heightened polyol and hexosamine fluxes, AGEs, as well as NF-kB-induced vascular inflammation.184,185
Advanced glycation end products
AGEs, a diverse group of glycosylated adducts generated through a complex “Maillard reaction”, can be produced both internally within the body and externally from outside sources.186,187 The primary origin of AGEs is the endogenous process, whereby sugars react with amino groups of proteins, lipids, and nucleic acids through a series of non-enzymatic reactions.188 The physiological formation and accumulation of AGEs occur naturally during aging; however, insulin resistance, inflammation, and oxidative stress expedite this progression while also mediating cytopathic changes such as inflammation, fibrosis, and thrombotic reactions.189,190,191,192 The levels of AGEs have been found to be significantly higher in patients with MAFLD, diabetes mellitus, and its complications. These AGEs have also been linked to the risk of disease progression and mortality.193,194,195,196,197 Notably, due to their resistance to degradation, proteins modified by AGEs persist in the blood vessels, kidneys, and heart of diabetic patients even after achieving glycemic control. Thus, AGEs may exert important roles in metabolic memory.18,198
The deleterious impacts of AGEs could be attributed to three primary molecular mechanisms: modification of extracellular proteins, alteration of intracellular proteins, and activation of signaling cascades by binding to the receptor for AGE (RAGE) situated on the cell surface.
Modification of extracellular proteins
AGEs induce structural and functional abnormalities in tissues through the modification of extracellular proteins, resulting in the formation of stable and anomalous crosslinks. Specifically, collagen, laminin, and other extracellular matrix proteins can be altered through glycation by AGEs, leading to the formation of abnormal crosslinks that are resistant to proteolytic digestion.199 This process ultimately leads to the thickening of the basement membrane in vascular endothelial cells to impact the thickness and flexibility of blood vessel walls.200,201,202 In addition, AGEs can induce glycation in circulating factors like fibrinogen, low-density lipoprotein (LDL), etc., resulting in an intensified response to blood clot formation and an increased tendency for blood coagulation. Studies have shown that glycated LDL reduces tissue plasminogen activator (tPA) production in vascular endothelial cells and enhances thrombosis.202,203 Furthermore, the accumulation of AGEs resulting from hyperglycemia triggers the glycation and aggregation of lens α-crystallin. This process ultimately results in a decrease in lens clarity and an elevation in light dispersion, both of which are crucial in the development of diabetic cataracts.204
Modification of intracellular protein
AGEs trigger cellular damage through cross-linking or modifying various intracellular molecules, leading to the accumulation of improperly folded proteins within the endoplasmic reticulum (ER) through advanced glycation-mediated modification of molecular chaperones.205 This process triggers ER stress and disrupts cellular homeostasis.206,207 The persistence of ER stress and improperly folded proteins activates the unfolded protein response (UPR), ultimately leading to apoptosis.208,209 In addition, intracellular AGEs are able to bind to the mitochondrial respiratory chain complexes I and IV, which are involved in electron transport, and reduce their activity, decreasing ATP levels while increasing the production of superoxide and ROS, thereby inducing mitochondrial dysfunction.205,210,211 Furthermore, AGEs also diminish the activities of antioxidant enzymes like superoxide dismutase (SOD), catalase, glutathione peroxidase, and glutathione reductase. This acceleration further perpetuates the vicious cycle of ROS generation along with the accumulation of AGEs.194,212
AGE-mediated signaling cascades
The binding of AGE to RAGE is currently considered the primary pathogenic mechanism. RAGE, a pattern-recognition receptor belonging to the immunoglobulin superfamily, exhibits recognition and binding capabilities toward various ligands, including AGEs, Amyloid β (Aβ), S100/calgranulin, and the high mobility group box (HMGB)-1.213,214 The interactions between AGEs and RAGE trigger a series of intricate signaling cascades that culminate in producing pro-inflammatory factors and reactive oxygen species, ultimately fostering inflammation and tissue damage.215 It has been found that AGE-RAGE signaling triggers multiple downstream pathways, including mitogen-activated protein kinases (MAPK), AMP-activated protein kinase (AMPK), extracellular-signal-regulated kinases (ERK), and activates the NF-κB pathway, leading to a range of overexpressed cytokines and adhesion molecules, like IL-6, TNFα, TGF-β, and VCAM-1, etc.212,216,217 Deficiencies in RAGE lead to suppression of immune cell recruitment and attenuation of inflammation.218 RAGE activation also enhanced Janus kinase (JAK)/STAT signaling pathway activity and upregulated interferon-responsive gene expression.219 Since the promoter region of the RAGE gene contains an NF-κB-binding structural domain, interactions between AGE and RAGE interactions can elicit an upregulation in RAGE expression by enhancing inflammatory responses, which creates a positive feedback loop.218 Moreover, activation of AGE-RAGE has been observed to consistently upregulate NADPH and nitric oxide synthase, causing impaired mitochondrial function and elevated levels of oxidative stress.219 Sustained inflammatory responses and oxidative stress induce cellular fibrosis as well as apoptosis, which ultimately leads to vascular inflammation and diabetic vascular complications.198,220,221
Low-grade inflammation
Low-grade inflammation is a significant determinant that potentially plays an integral part in metabolic memory. Inflammation has been identified as the major contributor to diabetes mellitus and its vascular complications. Extended inflammatory responses may act as agents responsible for metabolic memory. Reactive oxygen species (ROS) production is enhanced by metabolic reprogramming in response to modifications in the metabolic environment, which is crucial in linking epigenetic changes and the external surroundings, resulting in the upregulation of inflammatory factor expression and secretion. Enhanced inflammatory responses drive diverse epigenetic modifications in cells to adapt to environmental alterations. A positive feedback loop involving ROS, metabolic end products, inflammation, and epigenetic modifications leads to the progression of metabolic disorders.222,223,224 It has been well elucidated that activation of NF-κB is particularly instrumental in facilitating pro-inflammatory gene expression. In individuals with diabetes, inflammatory gene expression can be enhanced through the activation of NF-κB. The activation results in an increase of inflammatory cytokines associated with vascular inflammation, thereby promoting the synthesis of endothelial adhesion molecules, proteases, and other mediators. Monocytes are recruited and adhere to endothelial cells and vascular smooth muscle cells, ultimately leading to differentiation into macrophages.225 Toll-like receptors are also of great importance in connecting inflammation and oxidative stress, being identified as a pathogenic contributor to obesity and insulin resistance.223
In conclusion, oxidative stress and AGEs are pivotal factors implicated in metabolic memory and complications development. Oxidative stress, characterized by an excessive production of reactive oxygen species, plays a vital role in cellular damage and perpetuation of harmful effects even after metabolism returns to normal. AGEs, formed through nonenzymatic reactions, modify extracellular and intracellular proteins, triggering inflammation and impairing normal cellular functions. These processes contribute to vascular stiffness, thrombosis, atherosclerosis, and other pathological mechanisms. The inflammation and oxidative stress are further intensified by the activation of NF-κB and the interaction between AGEs and the RAGE receptor. A comprehensive understanding of these processes is crucial for developing strategies to mitigate metabolic memory and prevent complications associated with metabolic disorders.
Cells involved in metabolic memory
Immune cells
Immune cells are key players in the formation and maintenance of metabolic memory. Hyperglycemia induces innate immune cells (e.g., monocytes and macrophages) to acquire metabolic memory and promote pro-inflammatory responses.78,226 Monocytes and macrophages are thought to undergo metabolic reprogramming and form metabolic memories in response to hyperglycemia, resulting in sustained activation that contributes to the pathogenesis of atherosclerosis.227 It was found that hyperglycemia can drive an increase in H3K4me3 and H3K27ac on the promoters of genes related to inflammation in macrophages and bone marrow-derived macrophages (BMDM) from diabetic mice through a glycolysis-dependent mechanism, thereby promoting proinflammatory gene expression (Fig. 4).228 Switching BMDM from a high glucose environment to physiologic glucose for culture did not alter the epigenetic and pro-inflammatory changes. In addition, normoglycemic mice received bone marrow transplantation from diabetic mice also exhibited increased aortic root atherosclerosis, suggesting a long-term effect of hyperglycemia mediated through metabolic memory.111 Histone-modifying enzymes are thought to upregulate MCP-1 expression and activate inflammatory monocyte differentiation as well as macrophage M1 polarization, leading to diabetic wound healing.229,230 In addition, hyperglycemia has been reported to upregulate DNMT1 expression to stimulate the mammalian target of the rapamycin (mTOR) pathway and induce pathogenic activation of peripheral blood mononuclear cells.119 Moreover, elevated lipid concentrations can also contribute to insulin resistance in monocytes, and this effect continues to persist even after lipid levels normalize due to metabolic memory. THP-1 monocytes treated with the saturated fatty acid, palmitate, for 6–12 h exhibited resistance to insulin-mediated glucose uptake and persisted for at least 60 h after removal of palmitate.231

Different cell types involved in metabolic memory. Multiple types of cells are involved in the development of metabolic memory-mediated metabolic diseases and their complications. Different cells crosstalk with each other and work together to cause disease progression
Endothelial cells
Endothelial cells are key players in mediating the link between vascular function and metabolic circulation.232,233 Metabolic disorders can modulate endothelial dysfunction and vascular diseases through metabolic memory.234,235 Sustained activation of p66Shc by PKCβII has been observed in aortic endothelial cells of both humans and mice exposed to hyperglycemia, even after blood glucose has returned to normal.236 Activation of p66Shc is associated with reduced CpG methylation and increased histone acetylation at promoters, which leads to elevated mitochondrial ROS production and oxidative signals translating into apoptosis.237 ROS, in turn, upregulate PKCβII activity, creating a detrimental cycle that ultimately culminates in apoptosis and the onset of tissue damage. A growing number of evidence has demonstrated that excessive ROS-mediated metabolic memory leads to a pro-inflammatory milieu and endothelial dysfunction in the aorta.90,236,238 NADPH oxidase 4 (Nox4) and endothelial nitric oxide synthase (eNOS) are involved in mediating transient hyperglycemia-induced vascular ROS generation and are regulated by altered histone methylation.239 An increase in H3K4me1 at the promoters of Nox4 and eNOS was found in endothelial cells in the hyperglycemic state, which antagonized and downregulated H3K9me2 and H3K9me3. Transient hyperglycemia facilitates the recruitment of lysine-specific methyltransferase SET7 and specific demethylase LSD1 to the NF-κB-p65 promoter. The upregulation of H3K4me1 by Set7 synergistically combines with the downregulation of H3K9me2 and H3K9me3 by LSD1 to activate the NF-κB pathway and upregulate the expression of inflammatory factors such as MCP-1 and VCAM-1, ultimately leading to dysfunction of vascular endothelium.67,240
Recent studies have also found that hyperglycemia-induced enhancement of the NF-κB pathway, increase in miR-27a-3p, decrease in nuclear factor erythroid-2-related factor 2 (NRF2) expression, and ROS overproduction in endothelial cells were maintained after restoration of normoglycemia, resulting in perivascular fibrosis and cardiovascular dysfunction in the heart.241 It confirmed the presence of metabolic memory in endothelial cells, indicating that insulin alone fails to improve cardiac function, whereas the combined application of miR-27a-3p inhibitors reverses the adverse effects. Moreover, studies on retinal endothelial cells have revealed that hyperglycemia can upregulate miR-23b-3p expression by activating the NF-κB signaling and target SIRT1 to mediate NF-κB acetylation, creating a positive feedback loop to maintain metabolic memory.242
Epithelial cells
Metabolic memory is responsible for the accelerated accumulation of apoptosis and tissue damage in the epithelium. In mouse models with diabetic nephropathy, the reduction in the expression of Kruppel-like factor 4 (KLF4) was observed in podocytes, coinciding with the onset and exacerbation of proteinuria.243 The expression of KLF4 was associated with a decrease in methylation levels of renal unit promoters and promoters of other epithelial markers, simultaneously increasing the methylation of promoters of genes encoding mesenchymal markers, thereby reducing proteinuria. Further studies found that expression of DNMT1, NF-κB p65, and nuclear factor Sp1 was significantly increased in podocytes under diabetic state, promoting inflammatory responses and podocyte damage.118 The employment of DNA methylation inhibitors can downregulate DNMT1 expression and ameliorate the adverse effects of hyperglycemia through the Sp1/NF-κB p65-Dnmt1 pathway, attenuating albuminuria. In addition, podocyte apoptosis in diabetic nephropathy has been found to be closely associated with the regulation of various miRNAs.244 Disturbed lipid levels in circulation enhance FOXO1 activity and induce insulin resistance by modifying the abundance of H3K36me2 and H3K27me3 on the promoter region of FOXO1 in human urine-derived podocyte-like epithelial cells (HUPECs). These manifestations persist even after the normalization of circulating lipid levels.245
Fibroblasts
Altered fibroblast function is strongly associated with delayed healing of diabetic wounds. Compared to nondiabetic donors, fibroblasts from T2DM donors exhibit changes in TNF-α-induced resistance to the inflammatory milieu, characterized by diminished synthesis of extracellular matrix (ECM), as well as impaired migratory and proliferative capacities. Such changes may be associated with chronic, nonhealing diabetic foot ulcers after restoration of normoglycemia.246 It was found that interferon-beta (IFN-β) stimulation allowed fibroblasts to acquire histone H3.3 and H3K36me3 chromatin marks, thereby establishing epigenetic memory.247 Genome-wide DNA methylation analysis of fibroblasts derived from diabetic foot ulcers also showed a significant decrease in global DNA methylation levels compared to non-diabetic foot fibroblasts.120 Fibroblasts of diabetic foot ulcer origin were identified as having sustained altered levels of DNA methylation even after prolonged passaging through normoglycemic conditions.248 Hyperglycemia also promotes diabetic cardiac fibrosis by regulating DNA methylation levels.249 5, 10 methylenetetrahydrofolate reductase (MTHFR) is a crucial regulatory enzyme in cardiac fibroblasts that modulates fibrosis and pyroptosis.250 Significant increase of DNMT3a and decrease of MTHFR were observed in cardiac fibrosis tissues from both humans and mice with diabetes, accompanied by CpG hypermethylation of the MTHFR promoters. The enhanced pyroptosis mediated by MTHFR knockdown in cardiac fibroblasts was reversed upon knockdown of DNMT3a. Moreover, a high-fat diet could promote the resistance of lung fibroblasts to apoptosis by inhibiting the expression of the death receptor Fas (also called CD95), leading to the progression of pulmonary fibrosis.251 The Fas promoter in fibroblasts from the murine model of pulmonary fibrosis displayed a reduction in histone acetylation and an increase in H3K9me3, which correlated with elevated expression of HDAC2 and HDAC4.252 A high-fat diet has also been shown to induce H4K16ac accumulation, leading to pro-fibrotic gene overexpression and collagen deposition in lung fibroblasts.253,254
