
Abstract
Deciphering the effects of historical and recent demographic processes responsible for the spatial patterns of genetic diversity and structure is a key objective in evolutionary and conservation biology. Using population genetic analyses, we investigated the demographic history, the contemporary genetic diversity and structure, and the occurrence of hybridization and introgression of two species of anadromous fish with contrasting life history strategies and which have undergone recent demographic declines, the allis shad (Alosa alosa) and the twaite shad (Alosa fallax). We genotyped 706 individuals from 20 rivers and 5 sites at sea in Southern Europe at thirteen microsatellite markers. Genetic structure between populations was lower for the nearly semelparous species A. alosa, which disperses greater distances compared to the iteroparous species, A. fallax. Individuals caught at sea were assigned at the river level for A. fallax and at the region level for A. alosa. Using an approximate Bayesian computation framework, we inferred that the most likely long term historical divergence scenario between both species and lineages involved historical separation followed by secondary contact accompanied by strong population size decline. Accordingly, we found evidence for contemporary hybridization and bidirectional introgression due to gene flow between both species and lineages. Moreover, our results support the existence of at least one distinct species in the Mediterrannean sea: A. agone in Golfe du Lion area, and another divergent lineage in Corsica. Overall, our results shed light on the interplay between historical and recent demographic processes and life history strategies in shaping population genetic diversity and structure of closely related species. The recent demographic decline of these species’ populations and their hybridization should be carefully considered while implementing conservation programs.
Similar content being viewed by others

Introduction
Reconstructing how historical and recent processes shape present genetic diversity is an important step in evolutionary biology. In particular, long-term processes such as vicariance events, long term contraction during the last ice age or postglacial recolonization are well known to shape current patterns of genetic diversity (Hewitt 1996). In addition, several species and/or populations are currently declining at a fast pace due to human activity or climate change (Ceballos et al. 2020; Ryan et al. 2018). The interplay of these processes may leave counter-intuitive signatures on contemporary patterns of genetic diversity, making it difficult to decorrelate them and quantify their respective roles. In addition, populations within species or closely related species may differ in terms of life history traits, which can result in contrasting levels of population genetic structure and diversity.
In particular, the movement of species or dispersal processes are key factors affecting genetic structure through space and time (Cayuela et al. 2018). Many migratory species have undergone sharp declines in population sizes worldwide (Wilcove and Wikelski 2008; Limburg and Waldman 2009), which can have evolutionary consequences for these species. Indeed, small populations can display higher rates of genetic drift, accumulation of deleterious alleles and loss of genetic variability, which can ultimately threaten their genetic integrity and adaptive potential (Frankham 2005). Therefore, quantifying a species’ evolutionary potential is vital for the conservation of wild and managed populations. This task can benefit from reconstruction of demographic history from genetic data (Rougemont et al. 2020), monitoring of population genetic parameters including genetic diversity, effective population size (Ne), and quantifying hybridization between closely related groups or species (Barton and Hewitt 1985) as well as their positive (Abbott et al. 2013) or negative consequences on hybrid fitness (Mikkelsen and Irwin 2021).
Diadromous fish are keystone species that move between freshwater and marine environments. Many of these species, including salmonids, eels, sturgeons or shads, have undergone steep declines in their population size due to human activities such as dams building, over-harvesting, habitat degradation, and pollution (Parrish et al. 1998; Waters et al. 2000; Limburg and Waldman 2009). Some of them (e.g., salmonids and shads) tend to return to their natal river for breeding (i.e “homing” behavior), as demonstrated by otolith analysis (Tomás et al. 2005; Walther and Thorrold 2008; Perrier et al. 2011; Martin et al. 2015; Randon et al. 2017). This homing behavior fosters local adaptation, which is beneficial in stable environmental conditions (Keefer and Caudill 2014), but can also isolate some populations and result in smaller local effective population sizes. Consequently, in the context of environmental changes, this strategy may lead to less stable populations as compared to those displaying higher levels of gene flow and effective population size (Frankham 1997, 2005), although this question has given rise to recent debates (e.g. Teixeira and Huber 2021; García-Dorado and Caballero 2021).
The allis shad, Alosa alosa (Limnaeus 1758), and the twaite shad, Alosa fallax (Lacépède 1803), are two closely related anadromous Clupeidae species that have undergone steep declines in abundance and distribution range (Baglinière et al. 2003a). Since the middle of the 20th century, declines for both species have mostly been attributed to freshwater habitat degradation inducing loss of spawning grounds and over-harvesting (Aprahamian et al. 2003; Limburg and Waldman 2009). Both species are now mostly restricted to large rivers in Portugal, Spain, France and the United Kingdom (mainly A. fallax). Presently, this decline appears more substantial for A. alosa; according to the last assessment of IUCN status for France, A. alosa is now considered as critically endangered (Baglinière et al. 2020a), while A. fallax is considered as vulnerable (Baglinière et al. 2020b). It is expected that this decrease in abundance has had important repercussions on the genetic diversity of both species. Importantly, a decrease in census population size will reduce effective population size and decrease selection efficacy, but the effects on either species may differ given differences in life history strategies despite their close phylogenetic relationship (Faria et al. 2012). For example, A. alosa is nearly-semelparous while A. fallax is iteroparous (Mennesson-Boisneau et al. 2000a) and A. alosa displays a stronger dispersal behavior than A. fallax (Tavernie and Elie 2001; Jolly et al. 2012; Martin et al. 2015; Nachón et al. 2020). Following the study of Hasselman et al. (2013) we predict that iteroparity sets a constraint on A. fallax dispersal and migratory distance due to stronger selection for homing every year for breeding (Jolly et al. 2012). In contrast, longer migratory distance may facilitate gene flow in the semelparous A. alosa (Martin et al. 2015; Randon et al. 2017).
Because of these differences in dispersal and parity mode, we expect stronger gene flow and weaker population genetic structure in A. alosa compared to A. fallax. The genetic distinctiveness of A. alosa and A. fallax has been confirmed (Faria et al. 2004; Alexandrino et al. 2006; Jolly et al. 2012), but the details of their demographic history of prior isolation and subsequent contact has not been fully resolved (Faria et al. 2012; Taillebois et al. 2020). This issue could benefit from demographic modeling based on genetic data. Such an approach would help to decipher whether these species evolved separately before a secondary contact, or whether they could have evolved in sympatry, which has important implications for understanding speciation processes, as well as for the conservation of the species. This might especially influence the rate at which hybridization could result in a generalized meltdown of both species into one. In fact, hybrids between both species have been documented in some geographic areas like Portugal, United Kingdom and France (Alexandrino et al. 2006; Coscia et al. 2010; Jolly et al. 2011; Faria et al. 2012; Taillebois et al. 2020), suggesting that barriers to gene flow between species are permeable. In the context of river fragmentation, barriers (e.g. dams) generate “forced” common spawning grounds between A. alosa and A. fallax (Alexandrino et al. 2006), which would otherwise be spatially segregated along the river network (Mennesson-Boisneau et al. 2000a). It is therefore expected that both species hybridize frequently in rivers with high levels of fragmentation (see also Taillebois et al. 2020). In addition, the taxonomic status of A. fallax within the Mediterranean Sea is debated (Chiesa et al. 2014; Baglinière et al. 2020c), despite genetic and morphological support for the existence of two lineages (Le Corre et al. 2005; Bianco 2005). Analysis of a broader geographic sample such as that described herein may help to resolve this taxonomic conundrum.
In this study we used 13 microsatellite markers (Rougemont et al. 2015) to (i) determine the extent of genetic diversity and structure among Alosa alosa and Alosa fallax along the Atlantic and Mediterranean coasts, (ii) determine the historical process of divergence between species using approximate Bayesian computations, (iii) contrast the patterns of long-term historical gene flow between species and among major genetic groups versus contemporary dispersal at sea in each species, and (iv) test the power of this marker set to assign individuals captured at sea. In doing so, we tested the hypotheses that (i) both species were formerly isolated and came into secondary contact, (ii) both species differ in their genetic structure, with the more dispersive, semelparous species showing weaker genetic structure than the iteroparous species, (iii) hybrids are frequently found, potentially as a result of frequent “forced” common spawning grounds and (iv) individuals caught at sea can be confidently assigned to rivers or geographic region, depending on the species.
Materials and methods
Study area and sampling
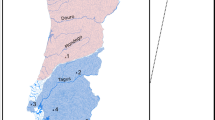
A total of 706 individuals from both species (367 A. alosa and 339 A. fallax) were sampled from 20 rivers distributed along the French Atlantic coast (N = 514), Spanish coast (N = 56 A. fallax), Mediterranean coast (N = 92 A. fallax) and Corsica (N = 65 A. fallax). A subset of these (40 A. alosa and 65 A. fallax) were captured at sea along the Atlantic coast and were also used for population assignment (Fig. 1, Tables 1 and S1). Individuals collected in rivers were captured between 2009 and 2012 either by nets or trapping and a fin clip was stored in 95% ethanol, while individuals captured at sea were collected by professional fishermen and identified phenotypically. Scales were also collected from carcasses after breeding migration and conserved in paper envelopes. Collections of scales (stored at INRAE Rennes, France) from cohorts 1997 to 1999 and 2001 (Aulne and Charente rivers) were also used (Table 1).

