
Abstract
Absence of genetic differentiation is usually taken as an evidence of panmixia, but can also reflect other situations, including even nearly complete demographic independence among large-sized populations. Deciphering which situation applies has major practical implications (e.g., in conservation biology). The endangered harbor porpoises in the Black Sea illustrates this point well. While morphological heterogeneity suggested that population differentiation may exist between individuals from the Black and Azov seas, no genetic study provided conclusive evidence or covered the entire subspecies range. Here, we assessed the genetic structure at ten microsatellite loci and a 3904 base-pairs mitochondrial fragment in 144 porpoises across the subspecies range (i.e., Aegean, Marmara, Black, and Azov seas). Analyses of the genetic structure, including FST, Bayesian clustering, and multivariate analyses revealed a nearly complete genetic homogeneity. Power analyses rejected the possibility of underpowered analyses (power to detect FST ≥ 0.008 at microsatellite loci). Simulations under various demographic models, evaluating the evolution of FST, showed that a time-lag effect between demographic and genetic subdivision is also unlikely. With a realistic effective population size of 1000 individuals, the expected “gray zone” would be at most 20 generations under moderate levels of gene flow (≤10 migrants per generation). After excluding alternative hypotheses, panmixia remains the most likely hypothesis explaining the genetic homogeneity in the Black Sea porpoises. Morphological heterogeneity may thus reflect other processes than population subdivision (e.g., plasticity, selection). This study illustrates how combining empirical and theoretical approaches can contribute to understanding patterns of weak population structure in highly mobile marine species.
Similar content being viewed by others


Introduction
Delineating populations and their connectivity is of primary importance for the management of endangered and exploited species (Begg and Waldman 1999). In marine species, it facilitates identification of stocks, assessing exploitation status, and preserving the population genetic diversity underlying ecological resilience and adaptability (Begg and Waldman 1999; Palumbi 2003). Once distinct groups are identified, estimates of their effective size and migration rates is needed to assess their viability and resilience (Frankham 2010). These population parameters are particularly difficult to estimate for highly mobile species (e.g., marine mammals, turtles, and fishes) using direct field-based methods (e.g., sightings, tracking, or mark-recapture). Yet they are crucially needed to understand the impact of anthropogenic pressures (Payne et al. 2016) and the key roles many of these species play within food webs (Bowen 1997). Population genetic approaches provide a powerful alternative framework for estimating indirectly those parameters (Gagnaire et al. 2015).
Life-history traits of many marine species, such as high fecundity, large population sizes and high-dispersal potential, can lead to weak or no genetic differentiation over entire ocean basins (Waples 1998; Gagnaire et al. 2015). Indeed, the accumulation of genetic differentiation among populations by genetic drift depends on the effective population size (Ne) and the effective number of migrants (m) exchanged per generation (Ne x m), whereas the level of demographic interdependency depends only on the rate of migrants (m) exchanged (Lowe and Allendorf 2010). In other words, the genetic and demographic connectivity exhibit, in some conditions, a phase difference that prevents the former from being a good proxy of the latter. Such lag is proportional to Ne, which conditions the strength of the genetic drift. Common situations involving homogeneous distribution of genetic polymorphism can thus derive from a wide range of distinct demographic scenarios, depending on the relative weight of Ne and m. These scenarios range from a rate of migratory exchange high enough to lead to both genetic and demographic homogeneity among (sub-)populations (i.e., panmixia), even with limited effective population sizes, to nearly negligible migratory exchanges among populations exhibiting large effective sizes. Gagnaire et al. (2015) and Bailleul et al. (2018) described these effects and showed that the incomplete lineage sorting of populations can be considered as the homologous version at an intraspecific level of the “gray zone” of speciation described by De Queiroz (2007). This “gray zone” represents the time-lag during which, lineage sorting being incomplete, species delimitation is not possible based solely on the genetic information (Gagnaire et al. 2015; Bailleul et al. 2018). This concept of “gray zone” of population differentiation was coined by Bailleul et al. (2018) as the number of generations after a population split for genetic drift to change the allele frequencies in each diverging population and reach an equilibrium between migration and genetic drift (Epps and Keyghobadi 2015). During that period, a time-lag between genetic and demographic structure occurs, and no decision can be made from genetic data to assess whether two groups are demographically independent based solely on genetic data. The length of that period increases with Ne. It is therefore critical to assess whether the lack of genetic structure observed in a particular biological system results from such a time-lag effect, from a lack of genetic power, or from an actual demographic and genetic homogeneity. Marine species with large Ne, high fecundity, and high-dispersal abilities, such as fishes or invertebrates, are the primary species where such a lag between genetic and demographic processes is expected (see for example Waples 1998; Palumbi 2003; Gagnaire et al. 2015). In contrast, theory predicts that species with smaller Ne, lower fecundity, but high-dispersal abilities, such as marine mammals, should have a shorter “gray zone” period. Observing genetic panmixia in those species is thus more likely to reflect an actual absence of genetic and demographic population structure, rather than a demographic independence not yet captured by genetic data. However, even if the “gray zone” of population differentiation is expected to be short, it is important to consider this hypothesis to fully rule out this effect, especially when conservation and management of the focal group is at stake.