Nerve cells
Neurons, Schwann cells, and glial cells are important participants in metabolic memory-mediated neuropathy.255 Metabotropic glutamate receptor (mGluR) 1 and mGluR5 are important proteins mediating neuropathic pain, with their expression significantly increased in diabetic neuropathic rats.256 The acetylation level of H3 in the promoter region of the gene encoding mGluR1/5 in spinal cord neurons was increased upon exposure to high glucose or high-fat stimulation, concomitant with a reduction in SIRT1 expression and activity. The SIRT1 activator, SRT1720, reversed the overexpression of mGluR1/5 and attenuated neuropathic pain in diabetic rats. Autophagy inhibition and dysfunction of Schwann cells are important pathogenic mechanisms in diabetic peripheral neuropathy (DPN). Reduced autophagy markers and brain-derived neurotrophic factor (BDNF) with increased expression of thioredoxin-interacting protein (TXNIP) in Schwann cells were observed in the sciatic nerves of diabetic mice.257,258,259 High glucose levels triggered the activation of the JAK/STAT signaling pathway, which induced HDAC1-dependent inhibition of autophagy and myelin abnormalities in Schwann cells.258 Expression of DNMT1 and DNMT3a in Schwann cells exposed to high glucose is regulated by the PI3K/Akt/mTOR pathway to mediate the progression of DPN.257,259
Endocrine cells
Metabolic memory of endocrine cells is intimately linked to the continued progression of metabolic diseases. β-cells, crucial constituents of the pancreas, participate in insulin secretion upon hyperglycemia condition and facilitate glucose uptake by peripheral tissues.260 Dysfunction of β-cells serves as the central mechanism underlying diabetes development and is intricately linked with metabolic disorder-mediated epigenetic modifications.261,262 The results of several genome-wide DNA methylation sequencing of human islets from T2DM patients and healthy controls identified multiple genes associated with β-cell insulin secretion as being located in T2DM differentially methylated regions.263,264 Metabolic disturbances during pregnancy mediate the risk of T2DM in the offspring through CpG methylation modifications in insulin signaling-related genes.265 Hypomethylation in the promoter region of cyclin-dependent kinase inhibitor 2 A (CDKN2A/B) and pro-apoptotic genes has been reported in the offspring of rats with gestational diabetes, contributing to β-cell apoptosis and increased susceptibility to T2DM.266 In addition, obesity can also modulate β-cell function through activation of histone modifications. Mice fed with a high-fat diet exhibited an upregulation of H3K27ac in the binding regions of transcription factors such as NRF1, GA-binding protein transcription factor alpha subunit (GABPA), and MEF2A, while a downregulation of H3K27ac was observed in the binding region of MAFK, which is implicated in the negative regulation of β-cell function.267,268
Metabolic memory and diseases
Circulatory system
Diabetes significantly increases the incidence of cardiovascular diseases and mortality.269 Growing evidence suggests that metabolic memory is an essential factor contributing to the prolonged deleterious consequences of high glucose on the circulatory system (Fig. 5).270 The findings of the UKPDS showed that the deleterious influence of hyperglycemia on the microvascular and macrovascular complications in those with diabetes persisted after glycemic control.10,31 Hyperglycemia may increase methylation of the promoter region of sarcoplasmic/endoplasmic reticulum calcium-ATPase 2a (SERCA2a) by upregulating the expression of pro-inflammatory TNF-α.271,272 Decreased expression of SERCA2a leads to myocardial diastolic function disorder, thus triggering diabetic cardiomyopathy. Epigenetic modifications facilitated by hyperglycemia interfere with lengthy and relatively stable alterations in gene expression. For instance, aortic endothelial cells cultured in transient hyperglycemia show a sustained increase in H3K4me1 at the NF-κB p65 promoter, which persists under normoglycemia conditions.66 Transient hyperglycemia has also been shown to maintain hyperacetylation and mediate persistent endothelial senescence by regulating deacetylase and acetyltransferase activities in vascular endothelial cells.273 In addition, altered levels of some miRNAs have been proposed to be closely correlated to diabetic macrovascular complications, like miR-133a, miR-195-5p, and miR-457a, among others.274,275,276,277

Complex interplay between metabolic memory and metabolic memory-regulated diseases. Metabolic disorders (including hyperglycemia, hyperlipidemia, hyperuricemia, hypoxia, and malnutrition) may induce epigenetic modifications and metabolic reprogramming at molecular and cellular levels, which take a toll on chronic inflammation and oxidative stress. Some metabolic end products, inflammatory cytokines, and reactive oxygen species could affect epigenetic regulations in return. Then, the epigenetic landscape and metabolic reprogramming could destroy the structure and function of different cells and tissues, manifesting as cell proliferation, immunocyte recruitment, cellular apoptosis, fibrosis, and senescence. The long-term accumulation of cellular and tissue dysregulation could give rise to metabolic memory-associated diseases, even after the elimination of metabolic disorders. The figure was created with BioRender.com (https://biorender.com/)
Epigenetic modifications driven by metabolic environmental alterations promote the expression of inflammation- and fibrosis-associated genes, resulting in endothelial dysfunction.278 Endothelial dysfunction can induce functional impairment of the vasculature and heart by promoting the secretion of vasoconstrictor agents, elevated endothelial permeability, and pathologic angiogenesis.279 As previously described, high glucose can mediate a significant elevation of p66Shc through decreased DNA methylation and increased histone acetylation, contributing to sustained oxidative stress and inflammation.236 p66Shc activation can also upregulate miR-34a in endothelial cells, which can lead to increased p53 acetylation and apoptosis by targeting downregulation of SIRT1.280,281 Silambarasan et al. revealed a correspondence between the expression levels of several miRNAs and endothelial dysfunction with miRNA microarray analysis.282 Among them, miR-130b-3p, miR-140-5p, and miR-221-3p exhibited a positive correlation with endogenous glucose levels, triggering endothelial function disorders through targeting genes involved in inflammation, senescence, as well as apoptosis. Another study conducting ingenuity pathway analysis of miRNA variants in mice hearts with diabetes revealed that various dysregulated miRNAs are associated with transcriptional regulation of apoptosis, fibrosis, hypertrophy, and heart failure and would not be reverted by intensive glycemic control.283 In addition, microarray analysis of lncRNAs and circRNAs from endothelial cells in the hyperglycemic milieu also identified multiple changes of ncRNAs associated with vascular endothelial damage, further demonstrating that metabolic disorders epigenetically modulate long-term deleterious cardiovascular effects.284,285 Intervention and therapy targeting metabolic memory and epigenetic modifications can effectively reduce the long-term damage of metabolic disorders on the circulatory system.286 For instance, metformin, a well-established antidiabetic drug, has been shown to effectively ameliorate the negative cardiovascular impacts of hyperglycemia by affecting the activity of numerous epigenetically modified enzymes.287,288
In addition to hyperglycemia, other metabolic disorders can mediate sustained cardiovascular injury through metabolic memory. Obesity could persistently affect ROS generation and cause vascular endothelial dysfunction through epigenetic modifications.289 Methyltransferase SUV39H1, demethylase JMJD2C, and acetyltransferase SRC-1 expression were significantly dysregulated in visceral adipose arteries of obese patients compared with normal controls.289,290 H3K9me2/3 and H3K9ac were correspondingly downregulated on the p66Shc promoter in obese patients, increasing ROS production and decreasing NO levels. Furthermore, a hypoxic environment can trigger metabolic memory in cardiac fibroblasts, leading to cardiac tissue fibrosis.291 Hypoxia increases DNMT1 and DNMT3B expression by upregulating HIF-1α, which causes sustained transcriptional repression of genes, including SOD2, in fibroblasts, promoting the expression of fibrosis factors and HIF-1α.44 Further studies have shown that dysregulation of DNMT1 leads to normoxic HIF-1α activation by affecting SOD2.45,292 In addition, exposure to maternal chronic hypoxia upregulates the CpG methylation of the promoter of PKC epsilon (PKCε) in the myocardium.293 The expression level of PKCε is subsequently downregulated and causes increased susceptibility to ischemia and reperfusion injury in the male heart in a sex-dependent manner, which persists into adulthood.
Endocrine system
Accumulated evidence has demonstrated the involvement of metabolic memory in the pathogenesis of various endocrine metabolic diseases. Internal and external environmental elements influence the development of endocrine tissues and organs through epigenetic alterations.294 These alterations can disrupt hormone secretion and action, which mediate disease progression, including metabolic syndrome, diabetes mellitus, and MAFLD.
Insulin, a crucial endocrine peptide hormone, is of great significance in modulating nutrient intake, utilization, and storage within the body. Impaired insulin secretion or reduced responsiveness of target cells to insulin serves as a causative driver of several diseases. Extensive research has shown that elevated lipid levels may contribute to the pathogenesis of insulin resistance, primarily through chronic tissue inflammation, oxidative stress, and epigenetic modifications with long-lasting effects.295,296 Metabolic syndrome represents a constellation of intricate metabolic disorders characterized by insulin resistance, hyperglycemia, hyperlipidemia, hypertension, and central obesity.297 Moreover, T2DM is closely related to impaired insulin sensitivity and insufficient insulin secretion, primarily resulting from defective pancreatic β-cell function.
Numerous epigenetic modifications have been identified in target tissues of insulin, such as skeletal muscle, adipose tissue, and liver, in case-control cohort studies on T2DM.298,299,300,301,302,303,304,305 These modifications include alternations in DNA methylation patterns within candidate genes correlated with T2DM like PPARG, transcription factor 7-like 2 (TCF7L2), potassium voltage-gated channel subfamily Q member 1 (KCNQ1), as well as insulin receptor substrate 1 (IRS1).306,307,308 Additionally, changes in DNA methylation were observed in obesity-induced insulin resistance compared to their non-obese counterparts. Obese individuals undergoing metabolic surgery appear to have epigenetic and metabolic changes in skeletal muscle that accompany the process of improved insulin sensitivity.309 You et al. identified DNMT3a as essential for mediating insulin resistance of both murine and human adipocytes.310 Knockout of the DNMT3a gene in mice adipose tissue avoids diet-associated insulin resistance yet does not result in altered adiposity.
Several investigations examined how human epigenetic modifications relate to insulin production.263,264,311,312,313,314,315,316,317,318 Researches unveiled the enhanced DNA methylation of PPARGC1A, insulin gene (INS), along with pancreatic and duodenum homeobox 1 (PDX1) in pancreatic islets of individuals suffering from T2DM in comparison to nondiabetic subjects.312,313 Barres et al. observed upregulated methylation and downregulation of PPARGC1A in skeletal muscle from T2DM patiens.300 PDX1 is associated with regulating early exocrine and endocrine pancreatogenesis, β cell development, as well as INS expression in mature β cells.319,320 Whole-genome bisulfite sequencing identified seven differentially methylated regions (DMRs) associated with T2DM islets, which supports a pivotal epigenetic regulatory role of PDX1 transcription factor in diabetes.264 Mice lacking PDX1 are prone to diabetes, while individuals mutated in PDX1 undergo one specific diabetes type known as MODY4.321,322,323
Maternal obesity or overnutrition during pregnancy can result in epigenetic changes in organs such as the fetal hypothalamus and liver, enhancing metabolic disease risk in offspring. In addition, studies of the prolonged effects of abnormal glucocorticoid secretion have also shown that an organism’s reactivity to stress can be altered continuously through epigenetic mechanisms but that it is not possible to “turn back the clock” after the stress relieved.324 Early life stress results in post-stress phosphorylation of methyl-CpG-binding protein 2 (Mecp2), which diminishes the combination of MeCp2 with methylated DNA, leads to hypomethylation of arginine vasopressin (AVP) in neurons of the paraventricular nucleus of the hypothalamus and promotes Avp expression.325 Such alterations in gene expression last into adulthood, suggesting that stress from earlier life may have a persistent influence, altering how your brain responds to pressure.326
Nervous system
Diabetic neuropathy is a group of clinical syndromes with diverse manifestations caused by different pathophysiologic mechanisms and widely distributed in various tissues.327 In general, these syndromes occurring in the peripheral nervous system can be categorized into diffuse and focal neuropathies. Diffuse neuropathies are more common and include DPN and diabetic autonomic neuropathy (DAN), which tend to be chronic and progressive. DPN is the most common type and typically presents as symmetric polyneuropathy that initially affects the distal lower extremities and progresses gradually upward as the disease progresses. Focal neuropathy is less common and is usually characterized by acute onset and self-limiting. In addition, diabetes causes degenerative changes in the central nervous system, resulting in an increased risk of cognitive impairment and Alzheimer’s disease (AD), Parkinson’s disease (PD), etc., also called diabetic encephalopathy (DE). Multiple mechanisms, including oxidative stress, chronic inflammation, and epigenetic dysregulation, are attributed to the ongoing metabolic changes in diabetic neuropathy.327,328
The engagement of epigenetic modifications in regulating the progression of DPN has been extensively investigated. Hyperglycemia continuously changes the state of DNA methylation and induces altered expression of genes associated with DPN. Whole genomic DNA methylation levels are dramatically reduced in leukocytes from DPN patients and may mediate insulin resistance along with low-grade inflammation to exacerbate neurologic lesions.329,330,331 Thorough analysis of methylome and transcriptome of the peroneal nerve in patients with T2D and DPN demonstrated that blood glucose levels independently and significantly affected the differential expression of the transcriptome engaged in the modulation of key aspects of DPN progression like an inflammatory response, insulin resistance, and ECM remodeling.332,333 For example, prostaglandin-endoperoxide synthase 1 (PTGS1), which encodes cyclooxygenase 1 (COX-1) that can promote inflammatory response and neuropathic pain, was found to be reduced methylated in patients with high HbA1c, indicating that it may function in the development of DPN.332,334 Increased demethylation of the promoter region of purinergic receptor P2X ligand-gated ion channel 3 (P2X3R) was found in dorsal root ganglion (DRG) of diabetic rats, being correlated with downregulated DNMT3a and DNMT3b.335,336 P2X3R can bind and activate NF-κB p65 to induce diabetic pain hypersensitivity. High glucose also induces the dysfunction of Schwann cells through DNA methylation, leading to myelin structural disorders as well as abnormalities in peripheral nerve conduction and action amplitude.259,337 DNMT1 and DNMT3a are upregulated in Schwann cells by the PI3K/Akt pathway suppressed by elevated glucose, thus upregulating thioredoxin-interacting protein (TXNIP) to adversely affect cellular function and metabolism.338,339 Treatment with DNMT inhibitor 5-Aza effectively downregulated TXNIP in the diabetic sciatic nerve, along with inhibited dysfunction and apoptosis of Schwann cells induced by hyperglycemia.