Sample site numbers correspond to those given in Table 1.
Molecular methods
Genomic DNA was extracted using a Chelex protocol (modified from Estoup et al. 1996). We genotyped each individual at 13 microsatellite loci specifically developed for A. alosa and A. fallax (Rougemont et al. 2015). Occurrence of null alleles and scoring errors due to large allelic dropout or stuttering were checked using Micro-Checker v2.2.3 (Oosterhout et al. 2004). Linkage disequilibrium (LD) was inspected using Genepop 4.0 (Rousset 2008).
Genetic diversity and HWE within populations
Fish captured at sea for which the river of origin was unknown and hybrids were excluded from genetic diversity and Hardy-Weinberg equilibrium (HWE) analyses, leaving a total of 586 fish (315 A. alosa and 271 A. fallax). For each population (sampled in each river) of each species, diversity indices (observed heterozygosity Ho and expected Heterozygosity He under HWE) were calculated with Genetix 4.0.5 (Belkhir et al. 1996). Significance of heterozygosity comparisons was assessed with a Wilcoxon sign rank test. Deviations from HWE and linkage disequilibrium were tested for each locus within each population and globally with Genepop 4.0 (Rousset 2008). Fstat version 2.9.3 (Goudet 1995) was used to calculate inbreeding coefficient (FIS), allele number, and allelic richness (Ar), with Ar differences between species tested using 4000 permutations. We tested the significance of the differences in genetic diversity among each river within species using a linear mixed model. First we tested the effect of the “River” factor considered as a fixed effect on levels of Allelic Richness considered as the response variable. We included the locus as a random effect to account for their variation and for their random representation of the genetic variability. An Anova was used to test the effect of the River and the amount of variance was quantified using the conditional and marginal coefficient of determination (R2c and R2m) respectively describing the variance accounted for by locus variability and by “River” alone. Finally differences among rivers were tested using TukeyHSD post-hoc comparisons on the models. Tests were implemented using the lme4 package (Bates et al. 2015), MuMIn (Barton 2020) and multComp (Hothorn et al. 2008).
Hybrid identification
We were interested in accurately distinguishing purebred individuals from hybrids. We used a similar framework to the one of Vähä and Primmer (2006) using Structure 2.3.3 (Pritchard et al. 2000) and NewHybrids 1.1 (Anderson and Thompson 2002). For the purpose of hybrid identification we assumed K = 2 where each cluster is composed of a single species contributing to the total gene pool of the sample. Structure was used without prior information about the species classification using an admixture model with (i) correlated allele frequency (Falush et al. 2003) and (ii) 500,000 burn-in steps followed by 1,000,000 MCMC iterations, replicated 10 times. Individual admixture proportions (q-values) and their 90% confidence intervals were averaged over the ten replicates and used to assign individuals to their respective genetic clusters. NewHybrids assumes that the sample is drawn from a mixture of pure individuals and hybrids (F1, F2 or backcrosses) so that the q-value inferred with this method is a discrete variable (Anderson and Thompson 2002). NewHybrids was used to calculate the posterior probability for an individual to belong to one of the following classes: purebred A. alosa, purebred A. fallax, hybrid F1, hybrid F2 or backcross. The q-values were summed over all hybrid categories (F1, F2 and backcross) because preliminary tests showed that performance was greater under these conditions. Hence, hybrids, regardless of their hybrid class, were distinguished from purebreds. Uniform priors were used for allele frequency and admixture estimations were performed with a burn in of 1,000,000 steps followed by 1,000,000 iterations. Tests using Jeffrey priors, instead of uniform, yielded similar results. Results are based on the average of 10 runs performed with random seed.
Performance of admixture analysis
To test the correctness of assignment of Structure and NewHybrids, the software HybridLab (Nielsen et al. 2006) was used. Individuals showing q-value >0.9 with both methods were chosen randomly and 3500 simulated genotypes of each parental species were generated and divided into 10 datasets of 350 individuals. The sample size was chosen to be close to our real dataset. Then 10 other datasets were created containing both parental and hybrid individuals (F1, F2, backcrosses). Ten individuals of each hybrid category were incorporated into the datasets of 350 individuals to represent the actual sample size of our empirical dataset and to incorporate a small proportion of hybrids. The simulated dataset was analyzed with Structure and Newhybrids to calculate (1) the hybrid proportion, which is the number of individuals classified as hybrid divided by the total number of individuals in the sample, (2) efficiency, (3) accuracy and (4) overall performance of these methods following the definition of Vähä and Primmer (2006). We used a q-value threshold of 0.90 for hybrid classification.
Population genetic structure
Genetic structure was assessed excluding hybrids (based on their genotype) and fish captured at sea. The extent of genetic structure was quantified using the pairwise FST estimator θST of (Weir and Cockerham 1984) between sample pairs with Fstat, and with significance assessed using 10,000 permutations. We tested for a signal of isolation by distance (IBD) in our data in both species separately using a mantel test on matrices of linearized FST using FST/(1–FST) against the logarithm of the waterway distances measured between river mouths manually in ArcGIS following the coastline. We next plotted the signal of isolation by distance (Fig. 3a) in each species separately with the ggplot2 package and tested the strength of the relationship (R2) and its significance using a simple linear model.
The chord distance (Cavalli-Sforza and Edwards 1967) was used to quantify genetic differentiation between sample pairs and to construct neighbor-joining phylograms (Saitou and Nei 1987) with MSA 4.05 (Dieringer and Schlötterer 2003). Trees were computed using the software Phylip 3.6 using a maximum likelihood optimality criterion (Felsenstein 1995) and 10,000 permutations were conducted to establish bootstrap support for the nodes. The results were then visualized using Tree view 1.6 (Page 1996). Individual genetic clustering was further investigated without a priori definition of population boundaries using Structure. The admixture and correlated allele frequency model was used to detect a number of genetic clusters (K) varying from 1 to 14 separately for A. alosa and A. fallax. Fifteen replicates were run for each K with a burn-in period of 400,000 followed by 400,000 MCMC iterations. The optimal number of clusters was evaluated using the likelihood distribution (Pritchard et al. 2000) and the ∆K method (Evanno et al. 2005). Results were combined with Structure harvester (Earl and vonHoldt 2012) and graphs were plotted in R using Pophelper (Francis 2017). Finally, we used a multivariate method, the Discriminant Analysis of Principal Components implemented in the Adegenet Package (Jombart 2008) in R (R Development Core Team 2015), to inspect structure between populations for each species. We used the function find.cluster, which uses k-means to find the optimal number of clusters and selected the number of groups with the lowest Bayesian information criteria (BIC). We also used the alpha score to choose the optimal number of principal components to retain.
Genetic stock assignment
Genetic assignment of fish captured at sea was performed using three different methods. First Structure was run with fish captured at sea included in the dataset. The same settings as described above were used. Second, DAPC assignments were performed with fish from the sea as supplementary individuals using the previously defined parameters. Third, we used the Bayesian method implemented in GeneClass 2 (Piry et al. 2004) specifically designed for assignment tests. The likelihood that any fish captured at sea came from one of the sampled populations was tested using the resampling algorithm described by Paetkau et al. (2004) with 100,000 simulated individuals. Fish that displayed a probability <0.01 were assumed to come from an un-sampled reference population and were excluded from the assignment tests.
Demographic history: approximate Bayesian computations
The demographic history of divergence between A. alosa and A. fallax, as well as between A. fallax from Atlantic vs A. fallax from the Mediterranean sea, was investigated using an ABC framework. To avoid any bias due to sparse sampling, intra population structure or isolation by distance (e.g. Mason et al. 2020), we focused our between species comparison on A. fallax and A. alosa sampled along the Atlantic coast. Our between lineage comparison included all individuals from the Rhone river and all individuals from Corsica.