The endangered subspecies of harbor porpoise inhabiting the Black Sea (Phocoena phocoena relicta) is a good example to illustrate this point. The harbor porpoise is one of the three extant cetacean species crowning the Black Sea marine trophic food-web. P. p. relicta became isolated ca. 7000 years ago from the rest of the species range in the North Atlantic during the postglacial warming of the Mediterranean Sea, which became unsuitable for temperate species like porpoises (Fontaine et al. 2010; 2012; 2014 and reviewed in Fontaine 2016). Black Sea porpoises are recognized as a distinct subspecies based on morphological and genetic differences as compared to the North Atlantic porpoises (P. p. phocoena) (Viaud-Martinez et al. 2007; Fontaine et al. 2007; 2014; Galatius and Gol’din 2011; and reviewed in Fontaine 2016). In the Black Sea and adjacent waters (Fig. 1), porpoises are observed in the northern Aegean Sea, Marmara Sea, Black Sea, Kerch Strait, and Azov Sea (Fontaine 2016). The Black Sea harbor porpoise is listed as “endangered” by the IUCN (Birkun and Frantzis 2008). These porpoises were hunted to near extinction between the 1930s and the 1980s, causing a ~90% population decline (Birkun 2002; Fontaine et al. 2012; Vishnyakova 2017). Subsequent incidental catches in fisheries reached thousands of porpoise casualties annually through the 1980s and are likely to have increased since then (Birkun and Frantzis 2008; Vishnyakova and Gol’din 2015; Vishnyakova 2017). Having a clear understanding of their genetic structure is thus crucial for devising conservation strategies (Allendorf et al. 2012).

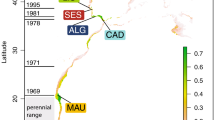
Map showing the sampling locations. The radius of the circles is proportional to the sample size. Rectangles and circles represent individuals sampled, respectively, in Fontaine et al. (2012) and this study. AG, Aegean Sea; MS, Marmara Sea; BS, Black Sea; KS, Kerch Strait; AZ, Azov Sea
It is still unclear whether the Black Sea porpoises are composed of a single-homogeneous demographic and genetic unit or multiple interconnected demes, but differentiated enough to be considered as distinct populations. Some authors suggested that population subdivision might exist (Rosel et al. 2003; Gol’din 2004; Tonay et al. 2017). For example, morphological differences between porpoises from the Black and Azov seas suggested that they may belong to differentiated subpopulations (Gol’din 2004; Gol’din and Vishnyakova 2015, 2016). For instance, compared to animals from the Black Sea, porpoises from the Azov Sea display slightly larger body sizes (Gol’din 2004) and distinct skull sizes and shapes (Gol’din and Vishnyakova 2015, 2016). The authors suggested that these differences may reflect distinct feeding ecology, ontogeny, and thus possibly demographically and genetically distinct units. However, so far, no genetic analysis has been conducted to test whether porpoises from the Azov Sea were genetically differentiated from those in the Black Sea. Population genetic studies were conducted on other populations from the Black Sea and adjacent waters (i.e., Turkish Straits System and Aegean Sea) with contradicting results. For example, shallow but statistically significant differences in haplotype frequencies of the mitochondrial control-region (mtDNA-CR) were interpreted by Viaud-Martinez et al. (2007) and Tonay et al. (2017) as evidence of population subdivision between groups from the Marmara Sea and the Black Sea. However, their analyses were limited by a small sample size in the Marmara Sea (respectively, n = 3 and n = 5) and the analysis of a single locus consisting of a short fragment of the mtDNA-CR (≤364 bps). Furthermore, given that the authors only considered the mitochondrial locus, they could not test whether such differentiation of the Marmara porpoises could result from