Different histone PTMs of genes associated with nerve regeneration and function are essential in DPN development. For instance, HDAC1 and HDAC5 were downregulated in rat Schwann cells stimulated by high glucose, accompanied by a decrease in autophagy markers (LC3-II/LC3-I) and abnormal myelination.258 Taking trichostatin A and small hairpin RNA vector as HDAC inhibitors to treat high glucose-cultured Schwann cells resulted in a significant improvement of LC3-II/LC3-I. Hyperglycemic mice showed persistent neuronal damage even after normalization of blood glucose, which could be reversed through combination therapy of glucagon-like peptide-1 receptor agonist (GLP-1RA) and metformin.340,341 Chen et al. revealed that GLP-1 and its analogs may attenuate FoxO-mediated oxidative stress and neurotoxicity in the hyperglycemic environment by deacetylating and phosphorylating FoxO1 through the SIRT1 and Akt pathways, respectively.342,343,344
Furthermore, ncRNAs are demonstrated to be involved in DPN progression. Various miRNAs have been identified to induce neuropathic pain and neuronal inflammation in DPN by activating the expression of pain-recognizing and pro-inflammatory-related proteins, including miR-9, miR-23, miR-146a, and miR-182, among others.345,346,347,348,349,350,351 miR-9 expression in DPN rats was reduced and accompanied by upregulation of calcium homeostasis modulator protein 1 (CALHM1), which controls extracellular Ca2+ influx and ATP production based on neuronal excitability.345,352,353 miR-9 interacts with CALHM1 to activate ATP-P2X7R signaling, which shows high expression in spinal cord and neurons, leading to neuropathic pain.354 In addition, DRG neuronal apoptosis in diabetic patients was related to downregulation of miR-146a.348 Downregulated miR-146a can upregulate the expression of IL-1 receptor-activated kinase (IRAK1) and tumor necrosis factor receptor-associated factor-6 (TRAF6) to mediate NF-κB overactivation, upregulating pro-inflammatory factors such as TNF-α and IL-1β along with DPN progression.349 Several recent studies have highlighted specific lncRNAs (NONRATT021972, uc.48+, and BC168687) as key factors in the pathogenesis of diabetic neuropathy and neuropathic pain.355,356,357 Targeting these particular lncRNAs in diabetic rat models attenuates DPN and may help ameliorate neuronal impairment potentially through modulation of inflammatory factor production.358,359,360,361
Diabetic encephalopathy is one complication of diabetes related to the central nervous system, usually associated with neurodegenerative lesions and cognitive impairment.362,363 The interaction between diabetes and cognition is complicated by the influence of epigenetic factors.364,365 On the one hand, persistent insulin resistance in peripheral tissues of metabolic syndrome leads to dysregulation of insulin receptor pathway activity in the brain, which ultimately results in brain insulin resistance.366 Brain insulin resistance increases the accumulation of Aβ and tau hyperphosphorylation, causing intense inflammation and neurodegeneration of nerve cells, which is considered to be an important pathogenetic component of AD.341,366,367 Histone modifications have been shown to intervene in the progression of AD by modulating autophagy in neuronal cells. On the other hand, an aberrant metabolic environment itself leads to changes in epigenetic regulation, which mediates the continued progression of metabolism-related neurodegenerative diseases. HDAC4 is linked to the pathologic process of several neurodegenerative ailments, and its expression is elevated in high glucose-treated hippocampal neurons.368 Radix polygoni multiflori downregulates HDAC4 in diabetic encephalopathic rats and reduces the apoptosis of hippocampal neurons by suppressing the activity of c-Jun aminoterminal kinase (JNK) pathway, which ultimately improves the cognitive function.369 In addition, miRNAs play important regulatory roles in metabolic memory-mediated neurodegenerative diseases.370,371,372,373 For example, miR-132 is downregulated in hippocampal neurons in rats with diabetic encephalopathy, accompanied by upregulated glycogen synthase kinase (GSK)-3β and tau. miR-132 could alleviate the impairment of diabetic encephalopathy via inhibiting GSK-3β expression and attenuating Tau hyperphosphorylation.374 A recent study also revealed significant alterations of multiple circRNAs in hippocampal neurons of diabetic mice.375 circ-Nbea is significantly downregulated by hyperglycemia and may facilitate the progression of DE by sponging miR-128-3p.376,377
Urogenital system
Diabetic kidney disease (DKD) is one high-risk complication of diabetes.378 Several experimental and clinical research revealed the association between CpG DNA methylation and DKD.379 Differential methylation of several genes, including UNC13B, has been identified in diabetic individuals with DKD in comparison to those without DKD, with UNC13B being associated with hyperglycemia-induced apoptosis in glomerular cells.116 The persistence of differential methylation of TXNIP loci was detected in whole blood and monocyte samples from the same DCCT/EDIC patients collected 16–17 years apart, which is thought to deliver glucose stress to promote oxidative stress and apoptosis, playing a pivotal role in DKD progression.113,380,381 Upregulation of TGF-β1 enhances Ras activation in fibroblasts by inducing hypermethylation of RAS protein activator like-1 (Rasal1) promoter, which promotes cell proliferation and fibrosis.382 TET3-mediated hydroxymethylation reverses the changes in Rsasl1 promoter DNA methylation pattern to reduce fibrosis.383
Studies investigating histone PTMs at critical metabolic genes suggested the critical role of epigenetic regulation in DKD pathogenesis. Histone modifications engage in DKD progression by affecting aspects of fibrosis, oxidative stress, and inflammation. For example, histone methylation at different locations in diabetic glomerular thylakoid cells is altered, exhibiting an increase in H3K4me1/2/3, H3K36me2/3, and H3K79me2 which promotes inflammatory responses and ECM accumulation, while accompanied by a decrease in H3K9me2/3 and H3K27me3 which are involved in the inhibition of renal fibrosis process.141,384,385 In addition, renal inflammation-mediated DKD progression is associated with histone modifications, which are involved in activating inflammatory factors like NF-κB by macrophages and monocyte infiltration under hyperglycemia.386 Epigenome profiling investigations revealed variations in H3K4me2 and H3K9me2 for genes relevant to diabetes and inflammation in monocytes exposed to hyperglycemia versus normoglycemic controls.387,388 Similar epigenetic changes were found in blood monocytes and lymphocytes of T1DM patients. HMT SET7 has been identified as a promoter of H3K4 methylation, which could co-activate pro-inflammatory genes downstream of NF-κB in monocytes.389 The association between histone acetylation modifications and DKD has been increasingly studied.390 p300, CBP and other HATs expression is upregulated in DKD and mediates increased transcription of pro-inflammatory and pro-fibrotic factors that exacerbate glomerular dysfunction associated with DKD.391,392,393 Inhibition of p300/CBP effectively reduces H3K9/14Ac levels, offering a potential therapeutic approach for managing hyperglycemia-mediated long-term kidney injury.394
ncRNAs also serve as essential regulators in the progression of DKD. miR-192 was first found to be downregulated in DKD, which targets TGF-β/Smad3 to upregulate ECM and collagen, as well as to promote renal fibrosis.395,396,397,398,399 Several miRNAs are believed to modulate vital features of DKD, such as apoptosis, fibrosis, and hypertrophy.244 miR21, miR-34a-5p, miR217, and others were found to be elevated under high glucose induction, combined with the promotion of inflammatory pathway signaling and induction of oxidative stress along with tissue fibrosis.400,401,402 Studies in recent years have also implicated lncRNAs in DKD.403 LncRNA plasmacytoma variant translocation 1 (PVT1) has been found to contribute to fibrosis and DKD pathogenesis.404 Upregulated PVT1 of glomerular thylakoid cells is correlated with increased cellular expression of pro-fibrotic elements under hpyerglycemia.404,405,406
Furthermore, as previously mentioned, metabolic disorders such as dietary factors have been implicated in the reduction of sperm quality in men and would be inherited intergenerationally through epigenetic modifications.37 High-fat diet during early paternal years induces permanent alterations in testicular lipid content and leads to irreversible sperm quality damage, which can persist for up to two generations.38,407
Immune system
Cancer is intimately tied to dysregulated immune homeostasis. There is substantial evidence indicating that metabolic disorders such as obesity and diabetes are correlated to an increased susceptibility to a number of cancers, including endometrial, liver, pancreatic, colorectal, and breast cancers.408 These metabolic abnormalities associated with obesity and diabetes have been widely recognized as risk factors for cancer morbidity and mortality. Further studies have demonstrated that cancer cell proliferation, migration, invasion, and resistance to chemotherapy are enhanced in a hyperglycemic environment, and this enhancement persists after blood glucose level is normalized.409 This phenomenon suggests that sustained regulation of metabolic memory has an essential impact on metabolic diseases and cancer.410
Recent studies have pointed out the occurrence of diverse epigenetic modifications of cancer cells under an environment of metabolic abnormalities. For instance, in breast cancer cell line MDA-MB-231, exposure to a hyperglycemic environment triggers dephosphorylation and activation of GSK-3β, which is vital in mediating insulin-dependent glycogen synthesis.411 Upregulation of GSK-3β enhances phosphorylated imprinting of histone H3, which mediates the upregulation of metastatic genes along with promoting epithelial-mesenchymal transition (EMT) and cancer cell proliferation. Additional studies have revealed that high glucose mediates overexpressed neuromodulin 1 (Nrg1) in breast cancer, fueling malignant cancer growth via the epidermal growth factor receptor (EGFR) pathway.412 Subsequent investigations have identified increased active enhancer modifications, including H3K4me1 and H3K27ac, in Nrg1 enhancers after hyperglycemia treatment and were consistent with the upregulation of Nrg1 expression.413 These findings indicate the persistence of this epigenetic imprint even after cancer cells return to normoglycemic conditions, facilitating aggressive tumor growth.414
A recent study has revealed an intriguing discovery that drug-resistant residual cells in microscopic residual disease of breast cancer exhibit similar DNA methylation status as cancer cells, regardless of lacking proliferative phenotypes and oncogenic signals.415 Notably, the promoter regions of HIF-1α, which is commonly recognized as an active master modulator of tumor glycolysis, displayed a comparable DNA methylation profile. Furthermore, both glycolytic co-activator proteins and glycolytic target genes of HIF-1α exhibited consistent demethylation patterns in cancer as well as residual cells, and isozyme-specific demethylation was present in either type of cells. These findings suggest that even after the removal of metabolically aberrant oncogenic signals from the environment, residual cells retain cancer-associated metabolic traits through epigenetic modifications and favor cancer resistance and recurrence.
In addition, metabolic memory can modulate cancer development through the influence of AGEs, oxidative stress, and inflammation. Pan et al. demonstrated that in diabetic patients, glycosylation and oxidation for high-density lipoprotein (HDL) may contribute to abnormal enhanced adhesion of breast cancer cells to human umbilical vein endothelial cells or ECM, thereby facilitating breast cancer metastasis.416 Moreover, Nε-(1-carboxymethyl)-L-lysine (CML), an AGE, was found to enhance the formation of chondrosarcoma tumor spheroids and upregulate cancer stem cell activity.417 CML treatment also augmented the migratory and invasive capability of tumor cells by advancing the EMT process. These observations suggest a potential promotional effect of AGEs on the stemness and metastatic properties of cancer.
The discovery of metabolic memory mechanisms opens up new possibilities to explain the higher risk and difficulty in treating cancers coupled with metabolic disorders. Targeting metabolic memory to develop assays and therapeutic agents in conjunction with conventional anti-tumor therapies could be a promising cancer therapeutic strategy for the future.
Prospects for metabolic memory applications
Given that the molecular and cellular mechanisms of metabolic memory mediate the long-term adverse effects of metabolic disorders, simply controlling the initial metabolic disorders is clearly insufficient. The development of disease diagnostic and therapeutic measures based on the molecular mechanisms of metabolic memory, especially epigenetic modulation, is a highly promising direction. Here, we describe current research progress in applying metabolic memory for disease diagnosis, biomarker identification, treatment, prevention, and prognosis. In addition, we also summarize clinical trials in metabolic diseases that have investigated metabolic memory-related mechanisms for screening, treatment, and prevention of diseases and complications in Table 1.
Disease biomarkers
Due to the presence of metabolic memory, the onset and progression of complications may occur when early symptoms of metabolic disorders are not apparent in the patient and may persist after the metabolic disorder has been controlled.418,419 Early diagnosis and treatment of metabolic disorders is critical to the management and prognosis of metabolic diseases. Mounting evidence suggests that various epigenetic modifications are intimately associated with disease development as primary molecular mechanisms of metabolic memory.286,420 Epigenetic changes have been recognized to hold promise as sensitive, reliable, and easily accessible biomarkers to predict the risk of disease progression, playing a crucial role in early diagnosis and prognosis of metabolic disorders.
Research on DNA methylation biomarkers in metabolic memory-related diseases has flourished with considerable progress. For example, with the progress of a high-throughput epigenome-wide association study (EWAS), the results of differentially methylated genes were identified by microarray chip methylation analysis and microbead array chip methylation analysis revealed differentially methylated sites related to the risk of developing diabetes, and its complications, including TCF7L2, KCNQ1, TXNIP, ATP-binding cassette subfamily G (ABCG1), phosphoethanolamine/phosphocholine phosphatase (PHOSPHO1) and others.259,421,422,423,424,425,426 It has been shown that DNA methylation status in peripheral blood can be detected noninvasively from cells by methylation-specific PCR and is a promising marker for the detection of metabolic disorder-related diseases.427,428 In addition, DNA methylation modifications have been shown to correlate with β-cell dysfunction or death.429 The INS gene, which encodes the insulin precursor, is expressed chiefly in the β-cells of the pancreatic islets and exhibits unmethylation, whereas it is methylated in other tissues.430 When β-cells are disrupted by metabolic diseases, unmethylated INS DNA is shed into the circulation and can be detected by a variety of PCR-based molecular methods.431,432,433,434,435 Individuals with recent-onset type 1 diabetes (T1D) and those at-risk groups demonstrated notably elevated levels of unmethylated INS DNA in comparison to nondiabetic controls.436 Detection of unmethylated INS DNA by blood samples can be effective for early diagnosis of T1D and screening of high-risk subjects.437 Unmethylated INS DNA can also be used to monitor rejection and prognosis of islet transplantation. Immediate elevation of INS DNA levels occurs after islet transplantation and its persistent elevation may indicate a greater likelihood of long-term functional loss of the graft.438 Several epigenomic association studies of hepatic fat have shown that DNA methylation changes of peripheral blood origin are strongly associated with hepatic fat accumulation and could be attractive biomarkers for the detection of MAFLD.439,440
In addition, alternations in the levels of numerous miRNAs and lncRNAs are correlated with disease progression, being potential targets for disease diagnosis as well.98,441,442 For instance, examination of plasma circulating miRNA profiles, particularly changes in plasma levels of miR-150, miR-30a-5p, miR-15a, and miR-375, in the years prior to the onset of T2DM and prodromal DM can be utilized to assess the risk of developing T2DM.443 This may improve prediction and prevention in people at risk for T2DM. In addition, recent findings have identified a series of lncRNAs that are dysregulated in the adipocytes of obese mice fed with a high-fat diet, which showed dynamic alternations in the fed versus fasted state.444 Among them, lnc-ORIA9 (lnc-leptin) is strongly associated with of leptin, which is essential for adipogenesis and can potentially serve as a novel molecular indicator of adipose energy status.
Therapeutic targets
Behavioral interventions
Various lifestyle or environmental exposures (including malnutrition, high-fat diets, and physical inactivity) are important factors influencing the formation of metabolic memory.445 Lifestyle interventions such as appropriate dietary modifications and exercise are known to have important health benefits for metabolic homeostasis and the immune system and may be beneficial for preventing or mitigating the risk of metabolic diseases.446 Recent studies have identified ways in which dietary modifications can intervene in the possible long-term adverse effects mediated by metabolic memory by influencing epigenetic modifications.447,448 Dietary bioactive compounds such as polyphenols and terpenoids serve as epigenetic modifiers, and appropriately increasing intake of them may reverse epigenetic alterations in metabolism-related genes in offspring from a poor maternal diet.449,450 Specifics on increasing the intake of dietary bioactive compounds for the intervention of metabolic memory-related disorders will be presented in the next section.
A sedentary lifestyle or lack of exercise is one of the main risk factors for the progression of diseases related to metabolic disorders.451 Past studies have shown that exercise is beneficial in ameliorating insulin resistance, which has recently been shown to be possibly correlated with altered epigenetic modifications.452 It was shown that promoter methylation levels of genes, including peroxisome proliferator-activated receptor gamma coactivator 1 (PGC1), MEF2, and PPARγ, are reduced immediately after acute exercise and lead to a subsequent increase in the expression of their genes.453 PGC1 is decreased in insulin-resistant skeletal muscle, which induces mitochondrial dysfunction, leading to increased intracellular lipid levels in myocytes and further insulin resistance.454 Exercise may improve the persistence and progression of insulin resistance by upregulating PGC1 expression and activity in skeletal muscle. A study analyzing DNA methylation changes in adipose tissue of healthy subjects before and after a six-month exercise intervention showed significant alterations in CpG methylation levels of several obesity- and diabetes-related genes, accompanied by simultaneous changes in mRNA expression.455 Sedentary patients with metabolic diseases also showed hypomethylation of the promoter regions of genes such as NRF1, solute carrier family 27 member 4 (SLC27A4), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), and GSK3A in skeletal muscle after 16 weeks of aerobic training, which is thought to contribute to the reduction of circulating lipids and improved glucose metabolism.456 In addition, multiple types of physical exercise modulate miRNA expression in healthy and metabolic disease patients.457,458,459 Aerobic or resistance exercise training was associated with substantial circulating mi-RNA alterations in diabetic patients and significantly upregulated miR-423-3p, miR-451a, and miR-766-3p. miR-451a and miR-423-3p target fatty acid biosynthesis and metabolism regulation and were significantly associated with fat loss. Physical exercise also reversed some miRNAs that are aberrantly expressed in T2DM.460 Chronic physical exercise downregulated miR-144, which is overexpressed in diabetes, and ameliorated miR-144-mediated insulin resistance through altered IRS1 and IRS2 expression.461,462,463 In contrast, downregulation of miR-15a and miR-192 in the plasma of T2DM patients promoted insulin resistance and disease progression.443,464,465 Physical exercise leads to increased expression of miR-15a and miR-192 to ameliorate insulin resistance and lipid accumulation.466,467
Natural products
Despite the alarming long-term effects of metabolic disorders and their complications, advances in research on natural products of foods and drugs offer new insights on the treatment of metabolic diseases.