We excluded all putative hybrids to avoid favoring models of ongoing gene flow because our focus was on long-term patterns of gene flow. A total of four scenarios of divergence were compared (Fig S1). The model of strict isolation (SI) assumes that an ancestral population of size NANC splits instantaneously at time Tsplit into two daughter populations of constant and independent size Npop1 and Npop2. The split is not followed by any gene flow and the population can either undergo an instantaneous bottleneck or an expansion at Tsplit. In contrast, the three other models assume various rates of gene flow following the instantaneous split. This migration occurs at a rate M = 4 N0.m. with M 1←2 being the number of migrants from population 2 to population 1 and M2←1being the reciprocal. In the model of ancestral migration (AM), the first generations of divergence are followed by gene flow until time Tam (Fig S1), at which point there will be no further gene flow (going forward in time). In the model of isolation with migration (IM), gene flow occurs continuously from Tsplit to the present and at a constant rate each generation. Under the secondary contact (SC) model, Tsplit is followed by a period of strict isolation and then by a period of secondary contact at Tsc generations ago that is still ongoing. We used uniform priors for model choice and parameter estimation. Simulation of microsatellites strictly followed the procedure of Illera et al. (2014) and later modified by Rougemont et al. (2016). In detail, the ms software (Hudson 2002) was used to perform coalescent simulations under an infinite-site model of mutation. Binary simulated data from ms were converted into microsatellite data using a generalized stepwise mutation model (GSM) in which the probability of changes of the repeat number in each mutation event was modeled by a geometrical parameter α following a uniform prior distribution sampled on the interval 0–0.5. Each effective population size Npop1, Npop2, NANC was scale by the parameter θ = 4Nref *µ, with Nref representing the effective population size of an arbitrarily chosen reference population (Nref, here set to 50,000) and µ representing the mutation rate per generation (chosen as µ = 2.5e−4 bp/generations). We chose µ according to values frequently observed in fishes (Shimoda et al. 1999; Yue et al. 2006). All divergence time parameters (Tsplit, Tsc, Tam) were also scaled by the parameter 4Nref. Priors for the effective population size were set to [0–500,000], [0–2,500,000] for the ancestral population size and [0–1,000,000] generations for the split time (Table S7). These correspond to large and uniforms priors.
Given (i) the arbitrarily fixed mutation rate and (ii) the interspecific variability in age at maturity, we did not attempt to convert the inferred divergence time or timing of secondary contact into a number of years to avoid over-interpretation of these parameters. Yet, the ratios of divergence time or symmetry in effective population size between species remained biologically relevant since the effect of the assumed mutation rates cancels out.
All computations took into account differences in sample size for each of the thirteen loci. four millions simulations composed of the thirteen microsatellite loci were computed under each demographic model.
Summary statistics were computed from the transformed microsatellite data and included the average and standard deviation values of: the number of alleles (A), Allelic richness (Ar), observed and expected heterozygosity (Ho and He, respectively), allele size in base pairs, the Garza-Williamson index (GW, Garza and Williamson 2001), GST (Nei 1973) and δµ2 (Goldstein et al. 1995). All statistics were computed using R scripts (R Development Core Team 2015).
Model selection
We evaluated the posterior probabilities of each demographic model using an ABC framework implemented in the abc package in R (Csilléry et al. 2012). We computed posterior probabilities using a feed forward neural network based on a nonlinear conditional heteroscedastic regression in which the model is considered as an additional parameter to be inferred. In the rejection step, we retained the 0.02% of simulations closest to the observed summary statistics, which were subsequently weighted by an Epanechnikov kernel. The regression step was performed using 50 neural networks and 15 hidden layers.
Parameter estimation and cross-validation
Parameter estimation was performed for the best models using nonlinear regressions. We used a logit transformation of the parameters on the 4000 best replicate simulations providing the smallest Euclidean distance to the observed data and then jointly estimated the posterior probability of each parameter using the neural network procedure implemented in the abc package using 50 feed-forward neural networks and 15 hidden layers.
Cross-validation was performed by computing the robustness of the model choice using a total of 4000 pseudo observed datasets (PODS) sampled randomly from each model. The same ABC procedure as for the empirical dataset was performed but this time considering a given model M instead of the empirical data. Then the robustness between two given models M1 and M2 was computed as:
Where P(PM1 = P|M1) represents the probability of correctly supporting M1 given the observed posterior probability P, and P(PM1 = P|M2) is the probability of erroneously supporting M1 given that the true model is M2 (Fagundes et al. 2007). Parameter estimation was performed for the best model by running another round of simulations for a total of 4 million simulations. The whole pipeline used for ABC can be found at: https://github.com/QuentinRougemont/MicrosatDemogInference.
Results
A total of 706 individuals were genotyped at 13 microsatellite loci and 180 different alleles were found, with 153 in A. alosa and 138 in A. fallax. No evidence of null alleles was detected. In A. alosa one test of linkage disequilibrium (LD) out of 936 comparisons was significant. Similarly, one LD test out of 858 comparisons was significant in A. fallax.
Genetic diversity
Fis was significant for only one marker (Alo29) in the Loire River population of A. alosa. All other markers across all populations did not show deviations from HWE or significant Fis values. The total number of alleles varied from 4 to 20 in A. alosa and from 4 to 17 in A. fallax. The two species shared 62% of alleles. Mean adjusted allelic richness (Ar) per population ranged from 3.74 to 5.94 in A. alosa and from 3.44 to 5.70 in A. fallax (Table 1 for details by river and Tables S2, S3 for details for each river and each marker). Mean observed heterozygosity was 0.596 (range: 0.49–0.62) in A. alosa and 0.523 (range: 0.44–0.59) in A. fallax and differed significantly between species (P < 0.005, 5000 permutations). In contrast, the difference in Ar between species was not significant (P = 0.061, 5000 permutations). For A. alosa, Ar and He were greatest in the Dordogne, Minho and Loire River and lowest in the Vire, Aulne and Trieux rivers. Accordingly, our linear models indicated a significant effect of the River factor (p = 0.0010**, Table S4, R2m = 0.065, R2c = 0.700). Yet, none of the observed differences between rivers were significant according to our TukeyHSD comparisons (p > 0.05, Table S5). For A. fallax, Ar and He were highest in the Rhône (Mediterranean) and lowest in the Ulla, Orne and Dordogne (Atlantic). Accordingly, our linear models indicated a significant effect of the River factor (p = 8e–5***, Table S4, R2m = 0.147, R2c = 0.496, Table S6). In this case the Ulla was the only population with significantly less allelic richness (p < 0.01) than the Rhône. None of the remaining comparisons were significant (Table S6).
Hybrid identification
Results of admixture analyses in Newhybrids and Structure using simulated genotypes from Hybridlab varied depending on the presence or absence of hybrids (Table S6). Both Structure and Newhybrids produced some false hybrids in the purebreds’ dataset and this overestimation was greater in Structure than in Newhybrids (Table S6). In brief, both software displayed a high accuracy and efficiency at a q-value threshold of 0.90, although Newhybrids displayed a slightly higher efficiency and accuracy compared to STRUCTURE with q > 0.90. In brief, Newhybrids displayed slightly higher efficiency and accuracy at a q-value threshold of 0.90 in the presence of hybrids with both software displaying accuracy and efficiency above 0.90.
Regarding our empirical data, both species were separated in two fully distinct clusters with average q-values greater than 0.99 for A. alosa and A. fallax with both Structure and Newhybrids. Both methods reclassified 13 individuals captured at sea that were morphologically identified as A. alosa but genotypically classified as A. fallax and 1 individual wrongly identified as A. fallax but genotypically classified as A. alosa. A total of 25 individuals displayed a q-value less than 0.9 with either of the methods and were not classified as purebreds. All hybrids identified by Newhybrids had a q-value greater than 0.8 and were all identified as hybrids in Structure (Table S7). From a geographic standpoint, the majority (80%) of the 25 individuals came from three major areas: namely the Charente River and Pertuis Charentais (28%), the Loire River (28%) and South Brittany/Scorff River (24%).
Population genetic structure
Global inter-species FST was 0.240 (95% CI: 0.194–0.286) and significant (P < 0.00087. 15,000 permutations). Global FST for A. alosa was 0.046 (95% CI: 0.034–0.058) (P < 0.0001. 10,000 permutations). Global FST for A. fallax was higher with a value of 0.219 (95% CI: 0.175–0.263) (P < 0.0001. 10,000 permutations). In A. alosa, 89% of pairwise FST comparisons were significant after Bonferroni corrections (Table 2) compared to 82% in A. fallax. Levels of differentiation between sampling localities were lower in A. alosa than in A. fallax. Populations sampled from the same river basin (i.e Rhône, or Tavignano) were not significantly differentiated from each other (Table 2), but were significantly differentiated from all other rivers. Similarly, Mediterranean A. fallax populations (Aude, Rhône, Vidourle) were not differentiated among one another but were strongly differentiated from Atlantic populations. The Tavignano (Corsica) population was significantly differentiated from all other rivers (FST ranged between 0.233 and 0.358). A. fallax populations from the Minho, Ulla and Orne were also distinct from all other populations (FST ranged between 0.08 and 0.289), but comparisons with the Tavignano were the highest (Table 2, Table S8 for interspecific comparison).