processes other than population subdivision. For example, a high degree of relatedness among samples (e.g., members of the same family) can generate spurious signals of genetic differentiation (Anderson and Dunham 2008; Rodriguez Ramilio and Wang 2012). In contrast, another study combining ten highly polymorphic nuclear microsatellites and mtDNA-CR loci to screen for genetic variation across the subspecies range (i.e., Black, Marmara and Aegean seas), excluding the Azov Sea, failed to detect any significant evidence of genetic structure (Fontaine et al. 2012). This study suggested that porpoises from the Black Sea and adjacent waters formed a panmictic population. Clarifying conflicting genetic evidences in this endangered subspecies is thus needed. Furthermore, in case of an absence of genetic structure, determining whether it results from underpowered analyses, a population “gray zone” effect, or from actual panmixia is of paramount importance to provide a meaningful biological interpretation of this genetic homogeneity and to design efficient conservation strategies.
In this study, we aimed to provide a comprehensive picture of the genetic structure of the harbor porpoise in the Black Sea and adjacent waters. We augmented the previous microsatellite data set of Fontaine et al. (2012) obtained for 89 porpoises from the Aegean, Marmara, and Black seas, with 55 new samples from the Black Sea, Azov Sea, and Kerch Strait (Fig. 1 and Supplementary Table S1). For a subset of the sampling in the Black Sea and Azov Sea, we also sequenced a 3904 bps long mitochondrial fragment encompassing five genes, since Fontaine et al. (2014) showed it had a higher power than the mtDNA-CR to discriminate among distinct lineages. Using this data set, we reassessed the genetic evidence of population subdivision previously reported based on phenotypic (Gol’din and Vishnyakova 2015, 2016) or mtDNA-CR variation (Viaud-Martinez et al. 2007; Tonay et al. 2017). Using the microsatellite data set, we also tested whether relatedness rather than population subdivision could account for the previously reported genetic distinctiveness of the Marmara porpoises compared to the others (Viaud-Martinez et al. 2007; Tonay et al. 2017). Finally, we built a theoretical framework to interpret an absence of genetic structure, and decipher the possible hypotheses, which would explain such panmixia (i.e., limited power, population “gray zone” effect, or panmixia). Specifically, we used power analyses and simulations under various demographic and migration models to evaluate the evolution of FST by genetic drift through time.
Materials and methods
Sampling and data collection
The samples used in this study originated from five geographic locations: the Aegean Sea, Marmara Sea, Black Sea, Kerch Strait, and Azov Sea (Fig. 1, Table 1, and Supplementary Table S1). Genotypes at ten microsatellite loci for 89 porpoises from the Aegean, Marmara, and Black seas were taken from Fontaine et al. (2012). We added 55 newly genotyped individuals from the Azov Sea, Kerch Strait, and Black Sea. The final data set included 144 individuals covering the complete subspecies range (Azov Sea: N = 32, Black Sea: N = 87, Marmara Sea: N = 3, Aegean Sea: N = 11, Kerch Strait: N = 3, and four individuals of unknown locations) (Fig. 1 and Table 1, and for details see Table S1). The new tissue samples were collected from dead animals stranded along the coasts of the Black Sea, the Crimea peninsula (Ukraine), the Kerch Strait, and the Azov Sea, and kept in dymethyl sulfoxide (DMSO) saturated with sodium chloride (NaCl) until analyses. Total genomic DNA was extracted from tissues using a PureGene and DNeasy Tissue kit (Qiagen), following the manufacturer’s recommendations. The microsatellite genotyping procedure followed the protocol described in Fontaine et al. (2006; 2007).