The monoterpenes, sesquiterpenes, phenolics, and diarylheptaterpenes (curcumin) extracted from ginger, widely used as food and medicine in daily life, are involved in the regulation of apoptosis, cell cycle/DNA damage, chromatin/epigenetic regulation, cytoskeletal regulation, and adhesion through specific signaling pathways, which are beneficial for diabetes and metabolic syndrome, cholesterol levels and lipid metabolism, and inflammation.468 Curcumin is a natural product and a p300/CBP-specific HAT inhibitor involved in suppressing cellular histone acetylation levels.469 In endothelial cells, curcumin has shown the ability to prevent hyperglycemia-induced histone acetylation in the promoter regions with important regulatory roles in vascular structure and function under diabetic conditions.470 Garcinol, which can also act as a HAT inhibitor, reduces the transcription of inflammatory factors by inhibiting the hyperglycemia-induced enhancement of histone H3 acetylation.471
Astragalus is also considered a promising drug containing prominent bioactive natural saponins, such as Astragaloside IV (AST) and cyclosiversioside F (CSF). AST decreased RSC96 cell apoptosis and alleviated DPN in rats through the miR-155-mediated PI3K/Akt/mTOR signaling pathway.472 CSF attenuates insulin resistance, adipocyte inflammation, and apoptosis through PI3K/Akt, NF-κB, and MAPK signaling.473
What’s more, long-term dysfunction of pancreatic β-cells induces insulin resistance and chronic complications. Genistein with structural specificity directly regulates β-cell proliferation and insulin secretion and protects against apoptosis through the cAMP/PKA pathway.474,475 Polyphenolic compounds in fruits and grains protect β-cells under high glucose conditions through the inhibition of histone acetylation.476,477,478,479,480 Furthermore, resveratrol and sirtuins treat metabolic disorders in epigenetic regulation of DNMT, HDAC, and lysine-specific demethylase-1 (LSD1).481,482 Resveratrol can reverse hyperglycemia-induced AMPK inactivation by activating HDAC SIRT1, inhibiting the NF-κB pathway and pro-inflammatory factors.483 Glucose metabolism and insulin sensitivity were significantly improved in T2DM patients receiving resveratrol treatment.484
Epigenetic modulation of cell function and metabolism by various natural products provides a new therapeutic approach to eliminating the long-term effects of metabolic disorders. The effect of the dose of various natural ingredients on metabolic memory, as well as synergistic or antagonistic effects between components, warrant further study.
Targeted drugs
A number of novel drugs that target epigenetic changes to eliminate the long-term effects of metabolic memory are being developed.485 These drugs are named epigenetic drugs (or epidrugs) and mainly include HAT inhibitors (HATi), HDAC inhibitors (HDACi), HDAC activators, and miRNA inhibitors.486,487 Epigenetic drugs have not been formally approved for metabolic disease treatment, but there have been several epigenetic drugs approved for use in other diseases related to metabolic diseases, including decitabine, valproic acid (VPA), vorinostat, and so on.488,489,490 It is reasonable to believe in the promising efficacy of drugs targeting epigenetic regulation for metabolic diseases and metabolic memory, but there is still a need to address the systemic and low specificity of current epigenetic drug targets to make epigenetic therapies fittingly appropriate for metabolic memory.491,492,493 Targeting miRNAs for inhibition is also considered a promising therapy, such as locked nucleic acid (LNA) modification. In diabetic mice, the specific inhibition of miR-192 using LNA-anti miR-192 demonstrated a reduction in the expression of pro-fibrotic genes, leading to the reversal of persistent renal fibrosis.396 In addition, miRNA delivery via extracellular vesicles (EVs) has been shown to be effective in preventing the progression of metabolic complications in animal models.494 Adipose-derived stem cells (ADSC) secreted EVs with high expression of miR-130a-3p compared with DPN patients and rats. Treatment with ADSC-derived EVs in DPN rats resulted in a targeted decrease of DNMT1 expression and activation of the NRF2/HIF-1α/skeletal muscle actin alpha 1 (ACTA1) pathway, which ultimately contributed to the repair of peripheral vascular injury.495 miR-20b-3p is specifically enriched in plasma-derived exosomes of healthy rats (hplasma-exos).495,496 The injection of hplasma-exos into rats with diabetic peripheral neuropathy (DPN) resulted in an upregulation of miR-20b-3p levels in the sciatic nerve, in turn, mitigated autophagic damage induced by high glucose in Schwann cells and improved DPN symptoms.497
The development of novel drugs targeting metabolism-related epigenetic changes in cancer and combining them with conventional cancer therapies will help to inhibit cancer proliferation, metastasis, and drug resistance. As mentioned earlier, metabolic memory ties metabolism-related diseases to cancer, and the development of drugs targeting epigenetic modulators has gathered significant interest in the field of cancer research.498 Restoring chromatin structure to its original state through epigenetic therapies is a major research focus in cancer treatment. Epigenetic therapies hold promise in reversing abnormal imprinting events in chromatin and may provide a novel approach to treating the memory effects of hyperglycemia. HMT inhibitors are reported as a key candidate for cancer therapies by recent preclinical studies.499 The administration of the lysine inhibitors tazemetostat, SHR2554, and pinometostat exhibited satisfactory therapeutic efficacy in several cancers with an acceptable safety profile.500 Further studies are still required to explore possible avenues for targeting metabolic memory in combination with cancer treatment.
Conclusion
“Metabolic memory” is originally defined as the long-term adverse effects of hyperglycemia even after glycemic normalization. In this review, we outline the research history and put novel insights to the salient features of metabolic memory from molecular cellular aspects to different organs and systems levels. The early metabolic abnormalities leave memory imprints on target cells, which persistently affect cellular function at multiple levels and finally induce multisystem diseases. The persistent harmful effects include inflammatory changes, premature cell senescence, and apoptosis. The enduring adverse effects eventuate metabolic diseases on different tissues and organs. Early intensive metabolic control has long-lasting benefits in reducing the risk of these complications, even after patients return to standard metabolic control. we further summarize recent advances in metabolic memory phenomena and their associated molecular mechanisms as indicators for detecting metabolic diseases and targets for therapeutic interventions. This phenomenon probably implies the potential reasons for metabolic diseases and their chronic progressive complications.
In addition, we emphasize the presence and impact of metabolic memory in a variety of metabolic diseases and establish a link between molecular mechanisms and disease progression. Metabolic reprogramming and epigenetic modifications are critically involved in the of biological mechanism of metabolic memory. They both interact and work together. Long-lasting epigenetic changes induced by metabolic stimuli could be the pivotal mechanism underlying metabolic memory. Epigenetic modifications could induce persistent expression of metabolic disease-related genes and pro-inflammatory genes, while the accumulation of some products may be the source of epigenetic changes. DNA methylation, as well as histone modification and non-coding RNA, have been implicated in the progression of metabolic complications. Early metabolic control can prevent the development of epigenetic changes that contribute to the alleviation of excessive oxidative stress and inflammation. The evaluation of epigenetic changes and metabolic reprogramming will lead to the discovery of the pathogenesis of metabolic diseases and complications.
Although the concept of metabolic memory initiates from the progression of diabetic complications, it is nowadays extended to the legacy effects of not only hyperglycemia but also hyperlipidemia and other detrimental metabolic environments. The existence of the metabolic memory phenomenon has challenged conventional therapeutic approaches to metabolic diseases. Based on the importance of early intervention in metabolic disease management, metabolic memory phenomena, and their associated molecular mechanisms might indicate new ways and targets for therapeutic interventions. Variations in circulating DNA methylation levels may be a potential tool to aid in the early screening and prognostic assessment of metabolic diseases. Targeting epigenetic modifications for behavioral interventions, dietary supplementation, and pharmacotherapy are promising future directions for investigation and development. At the same time, there are still confusions that need to be clarified in the field of metabolic memory. For example, more analysis is needed to precisely detect the persistent time and severity in different metabolic abnormalities. It is also of great significance to investigate the potential differences in metabolic memory according to age variations. For patients with metabolic syndrome comorbid with AD, cancer, or other age-related diseases, it is necessary to develop combined therapies targeting epigenetic changes to help these patients achieve better outcomes and prognosis. Also, more clinical trials and biological evidence are needed to explore the mechanisms of metabolic memory and its association with diseases in the future.
References
Wu, Y. L. et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduct. Target Ther. 8, 98 (2023).
Sun, H. et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119 (2022).
Afshin, A. et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017).
Koenen, M., Hill, M. A., Cohen, P. & Sowers, J. R. Obesity, adipose tissue and vascular dysfunction. Circ. Res. 128, 951–968 (2021).
Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20 (2018).
Eslam, M., Sanyal, A. J. & George, J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1991 (2020).
Hoffman, D. J., Powell, T. L., Barrett, E. S. & Hardy, D. B. Developmental origins of metabolic diseases. Physiol. Rev. 101, 739–795 (2021).
Li, D. et al. Diet-gut microbiota-epigenetics in metabolic diseases: from mechanisms to therapeutics. Biomed. Pharmacother. 153, 113290 (2022).
Pillai, U. J., Ray, A., Maan, M. & Dutta, M. Repurposing drugs targeting metabolic diseases for cancer therapeutics. Drug Discov. Today 28, 103684 (2023).
Holman, R. R. et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 359, 1577–1589 (2008).
Lachin, J. M. & Nathan, D. M. Understanding metabolic memory: the prolonged influence of glycemia during the diabetes control and complications trial (DCCT) on future risks of complications during the study of the epidemiology of diabetes interventions and complications (EDIC). Diabetes Care 44, 2216–2224 (2021).
Crisóstomo, L. et al. Inheritable testicular metabolic memory of high-fat diet causes transgenerational sperm defects in mice. Sci. Rep. 11, 9444 (2021).
Smith, B. W. & Adams, L. A. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat. Rev. Endocrinol. 7, 456–465 (2011).
Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. J. Am. Med. Assoc. 290, 2159–2167 (2003).
Holman, R. R. Type 2 diabetes mellitus in 2012: optimal management of T2DM remains elusive. Nat. Rev. Endocrinol. 9, 67–68 (2013).
Singh, R. et al. Epigenetic modification and therapeutic targets of diabetes mellitus. Biosci. Rep. 40, BSR20202160 (2020).
Vasishta, S., Umakanth, S., Adiga, P. & Joshi, M. B. Extrinsic and intrinsic factors influencing metabolic memory in type 2 diabetes. Vasc. Pharmacol. 142, 106933 (2022).
Yamagishi, S. I., Nakamura, N. & Matsui, T. Glycation and cardiovascular disease in diabetes: a perspective on the concept of metabolic memory. J. Diabetes 9, 141–148 (2017).
Ihnat, M. A. et al. Reactive oxygen species mediate a cellular ‘memory’ of high glucose stress signalling. Diabetologia 50, 1523–1531 (2007).
Nathan, D. M. et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986 (1993).
Nathan, D. M. The pathophysiology of diabetic complications: how much does the glucose hypothesis explain? Ann. Intern Med. 124, 86–89 (1996).
The Diabetes Control and Complications Trial (DCCT). Design and methodologic considerations for the feasibility phase. The DCCT Research Group. Diabetes 35, 530–545 (1986)..
Diabetes Control and Complications Trial (DCCT): results of feasibility study. The DCCT Research Group. Diabetes Care 10, 1–19 (1987).
Implementation of treatment protocols in the Diabetes Control and Complications Trial. Diabetes Care 18, 361–376 (1995).
Control, T. D. & Group, C. T. R. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 44, 968–983 (1995).
Control, D. & Group, C. T. R. The absence of a glycemic threshold for the development of long-term complications: the perspective of the diabetes control and complications trial. Diabetes 45, 1289–1298 (1996).
Nathan, D. M. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 37, 9–16 (2014).
Epidemiology of Diabetes Interventions and Complications (EDIC). Design, implementation, and preliminary results of a long-term follow-up of the Diabetes Control and Complications Trial cohort. Diabetes Care 22, 99–111 (1999)..
Lachin, J. M. et al. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N. Engl. J. Med. 342, 381–389 (2000).
Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 352, 837–853 (1998).
Chalmers, J. & Cooper, M. E. UKPDS and the legacy effect. N. Engl. J. Med. 359, 1618–1620 (2008).
Gaede, P., Vedel, P., Parving, H. H. & Pedersen, O. Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. Lancet 353, 617–622 (1999).
Gaede, P. et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N. Engl. J. Med. 348, 383–393 (2003).
Gaede, P., Lund-Andersen, H., Parving, H. H. & Pedersen, O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 358, 580–591 (2008).
Lu, Q. et al. The role of long noncoding RNA in lipid, cholesterol, and glucose metabolism and treatment of obesity syndrome. Med. Res. Rev. 41, 1751–1774 (2021).
Duan, Y. et al. Regulation of cholesterol homeostasis in health and diseases: from mechanisms to targeted therapeutics. Signal. Transduct. Target Ther. 7, 265 (2022).
Crisóstomo, L. et al. A switch from high-fat to normal diet does not restore sperm quality but prevents metabolic syndrome. Reproduction 158, 377–387 (2019).
Crisóstomo, L. et al. Diet during early life defines testicular lipid content and sperm quality in adulthood. Am. J. Physiol. Endocrinol. Metab. 319, E1061–e1073, (2020).
Crisóstomo, L. et al. Inherited metabolic memory of high-fat diet impairs testicular fatty acid content and sperm parameters. Mol. Nutr. Food Res. 66, e2100680 (2022).
Crisóstomo, L. et al. Testicular “Inherited Metabolic Memory” of ancestral high-fat diet is associated with sperm sncRNA Content. Biomedicines 10, 909 (2022).
Leung, A. et al. Persistent chromatin modifications induced by high fat diet. J. Biol. Chem. 291, 10446–10455 (2016).
Inoue, Y. et al. High-fat diet in early life triggers both reversible and persistent epigenetic changes in the medaka fish (Oryzias latipes). BMC Genomics 24, 472 (2023).
Li, M. et al. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation 134, 1105–1121 (2016).
Watson, C. J. et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 23, 2176–2188 (2014).
Tian, L. et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: a pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ. Res. 126, 1723–1745 (2020).
Vaupel, P. & Mayer, A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 26, 225–239 (2007).
Semenza, G. L. & Wang, G. L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 12, 5447–5454 (1992).
Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411–421 (2000).
Luo, W. & Wang, Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-α protein stability and activity. Cell Mol. Life Sci. 75, 1043–1056 (2018).
Chen, Y., Liu, M., Niu, Y. & Wang, Y. Romance of the three kingdoms in hypoxia: HIFs, epigenetic regulators, and chromatin reprogramming. Cancer Lett. 495, 211–223 (2020).
Zhang, F. et al. Crosstalk among m(6)A RNA methylation, hypoxia and metabolic reprogramming in TME: from immunosuppressive microenvironment to clinical application. J. Hematol. Oncol. 15, 84 (2022).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 (2008).
Mukherjee, A., Bilecz, A. J. & Lengyel, E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 41, 575–587 (2022).
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Fan, P. et al. Alleviating hypoxia to improve cancer immunotherapy. Oncogene 42, 3591–3604 (2023).
Li, B. et al. CCL2 promoter hypomethylation is associated with gout risk in Chinese Han male population. Immunol. Lett. 190, 15–19 (2017).
Wang, Z. et al. Differential DNA methylation of networked signaling, transcriptional, innate and adaptive immunity, and osteoclastogenesis genes and pathways in gout. Arthritis Rheumatol. 72, 802–814 (2020).
Crișan, T. O. et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann. Rheum. Dis. 75, 755–762 (2016).
Cleophas, M. C. P. et al. Romidepsin suppresses monosodium urate crystal-induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res. Ther. 21, 50 (2019).
Li, S. L. et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes 55, 2611–2619 (2006).
Reddy, M. A. et al. Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler Thromb. Vasc. Biol. 32, 721–729 (2012).
Roy, S., Sala, R., Cagliero, E. & Lorenzi, M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc. Natl Acad. Sci. USA 87, 404–408 (1990).
Kowluru, R. A. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes 52, 818–823 (2003).
Kowluru, R. A., Abbas, S. N. & Odenbach, S. Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J. Diabetes Complic. 18, 282–288 (2004).