In A. alosa the highest likelihood (Ln(K)) was obtained for K = 6 (Fig. S2), while ΔK showed a strong peak for K = 3 and another for K = 6 (Fig. S2). Therefore we present the clustering results for both of these two values. For K = 3 the clustering separated the populations into three geographic regions (Atlantic, Brittany, and Normandy/Nivelle, Fig. 2A). The Nivelle individuals (Southern France) clustered with individuals from Normandy in Northern France (Vire and Orne river in Fig. 2A) and this cluster was itself admixed with Brittany. The clusters from Brittany and Atlantic were themselves highly admixed between each other. For K = 6 (Fig. 2A), the Nivelle individuals were separated from Normandy and formed a single cluster (contribution = 0.851). Normandy individuals formed a separated cluster, as well as Brittany individuals. Populations from the Atlantic were separated into three admixed clusters. Overall, most clusters were admixed with individuals from other genetic groups.

A Bayesian individual clustering performed with Structure for each species separately. In A. alosa K = 3 and K = 6 clusters are identified according to the Ln(K) and ΔK methods presented in Fig S2. B In A. fallax 3 and 5 clusters are identified according to the Ln(K) and ΔK methods presented in Fig S2. Individuals are represented by a bar with each color representing its membership proportion to each cluster. C plot of the Discriminant Analysis of Principal Components (DAPC) performed in A. alosa. The “best” number of clusters was inferred using the Bayesian Information Criteria (BIC) (Fig S3). Clusters are numbered from one to six given that mixed membership probability was often attributed to different rivers. Membership assignment of individuals averaged by river to each cluster can be found in (Table S8). D Plot of the DAPC performed in A. fallax. Membership assignment of individuals averaged by river to each cluster can be found in (Table S9).
In A. fallax, the highest likelihood plateaued between K = 5 and K = 8, while ΔK showed a single strong peak for K = 3 (Fig. S3). Therefore we present the results for K = 3 and K = 5 since other values were less supported. At K = 3, individuals from the Atlantic, the North Mediterranean coast and Corsica were separated. In contrast to patterns observed in A. alosa, no sign of admixture was observed in A. fallax populations (Fig. 2B). Indeed, all individuals except those from the Orne River had >0.95 membership probability to a separate cluster (individuals in the Orne River still had membership probabilities to a distinct cluster greater than 0.86). With K = 5, a separation of samples along the Atlantic Coast into 3 groups was revealed (Fig. 2B). The Orne (Normandy) river formed one cluster and the Ulla (Galicia, Spain) formed a separate cluster. Increasing the number of groups to 6 separated the Minho from the remaining groups. The remaining populations from the French Atlantic Coast formed a single cluster on the Rhône River. One individual was assigned as a migrant from the Tavignano (individual q-value = 0.981 CI: 0.894–1.00). Although some individuals displayed admixed ancestry, only one individual from the Ulla River was assigned with probability greater than 0.8 to the Minho suggesting relatively low dispersal among clusters as opposed to the pattern observed in A. alosa. The DAPC approach revealed a similar level of population structure in both A. alosa (Fig. 2C) and A. fallax (Fig. 2D). Indeed, according to the BIC, a total of 6 and 5 groups were present in each species respectively (Fig S4, Fig S5). However, the results for A. alosa were difficult to interpret (Table S9). Indeed, we find little congruence in the assignment of individuals to different groups in the DAPC as compared to structure. Only individuals from the Minho and from the Nivelle were assigned to a discrete cluster (Table S10) but these clusters displayed a very close relationship to other clusters in the DAPC plot (Fig. 2).
To reveal potential hierarchical structure and fine scale genetic structure we replicated the analysis in A. fallax including only populations from the Atlantic coast. This revealed the existence of 4 clusters (Fig. S3B).
The neighbor-joining tree revealed a clear separation between A. alosa and A. fallax (Fig. 3B). In A. alosa 5 clusters can be delineated, with the Adour being separate and the Vilaine grouping with the Minho. In A. fallax three main clusters can be distinguished corresponding to a geographic clustering pattern. Separation of A. fallax from the Tavignano was as strong as the separation between the two cryptic lineages from the Mediterranean Sea and the Atlantic coast. Finally, tests for IBD were significant in both species (Mantel tests: P < 0.001. r = 0.621 in A. alosa and P < 0.0001. r = 0.554 in A. fallax. Figure 3A). These results were also supported by significant linear models (Fig. 3A).

A Relationship between genetic distance as measured by FST/(1-FST) and ln(distance) in kilometers between populations of each species. All correlations were significant based on linear models and Mantel tests. B Neighbor-joining tree based on Cavalli-Sforza and Edwards (1967) genetic distances. Only bootstrap values >50% are indicated. Sample abbreviations match those given in Table 1. Aa = A. alosa. Af = A. fallax.
Assignment tests
According to GeneClass three individuals of A. fallax captured at sea displayed a probability <0.01 to originate from one of the sampled rivers and were excluded from further analyses with the three assignment methods. The forty A. alosa had a probability >0.01 to originate from one of the rivers. Contrasting results were obtained for each species; A. fallax were assigned with higher probability than A. alosa (only fish with score >0.9 were kept for assignment), with a total of 34 to 45 and of 18 to 34 fish respectively assigned depending on the method (Table 3). All methods assigned the majority of A. alosa to the Atlantic cluster. The DAPC approach classified 6 A. alosa from South Brittany in Normandy and 4 in the Minho, while these fish were either assigned to the Atlantic cluster or left unassigned by the other methods. Similarly, one A. alosa from the Pertuis Charentais was assigned to the Minho by DAPC while the two other methods classified this fish as belonging to the Atlantic cluster. In A. fallax the majority of fish from the North Sea, the South Brittany and the Southern part of the Bay of Biscay were assigned to the Orne cluster. Most fish from the Pertuis Charentais were assigned to the Atlantic cluster. One fish was assigned to the Ulla by the DAPC approach but was left unassigned by the two other methods. Similarly, two fish from Southern Brittany were assigned to the Minho with DAPC, whereas both were left unassigned by Structure and one was unassigned by GeneClass, while the other was assigned to the Atlantic cluster.
Demographic history
Reconstruction of demographic history was performed (i) between the two species along the Atlantic coast, as well as between the divergent lineages of A. fallax, namely between (ii) A. fallax “Atlantic” and A. fallax “Mediterranean Sea” and between (iii) A. fallax “Mediterranean Sea” and A. fallax “Corsica”. In all three cases, ABC model choice rejected SI and AM in favor of models with ongoing gene flow, with support for secondary contact (SC) (Table S11). For instance, in between species comparisons, posterior probabilities were P(SC) = 0.980 versus P(SI) = 0.02; P(IM) = 0.851 versus P(SI) = 0.149 and. P(AM) = 0.720 versus P(SI) = 0.20. For the sake of conciseness we only present results for between species comparisons, but the others are shown in Table S11. In each between species pairwise comparison, our cross-validation procedure revealed that the robustness was 1 for all comparisons (Fig S6 A-F). Comparison of the model with ancient gene flow against ongoing gene flow led to a rejection of the AM model (P(SC) = 0.669 vs P(AM) = 0.331; P(IM) = 0.599 vs P(AM) = 0.401) with a cross-validation procedure revealing a robustness of 1 (Fig S6A, B). Finally, comparing IM and SC leads to a higher posterior probability of the latter with P(SC) = 0.738 vs P(IM) = 0.262 and a robustness of 1 (Fig. 4a, Fig S7C, D).

Schematic of the best model of divergence (A) and distributions of prior and posterior parameter estimates based on the model of best fit associated with divergence and diversity data (B). A Simplified representation of the best inferred scenario of divergence between Alosa alosa and A. fallax. B Prior (gray) and Posterior (orange) parameter distributions inferred by Approximate Bayesian Computation. θ A. alosa = Scaled Effective population size of A. Alosa. θ A. alosa = Scaled Effective population size of A. fallax. θancestral = Effective population size of the ancestral population. All populations size are scaled by 4Nref*µ. Tsplit = split time (here Tsplit = tau* 4Nref). Tsc = Time of Secondary Contact. M12 = 4Nref*m12 and M21 = 4Nref*m21 corresponds to the scaled migration rate where mij represents the fraction of subpopulation i that is made up of migrants from subpopulation j each generation. All values are provided in coalescent units and scaled by the effective reference population size (Nref = 50,000).