In addition to the nuclear microsatellite data set, we sequenced a 3904 base-pair fragment of the mtDNA genome encompassing five coding regions (CytB, ATP6, ATP8, ND5, and COXI) for ten individuals (Azov Sea: N = 6, Black Sea: N = 3, and Kerch Strait: N = 1) and combined it with the 12 sequences previously obtained for porpoises from the Black Sea in Fontaine et al. (2014), following the same protocol. Since the porpoises from the other locations were surveyed using the mtDNA-CR in previous studies (Viaud-Martinez et al. 2007; Fontaine et al. 2012; Tonay et al. 2017), we focused here only on comparing porpoises from the Azov and Black seas. We used Geneious v.10.0.9 (Kearse et al. 2012) to visually inspect raw sequences, assemble contigs, and perform multiple sequence alignments using MUSCLE (Edgar 2004) with the default settings.
Genetic diversity at the microsatellite and mitochondrial loci
Genetic diversity at the microsatellite loci was quantified over the entire sampling (global) and per geographic location (local) using allelic richness (Ar), expected heterozygosity (He), and observed heterozygosity (Ho). Global Ar was calculated using Fstat v.2.9.3.2 (Goudet 1995). Global and local Ho and He were calculated using GenAlEx v.6.5 (Peakall and Smouse 2012). Local Ar and private Ar (pAr) were estimated using ADZE (Szpiech et al. 2008), assuming a standardized sample size of two individuals to account for differences in sample size among localities and align the values on the smallest sample (Szpiech et al. 2008). We tested for significant differences in Ar, pAr, Ho, and He among locations using Wilcoxon signed-ranked tests. Adjustment for multiple comparisons was performed using a Bonferroni correction (error rate α = 0.05). Overall departure from Hardy–Weinberg Expectation (HWE) was tested using an exact test (Guo and Thompson 1992), implemented in Genepop v.4.7.0 (Rousset 2008) and we quantified this departure using the FIS estimator of Weir and Cockerham (1984) in GenAlEx v.6.5.
The variation among mitochondrial sequences was assessed using various statistics, including the number of segregating sites (S), number of singletons, number of shared polymorphisms (Shared P), number of haplotypes (#hap), haplotype diversity (Hd), two estimators of population genetic diversity θπ (Tajima 1983) based on the average number of pair-wise differences (K), and θw (Watterson 1975) based on the number of segregating sites. Tajima’s D was also estimated to assess departure from neutral expectations, such as change in population size or selective processes. The significance level of D was estimated using 10,000 coalescent simulations. All these statistics were computed using DnaSP v.5.10.01 (Librado and Rozas 2009).
Mitochondrial phylogenetic relationships
Phylogenetic relationships among mtDNA haplotypes were estimated using the maximum-likelihood approach of PhyML v3.0 (Guindon et al. 2010), implemented as a plug-in in Geneious v.10.0.9 (Kearse et al. 2012). We used jModelTest2 (Darriba et al. 2012) to select the model of nucleotide substitution best fitting with our sequence alignment. The tree was rooted with two mitochondrial sequences of Dall’s porpoise (Phocoenoides dalli) from Fontaine et al. (2014). We drew the phylogenetic trees using FigTree v.1.4.3 (Rambaut and Drummond 2012). Node support was estimated using 1 × 104 bootstrap replicates. As a complementary visualization of phylogenetic relationships among haplotypes, we also reconstructed a Median-Joining haplotype network (Bandelt et al. 1999) using PopART (http://popart.otago.ac.nz).