Kowluru, R. A., Kanwar, M. & Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 21976 (2007).
El-Osta, A. et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 205, 2409–2417 (2008).
Brasacchio, D. et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236 (2009).
Zhong, Q. & Kowluru, R. A. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 60, 1304–1313 (2011).
Zhong, Q. & Kowluru, R. A. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 62, 2559–2568 (2013).
Yokoi, T. et al. Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 55, 1660–1665 (2006).
Ksiazek, K., Passos, J. F., Olijslagers, S. & von Zglinicki, T. Mitochondrial dysfunction is a possible cause of accelerated senescence of mesothelial cells exposed to high glucose. Biochem. Biophys. Res. Commun. 366, 793–799 (2008).
Cramer, C. et al. Persistent high glucose concentrations alter the regenerative potential of mesenchymal stem cells. Stem Cells Dev. 19, 1875–1884 (2010).
Liu, J. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 26, 110–121 (2014).
Kuilman, T. & Peeper, D. S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 9, 81–94 (2009).
Zhu, Y., Armstrong, J. L., Tchkonia, T. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care 17, 324–328 (2014).
Palmer, A. K. et al. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes 64, 2289–2298 (2015).
Zhang, P. et al. Hyperglycemia-induced inflamm-aging accelerates gingival senescence via NLRC4 phosphorylation. J. Biol. Chem. 294, 18807–18819 (2019).
Thiem, K. et al. Hyperglycemic memory of innate immune cells promotes in vitro proinflammatory responses of human monocytes and murine macrophages. J. Immunol. 206, 807–813 (2021).
Kowluru, R. A. & Chan, P. S. Metabolic memory in diabetes—from in vitro oddity to in vivo problem: role of apoptosis. Brain Res. Bull. 81, 297–302 (2010).
Zhong, Q. & Kowluru, R. A. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J. Cell Biochem. 110, 1306–1313 (2010).
Engerman, R. L. & Kern, T. S. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36, 808–812 (1987).
Patel, A. et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 358, 2560–2572 (2008).
Duckworth, W. et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 360, 129–139 (2009).
Del Prato, S. Megatrials in type 2 diabetes. From excitement to frustration? Diabetologia 52, 1219–1226 (2009).
Newman, T. M. et al. Early-life dietary exposures mediate persistent shifts in the gut microbiome and visceral fat metabolism. Am. J. Physiol. Cell Physiol. 324, C644–c657 (2023).
Summerfield, M. et al. A long-term maternal diet transition from high-fat diet to normal fat diet during pre-pregnancy avoids adipose tissue inflammation in next generation. PLoS ONE 13, e0209053 (2018).
Xu, H. et al. A long-term maternal diet intervention is necessary to avoid the obesogenic effect of maternal high-fat diet in the offspring. J. Nutr. Biochem. 62, 210–220 (2018).
Fu, Q. et al. A short-term transition from a high-fat diet to a normal-fat diet before pregnancy exacerbates female mouse offspring obesity. Int. J. Obes. 40, 564–572 (2016).
Tewari, S., Zhong, Q., Santos, J. M. & Kowluru, R. A. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 53, 4881–4888 (2012).
Zhong, Q. & Kowluru, R. A. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest. Ophthalmol. Vis. Sci. 54, 244–250 (2013).
Magri-Tomaz, L. et al. Two weeks of high-fat feeding disturb lipid and cholesterol molecular markers. Cell Biochem. Funct. 36, 387–393 (2018).
Welles, J. E. et al. An integrative approach to assessing effects of a short-term Western diet on gene expression in rat liver. Front. Endocrinol. 13, 1032293 (2022).
Suter, M. A. et al. In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am. J. Obstet. Gynecol. 210, 463.e461–463.e411 (2014).
Wankhade, U. D. et al. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 12, e0175675 (2017).
Reddy, M. A., Zhang, E. & Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 58, 443–455 (2015).
Li, K. Y. et al. DNA methylation markers for kidney function and progression of diabetic kidney disease. Nat. Commun. 14, 2543 (2023).
Sun, G. et al. Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 21, 2069–2080 (2010).
Alvarez, M. L. & Distefano, J. K. The role of non-coding RNAs in diabetic nephropathy: potential applications as biomarkers for disease development and progression. Diabetes Res. Clin. Pract. 99, 1–11 (2013).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Bošković, A. & Rando, O. J. Transgenerational epigenetic inheritance. Annu. Rev. Genet. 52, 21–41 (2018).
Kato, M. & Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 15, 327–345 (2019).
Aranyi, T. & Susztak, K. Cytosine methylation studies in patients with diabetic kidney disease. Curr. Diab. Rep. 19, 91 (2019).
Whitelaw, N. C. & Whitelaw, E. Transgenerational epigenetic inheritance in health and disease. Curr. Opin. Genet. Dev. 18, 273–279 (2008).
Breton, C. V. et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun. Biol. 4, 769 (2021).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Portela, A. & Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068 (2010).
Chen, Z. X. & Riggs, A. D. DNA methylation and demethylation in mammals. J. Biol. Chem. 286, 18347–18353 (2011).
Jirtle, R. L. & Skinner, M. K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8, 253–262 (2007).
Wang, J. et al. Nutrition, epigenetics, and metabolic syndrome. Antioxid. Redox Signal. 17, 282–301 (2012).
Chen, Z. et al. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat. Metab. 2, 744–762 (2020).
Avellino, R. & Delwel, R. Expression and regulation of C/EBPα in normal myelopoiesis and in malignant transformation. Blood 129, 2083–2091 (2017).
Chen, Z. et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc. Natl Acad. Sci. USA 113, E3002–E3011 (2016).
Fradin, D. et al. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS ONE 7, e36278 (2012).
Maghbooli, Z. et al. Aberrant DNA methylation patterns in diabetic nephropathy. J. Diabetes Metab. Disord. 13, 69 (2014).
Bell, C. G. et al. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genomics 3, 33 (2010).
Zhu, Y. et al. DNMT1-mediated PPARα methylation aggravates damage of retinal tissues in diabetic retinopathy mice. Biol. Res. 54, 25 (2021).
Zhang, L. et al. DNA methyltransferase 1 may be a therapy target for attenuating diabetic nephropathy and podocyte injury. Kidney Int. 92, 140–153 (2017).
Chen, G. et al. Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int. 96, 409–420 (2019).
Park, L. K. et al. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics 9, 1339–1349 (2014).
Olsen, A. S., Sarras, M. P. Jr., Leontovich, A. & Intine, R. V. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 61, 485–491 (2012).
Wang, Y., Tar, M. T. & Davies, K. P. Hyperglycemic memory in the rat bladder detrusor is associated with a persistent hypomethylated state. Physiol. Rep. 8, e14614 (2020).
Kim, H. et al. Persistent changes in liver methylation and microbiome composition following reversal of diet-induced non-alcoholic-fatty liver disease. Cell Mol. Life Sci. 76, 4341–4354 (2019).
Gomez-Arango, L. F. et al. Increased systolic and diastolic blood pressure is associated with altered gut microbiota composition and butyrate production in early pregnancy. Hypertension 68, 974–981 (2016).
Peng, K. et al. Butyrate and obesity: current research status and future prospect. Front. Endocrinol. 14, 1098881 (2023).
Čugalj Kern, B. et al. The role of epigenetic modifications in late complications in type 1 diabetes. Genes (Basel). 13, 705 (2022).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014).
Li, X., Li, C. & Sun, G. Histone acetylation and its modifiers in the pathogenesis of diabetic nephropathy. J. Diabetes Res. 2016, 4065382 (2016).
Yun, J. M., Jialal, I. & Devaraj, S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J. Nutr. Biochem. 22, 450–458 (2011).
Zheng, Z. et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 61, 217–228 (2012).
Tu, Y. et al. Melatonin attenuates oxidative stress and inflammation of Müller cells in diabetic retinopathy via activating the Sirt1 pathway. Biomed. Pharmacother. 137, 111274 (2021).
Chen, F. et al. Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. Cell Death Differ. 28, 1001–1012 (2021).
Wang, X. et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 86, 712–725 (2014).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Mishra, M., Duraisamy, A. J. & Kowluru, R. A. Sirt1: a guardian of the development of diabetic retinopathy. Diabetes 67, 745–754 (2018).
Brooks, W. H. et al. Epigenetics and autoimmunity. J. Autoimmun. 34, J207–J219 (2010).
Klose, R. J. & Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318 (2007).
Villeneuve, L. M. et al. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl Acad. Sci. USA 105, 9047–9052 (2008).
Komers, R. et al. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab Invest. 93, 543–552 (2013).
Jia, Y. et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 294, 12695–12707 (2019).
Cedar, H. & Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304 (2009).
Hashimoto, H., Vertino, P. M. & Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2, 657–669 (2010).
Gowher, H. & Jeltsch, A. Mammalian DNA methyltransferases: new discoveries and open questions. Biochem. Soc. Trans. 46, 1191–1202 (2018).
Alghamdi, T. A. et al. Histone H3 serine 10 phosphorylation facilitates endothelial activation in diabetic kidney disease. Diabetes 67, 2668–2681 (2018).
Liu, J. J. et al. Vascular cell adhesion molecule-1, but not intercellular adhesion molecule-1, is associated with diabetic kidney disease in Asians with type 2 diabetes. J. Diabetes Complic. 29, 707–712 (2015).
Svikle, Z. et al. Ubiquitin-proteasome system in diabetic retinopathy. PeerJ 10, e13715 (2022).
Gao, C. et al. Impact of high glucose and proteasome inhibitor MG132 on histone H2A and H2B ubiquitination in rat glomerular mesangial cells. J. Diabetes Res. 2013, 589474 (2013).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Taft, R. J. et al. Non-coding RNAs: regulators of disease. J. Pathol. 220, 126–139 (2010).
Palazzo, A. F. & Lee, E. S. Non-coding RNA: what is functional and what is junk? Front. Genet. 6, 2 (2015).
Wang, J. et al. Regulatory roles of non-coding RNAs in colorectal cancer. Int. J. Mol. Sci. 16, 19886–19919 (2015).
Nojima, T. & Proudfoot, N. J. Mechanisms of lncRNA biogenesis as revealed by nascent transcriptomics. Nat. Rev. Mol. Cell Biol. 23, 389–406 (2022).
Shantikumar, S., Caporali, A. & Emanueli, C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc. Res. 93, 583–593 (2012).
Esguerra, J. L., Bolmeson, C., Cilio, C. M. & Eliasson, L. Differential glucose-regulation of microRNAs in pancreatic islets of non-obese type 2 diabetes model Goto-Kakizaki rat. PLoS ONE 6, e18613 (2011).
Trajkovski, M. et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474, 649–653 (2011).
Long, J. et al. MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J. Biol. Chem. 286, 11837–11848 (2011).
Hartig, S. M., Hamilton, M. P., Bader, D. A. & McGuire, S. E. The miRNA interactome in metabolic homeostasis. Trends Endocrinol. Metab. 26, 733–745 (2015).
Agbu, P. & Carthew, R. W. MicroRNA-mediated regulation of glucose and lipid metabolism. Nat. Rev. Mol. Cell Biol. 22, 425–438 (2021).
Macvanin, M. et al. The role of miRNAs in metabolic diseases. Curr. Med. Chem. 30, 1922–1944 (2023).
Zhong, X. et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 56, 663–674 (2013).
Kato, M. & Natarajan, R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat. Rev. Nephrol. 10, 517–530 (2014).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011).
Zhao, X. Y. & Lin, J. D. Long noncoding RNAs: a new regulatory code in metabolic control. Trends Biochem. Sci. 40, 586–596 (2015).
Schmitz, S. U., Grote, P. & Herrmann, B. G. Mechanisms of long noncoding RNA function in development and disease. Cell Mol. Life Sci. 73, 2491–2509 (2016).
Thomas, A. A., Feng, B. & Chakrabarti, S. ANRIL: a regulator of VEGF in diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 58, 470–480 (2017).
Forrest, M. E. & Khalil, A. M. Review: regulation of the cancer epigenome by long non-coding RNAs. Cancer Lett. 407, 106–112 (2017).
Yang, J. et al. LncRNAs in tumor metabolic reprogramming and immune microenvironment remodeling. Cancer Lett. 543, 215798 (2022).
Goyal, N., Kesharwani, D. & Datta, M. Lnc-ing non-coding RNAs with metabolism and diabetes: roles of lncRNAs. Cell Mol. Life Sci. 75, 1827–1837 (2018).
Qiu, G. Z. et al. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 471, 135–141 (2016).
He, Y. et al. DNMT1-mediated lncRNA MEG3 methylation accelerates endothelial-mesenchymal transition in diabetic retinopathy through the PI3K/Akt/mTOR signaling pathway. Am. J. Physiol. Endocrinol. Metab. 320, E598–e608, (2021).
Russell, N. D. & Cooper, M. E. 50 years forward: mechanisms of hyperglycaemia-driven diabetic complications. Diabetologia 58, 1708–1714 (2015).
Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 (2001).
Trumpower, B. L. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 265, 11409–11412 (1990).
Creager, M. A., Lüscher, T. F., Cosentino, F. & Beckman, J. A. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108, 1527–1532 (2003).
Yerneni, K. K. et al. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes 48, 855–864 (1999).
Inoguchi, T. et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945 (2000).
Geraldes, P. & King, G. L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106, 1319–1331 (2010).
Foury, F., Hu, J. & Vanderstraeten, S. Mitochondrial DNA mutators. Cell Mol. Life Sci. 61, 2799–2811 (2004).
Murray, P., Chune, G. W. & Raghavan, V. A. Legacy effects from DCCT and UKPDS: what they mean and implications for future diabetes trials. Curr. Atheroscler. Rep. 12, 432–439 (2010).
Kowluru, R. A., Chakrabarti, S. & Chen, S. Re-institution of good metabolic control in diabetic rats and activation of caspase-3 and nuclear transcriptional factor (NF-kappaB) in the retina. Acta Diabetol. 41, 194–199 (2004).
Savitha, S., Tamilselvan, J., Anusuyadevi, M. & Panneerselvam, C. Oxidative stress on mitochondrial antioxidant defense system in the aging process: role of DL-alpha-lipoic acid and L-carnitine. Clin. Chim. Acta 355, 173–180 (2005).
Brownlee, M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625 (2005).
Giacco, F. & Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070 (2010).
Singh, R., Barden, A., Mori, T. & Beilin, L. Advanced glycation end-products: a review. Diabetologia 44, 129–146 (2001).
Garay-Sevilla, M. E., Rojas, A., Portero-Otin, M. & Uribarri, J. Dietary AGEs as exogenous boosters of inflammation. Nutrients 13, 2802 (2021).
Henning, C. & Glomb, M. A. Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 33, 499–512 (2016).
Monnier, V. M. Nonenzymatic glycosylation, the Maillard reaction and the aging process. J. Gerontol. 45, B105–111, (1990).
Vlassara, H. & Bucala, R. Recent progress in advanced glycation and diabetic vascular disease: role of advanced glycation end product receptors. Diabetes 45(Suppl 3), S65–S66 (1996).
Tan, A. L., Forbes, J. M. & Cooper, M. E. AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 27, 130–143 (2007).
Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 46, 217–224 (2011).
Hyogo, H. & Yamagishi, S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr. Pharm. Des. 14, 969–972 (2008).
Yamagishi, S. & Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 20, 15 (2015).
Arsov, S. et al. Advanced glycation end-products and skin autofluorescence in end-stage renal disease: a review. Clin. Chem. Lab Med. 52, 11–20 (2014).
Genuth, S. et al. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 64, 266–278 (2015).
de Vos, L. C. et al. Skin autofluorescence is associated with 5-year mortality and cardiovascular events in patients with peripheral artery disease. Arterioscler. Thromb. Vasc. Biol. 34, 933–938 (2014).
Brownlee, M., Cerami, A. & Vlassara, H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 318, 1315–1321 (1988).
Goldin, A., Beckman, J. A., Schmidt, A. M. & Creager, M. A. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114, 597–605 (2006).