The posterior distribution of parameter estimates associated with effective population size and divergence time under SC were well differentiated from the prior yielding confidence for interpretations of the mean values and credible intervals in between species comparisons (Fig. 4b) and between A. fallax lineages (Figs. S8, S9). These indicated population size reduction compared to the estimated ancestral effective population size (NANC). The median estimated effective population size was N1 = 209 [IC = 47–646] for A. alosa, N2 = 2573 [940–5649] for A. fallax, and NANC = 617,000 [39,074–1,228,000] for the ancestral population (Fig. 4b). Credible intervals around the split time and the time of secondary contact overlapped. Median estimate of Tsplit was ~774,000 generations but with large credible intervals [119,000–1,000,000]. Given the difference in life history, we did not convert these estimates into years. The time of secondary contact would be 157,000 generations [24,000–620,000] (Fig. 4b). Considering effective population size within A. fallax, we found that the Corsican lineage displayed the highest Ne (median = 19,500 [6,600–51,800]) and the Mediterranean lineage displayed a similar value to the Atlantic one (median = 3000 [CI = 100–9600] depending on the comparison). Our inference further suggested that the divergence of A. fallax from the Atlantic and Mediterranean Sea was slightly more recent than that observed with A. alosa (median = 630,000 [CI = 69,000–970,000] generations) followed by a more recent divergence from A. fallax “Corsica” (median = 350,000) [CI = 35,000–960,000]. Additional parameter estimates such as migration rate can be found in Table S12 and Figs. S8 and S9.
Discussion
Our results shed light on evolutionary processes affecting the rate of speciation and provide key information for the management of two declining fish species. We found that genetic structure among populations was lower for the nearly semelparous species, A. alosa, compared to the iteroparous species, A. fallax. Moreover, individuals captured at sea could be assigned at the region level for A. alosa and at the river level in A. fallax. We inferred that the most likely long term historical divergence scenario between both species implicated historical separation followed by a secondary contact accompanied by contemporary hybridization and strong population size declines. These observations regarding hybridization, demographic history and gene flow are in large agreement with those from Faria et al. (2012) which is the most recent broad scale study but uses mtDNA, thus bringing complementary insights into the species evolutionary dynamics. Our results also corroborate the hypothesis of a divergent lineage (A. agone) present along the Mediterranean coast and suggest a possible undocumented new lineage present in the Island of Corsica, which were both formerly grouped with A. fallax. These observations have strong conservation implications showing the importance of combining catchment and region-based management.
Level of species differentiation and hybridization
Our results confirm previous estimates of introgression and hybridization between both species (Alexandrino et al. 2006; Coscia et al. 2010; Jolly et al. 2011; Faria et al. 2012; Taillebois et al. 2020). Normally, spatio-temporal segregation mechanisms exist and maintain or minimize the contact between the two species during reproduction. In particular, A. fallax generally spawns in lower areas of the watershed than A. alosa, which normally spawns in the upper parts of rivers (Aprahamian et al. 2003; Baglinière et al. 2003b), thereby minimizing hybridization opportunities between species. Yet, human river fragmentation by dams and various obstacles is increasingly restricting shad migration to downstream areas. This favors sympatry and leads to increased opportunities for hybridization. The removal of this premating reproductive barrier thus promotes hybridization and introgression (Alexandrino et al. 2006; Taillebois et al. 2020). Accordingly, we find strong support for ongoing hybridization between both species in populations distributed in the same watershed and reaching similar spawning sites (e.g. Loire and Charente River). The preferential occurrence of hybrids in these areas is unclear and may be related to the disruption of spawning grounds due to human activity, but this speculation would require further investigations. However, the two species seem to remain genetically distinct despite hybridization. This may indicate the existence of pre-zygotic (e.g. behavioral) and post-zygotic genetic barriers that are likely involved in the maintenance of reproductive isolation despite hybridization that seems to have existed over relatively long historical times (Ravinet et al. 2017, Barth et al. 2020). Yet, genetic barriers are often semi-permeable (Wu 2001), resulting in heterogeneous differentiation across the genome (Ravinet et al. 2017; Rougemont et al. 2017). The porosity of the species barrier is well illustrated by the numerous backcrosses found recently by Taillebois et al. (2020) in a set of partially overlapping rivers using a SNP array as well as by the mtDNA results from Faria et al. (2012) on another set of overlapping rivers, thus providing support for our results. For instance, they identified bidirectional mitochondrial introgression ranging from 25 to 63% depending on the river considered (note that this percentage derived from a small number of individuals however). Whole genome re-sequencing of these hybridizing populations would be needed to (i) quantify the mosaic of local ancestry across the genome (Duranton et al. 2018) (ii) identify the regions involved in adaptive introgression from one species to the other (Hedrick 2013) and (iii) determine the deleterious effect of introgression and extent of selection against introgression (Kim et al. 2018). The hybridization of these two species is hence an interesting context to study speciation and also highlights the consequences of river fragmentation on the genetic integrity of these declining species (Harris et al. 2018; Rougemont et al. 2020).
Influence of life history strategy on the population genetic structure
As expected from theory and from previous research, the species that disperses more readily (and semelparous), A. alosa, displays lower levels of genetic structure and higher levels of genetic diversity compared to A. fallax. A limit here is that the inference of the number of clusters in A. alosa was difficult with structure analysis pointing to either three or six clusters. Therefore, it is likely that the number of real ancestral groups for A. alosa is below the 3 or 6 groups inferred here. Another complicating factor was the significant signal of isolation by distance which itself can generate genetic clusters (Meirmans 2012; Battey et al. 2020). Regardless of the exact number of clusters, A. alosa is likely to derive from one major group with signals of admixture being inflated due to IBD. These variations in dispersal syndrome are well documented and are associated with variation in life history traits across many species (Stevens et al. 2013; Cayuela et al. 2016, 2018). Here, this suggests a substantial trade-off associated with these life history traits. Since A. fallax is iteroparous, we hypothesized that they maximize survival probability by dispersing less and reducing the duration of their marine phase due to an earlier age of sexual maturity than A. alosa (Bagliniere et al. 2020a, b). Other hypotheses have been made to explain the weaker genetic structure of the semelparous American shad (A. sapidissima) as compared to its iteroparous form and relate to environmental differences along a latitudinal gradient (discussed in Hasselman et al. 2013). Here, however, both species reproduce in the same rivers along the Atlantic coast, so the proposed environmental mechanisms are less likely to apply. A meta-analysis in terrestrial and semi-terrestrial animals revealed that higher dispersal was associated with higher fecundity and survival (Stevens et al. 2014). In a less stable environment, higher dispersal, higher fecundity and shorter lifespan can be favored (Cayuela et al. 2016). This leads us to hypothesize that A. alosa populations may be more resilient under climate change, but this hypothesis remains to be investigated.
Demography and speciation
The differentiation levels among A. fallax lineages are similar to those observed between species. This led us to identify two putative cryptic species in addition to the already documented A. fallax located along the Atlantic coast. The first putative lineage occurs in the continental coast of the Mediterranean and the second one in Corsica. One caveat stems from our limited sampling of A. fallax between the Vidourle and Southern Spain. With such sparse sampling, genetic clustering methods can be confounded by isolation by distance as well as coalescent based species delimitation methods (Meirmans 2012, Mason et al. 2020). Yet, our results are also supported by the observed increased differentiation, as well as the genetic differences between Atlantic and Mediterranean A. fallax based on protein markers and morphological differences observed by Le Corre et al. (2005). They also suggested that the Mediterranean A. fallax may be considered as an independent species, Alosa agone (Scopoli 1786). Indeed, based on the genetic studies of Le Corre et al. (2005) and morphological observations of Bianco (2005), Mediterranean A. fallax was considered as a separate species (A. agone) in the recent atlas of freshwater fish from France. Overall the taxonomic status of this species is not yet firmly established (Chiesa et al. 2014; Baglinière et al. 2020c). While it is considered a valid species in the atlas of fish, previous genetic studies failed to distinguish A. agone from A. fallax (Faria et al. 2006; Chiesa et al. 2014) and our study is the first to provide evidence for differentiation between A. fallax and A. agone. Additional examination of phenotypic and ecological differences between this lineage and the other A. fallax lineages, along with sampling of remote lineages (e.g. Alosa immaculata) would enable a better description of this species. The identification of a putative lineage in Corsica is a new finding. A parallel can be drawn with Salmo trutta, which is also represented with many phylogeographic lineages (Bernatchez 2001) and locally differentiated populations in the Tyrrhenian area, including Corsica, that are attached to the Adriatic lineage rather than the Mediterranean lineage (Berrebi et al. 2019). Unfortunately, microsatellite data are poorly suited to disentangle recently high genetic drift from long divergence time. A whole genome approach would be necessary to better reconstruct the divergence history of all the species and sub-populations or cryptic lineages using explicit modeling and additional measures such as absolute divergence (DXY; Nei 1987). Presently, the systematics of the genus Alosa remain unclear because of the large number of subspecies of A. fallax (Chiesa et al. 2014; Coscia et al. 2010; Baglinière et al. 2020c).