Relatedness
Considering closely related individuals to delineate population genetic structure can generate spurious signals of population structure and violates the assumptions of population genetic approaches, such as the model-based Bayesian clustering (Anderson and Dunham 2008; Rodriguez Ramilio and Wang 2012). Therefore, we used the microsatellite data set to analyze patterns of relatedness among individuals using the R package related v.1.0 (Pew et al. 2015) in the R statistical environment v.3.5.3 (R Core Team 2019). Specifically, we estimated the relatedness coefficient (r) among individuals and tested whether it was greater within each location than expected by chance. As the performance depends on the characteristics of the data set (Csilléry et al. 2006) and on the estimators, we compared seven estimators implemented in the related package following the user-guide recommendation using the function “compareestimators()”. This approach generates 1000 simulated data sets with the same characteristics as the observed microsatellite data set. Then, for each estimator, a Pearson’s r correlation coefficient is computed between the observed and simulated values. Wang’s (2002) r estimator provided the best performance for our data set (i.e., highest correlation coefficient) and was thus chosen for the analysis. We assessed whether individuals within each location were more closely related to each other than expected by chance. To do so, we compared the observed r value in each location against the null distribution of pair-wise average r generated by randomly shuffling individuals among populations for 1000 permutations while keeping the population size constant. If some individuals are highly related within a population, the observed r value is expected to be higher than the simulated r values obtained by permutations. To assess the significance of the test, an empirical p-value was obtained by comparing the observed average r value for each population with the null distribution by counting the number of times the observed value was greater than those obtained from permuted data. We applied a Bonferroni correction to adjust for multiple comparisons with a significance threshold of 0.01.
Population genetic structure
We assessed the genetic structure among porpoises with the Bayesian clustering approach of STRUCTURE v.2.3.4 (Pritchard et al. 2000; Hubisz et al. 2009), using an admixture “locprior” model and correlated allele frequencies among clusters (Hubisz et al. 2009). This parametrization is suitable for detecting weak genetic structure when it exists, yet without forcing it (Hubisz et al. 2009). The sampling location of each individual was used as prior information in the locpior model. We conducted a series of independent runs with different numbers of clusters (K) ranging from 1 to 7. Each run used 1 × 106 iterations after a burn-in of 1 × 105 iterations with 10 replicates per K value. We assessed convergence of the Monte Carlo Markov Chains (MCMC) using CLUMPAK (Kopelman et al. 2015). We determined the best K value using (1) the log likelihood of the data for each K value, (2) the rate of change of K with increasing K (Evanno et al. 2005), and (3) the visual inspection of newly created cluster as K increased. For steps (1) and (2) we used STRUCTURE HARVESTER v.0.6.94 (Earl and vonHoldt 2011).
We also investigated genetic structure using a principal component analysis (PCA) on the allele frequencies (Jombart et al. 2009). This analysis does not rely on any model assumptions and provides a complementary visualization of the genetic structure. This analysis was conducted in R (R Core Team 2019) using the adegenet v.2.1 package (Jombart 2008; Jombart and Ahmed 2011) on centered data (i.e., centering the mean allele frequency on zero), with missing data replaced by the mean value as recommended by the authors. We also conducted a discriminant analysis of principal components (DAPC) (Jombart et al. 2010). The analysis uses the principal components (PCs) of the PCA to maximize differences among predefined groups using a discriminant analysis. We used the sampling locations as putative grouping. The number of PCs retained and the reliability of the DAPC were assessed using the a-score approach, following the user-guide recommendation. As a result, a total of 21 PCs and 4 discriminant functions were retained to describe the relationship between the clusters, which captured 91% of the total genetic variation.
Genetic differentiation among populations
For microsatellites data, we estimated the overall and pair-wise departure from HWE due to population subdivision using the Weir and Cockerham’s (1984) FST estimator. The pair-wise comparisons were carried among pairs of geographical locations (i.e., the Aegean Sea, Marmara Sea, Black Sea, Kerch Strait, and Azov Sea). The 95% confidence interval (95% CI) was estimated using 5000 bootstrap resampling with the DiveRsity v1.9.90 R package (Keenan et al. 2013). The significance was tested using an exact G-test (Goudet et al. 1996) implemented in Genepop v.4.7.0 (Rousset 2008), with default options. We used a Bonferroni correction to adjust the p-value to 0.05 of the pair-wise comparisons to account for multiple comparisons.
For mtDNA data, due to the absence of samples for other locations, we only quantified the genetic differentiation between porpoises from the Black and Azov seas using the Hudson’s estimator of FST (Hudson et al. 1992) in DnaSP v.5.10.01. Significance was tested with 10,000 permutations of Hudson’s nearest neighbor distance Snn statistics (Hudson 2000) in DnaSP. We also conducted an exact test on the mtDNA haplotype frequencies using Arlequin v.3.5.2.2 (Excoffier and Lischer 2010).