Bailey, A. J. Molecular mechanisms of ageing in connective tissues. Mech. Ageing Dev. 122, 735–755 (2001).
Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 21, 3–12 (2003).
Fishman, S. L. et al. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Mol. Med. 24, 59 (2018).
Zhang, J., Ren, S., Sun, D. & Shen, G. X. Influence of glycation on LDL-induced generation of fibrinolytic regulators in vascular endothelial cells. Arterioscler Thromb. Vasc. Biol. 18, 1140–1148 (1998).
Bahmani, F., Bathaie, S. Z., Aldavood, S. J. & Ghahghaei, A. Inhibitory effect of crocin(s) on lens α-crystallin glycation and aggregation, results in the decrease of the risk of diabetic cataract. Molecules 21, 143 (2016).
Pugliese, G. Do advanced glycation end products contribute to the development of long-term diabetic complications? Nutr. Metab. Cardiovasc Dis. 18, 457–460 (2008).
Yamabe, S. et al. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 280, 1617–1629 (2013).
Suzuki, R. et al. Intracellular accumulation of advanced glycation end products induces osteoblast apoptosis via endoplasmic reticulum stress. J. Bone Min. Res. 35, 1992–2003 (2020).
Adamopoulos, C. et al. Advanced glycation end-products induce endoplasmic reticulum stress in human aortic endothelial cells. Clin. Chem. Lab Med. 52, 151–160 (2014).
Tabas, I. & Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 (2011).
Akhter, F. et al. High dietary advanced glycation end products impair mitochondrial and cognitive function. J. Alzheimers Dis. 76, 165–178 (2020).
Akhter, F. et al. Age-dependent accumulation of dicarbonyls and advanced glycation endproducts (AGEs) associates with mitochondrial stress. Free Radic. Biol. Med. 164, 429–438 (2021).
Ramasamy, R. et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15, 16r–28r (2005).
Morbini, P. et al. The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod. Pathol. 19, 1437–1445 (2006).
Fritz, G. RAGE: a single receptor fits multiple ligands. Trends Biochem. Sci. 36, 625–632 (2011).
Hudson, B. I. & Lippman, M. E. Targeting RAGE signaling in inflammatory disease. Annu. Rev. Med. 69, 349–364 (2018).
Schmidt, A. M., Yan, S. D., Yan, S. F. & Stern, D. M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 108, 949–955 (2001).
Ramasamy, R., Yan, S. F. & Schmidt, A. M. The RAGE axis and endothelial dysfunction: maladaptive roles in the diabetic vasculature and beyond. Trends Cardiovasc. Med. 15, 237–243 (2005).
Kumar Pasupulati, A., Chitra, P. S. & Reddy, G. B. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol. Concepts 7, 293–309 (2016).
Ott, C. et al. Role of advanced glycation end products in cellular signaling. Redox Biol. 2, 411–429 (2014).
Prasad, A., Bekker, P. & Tsimikas, S. Advanced glycation end products and diabetic cardiovascular disease. Cardiol. Rev. 20, 177–183 (2012).
Stitt, A. W., Jenkins, A. J. & Cooper, M. E. Advanced glycation end products and diabetic complications. Expert Opin. Investig. Drugs 11, 1205–1223 (2002).
Reddy, M. A. & Natarajan, R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res. 90, 421–429 (2011).
Thompson, J. A. & Webb, R. C. Potential role of Toll-like receptors in programming of vascular dysfunction. Clin. Sci. 125, 19–25 (2013).
Schroen, B. & Heymans, S. Small but smart–microRNAs in the centre of inflammatory processes during cardiovascular diseases, the metabolic syndrome, and ageing. Cardiovasc. Res. 93, 605–613 (2012).
Guarner, V. & Rubio-Ruiz, M. E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 40, 99–106 (2015).
Mossel, D. M. et al. Epigenetic regulation of S100A9 and S100A12 expression in monocyte-macrophage system in hyperglycemic conditions. Front Immunol. 11, 1071 (2020).
van Diepen, J. A. et al. Diabetes propels the risk for cardiovascular disease: sweet monocytes becoming aggressive? Cell Mol. Life Sci. 73, 4675–4684 (2016).
Edgar, L. et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation 144, 961–982 (2021).
Cabanel, M. et al. Epigenetic control of macrophage shape transition towards an atypical elongated phenotype by histone deacetylase activity. PLoS ONE 10, e0132984 (2015).
Davis, F. M. et al. Palmitate-TLR4 signaling regulates the histone demethylase, JMJD3, in macrophages and impairs diabetic wound healing. Eur. J. Immunol. 50, 1929–1940 (2020).
Gao, D., Bailey, C. J. & Griffiths, H. R. Metabolic memory effect of the saturated fatty acid, palmitate, in monocytes. Biochem. Biophys. Res. Commun. 388, 278–282 (2009).
Avogaro, A. et al. Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care 34(Suppl 2), S285–S290 (2011).
Walsh, L. K. et al. Increased endothelial shear stress improves insulin-stimulated vasodilatation in skeletal muscle. J. Physiol. 597, 57–69 (2019).
Dimassi, S. et al. Role of eNOS- and NOX-containing microparticles in endothelial dysfunction in patients with obesity. Obesity 24, 1305–1312 (2016).
Meza, C. A., La Favor, J. D., Kim, D. H. & Hickner, R. C. Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci. 20, 3775 (2019).
Paneni, F. et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 111, 278–289 (2012).
Cosentino, F. et al. Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler. Thromb. Vasc. Biol. 28, 622–628 (2008).
Cai, H. & Harrison, D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844 (2000).
Liao, Y. et al. NADPH oxidase 4 and endothelial nitric oxide synthase contribute to endothelial dysfunction mediated by histone methylations in metabolic memory. Free Radic. Biol. Med. 115, 383–394 (2018).
Rana, G. et al. Cortical spreading depression differentially affects lysine methylation of H3 histone at neuroprotective genes and retrotransposon sequences. Brain Res. 1467, 113–119 (2012).
Yao, Y. et al. Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc. Res. 118, 196–211 (2022).
Zhao, S. et al. miR-23b-3p induces the cellular metabolic memory of high glucose in diabetic retinopathy through a SIRT1-dependent signalling pathway. Diabetologia 59, 644–654 (2016).
Hayashi, K. et al. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Invest. 124, 2523–2537 (2014).
Kato, M. & Natarajan, R. MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann. N. Y. Acad. Sci. 1353, 72–88 (2015).
Kumar, S., Pamulapati, H. & Tikoo, K. Fatty acid induced metabolic memory involves alterations in renal histone H3K36me2 and H3K27me3. Mol. Cell Endocrinol. 422, 233–242 (2016).
Al-Rikabi, A. H. A., Tobin, D. J., Riches-Suman, K. & Thornton, M. J. Dermal fibroblasts cultured from donors with type 2 diabetes mellitus retain an epigenetic memory associated with poor wound healing responses. Sci. Rep. 11, 1474 (2021).
Kamada, R. et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl Acad. Sci. USA 115, E9162–e9171 (2018).
Bhamidipati, T. et al. Epigenetic basis of diabetic vasculopathy. Front. Endocrinol. 13, 989844 (2022).
Sun, H. et al. MTHFR epigenetic derepression protects against diabetes cardiac fibrosis. Free Radic. Biol. Med. 193, 330–341 (2022).
Raghubeer, S. & Matsha, T. E. Methylenetetrahydrofolate (MTHFR), the one-carbon cycle, and cardiovascular risks. Nutrients 13, 4562 (2021).
Yang, J. et al. High-fat diet related lung fibrosis-epigenetic regulation matters. Biomolecules 13, 558 (2023).
Huang, S. K. et al. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 4, e621 (2013).
Pessoa Rodrigues, C. et al. Histone H4 lysine 16 acetylation controls central carbon metabolism and diet-induced obesity in mice. Nat. Commun. 12, 6212 (2021).
Zhang, X. et al. Modulation of H4K16Ac levels reduces pro-fibrotic gene expression and mitigates lung fibrosis in aged mice. Theranostics 12, 530–541 (2022).
Eid, S. A. et al. New perspectives in diabetic neuropathy. Neuron 111, 2623–2641 (2023).
Zhou, C. H. et al. SIRT1 attenuates neuropathic pain by epigenetic regulation of mGluR1/5 expressions in type 2 diabetic rats. Pain 158, 130–139 (2017).
Zhang, C. H. et al. The Akt/mTOR cascade mediates high glucose-induced reductions in BDNF via DNMT1 in Schwann cells in diabetic peripheral neuropathy. Exp. Cell Res. 383, 111502 (2019).
Du, W. et al. STAT3 phosphorylation mediates high glucose-impaired cell autophagy in an HDAC1-dependent and -independent manner in Schwann cells of diabetic peripheral neuropathy. FASEB J. 33, 8008–8021 (2019).
Zhang, X. et al. TXNIP, a novel key factor to cause Schwann cell dysfunction in diabetic peripheral neuropathy, under the regulation of PI3K/Akt pathway inhibition-induced DNMT1 and DNMT3a overexpression. Cell Death Dis. 12, 642 (2021).
Vetere, A., Choudhary, A., Burns, S. M. & Wagner, B. K. Targeting the pancreatic β-cell to treat diabetes. Nat. Rev. Drug Discov. 13, 278–289 (2014).
Campbell, S. A. & Hoffman, B. G. Chromatin regulators in pancreas development and diabetes. Trends Endocrinol. Metab. 27, 142–152 (2016).
Saisho, Y. Importance of beta cell function for the treatment of type 2 diabetes. J. Clin. Med. 3, 923–943 (2014).
Dayeh, T. et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 10, e1004160 (2014).
Volkov, P. et al. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 66, 1074–1085 (2017).
Franzago, M., Fraticelli, F., Stuppia, L. & Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: consequences in mother and child. Epigenetics 14, 215–235 (2019).
Nazari, Z. et al. In utero exposure to gestational diabetes alters DNA methylation and gene expression of CDKN2A/B in Langerhans islets of rat offspring. Cell J. 22, 203–211 (2020).
Nammo, T. et al. Genome-wide profiling of histone H3K27 acetylation featured fatty acid signalling in pancreatic beta cells in diet-induced obesity in mice. Diabetologia 61, 2608–2620 (2018).
Nomoto, H. et al. Inhibition of small Maf function in pancreatic β-Cells improves glucose tolerance through the enhancement of insulin gene transcription and insulin secretion. Endocrinology 156, 3570–3580 (2015).
Kannel, W. B. & McGee, D. L. Diabetes and cardiovascular disease. The Framingham study. J. Am. Med. Assoc. 241, 2035–2038 (1979).
Prandi, F. R. et al. Epigenetic modifications and non-coding RNA in diabetes-mellitus-induced coronary artery disease: pathophysiological link and new therapeutic frontiers. Int. J. Mol. Sci. 23, 4589 (2022).
Kao, Y. H. et al. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 38, 217–222 (2010).
Asrih, M. & Steffens, S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc. Pathol. 22, 117–125 (2013).
Zhang, E. et al. Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/p53/p21 pathway. PLoS ONE 10, e0143814 (2015).
Liu, N. et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 22, 3242–3254 (2008).
Giordano, M. et al. Circulating MiRNA-195-5p and −451a in diabetic patients with transient and acute ischemic stroke in the emergency department. Int. J. Mol. Sci. 21, 7615 (2020).
Ortega, F. J. et al. Profiling of circulating microRNAs reveals common microRNAs linked to type 2 diabetes that change with insulin sensitization. Diabetes Care 37, 1375–1383 (2014).
Al-Hayali, M. A. et al. Clinical value of circulating microribonucleic acids miR-1 and miR-21 in evaluating the diagnosis of acute heart failure in asymptomatictype 2 diabetic patients. Biomolecules 9, 193 (2019).
Pasquier, J. et al. Epigenetics and cardiovascular disease in diabetes. Curr. Diabetes Rep. 15, 108 (2015).
Tabit, C. E., Chung, W. B., Hamburg, N. M. & Vita, J. A. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 11, 61–74 (2010).
Li, Q. et al. P66Shc-induced microRNA-34a causes diabetic endothelial dysfunction by downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 36, 2394–2403 (2016).
Wu, J. et al. Inhibition of P53/miR-34a improves diabetic endothelial dysfunction via activation of SIRT1. J. Cell Mol. Med. 23, 3538–3548 (2019).
Silambarasan, M. et al. MicroRNAs in hyperglycemia induced endothelial cell dysfunction. Int. J. Mol. Sci. 17, 518 (2016).
Costantino, S., Paneni, F., Lüscher, T. F. & Cosentino, F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur. Heart J. 37, 572–576 (2016).
Singh, K. K. et al. A global profile of glucose-sensitive endothelial-expressed long non-coding RNAs. Can. J. Physiol. Pharmacol. 94, 1007–1014 (2016).
Shang, F. F., Luo, S., Liang, X. & Xia, Y. Alterations of circular RNAs in hyperglycemic human endothelial cells. Biochem. Biophys. Res. Commun. 499, 551–555 (2018).
Sommese, L. et al. Clinical relevance of epigenetics in the onset and management of type 2 diabetes mellitus. Epigenetics 12, 401–415 (2017).
Li, P. et al. Sirt 1 activator inhibits the AGE-induced apoptosis and p53 acetylation in human vascular endothelial cells. J. Toxicol. Sci. 40, 615–624 (2015).
Bridgeman, S. C. et al. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes. Metab. 20, 1553–1562 (2018).
Costantino, S. et al. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur. Heart J. 40, 383–391 (2019).
Kluge, M. A., Fetterman, J. L. & Vita, J. A. Mitochondria and endothelial function. Circ. Res. 112, 1171–1188 (2013).
Dave, J., Jagana, V., Janostiak, R. & Bisserier, M. Unraveling the epigenetic landscape of pulmonary arterial hypertension: implications for personalized medicine development. J. Transl. Med. 21, 477 (2023).
Archer, S. L. et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671 (2010).
Patterson, A. J. et al. Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat hearts. Circ. Res. 107, 365–373 (2010).
Rosen, E. D. et al. Epigenetics and epigenomics: implications for diabetes and obesity. Diabetes 67, 1923–1931 (2018).
Glass, C. K. & Olefsky, J. M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 15, 635–645 (2012).
Lee, S. H., Park, S. Y. & Choi, C. S. Insulin resistance: from mechanisms to therapeutic strategies. Diabetes Metab. J. 46, 15–37 (2022).
Xu, H. et al. Etiology of metabolic syndrome and dietary intervention. Int. J. Mol. Sci. 20, 128 (2018).
Abderrahmani, A. et al. Increased hepatic PDGF-AA signaling mediates liver insulin resistance in obesity-associated type 2 diabetes. Diabetes 67, 1310–1321 (2018).
Baumeier, C. et al. Hepatic DPP4 DNA methylation associates with fatty liver. Diabetes 66, 25–35 (2017).
Barrès, R. et al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 10, 189–198 (2009).
Nilsson, E. et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63, 2962–2976 (2014).
Nilsson, E. et al. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J. Clin. Endocrinol. Metab. 100, E1491–1501, (2015).
Nitert, M. D. et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61, 3322–3332 (2012).
Ribel-Madsen, R. et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS ONE 7, e51302 (2012).
Kirchner, H. et al. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol. Metab. 5, 171–183 (2016).
Rönn, T. et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 24, 3792–3813 (2015).
Keller, M. et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol. Metab. 6, 86–100 (2017).
Davegårdh, C. et al. Abnormal epigenetic changes during differentiation of human skeletal muscle stem cells from obese subjects. BMC Med. 15, 39 (2017).
Gancheva, S. et al. Dynamic changes of muscle insulin sensitivity after metabolic surgery. Nat. Commun. 10, 4179 (2019).
You, D. et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 6, e30766 (2017).
Ling, C. et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51, 615–622 (2008).
Yang, B. T. et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia 54, 360–367 (2011).
Yang, B. T. et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 26, 1203–1212 (2012).
Volkmar, M. et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. Embo J. 31, 1405–1426 (2012).
Hall, E. et al. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 15, 522 (2014).