In addition, we have attempted to reconstruct the demographic history of the two species and the divergent A. fallax lineages, though we call attention to a few caveats. First, we had too few markers to highlight the semi-permeable nature of the genome and to discriminate local regions of the genome that are freely exchanged from those that are possibly impermeable to gene flow and may be involved in reproductive isolation (Duranton et al. 2018). Moreover, the mutation rate is not known and the generation time remains difficult to estimate given that A. alosa reproduces only once between 3 to 8 years, whereas A. fallax can reproduce multiple times between 2 to 8 years (Baglinière et al. 2020a, b). Assuming the same generation time as Faria et al. (2012), namely five years, we would obtain a divergence time of ~3 My [95%CI: 500 Ky–4.6 My], thus compatible with estimates [0.294–3.477 My] obtained in their study. Regardless of the timing of divergence, Faria et al. (2012) net sequence divergence (Da = 0.02) can be used to replace the species along the speciation continuum proposed by Roux et al. (2016). Their results thus indicate that the species fall exactly in the gray zone of speciation where reconstructing the divergence history is likely to be most difficult. Interestingly, the estimated time of divergence among species and lineages differ only by a small magnitude and, considering the uncertainty associated with these parameters, it is possible that all species and lineages have diverged simultaneously. More insights about the full species radiation history would require sampling all possible lineages within the Mediterranean and Black sea along with whole genome sequencing. Moreover, our estimates of the difference in effective population size between A. fallax and A. alosa as well as when compared to the ancestral population are also in broad agreement with those of Faria et al. (2012), although the exact value differs. This discrepancy may be due to the difference in marker resolution, difference in selective constraints undergone by mtDNA versus microsatellite markers or the difference in the methods used for demographic reconstruction. For instance, Faria et al. (2012) used IMa2, which is sensitive to confounding by linked selection (Cruickshank and Hahn 2014). Most importantly, we used individuals from a single river to avoid increasing local population structure, whereas Faria et al. (2012) included samples from several rivers, and thus estimated the metapopulation Ne, preventing any direct comparison with our estimates.
Among other limits, our sampling design did not include A. immaculata, a species from the Caspian and Black sea that is more closely related to A. fallax than to A. alosa according to mtDNA analyses (Faria et al. 2012). However, the node support for their grouping was weak and did not entirely allow us to conclude that A. fallax originate from the same area as A. immaculata (Alexandrino et al. 2006; Faria et al. 2006, 2012). Therefore, the hypothesis of a shared origin in the Mediterranean Sea for A. fallax and A. immaculata versus an Atlantic origin for A. alosa cannot be excluded. This would lend support to the secondary contact model that we inferred here. Moreover, the absence of A. immaculata in our samples (i.e. ghost species) can influence our inferences of gene-flow (Mason et al. 2020; Tricou et al. 2022). For instance, ancient gene flow between A. immaculata and A. alosa may have left some footprint in the genome of A. alosa that may complicate parameter estimation. Yet, A. alosa and A. fallax are also known to share spawning grounds (Maitland and Lyle 2005) and produce hybrids (Taillebois et al. 2020). Therefore, we also expect ongoing gene flow due to secondary contact to be a supported scenario.
With all these caveats in mind, the higher support for a model of secondary contact between the two species than for alternative models, indicates that isolation was necessary to initiate divergence between the two species. Most theories suggest that speciation is difficult to establish in the presence of continuous gene flow (Barton and Bengtsson 1986; Bierne et al. 2011). Only a few studies convincingly support speciation with gene flow and have explicitly attempted to discriminate among competing models (Martin et al. 2013; Malinsky et al. 2015; Tusso et al. 2021). In contrast, an increasing number of studies have reported evidence for divergence initiated in allopatry followed by gene flow (e.g. Roux et al. 2013, 2014; Rougemont et al. 2017; Rougemont and Bernatchez 2018; Leroy et al. 2019; Cayuela et al. 2020). Interestingly, Tine et al. (2014) and Duranton et al. (2018) have reported the existence of two cryptic species of sea bass (Dicentrarchus labrax), the Atlantic and Mediterranean Sea Bass with evidence of secondary contact (Tine et al. 2014), multiple islands of reproductive isolation (Duranton et al. 2018), and cryptic genetic and demographic connectivity (Robinet et al. 2020). Similarly, Riquet et al. (2019) reported the existence of divergent lineages of seahorses (Hippocampus guttulatus) on the Mediterranean Sea and the Atlantic coast as well as the existence of partially reproductively isolated cryptic lineages maintained in sympatry within the Mediterranean Sea. Shared origin in the Mediterranean sea for A. fallax and A. immaculata versus an Atlantic origin for A. alosa cannot be excluded. This would lend support to the secondary contact model that we inferred here.
Finally, our results have conservation implications. First, our estimates of long-term effective population size indicated that the Ne of A. fallax was higher than that of A. alosa. This long-term average is in line with previous phylogeographic knowledge (Alexandrino et al. 2006; Faria et al. 2012). Despite all the uncertainty associated with ancestral population size estimates, our results indicate that both A. fallax and A. alosa have undergone strong reductions following their divergence. Therefore, the ongoing decline of the two species (Aprahamian et al. 2003; Baglinière et al. 2003b; Rougier et al. 2012) is of further concern if they already have a reduced adaptive potential. These long-term trends further suggest a reduced evolutionary potential (Leroy et al. 2021). Finally, we were able to identify and assign individuals of unknown origin captured at sea to putative river or regional groups with modest power. These results suggest that our marker set could be improved (e.g. using additional markers or by moving to a SNP array) to identify individuals dispersing at sea and draw inferences about dispersal distances of the two species, an important problem when assessing the provenance of individuals in mixed-stocks fisheries (Beacham et al. 2019; Nachón et al. 2020).
Conclusion and future directions
Our results indicate that Alosa species constitute an interesting model to study speciation and hybridization between closely related species. Understanding the fitness effects of such ongoing hybridization will be of utmost importance to help manage the species. For instance, if the species are increasingly constrained to breed on overlapping spawning sites, the rate of hybridization is expected to increase, which may lead to increased fitness of the hybrid during the first few generations of hybridization, but may also increase selection against introgression if recessive deleterious alleles are expressed. This could negatively affect population size, by reducing the fitness of backcrosses and hence decrease the species’ evolutionary potential. However, the application of the EU Water Framework Directive on good ecological status could reverse the hybridization trend of these species if the ecological connectivity of rivers is restored and the natural, separate breeding grounds of both species are accessible again. Overall, our results revealed deep divergence followed by secondary contact and a prevailing role of gene flow between the two species as well as among new lineages or possible species of A. fallax. These results highlighted the consequences of contrasting life history strategies at the genetic level and suggest that the two species should be managed jointly given their porous reproductive boundaries. We propose that whole genome sequences will help address several questions that have been raised, especially the inference of historical divergence and demography of these species, and the inference of putative post-zygotic barriers across the genome.