We assessed the statistical power of our markers to detect genetic differentiation given the observed genetic diversity and sample sizes using the POWSIM v4.1 program (Ryman and Palm 2006). POWSIM assesses whether the observed data set carries enough statistical power (i.e., ≥80%) to detect a Nei’s FST (FST–Nei) value significantly larger than zero, using X2 and Fisher tests (Ryman and Palm 2006). Parameters of the Markov chains, including the burn-ins, batches and iterations per run were set, respectively, to 10,000, 200, and 5000. Allele frequencies were estimated with GenAlEx and haplotype frequencies with DnaSP. Sample sizes were divided by two for the mtDNA to reflect the sampling of haploid genes (Larsson et al. 2008). Observed FST–Nei for microsatellite and mitochondrial data were calculated using DiveRsity v1.9.90 (Keenan et al. 2013), and the mmod v.1.3.3R package (Winter 2012), respectively. Ne was fixed to 1000 and the number of generations (t) was adjusted to obtain FST–Nei values ranging from 0.001 to 0.15 for microsatellites and from 0.001 to 0.4 for mtDNA.
Simulations of population connectivity and “gray zone” of population differentiation
We assessed whether an absence of significant genetic structure could result from a time-lag effect between demographic and genetic processes, using the simulation approach of Bailleul et al. (2018) adapted to our system. Simulations were used to assess the number of generations required to overcome the population “gray zone” and detect FST values significantly >0 for a pair of diverging populations. Specifically, we used simuPOP v.1.1.7 (Peng and Amos 2008) to conduct forward-time simulations of two diverging populations of random mating individuals (recombination rate of 0.01) to generate genetic data sets with similar properties to the one observed (ten loci with ten allelic states). To mimic the founding event of the Black Sea subspecies 700 generations ago (or ca. 7000 years before present) (Fontaine et al. 2010; 2012; 2014), we simulated an initial population with an effective size Neini, that split into two daughter populations 700 generations ago, each diverging from each other with a constant effective population size Necur. As the time to overcome the population “gray zone” depends on Ne and m, we ran the simulations assuming three values for Neini=10, 100, or 1000 individuals, thus testing a gradient in the strength of the founding effect, one value of Necur=1000 individuals (based on previous Ne estimates ranging between 360 and 700, Fontaine et al. 2010; 2012), and four values of symmetrical migration rates m set in such a way that the effective number of migrants per generation (Necur × m) was equal to 0 (no migration), 1, 10, or 100. For each of the 12 parameter combinations, we sampled the FST values during the differentiation process every seven generations (100 data points). For each time point, FST values were estimated based on the 1000 individuals in each population. At each time point, 100 sub-FST values were estimated based on a subsample of 50 in each population to mirror a realistic field sampling of natural populations. Significance of the sub-FST was assessed by randomly shuffling 1000 times the individuals in the subsamples and computing the FST. A p-value was derived from this null randomized FST distribution and estimated as the proportion of randomized FST inferior or equal to the simulated sub-FST. Finally, our ability to detect a FST value significantly >0 (in percent) was estimated by counting, out of the 100 replicates, the proportion of sub-FST with p-values ≤ 0.05.
Results
Genetic diversity at the microsatellite and mitochondrial loci
Out of the 144 individuals genotyped for the ten microsatellite loci, the total level of observed missing data reached 6.93%. Across all geographic areas and loci (Table 1 and S2), we observed an average allelic richness (Ar) of 7.5 and a genetic diversity (He) of 0.50. No departure from Hardy–Weinberg Equilibrium (HWE) was observed (FIS = -0.01, p-value = 0.956), indicating no detectable departure from panmixia. Moreover, we observed a genetic homogeneity in genetic diversity of the porpoises across the five sampled areas, each one displaying no deviation from HWE and no detectable differences in genetic diversity among each other (Table 1 and S2). For a standardized sample size of two individuals, Ar and private Ar (PAr) values ranged from 1.49 to 1.51 and 0.21 to 0.30 among the five sampled areas, respectively, without any significant differences among them (Table 1; Wilcoxon signed-ranked (WSR) test with a p-value > 0.05). The observed and expected heterozygosity (Ho and He) were also comparable among geographic areas, ranging between 0.50 and 0.59 for Ho and between 0.37 and 0.50 for He (WSR test with a p-value > 0.05).