Khetan, S. et al. Type 2 diabetes-associated genetic variants regulate chromatin accessibility in human islets. Diabetes 67, 2466–2477 (2018).
Hall, E. et al. The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol. Cell Endocrinol. 472, 57–67 (2018).
Hall, E. et al. Glucolipotoxicity alters insulin secretion via epigenetic changes in human islets. Diabetes 68, 1965–1974 (2019).
Park, J. H., Stoffers, D. A., Nicholls, R. D. & Simmons, R. A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 118, 2316–2324 (2008).
Sultan, S. & AlMalki, S. Analysis of global DNA methylation and epigenetic modifiers (DNMTs and HDACs) in human foetal endothelium exposed to gestational and type 2 diabetes. Epigenetics 18, 2201714 (2023).
Apelqvist, A. et al. Notch signalling controls pancreatic cell differentiation. Nature 400, 877–881 (1999).
Ahlgren, U. et al. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 12, 1763–1768 (1998).
Stoffers, D. A., Ferrer, J., Clarke, W. L. & Habener, J. F. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17, 138–139 (1997).
Gray, J. D., Kogan, J. F., Marrocco, J. & McEwen, B. S. Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nat. Rev. Endocrinol. 13, 661–673 (2017).
Murgatroyd, C. & Spengler, D. Polycomb binding precedes early-life stress responsive DNA methylation at the Avp enhancer. PLoS One 9, e90277 (2014).
Murgatroyd, C. et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12, 1559–1566 (2009).
Dewanjee, S. et al. Molecular mechanism of diabetic neuropathy and its pharmacotherapeutic targets. Eur. J. Pharmacol. 833, 472–523 (2018).
Singh, R., Kishore, L. & Kaur, N. Diabetic peripheral neuropathy: current perspective and future directions. Pharm. Res. 80, 21–35 (2014).
Zhang, H. H. et al. The association between genomic DNA methylation and diabetic peripheral neuropathy in patients with type 2 diabetes mellitus. J. Diabetes Res. 2019, 2494057 (2019).
Román-Pintos, L. M. et al. Diabetic polyneuropathy in type 2 diabetes mellitus: inflammation, oxidative stress, and mitochondrial function. J. Diabetes Res. 2016, 3425617 (2016).
Ollikainen, M. et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenetics 7, 39 (2015).
Guo, K. et al. Genome-wide profiling of DNA methylation and gene expression identifies candidate genes for human diabetic neuropathy. Clin. Epigenetics 12, 123 (2020).
Guo, K. et al. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics 14, 766–779 (2019).
Feldman, E. L., Nave, K. A., Jensen, T. S. & Bennett, D. L. H. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron 93, 1296–1313 (2017).
Zhang, H. H. et al. Promoted interaction of nuclear factor-κB with demethylated cystathionine-β-synthetase gene contributes to gastric hypersensitivity in diabetic rats. J. Neurosci. 33, 9028–9038 (2013).
Zhang, H. H. et al. Promoted interaction of nuclear factor-κB with demethylated purinergic P2X3 receptor gene contributes to neuropathic pain in rats with diabetes. Diabetes 64, 4272–4284 (2015).
Liu, Y. P., Shao, S. J. & Guo, H. D. Schwann cells apoptosis is induced by high glucose in diabetic peripheral neuropathy. Life Sci. 248, 117459 (2020).
Thielen, L. A. et al. Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab. 32, 353–365.e358 (2020).
Hackett, A. R., Strickland, A. & Milbrandt, J. Disrupting insulin signaling in Schwann cells impairs myelination and induces a sensory neuropathy. Glia 68, 963–978 (2020).
Chen, S. et al. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience 256, 137–146 (2014).
Cheng, D., Yang, S., Zhao, X. & Wang, G. The role of glucagon-like peptide-1 receptor agonists (GLP-1 RA) in diabetes-related neurodegenerative diseases. Drug Des. Dev. Ther. 16, 665–684 (2022).
Chen, S. et al. Evidence of metabolic memory-induced neurodegeneration and the therapeutic effects of glucagon-like peptide-1 receptor agonists via Forkhead box class O. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 371–377 (2019).
Chen, C. J. et al. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem. Biophys. Res. Commun. 378, 389–393 (2009).
Liang, R. et al. Evidence for AKT-independent regulation of FOXO1 and FOXO3 in haematopoietic stem and progenitor cells. Cell Cycle 15, 861–867 (2016).
Liu, W. et al. miR-9 mediates CALHM1-activated ATP-P2X7R signal in painful diabetic neuropathy rats. Mol. Neurobiol. 54, 922–929 (2017).
Amin, K. N. et al. miR-23c regulates wound healing by targeting stromal cell-derived factor-1α (SDF-1α/CXCL12) among patients with diabetic foot ulcer. Microvasc. Res. 127, 103924 (2020).
Wang, Y. et al. microRNA-182 mediates Sirt1-induced diabetic corneal nerve regeneration. Diabetes 65, 2020–2031 (2016).
Wang, L. et al. The role of miR-146a in dorsal root ganglia neurons of experimental diabetic peripheral neuropathy. Neuroscience 259, 155–163 (2014).
Feng, Y. et al. Involvement of microRNA-146a in diabetic peripheral neuropathy through the regulation of inflammation. Drug Des. Devel Ther. 12, 171–177 (2018).
Su, S. et al. MiR-30b attenuates neuropathic pain by regulating voltage-gated sodium channel Nav1.3 in rats. Front. Mol. Neurosci. 10, 126 (2017).
Yan, X. T. et al. MicroRNA-93 alleviates neuropathic pain through targeting signal transducer and activator of transcription 3. Int. Immunopharmacol. 46, 156–162 (2017).
Coolen, M., Katz, S. & Bally-Cuif, L. miR-9: a versatile regulator of neurogenesis. Front. Cell Neurosci. 7, 220 (2013).
Ma, Z. et al. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl Acad. Sci. USA 109, E1963–E1971 (2012).
Anderson, C. M. & Nedergaard, M. Emerging challenges of assigning P2X7 receptor function and immunoreactivity in neurons. Trends Neurosci. 29, 257–262 (2006).
Yu, W. et al. LncRNA NONRATT021972 was associated with neuropathic pain scoring in patients with type 2 diabetes. Behav. Neurol. 2017, 2941297 (2017).
Wang, S. et al. LncRNA uc.48+ is involved in diabetic neuropathic pain mediated by the P2X3 receptor in the dorsal root ganglia. Purinergic Signal. 12, 139–148 (2016).
Liu, C. et al. Long non-coding RNA BC168687 is Involved in TRPV1-mediated diabetic neuropathic pain in rats. Neuroscience 374, 214–222 (2018).
Li, Z. et al. Emerging roles of long non-coding RNAs in neuropathic pain. Cell Prolif. 52, e12528 (2019).
Xu, H. et al. LncRNA NONRATT021972 siRNA attenuates P2X7 receptor expression and inflammatory cytokine production induced by combined high glucose and free fatty acids in PC12 cells. Purinergic Signal. 12, 259–268 (2016).
Wu, B. et al. LncRNA uc.48+ siRNA improved diabetic sympathetic neuropathy in type 2 diabetic rats mediated by P2X7 receptor in SCG. Auton. Neurosci. 197, 14–18 (2016).
Peng, H. et al. lncRNA NONRATT021972 siRNA decreases diabetic neuropathic pain mediated by the P2X(3) receptor in dorsal root ganglia. Mol. Neurobiol. 54, 511–523 (2017).
Chen, R. et al. Morphological and pathological characteristics of brain in diabetic encephalopathy. J. Alzheimers Dis. 65, 15–28 (2018).
Sima, A. A. Encephalopathies: the emerging diabetic complications. Acta Diabetol. 47, 279–293 (2010).
Jankovic, M. et al. Genetic and epigenomic modifiers of diabetic neuropathy. Int. J. Mol. Sci. 22, 4887 (2021).
Raj, S., Dsouza, L. A., Singh, S. P. & Kanwal, A. Sirt6 deacetylase: a potential key regulator in the prevention of obesity, diabetes and neurodegenerative disease. Front. Pharmacol. 11, 598326 (2020).
Kakoty, V. et al. Brain insulin resistance linked Alzheimer’s and Parkinson’s disease pathology: an undying implication of epigenetic and autophagy modulation. Inflammopharmacology 31, 699–716 (2023).
Kandimalla, R., Thirumala, V. & Reddy, P. H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1078–1089 (2017).
Mielcarek, M. et al. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11, e1001717 (2013).
Xu, Y. et al. Radix polygoni multiflori protects against hippocampal neuronal apoptosis in diabetic encephalopathy by inhibiting the HDAC4/JNK pathway. Biomed. Pharmacother. 153, 113427 (2022).
Jiang, L. et al. Advanced glycation end products induce Aβ(1-42) deposition and cognitive decline through H19/miR-15b/BACE1 axis in diabetic encephalopathy. Brain Res. Bull. 188, 187–196 (2022).
Zhang, R. et al. Advanced glycosylation end products induced synaptic deficits and cognitive decline through ROS-JNK-p53/miR-34c/SYT1 axis in diabetic encephalopathy. J. Alzheimers Dis. 87, 843–861 (2022).
Tu, Y. et al. MiR-702-5p ameliorates diabetic encephalopathy in db/db mice by regulating 12/15-LOX. Exp. Neurol. 358, 114212 (2022).
Li, J., Xu, S., Wang, L. & Wang, X. PHPB attenuated cognitive impairment in type 2 diabetic KK-Ay mice by modulating SIRT1/insulin signaling pathway and inhibiting generation of AGEs. Pharmceuticals 16, 305 (2023).
Shi, L. et al. Decreased miR-132 plays a crucial role in diabetic encephalopathy by regulating the GSK-3β/Tau pathway. Aging (Albany NY). 13, 4590–4604 (2020).
Liu, J. et al. The circRNA circ-Nbea participates in regulating diabetic encephalopathy. Brain Res. 1774, 147702 (2022).
Chen, J. et al. MicroRNA-128-3p impaired water maze learning by suppressing Doublecortin expression in both wild type and Aβ-42 infused mice. Neurosci. Lett. 626, 79–85 (2016).
Wang, X. Y., Zhang, X. Z., Li, F. & Ji, Q. R. MiR-128-3p accelerates cardiovascular calcification and insulin resistance through ISL1-dependent Wnt pathway in type 2 diabetes mellitus rats. J. Cell Physiol. 234, 4997–5010 (2019).
Kanwar, Y. S. et al. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 6, 395–423 (2011).
Qiu, C. et al. Cytosine methylation predicts renal function decline in American Indians. Kidney Int. 93, 1417–1431 (2018).
Shah, A. et al. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J. Biol. Chem. 288, 6835–6848 (2013).
Shah, A. et al. Thioredoxin-interacting protein deficiency protects against diabetic nephropathy. J. Am. Soc. Nephrol. 26, 2963–2977 (2015).
Bechtel, W. et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 16, 544–550 (2010).
Tampe, B. et al. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. 25, 905–912 (2014).
Reddy, M. A. et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 85, 362–373 (2014).
Sun, G. D., Cui, W. P., Guo, Q. Y. & Miao, L. N. Histone lysine methylation in diabetic nephropathy. J. Diabetes Res. 2014, 654148 (2014).
Miao, F., Gonzalo, I. G., Lanting, L. & Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 279, 18091–18097 (2004).
Miao, F. et al. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 282, 13854–13863 (2007).
Miao, F. et al. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 57, 3189–3198 (2008).
Li, Y. et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 283, 26771–26781 (2008).
Di Pietrantonio, N. et al. Diabetes and its cardiovascular complications: potential role of the acetyltransferase p300. Cells 12, 431 (2023).
Ghosh, A. K. & Varga, J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell Physiol. 213, 663–671 (2007).
Yuan, H. et al. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol. Ren. Physiol. 304, F601–F613 (2013).
Nie, L., Liu, Y., Zhang, B. & Zhao, J. Application of histone deacetylase inhibitors in renal interstitial fibrosis. Kidney Dis. (Basel). 6, 226–235 (2020).
Mladenov, M. et al. Efficacy of the monocarbonyl curcumin analog C66 in the reduction of diabetes-associated cardiovascular and kidney complications. Mol. Med. 28, 129 (2022).
Kato, M. et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl Acad. Sci. USA 104, 3432–3437 (2007).
Putta, S. et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 23, 458–469 (2012).
Kato, M. et al. TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal. 6, ra43 (2013).
Deshpande, S. D. et al. Transforming growth factor-β-induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes 62, 3151–3162 (2013).
Chung, A. C., Huang, X. R., Meng, X. & Lan, H. Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 21, 1317–1325 (2010).
Kölling, M. et al. Therapeutic miR-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther. 25, 165–180 (2017).
Xue, M. et al. High glucose up-regulates microRNA-34a-5p to aggravate fibrosis by targeting SIRT1 in HK-2 cells. Biochem. Biophys. Res. Commun. 498, 38–44 (2018).
Sun, J., Li, Z. P., Zhang, R. Q. & Zhang, H. M. Repression of miR-217 protects against high glucose-induced podocyte injury and insulin resistance by restoring PTEN-mediated autophagy pathway. Biochem. Biophys. Res. Commun. 483, 318–324 (2017).
Leung, A., Amaram, V. & Natarajan, R. Linking diabetic vascular complications with LncRNAs. Vasc. Pharmacol. 114, 139–144 (2019).
Alvarez, M. L. & DiStefano, J. K. Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS ONE 6, e18671 (2011).
Alvarez, M. L. et al. Correction: role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: implications for diabetic nephropathy. PLoS ONE 11, e0168353 (2016).
Wu, F. et al. Regulation mechanism and pathogenic role of lncRNA plasmacytoma variant translocation 1 (PVT1) in human diseases. Genes Dis. 10, 901–914 (2023).
Bhadsavle, S. S. & Golding, M. C. Paternal epigenetic influences on placental health and their impacts on offspring development and disease. Front. Genet. 13, 1068408 (2022).
Talib, W. H. et al. Diabetes and cancer: metabolic association, therapeutic challenges, and the role of natural products. Molecules 26, 2179 (2021).
Giri, B. et al. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: an update on glucose toxicity. Biomed. Pharmacother. 107, 306–328 (2018).
Lee, C., An, D. & Park, J. Hyperglycemic memory in metabolism and cancer. Horm. Mol. Biol. Clin. Investig. 26, 77–85 (2016).
Gupta, C., Kaur, J. & Tikoo, K. Regulation of MDA-MB-231 cell proliferation by GSK-3β involves epigenetic modifications under high glucose conditions. Exp. Cell Res. 324, 75–83 (2014).
Park, J. et al. Neuregulin 1-HER axis as a key mediator of hyperglycemic memory effects in breast cancer. Proc. Natl Acad. Sci. USA 109, 21058–21063 (2012).
Lee, C. et al. Epigenetic regulation of Neuregulin 1 promotes breast cancer progression associated to hyperglycemia. Nat. Commun. 14, 439 (2023).
Dong, S., Wang, Z., Shen, K. & Chen, X. Metabolic syndrome and breast cancer: prevalence, treatment response, and prognosis. Front Oncol. 11, 629666 (2021).
Radic Shechter, K. et al. Metabolic memory underlying minimal residual disease in breast cancer. Mol. Syst. Biol. 17, e10141 (2021).
Pan, B. et al. HDL of patients with type 2 diabetes mellitus elevates the capability of promoting breast cancer metastasis. Clin. Cancer Res. 18, 1246–1256 (2012).
Chang, T. Y. et al. Nε-(1-Carboxymethyl)-L-lysine, an advanced glycation end product, exerts malignancy on chondrosarcoma via the activation of cancer stemness. Arch. Toxicol. 97, 2231–2244 (2023).
Fontes-Carvalho, R. et al. Diastolic dysfunction in the diabetic continuum: association with insulin resistance, metabolic syndrome and type 2 diabetes. Cardiovasc. Diabetol. 14, 4 (2015).
Cai, C. et al. DNA methylation in diabetic retinopathy: pathogenetic role and potential therapeutic targets. Cell Biosci. 12, 186 (2022).