Data availability
Data are deposited on dryad (https://doi.org/10.5061/dryad.rn8pk0pdb) and code for ABC simulations are available on github https://github.com/QuentinRougemont/MicrosatDemogInference.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N et al. (2013) Hybridization and speciation. J Evolut Biol 26:229–246
Alexandrino P, Faria R, Linhares D, Castro F, Corre ML, Sabatié R et al. (2006) Interspecific differentiation and intraspecific substructure in two closely related clupeids with extensive hybridization, Alosa alosa and Alosa fallax. J Fish Biol 69:242–259
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229
Aprahamian MW, Baglinière J-LJ-L, Sabatié M-R, Alexandrino P, Theil R, Aprhamian CD (2003) Biology, Status and Conservation of the Anadromous Atlantic Twaite Shad Alosa fallax fallax. In: Biodiversity, status and conservation of world’s shads, American Fisheries Society Symposium. American Fisheries Society
Baglinière J, Sabatié M, Rochard E, Alexandrino P, Aprahamian M (2003a) The allis shad Alosa alosa: Biology, ecology, range, and status of populations. Am Fish Soc Symp 2003:85–102
Baglinière J-L, Sabatié MR, Rochard E, Alexandrino P, Aprahamian MW (2003b) The allis shad Alosa alosa: Biology, ecology, range, and status of populations. Am Fish Soc Symp 2003:85–102
Baglinière JL, Launey S, Beaulaton L (2020a) La grande Alose Alosa alosa, Linnaeus, 1758. In “ Les poissons d’eau douce de France. 2nde édition, Biotope Editions, Mèze; Muséum national d’Histoire naturelle, Keith P., Poulet N., Denys G., Changeux T., Feunteun E. et Persat H. J. (Coords.), Muséum national d’histoire naturelle, Paris, Collection inventaire et biodiversité, pp. 294–297
Baglinière JL, Launey S, Denys G, Beaulaton L (2020b) L’Alose feinte atlantique Alosa fallax, Lacépède 1803. In “ Les poissons d’eau douce de France. 2nde édition, Biotope Editions, Mèze; Muséum national d’Histoire naturelle, Keith P., Poulet N., Denys G., Changeux T., Feunteun E. et Persat H. J. (Coords.), Muséum national d’histoire naturelle, Paris, Collection inventaire et biodiversité, pp. 298–300
Baglinière JL, Launey S, Denys G, Beaulaton L (2020c) Ordre des Clupéiformes. Famille des Clupeidae. In “ Les poissons d’eau douce de France. 2nde édition, Biotope Editions, Mèze; Muséum national d’Histoire naturelle, Keith P., Poulet N., Denys G., Changeux T., Feunteun E. et Persat H. J. (Coords.), Muséum national d’histoire naturelle, Paris, Collection inventaire et biodiversité, pp. 291–292
Barth JMI, Gubili C, Matschiner M, Toressen O, Watanabe S, Egger B et al. (2020) Stable species boundaries despite 10 million years of hybridization in tropical eels. Nat Commun 11:1433
Barton N, Bengtsson BO (1986) The barrier to genetic exchange between hybridising populations. Heredity 57:357–376
Barton NH, Hewitt GM (1985) Analysis of Hybrid Zones. Annu Rev Ecol Syst 16:113–148
Barton K (2020) Mu-MIn: Multi-model inference. R Package Version 0.12.2/r18. http://R-Forge.R-project.org/projects/mumin/
Bates D, Maechler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
Battey CJ, Ralph PL, Kern AD (2020) Space is the place: effects of continuous spatial structure on analysis of population genetic data. Genetics 215:193–214
Beacham TD, Wallace C, Jonsen K, McIntosh B, Candy JR, Willis D et al. (2019) Variation in migration pattern, broodstock origin, and family productivity of coho salmon hatchery populations in British Columbia, Canada, derived from parentage-based tagging. Ecol Evolution 9:9891–9906
Belkhir K, Borsam P, Chikhi L, Raufaste N, Bonhomme F, Belkir K et al. (1996) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier (France)
Bernatchez L (2001) The Evolutionary History of Brown Trout (salmo Trutta L.) Inferred from Phylogeographic, Nested Clade, and Mismatch Analyses of Mitochondrial Dna Variation. Evolution 55:351–379
Berrebi P, Caputo Barucchi V, Splendiani A, Muracciole S, Sabatini A, Palmas F et al. (2019) Brown trout (Salmo trutta L.) high genetic diversity around the Tyrrhenian Sea as revealed by nuclear and mitochondrial markers. Hydrobiologia 826:209–231
Bianco PG (2005) The Status of the Twaite Shad, Alosa agone, in Italy and the Western Balkans. Mar Ecol 23:51–64
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–2072
Cavalli-Sforza LL, Edwards AW (1967) Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet 19:233–257
Cayuela H, Boualit L, Arsovski D, Bonnaire E, Pichenot J, Bellec A et al. (2016) Does habitat unpredictability promote the evolution of a colonizer syndrome in amphibian metapopulations? Ecology 97:2658–2670
Cayuela H, Rougemont Q, Laporte M, Mérot C, Normandeau E, Dorant Y et al. (2020) Shared ancestral polymorphisms and chromosomal rearrangements as potential drivers of local adaptation in a marine fish. Mol Ecol 29:2379–2398
Cayuela H, Rougemont Q, Prunier JG, Moore J-S, Clobert J, Besnard A et al. (2018) Demographic and genetic approaches to study dispersal in wild animal populations: A methodological review. Mol Ecol 27:3976–4010
Ceballos G, Ehrlich PR, Raven PH (2020) Vertebrates on the brink as indicators of biological annihilation and the sixth mass extinction. PNAS 117:13596–13602
Chiesa S, Piccinini A, Lucentini L, Filonzi L, Marzano FN (2014) Genetic data on endangered twaite shad (Clupeidae) assessed in landlocked and anadromous populations: one or more species? Rev Fish Biol Fish 24:659–670
Coscia I, Rountree V, King JJ, Roche WK, Mariani S (2010) A highly permeable species boundary between two anadromous fishes. J Fish Biol 77:1137–1149
Cruickshank TE, Hahn MW (2014) Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol 23:3133–3157
Csilléry K, François O, Blum MGB (2012) ABC: an R package for Approximate Bayesian computation (ABC). Methods Ecol Evolution 3:475–479
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Duranton M, Allal F, Fraïsse C, Bierne N, Bonhomme F, Gagnaire P-A (2018) The origin and remolding of genomic islands of differentiation in the European sea bass. Nat Commun 9:2518
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Estoup A, Largiader C, Perrot E, Chourrout D (1996) Rapid one-tube DNA extraction for reliable PCR detection of fish polymorphic markers and transgenes. Mol Mar Biol Biotechnol 5:295–298
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fagundes NJR, Ray N, Beaumont M, Neuenschwander S, Salzano FM, Bonatto SL et al. (2007) Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci 104:17614–17619
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Faria R, Wallner B, Weiss S, Alexandrino P (2004) Isolation and characterization of eight dinucleotide microsatellite loci from two closely related clupeid species (Alosa alosa and A. fallax). Mol Ecol Notes 4:586–588
Faria R, Weiss S, Alexandrino P (2006) A molecular phylogenetic perspective on the evolutionary history of Alosa spp. (Clupeidae). Mol Phylogenet Evol 40:298–304
Faria R, Weiss S, Alexandrino P (2012) Comparative phylogeography and demographic history of European shads (Alosa alosa and A. fallax) inferred from mitochondrial DNA. BMC Evolut Biol 12:194
Felsenstein J (1995) PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genome Sciences. University of Washington, Seattle. WA
Francis RM (2017) pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour 17:27–32
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Frankham R (2005) Genetics and extinction. Biol Conserv 126:131–140
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
García-Dorado A, Caballero A (2021) Neutral genetic diversity as a useful tool for conservation biology. Conserv Genet 22:541–545
Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW (1995) Genetic absolute dating based on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA 92:6723–6727
Goudet J (1995) FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. J Hered 86:485–486
Harris RB, Sackman A, Jensen JD (2018) On the unfounded enthusiasm for soft selective sweeps II: Examining recent evidence from humans, flies, and viruses. PLOS Genet 14:e1007859
Hasselman DJ, Ricard D, Bentzen P (2013) Genetic diversity and differentiation in a wide ranging anadromous fish, American shad (Alosa sapidissima), is correlated with latitude. Mol Ecol 22:1558–1573
Hedrick PW (2013) Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol 22:4606–4618
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hothorn T, Bretz F, Westfall P (2008) Simultaneous inference in general parametric models. Biometrical J 50:346–363
Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18:337–338. https://doi.org/10.1093/bioinformatics/18.2.337
Illera JC, Palmero AM, Laiolo P, Rodríguez F, Moreno ÁC, Navascués M(2014) genetic, morphological, and acoustic evidence reveals lack of diversification in the colonization process in an Island Bird Evolution 68:2259–2274
Jolly MT, Aprahamian MW, Hawkins SJ, Henderson PA, Hillman R, O’Maoiléidigh N et al. (2012) Population genetic structure of protected allis shad (Alosa alosa) and twaite shad (Alosa fallax). Mar Biol 159:675–687
Jolly MT, Maitland PS, Genner MJ (2011) Genetic monitoring of two decades of hybridization between allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet 12:1087–1100
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Keefer ML, Caudill CC (2014) Homing and straying by anadromous salmonids: a review of mechanisms and rates. Rev Fish Biol Fish 24:333–368
Kim BY, Huber CD, Lohmueller KE (2018) Deleterious variation shapes the genomic landscape of introgression. PLOS Genet 14:e1007741
Le Corre ML, Alexandrino P, Sabatie MR, Aprahamian MW, Baglinière JL (2005) Genetic characterisation of the Rhodanian twaite shad, Alosa fallax rhodanensis. Fish Manag Ecol 12:275–282
Leroy T, Rougemont Q, Dupouey J-L, Bodénès C, Lalanne C, Belser C et al. (2019) Massive postglacial gene flow between European white oaks uncovered genes underlying species barriers. N Phytologist 226:1183–1197