For the 3904 bp mtDNA fragment analyzed, a total of 25 segregating sites defined 15 distinct haplotypes, with a haplotypic diversity of 0.93 ± 0.05 and a nucleotide diversity of 8.9 × 10-4 ± 1.9 × 10-4 (Table 1). The phylogenetic relationships among haplotypes revealed a star-like topology on the maximum-likelihood phylogenetic tree (Fig. 2a) and haplotype network (Fig. 2b). Indeed, rare haplotypes were all closely related to a dominant haplotype, with only one or two mutations. This topology is consistent with significant excess of rare over shared variants, as captured by the significant negative value of Tajima’s D statistics (−2.19; p-value < 0.01; Table 1).

Population structure observed at the mtDNA and microsatellite loci. a Maximum-likelihood mitochondrial phylogeny rooted with Dall’s porpoise sequences (not shown). The labels’ colors indicate the sampling location. Red circles on nodes represent bootstrap support >70%. b Median-joining mitochondrial haplotype network. Each circle represents a haplotype and the size is proportional to the observed haplotype frequency. Pie-chart sectors indicate the number of haplotypes observed in each locality. Mutational steps between haplotypes are represented on the branch. c Barplots of the Bayesian clustering analyses of STRUCTURE for K from 1 to 5. Each individual is represented by a vertical line divided into K segments showing the admixture proportions for each cluster. Vertical black lines delimit the sampled localities. d Scatter plot displaying the individual scores along the first two components of the principal component analysis. The proportion of variance explained by each axis and the first Eigen values (bottom left inset) are provided. AG, Aegean Sea; MS, Marmara Sea; BS, Black Sea; KS, Kerch Strait; AZ, Azov Sea
Relatedness
Relatedness estimates (r) among porpoises within each sampled locality revealed that only the three individuals from the Marmara Sea displayed an r value significantly greater than expected by chance alone (p-value < 0.001). The average r for these individuals was 0.55 ± 0.15, which corresponds to a parent–offspring or full sibling relationship. For all other populations, r values ranged from 0.06 to 0.08 as expected for unrelated individuals (Supplementary Fig. S1 and Table S3).
Population genetic structure
The clustering analyses of STRUCTURE did not reveal any evidence of population subdivision, irrespective of the number of clusters (K) tested (Fig. 2c and Supplementary Fig. S2a). The highest posterior probability for the data (X) of containing K clusters, Ln(Pr(X|K), was observed for K = 1 and K = 2 and was much lower for higher K values (Supplementary Fig. S2b). Regardless of the K value tested, individual patterns of admixture were identical for all individuals, suggesting that harbor porpoises from the different localities behave as a panmictic population. The analysis provided consistent results over ten replicated runs performed for each K (Supplementary Fig. S2a).
The principal component analysis (PCA) supported the results of STRUCTURE by showing no evidence of population subdivision, as all multilocus genotypes grouped into a single cluster (Fig. 2d). The discriminant analysis of principal component (DAPC; Supplementary Fig. S3), which focuses on optimizing the differences between predefined clusters (here the sampled localities) while minimizing the differences within groups, showed globally similar results as STRUCTURE (Fig. 2c). No genetic subdivision could be observed between individuals from the Black Sea and the Azov Sea, which are located in the center of the DAPC (Supplementary Fig. S3). Similarly, there was no clear separation between the individuals from the Kerch Strait, Marmara Sea, and Aegean Sea. Repeating the clustering analyses (STRUCTURE, PCA, and DAPC) keeping only one individual from the Marmara Sea (thus excluding the two other related samples) did not change the results (results not shown).