Willmer, T., Johnson, R., Louw, J. & Pheiffer, C. Corrigendum: blood-based DNA methylation biomarkers for type 2 diabetes: potential for clinical applications. Front. Endocrinol. 10, 1 (2019).
Baek, S. J. et al. Genome-wide DNA methylation profiling reveals candidate biomarkers and probable molecular mechanism of metabolic syndrome. Genes Dis. 9, 833–836 (2022).
Aran, D., Toperoff, G., Rosenberg, M. & Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 20, 670–680 (2011).
Toperoff, G. et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet. 21, 371–383 (2012).
Toperoff, G. et al. Premature aging of leukocyte DNA methylation is associated with type 2 diabetes prevalence. Clin. Epigenetics 7, 35 (2015).
Chambers, J. C. et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol. 3, 526–534 (2015).
Qie, R. et al. Association of ABCG1 gene methylation and its dynamic change status with incident type 2 diabetes mellitus: the Rural Chinese Cohort Study. J. Hum. Genet. 66, 347–357 (2021).
Duraisamy, A. J. et al. Epigenetic modifications in peripheral blood as potential noninvasive biomarker of diabetic retinopathy. Transl. Vis. Sci. Technol. 8, 43 (2019).
Constâncio, V., Nunes, S. P., Henrique, R. & Jerónimo, C. DNA methylation-based testing in liquid biopsies as detection and prognostic biomarkers for the four major cancer types. Cells 9, 624 (2020).
Kahn, S. E. et al. The β cell in diabetes: integrating biomarkers with functional measures. Endocr. Rev. 42, 528–583 (2021).
Kuroda, A. et al. Insulin gene expression is regulated by DNA methylation. PLoS ONE 4, e6953 (2009).
Akirav, E. M. et al. Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc. Natl Acad. Sci. USA 108, 19018–19023 (2011).
Husseiny, M. I. et al. Development of a quantitative methylation-specific polymerase chain reaction method for monitoring beta cell death in type 1 diabetes. PLoS ONE 7, e47942 (2012).
Fisher, M. M. et al. Detection of islet β-cell death in vivo by multiplex PCR analysis of differentially methylated DNA. Endocrinology 154, 3476–3481 (2013).
Lehmann-Werman, R. et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl Acad. Sci. USA 113, E1826–E1834 (2016).
Lebastchi, J. et al. Immune therapy and β-cell death in type 1 diabetes. Diabetes 62, 1676–1680 (2013).
Usmani-Brown, S. et al. Analysis of β-cell death in type 1 diabetes by droplet digital PCR. Endocrinology 155, 3694–3698 (2014).
Zhang, K. et al. Circulating unmethylated insulin DNA as a potential non-invasive biomarker of beta cell death in type 1 Diabetes: a review and future prospect. Clin. Epigenetics 9, 44 (2017).
Bellin, M. D. et al. Unmethylated insulin DNA is elevated after total pancreatectomy with islet autotransplantation: assessment of a novel beta cell marker. Am. J. Transplant. 17, 1112–1118 (2017).
Ma, J. et al. A peripheral blood DNA methylation signature of hepatic fat reveals a potential causal pathway for nonalcoholic fatty liver disease. Diabetes 68, 1073–1083 (2019).
Loomba, R. et al. DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3, e96685 (2018).
Angelescu, M. A. et al. miRNAs as biomarkers in diabetes: moving towards precision medicine. Int. J. Mol. Sci. 23, 12843 (2022).
Dandare, A., Khan, M. J., Naeem, A. & Liaquat, A. Clinical relevance of circulating non-coding RNAs in metabolic diseases: Emphasis on obesity, diabetes, cardiovascular diseases and metabolic syndrome. Genes Dis. 10, 2393–2413 (2023).
Jiménez-Lucena, R. et al. A plasma circulating miRNAs profile predicts type 2 diabetes mellitus and prediabetes: from the CORDIOPREV study. Exp. Mol. Med. 50, 1–12 (2018).
Lo, K. A. et al. Adipocyte long-noncoding RNA transcriptome analysis of obese mice identified Lnc-Leptin, which regulates leptin. Diabetes 67, 1045–1056 (2018).
Mahmoud, A. M. An overview of epigenetics in obesity: the role of lifestyle and therapeutic interventions. Int. J. Mol. Sci. 23, 1341 (2022).
Barrès, R. & Zierath, J. R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 12, 441–451 (2016).
Hibler, E., Huang, L., Andrade, J. & Spring, B. Impact of a diet and activity health promotion intervention on regional patterns of DNA methylation. Clin. Epigenetics 11, 133 (2019).
Li, S., Chen, M., Li, Y. & Tollefsbol, T. O. Prenatal epigenetics diets play protective roles against environmental pollution. Clin. Epigenetics 11, 82 (2019).
Silva, L. et al. Bioactive food compounds, epigenetics and chronic disease prevention: focus on early-life interventions with polyphenols. Food Res. Int. 125, 108646 (2019).
Castillo-Ordoñez, W. O., Cajas-Salazar, N. & Velasco-Reyes, M. A. Genetic and epigenetic targets of natural dietary compounds as anti-Alzheimer’s agents. Neural Regen. Res. 19, 846–854 (2024).
Hawley, J. A., Hargreaves, M., Joyner, M. J. & Zierath, J. R. Integrative biology of exercise. Cell 159, 738–749 (2014).
Dos Santos, J. M., Moreli, M. L., Tewari, S. & Benite-Ribeiro, S. A. The effect of exercise on skeletal muscle glucose uptake in type 2 diabetes: an epigenetic perspective. Metabolism 64, 1619–1628 (2015).
Barrès, R. et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 15, 405–411 (2012).
Santos, J. M., Tewari, S. & Benite-Ribeiro, S. A. The effect of exercise on epigenetic modifications of PGC1: the impact on type 2 diabetes. Med. Hypotheses 82, 748–753 (2014).
Rönn, T. et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 9, e1003572 (2013).
Rowlands, D. S. et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in Type 2 diabetic obesity. Physiol. Genomics. 46, 747–765 (2014).
Improta-Caria, A. C. et al. MicroRNAs in type 2 diabetes mellitus: potential role of physical exercise. Rev. Cardiovasc. Med. 23, 29 (2022).
Silveira, A. et al. MicroRNAs in obesity-associated disorders: the role of exercise training. Obes. Facts 15, 105–117 (2022).
Dos Santos, J. A. C. et al. Physical exercise and the functions of microRNAs. Life Sci. 304, 120723 (2022).
Olioso, D. et al. Effects of aerobic and resistance training on circulating micro-RNA expression profile in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 104, 1119–1130 (2019).
Karolina, D. S. et al. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 6, e22839 (2011).
Souza, R. W. et al. Regulation of cardiac microRNAs induced by aerobic exercise training during heart failure. Am. J. Physiol. Heart Circ. Physiol. 309, H1629–H1641 (2015).
Fachim, H. A. et al. Circulating microRNA changes in patients with impaired glucose regulation. Adipocyte 9, 443–453 (2020).
Sun, L. L. et al. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res. Clin. Pract. 91, 94–100 (2011).
Wang, Z. et al. Hepatic miR-192-3p reactivation alleviates steatosis by targeting glucocorticoid receptor. JHEP Rep. 2, 100179 (2020).
Radom-Aizik, S. et al. Effects of exercise on microRNA expression in young males peripheral blood mononuclear cells. Clin. Transl. Sci. 5, 32–38 (2012).
Párrizas, M. et al. Circulating miR-192 and miR-193b are markers of prediabetes and are modulated by an exercise intervention. J. Clin. Endocrinol. Metab. 100, E407–E415 (2015).
Kiyama, R. Nutritional implications of ginger: chemistry, biological activities and signaling pathways. J. Nutr. Biochem. 86, 108486 (2020).
Maradana, M. R., Thomas, R. & O’Sullivan, B. J. Targeted delivery of curcumin for treating type 2 diabetes. Mol. Nutr. Food Res. 57, 1550–1556 (2013).
Chen, S. et al. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 298, E127–E137 (2010).
Kadiyala, C. S. et al. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J. Biol. Chem. 287, 25869–25880 (2012).
Yin, Y. et al. Astragaloside IV alleviates Schwann cell injury in diabetic peripheral neuropathy by regulating microRNA-155-mediated autophagy. Phytomedicine 92, 153749 (2021).
Qin, S. et al. Could cyclosiversioside F serve as a dietary supplement to prevent obesity and relevant disorders? Int. J. Mol. Sci. 24, 13762 (2023).
Gilbert, E. R. & Liu, D. Anti-diabetic functions of soy isoflavone genistein: mechanisms underlying its effects on pancreatic β-cell function. Food Funct. 4, 200–212 (2013).
Yang, S. C. et al. Bioactive agent discovery from the natural compounds for the treatment of type 2 diabetes rat model. Molecules 25, 5713 (2020).
Naz, R. et al. Food polyphenols and type II diabetes mellitus: pharmacology and mechanisms. Molecules 28, 3996 (2023).
Den Hartogh, D. J. & Tsiani, E. Antidiabetic properties of naringenin: a citrus fruit polyphenol. Biomolecules 9, 99 (2019).
Wang, S. W. et al. Inhibition of histone acetyltransferase by naringenin and hesperetin suppresses Txnip expression and protects pancreatic β cells in diabetic mice. Phytomedicine 88, 153454 (2021).
Gómez-Martínez, S. et al. Moringa oleifera leaf supplementation as a glycemic control strategy in subjects with prediabetes. Nutrients 14, 57 (2021).
Yang, J. et al. Magnolol effectively ameliorates diabetic peripheral neuropathy in mice. Phytomedicine 107, 154434 (2022).
Fernandes, G. F. S. et al. Epigenetic regulatory mechanisms induced by resveratrol. Nutrients 9, 1201 (2017).
Karaman Mayack, B., Sippl, W. & Ntie-Kang, F. Natural products as modulators of sirtuins. Molecules 25, 3287 (2020).
Novelle, M. G. et al. Resveratrol supplementation: Where are we now and where should we go? Ageing Res. Rev. 21, 1–15 (2015).
Liu, K., Zhou, R., Wang, B. & Mi, M. T. Effect of resveratrol on glucose control and insulin sensitivity: a meta-analysis of 11 randomized controlled trials. Am. J. Clin. Nutr. 99, 1510–1519 (2014).
Costantino, S., Paneni, F. & Cosentino, F. Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr. Pharm. Biotechnol. 16, 531–543 (2015).
Arguelles, A. O., Meruvu, S., Bowman, J. D. & Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov. Today 21, 499–509 (2016).
Altucci, L. & Rots, M. G. Epigenetic drugs: from chemistry via biology to medicine and back. Clin. Epigenetics 8, 56 (2016).
Wang, X. et al. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 1, e87748 (2016).
Khan, S. & Jena, G. Valproic acid improves glucose homeostasis by increasing beta-cell proliferation, function, and reducing its apoptosis through hdac inhibition in juvenile diabetic rat. J. Biochem. Mol. Toxicol. 30, 438–446 (2016).
Dudakovic, A. et al. Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. J. Biol. Chem. 288, 28783–28791 (2013).
Bayoumi, A., Grønbæk, H., George, J. & Eslam, M. The epigenetic drug discovery landscape for metabolic-associated fatty liver disease. Trends Genet. 36, 429–441 (2020).
Navada, S. C., Steinmann, J., Lübbert, M. & Silverman, L. R. Clinical development of demethylating agents in hematology. J. Clin. Invest. 124, 40–46 (2014).
Ueda, J., Yamazaki, T. & Funakoshi, H. Toward the development of epigenome editing-based therapeutics: potentials and challenges. Int. J. Mol. Sci. 24, 4778 (2023).
Negi, S., Rutman, A. K. & Paraskevas, S. Extracellular vesicles in type 1 diabetes: messengers and regulators. Curr. Diabetes Rep. 19, 69 (2019).
Shi, J. et al. Exosomes from miR-20b-3p-overexpressing stromal cells ameliorate calcium oxalate deposition in rat kidney. J. Cell Mol. Med. 23, 7268–7278 (2019).
Wang, S. et al. Elevated microRNA-20b-3p and reduced thioredoxin-interacting protein ameliorate diabetic retinopathy progression by suppressing the NLRP3 inflammasomes. IUBMB Life 72, 1433–1448 (2020).
Li, J. et al. Plasma exosomes improve peripheral neuropathy via miR-20b-3p/Stat3 in type I diabetic rats. J. Nanobiotechnol. 21, 447 (2023).
Baylin, S. B. & Ohm, J. E. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6, 107–116 (2006).
Zhang, T. et al. Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenetics 12, 72 (2020).
Marzochi, L. L. et al. Use of histone methyltransferase inhibitors in cancer treatment: a systematic review. Eur. J. Pharmacol. 944, 175590 (2023).
Jacobson, A. M. et al. Brain structure among middle-aged and older adults with long-standing type 1 diabetes in the DCCT/EDIC study. Diabetes Care 45, 1779–1787 (2022).
Boyko, E. J. et al. Risk of foot ulcer and lower-extremity amputation among participants in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 45, 357–364 (2022).
Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 39, 686–693, (2016).
Monnier, V. M. et al. Skin collagen advanced glycation endproducts (AGEs) and the long-term progression of sub-clinical cardiovascular disease in type 1 diabetes. Cardiovasc. Diabetol. 14, 118 (2015).
White, N. H. et al. Prolonged effect of intensive therapy on the risk of retinopathy complications in patients with type 1 diabetes mellitus: 10 years after the Diabetes Control and Complications Trial. Arch. Ophthalmol. 126, 1707–1715 (2008).
Ray, K. K. et al. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. J. Am. Med. Assoc. 323, 1565–1573 (2020).
Kalantar-Zadeh, K. et al. Effect of apabetalone on cardiovascular events in diabetes, CKD, and recent acute coronary syndrome: results from the BETonMACE randomized controlled trial. Clin. J. Am. Soc. Nephrol. 16, 705–716 (2021).
Cummings, J. et al. Cognitive effects of the BET protein inhibitor apabetalone: a prespecified montreal cognitive assessment analysis nested in the BETonMACE randomized controlled trial. J. Alzheimers Dis. 83, 1703–1715 (2021).
Bo, S. et al. Impact of sirtuin-1 expression on H3K56 acetylation and oxidative stress: a double-blind randomized controlled trial with resveratrol supplementation. Acta Diabetol. 55, 331–340 (2018).
Arpón, A. et al. Adherence to Mediterranean diet is associated with methylation changes in inflammation-related genes in peripheral blood cells. J. Physiol. Biochem. 73, 445–455 (2016).
Arpón, A. et al. Impact of consuming extra-virgin olive oil or nuts within a mediterranean diet on dna methylation in peripheral white blood cells within the predimed-navarra randomized controlled trial: a role for dietary lipids. Nutrients 10, 15 (2017).
Sun, D. et al. Genetic, epigenetic and transcriptional variations at NFATC2IP locus with weight loss in response to diet interventions: The POUNDS Lost Trial. Diabetes Obes. Metab. 20, 2298–2303 (2018).
Chen, S. et al. The clinical significance of long non-coding RNA ANRIL level in diabetic retinopathy. Acta Diabetol. 57, 409–418 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81870779 to Qi Wang), the Department of Science and Technology of Sichuan Province (2021JDRC0039 to Qi Wang), and the Chengdu Science and Technology Program (2022-YF05-01760-SN to Qi Wang).
Author information
Authors and Affiliations
Contributions
Conceptualization, basic structure and paper revision: Qi Wang; Literature review and paper draft: Hao Dong, Yuezhang Sun, Lulingxiao Nie; Figure design and draft: Qi Wang, Hao Dong, Aimin Cui; Clinical trials summary: Hao Dong, Aimin Cui, Pengfei Zhao; Manuscript revise: Pengfei Zhao, Wai Keung Leung; Funding acquisition: Qi Wang. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, H., Sun, Y., Nie, L. et al. Metabolic memory: mechanisms and diseases. Sig Transduct Target Ther 9, 38 (2024). https://doi.org/10.1038/s41392-024-01755-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01755-x