Leroy T, Rousselle M, Tilak M-K, Caizergues AE, Scornavacca C, Recuerda M et al. (2021) Island songbirds as windows into evolution in small populations. Curr Biol 31:303–1310
Limburg KE, Waldman JR (2009) Dramatic declines in North Atlantic diadromous fishes. BioScience 59:955–965
Maitland PS, Lyle AA (2005) Ecology of allis shad Alosa alosa and twaite shad Alosa fallax in the Solway Firth, Scotland. Hydrobiologia 534:205–221
Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP et al. (2015) Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 350:1493–1498
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F et al. (2013) Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res 23:1817–1828
Martin J, Bareille G, Berail S, Pecheyran C, Daverat F, Bru N et al. (2013) Spatial and temporal variations in otolith chemistry and relationships with water chemistry: a useful tool to distinguish Atlantic salmon Salmo salar parr from different natal streams. J Fish Biol 82:1556–1581
Martin J, Rougemont Q, Drouineau H, Launey S, Jatteau P, Bareille G et al. (2015) Dispersal capacities of anadromous Allis shad population inferred from a coupled genetic and otolith approach. Can J Fish Aquat Sci 72:991–1003
Mason NA, Fletcher NK, Gill BA, Funk WC, Zamudio KR (2020) Coalescent-based species delimitation is sensitive to geographic sampling and isolation by distance. Syst Biodivers 3:269–280
Meirmans PG (2012) The trouble with isolation by distance. Mol Ecol 21(12):2839–2846
Mennesson-Boisneau C, Aprahamian MW, Sabatié MR, Cassous-Leins JJ (2000a) Caractéristiques des adultes. In Les aloses Alosa alosa et Alosa fallax spp. (Bagliniè re. JL & Elie P eds). pp. 33–53. Paris: INRA-Cemagref
Mikkelsen EK, Irwin (2021) Ongoing production of low-fitness hybrids limits range overlap between divergent cryptic species. Mol Ecol 30:4090–4102
Nachón DJ, Bareille G, Drouineau H, Tabouret H, Taverny C, Boisneau C et al. (2020) 1980s population-specific compositions of two related anadromous shad species during the oceanic phase determined by microchemistry of archived otoliths. Can J Fish Aquat Sci 77:164–176
Nei, M (1987) Molecular Evolutionary Genetics, 9780231063210, Columbia University Press
Nei M (1973) Analysis of Gene Diversity in Subdivided Populations. Proc Natl Acad Sci USA 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nielsen EE, Bach LA, Kotlicki P (2006) hybridlab (version 1.0): a program for generating simulated hybrids from population samples. Mol Ecol Notes 6:971–973
Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004) micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Page RDM (1996) Tree View: An application to display phylogenetic trees on personal computers. Bioinformatics 12:357–358
Parrish DL, Behnke RJ, Gephard SR, McCormick SD, Reeves GH (1998) Why aren’t there more Atlantic salmon (Salmo salar)? 55: 7
Perrier C, Guyomard R, Bagliniere J-L, Evanno G (2011) Determinants of hierarchical genetic structure in Atlantic salmon populations: environmental factors vs. anthropogenic influences. Mol Ecol 20:4231–4245
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: A Software for Genetic Assignment and First-Generation Migrant Detection. J Heredity 95:536–539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of Population Structure Using Multilocus Genotype Data. Genetics 155:945–959
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Randon, M, Daverat, F, Bareille, G, Jatteau, PH, Martin, J, Pecheyran, C, Drouineau, H (2017) Quantifying exchanges of Allis shads between river catchments by combining otolith microchemistry and abundance indices in a Bayesian model. ICES Journal of Marine Science
Ravinet M, Faria R, Butlin RK, Galindo J, Bierne N, Rafajlović M et al. (2017) Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J Evolut Biol 30:1450–1477
Riquet F, Liautard-Haag C, Woodall L, Bouza C, Louisy P, Hamer B et al. (2019) Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. Evolution 73:817–835
Robinet T, Roussel V, Cheze K, Gagnaire P-A (2020) Spatial gradients of introgressed ancestry reveal cryptic connectivity patterns in a high gene flow marine fish. Mol Ecol 29:3857–3871
Rougemont Q, Bernatchez L (2018) The demographic history of Atlantic salmon (Salmo salar) across its distribution range reconstructed from approximate Bayesian computations*. Evolution 72:1261–1277
Rougemont Q, Besnard A-L, Baglinière J-L, Launey S (2015) Characterization of thirteen new microsatellite markers for allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet Resour 7:259–261
Rougemont Q, Gagnaire P-A, Perrier C, Genthon C, Besnard A-L, Launey S et al. (2017) Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and nonparasitic lamprey ecotypes. Mol Ecol 26:142–162
Rougemont Q, Moore J-S, Leroy T, Normandeau E, Rondeau EB, Withler RE et al. (2020) Demographic history shaped geographical patterns of deleterious mutation load in a broadly distributed Pacific Salmon. PLOS Genet 16:e1008348
Rougemont Q, Roux C, Neuenschwander S, Goudet J, Launey S, Evanno G (2016) Reconstructing the demographic history of divergence between European river and brook lampreys using approximate Bayesian computations. PeerJ 4:e1910
Rougier T, Lambert P, Drouineau H, Girardin M, Castelnaud G, Carry L et al. (2012) Collapse of allis shad, Alosa alosa, in the Gironde system (southwest France): environmental change, fishing mortality, or Allee effect? ICES J Mar Sci 69:1802–1811
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol 14:e2000234
Roux C, Fraïsse C, Castric V, Vekemans X, Pogson GH, Bierne N (2014) Can we continue to neglect genomic variation in introgression rates when inferring the history of speciation? A case study in a Mytilus hybrid zone. J Evolut Biol 27:1662–1675
Roux C, Tsagkogeorga G, Bierne N, Galtier N (2013) Crossing the species barrier: genomic hotspots of introgression between two highly divergent Ciona intestinalis species. Mol Biol Evolut 30:1574–1587
Ryan SF, Deines JM, Scriber JM, Pfrender ME, Jones SE, Emrich SJ et al. (2018) Climate-mediated hybrid zone movement revealed with genomics, museum collection, and simulation modeling. PNAS 115:E2284–E2291
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Shimoda N, Knapik EW, Ziniti J, Sim C, Yamada E, Kaplan S et al. (1999) Zebrafish genetic map with 2000 microsatellite markers. Genomics 58:219–232
Stevens VM, Whitmee S, Galliard J-FL, Clobert J, Böhning-Gaese K, Bonte D et al. (2014) A comparative analysis of dispersal syndromes in terrestrial and semi-terrestrial animals. Ecol Lett 17:1039–1052
Taillebois L, Sabatino S, Manicki A, Daverat F, Nachón DJ, Lepais O (2020) Variable outcomes of hybridization between declining Alosa alosa and Alosa fallax. Evolut Appl 13:636–651
Tavernie P, Elie E (2001) Répartition spatio-temporelle de la grande alose (Alosa alosa, Linné, 1766) et de l’alose feinte (Alosa fallax, Lacépéde, 1803) dans le golfe de gascogne. 803-821. Knowl Manag Aquat -Ecosyst 362:803–820
Teixeira JC, Huber CD (2021) The inflated significance of neutral genetic diversity in conservation genetics. PNAS 118:e2015096118
Tine M, Kuhl H, Gagnaire P-A, Louro B, Desmarais E, Martins RST et al. (2014) European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat Commun 5:5770
Tomás J, Augagneur S, Rochard E (2005) Discrimination of the natal origin of young-of-the-year Allis shad (Alosa alosa) in the Garonne–Dordogne basin (south-west France) using otolith chemistry. Ecol Freshw Fish 14:185–190
Tricou T, Tannier E, de Vienne D (2022) Ghost Lineages Highly Influence the Interpretation of Introgression Tests, Systematic Biology, syac011
Tusso S, Nieuwenhuis BPS, Weissensteiner B, Immler S, Wolf JBW (2021) Experimental evolution of adaptive divergence under varying degrees of gene flow. Nat Ecol Evol 5:338–349
Vähä J-P, Primmer CR (2006) Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15:63–72
Walther BD, Thorrold SR (2008) Continental-scale variation in otolith geochemistry of juvenile American shad (Alosa sapidissima). Can J Fish Aquat Sci 65:2623–2635
Waters JM, Epifanio JM, Gunter T, Brown BL (2000) Homing behaviour facilitates subtle genetic differentiation among river populations of Alosa sapidissima: microsatellites and mtDNA. J Fish Biol 56:622–636
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilcove DS, Wikelski M (2008) Going, going, gone: is animal migration disappearing. PLOS Biol 6:e188
Wu C-I (2001) The genic view of the process of speciation. J Evolut Biol 14:851–865
Yue GH, David L, Orban L (2006) Mutation rate and pattern of microsatellites in common carp (Cyprinus carpio L.). Genetica 129:329–331
Acknowledgements
We thank the many professional fishermen involved in gathering samples. We thank the institutions involved in collecting samples, namely people at INRAE, OFB and FDPPMA 14, 22, 50, and Migado association. We thank A. Xuereb for extensive revision of the manuscript grammar. This study was funded by the European Regional Development Fund (Transnational program Interreg IV. Atlantic Aquatic Resource Conservation Project).
Author information
Authors and Affiliations
Contributions
Conception: QR, GE, SL, JLB; Data Collection: QR, IL, YA, EL, EF, ER, FC, DJN, JLB; Molecular Laboratory: QR, ALB, SL; Data Analysis: QR, CP; Writing first draft: QR with help from SL and DJN; Reviewing: QR with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor Ben Evans
Supplementary information
Rights and permissions
About this article

Cite this article
Rougemont, Q., Perrier, C., Besnard, AL. et al. Population genetics reveals divergent lineages and ongoing hybridization in a declining migratory fish species complex. Heredity 129, 137–151 (2022). https://doi.org/10.1038/s41437-022-00547-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00547-9