The absence of genetic structure at the microsatellite loci was further supported by the very low global FST values (FST–WC = 0.009 and Nei’s FST–Nei = 0.022), not significantly departing from zero (p-value = 0.109). Similarly, all pair-wise comparisons displayed non-significant differences in allelic frequencies (Table 2). Only the FST value between Marmara and Aegean porpoises was slightly higher (FST–WC ≥ 0.095 and FST-Nei ≥ 0.036), but none departed significantly from zero, and only the FST–WC did not include 0 in the 95% CI (Table 2). There was no obvious clustering according to geography for the mtDNA locus as well. Out of the 15 haplotypes identified, three were unique to the porpoises from the Azov Sea, nine only found in the Black Sea porpoises, and three were shared between the two (Table 1 and Fig. 2a, b). Consistent with these results, we did not detect any signal of population differentiation at the mtDNA locus between the porpoises from the Azov and Black seas (Hudson’s FST =0.007, Nei’s FST = 0.013, Snn = 0.519, Snn’s p-value = 0.726, and exact test on mtDNA haplotype frequencies p-value = 0.95). This suggests no mitochondrial genetic subdivision between porpoises from the Black Sea and Azov Sea.
The simulation-based assessment of the statistical power to detect significant differentiation as performed in POWSIM (Supplementary Fig. S4) indicated that our microsatellites and mitochondrial data sets have the power to detect significant differentiation for FST–Nei > 0.008 and FST–Nei > 0.1, respectively (Spplementary Fig. S4). Therefore, the lack of genetic differentiation observed at these loci among the five sampled locations does not simply result from a lack of statistical power.
Simulations of “gray zone” of population differentiation
In agreement with Bailleul et al. (2018), FST values estimated either from the entire simulated populations or from subsamples of 50 individuals were very similar irrespective of the effective population size (Ne) or the number of migrants exchanged (Necur.m) (Fig. 3 and Supplementary Fig. S5). Simulations showed that with a constant contemporary effective size (Necur) of 1000 reproducing individuals, as the number of effective migrants (Necur.m) increases, the power to detect significant genetic differentiation decreases and the number of generations to overcome the population “gray zone” increases. With less than one migrant per generation (Necur.m ≤ 1), it takes at most seven generations to obtain a power of 100% to detect significant FST and to reach FST values ≥ 0.1 after 700 generations. With ten migrants per generation (Necur.m = 10), a high power (>80%) to detect significant FST is reached in the 20 first generations, then between 20 and 700 generations, the detection capacity varies between 80 and 100% and the FST values vary around 0.017. With a high connectivity between the two diverging populations (Necur.m = 100), the detection ability stays below 40% during the 700 generations and the simulated FST values are lower than 0.002 (Fig. 3 and Supplementary Fig. S5). Variation in the initial Ne of the founding ancestral population (Neini), which mirrored the founding event of the harbor porpoise population in the Black Sea 700 generations ago (Fontaine et al. 2012), had no effect on the detection capacity and on the FST values (Fig. 3 and Supplementary Fig. S5).

Impact of the “gray zone” of population differentiation, varying level of connectivity, and number of founders on the genetic differentiation between two hypothetical diverging populations, illustrated using simulations. Simulations correspond to two populations, each one with an effective size of 1000, splitting from a small ancestral population with variable initial sizes (Nini), and variable migration rates (m), and number of migrants (N.m) after the split. For each plot, the x-axis shows the number of generations since the split from the ancestral population. Only the first 200 generations out of the 700 are shown (see Fig. S5 for the entire simulations). The right y-axis displays the evolution of FST values. The median FST values and their 95% CI are displayed in blue plain and dashed lines, respectively. The left y-axis shows the proportion of FST values significantly different from zero (green line). The vertical gray shades represent the “gray zone” of population differentiation, defined as the number of generations since the split during which FST values are unlikely to be statistically different from 0 in >95% of the cases
Discussion
Highly mobile marine species can display combinations of life-history traits (e.g., high fecundity, large population sizes, high-dispersal potential) that produce weak pattern of genetic differentiation or even no differentiation at all across large geographic scales (Ward et al. 1994; Waples 1998; Palumbi 2003; Hedgecock et al. 2007; Gagnaire et al. 2015). For instance, due to high-dispersal abilities, species like the common dolphin (Delphinus delphis) or the deep-water squaloid shark, the Portuguese dogfish (Centroscymus coelolepis), form a single panmictic population across the eastern North Atlantic (Moura et al. 2013; Veríssimo et al. 2011). Even at world ocean scale, blue sharks (Prionace glauca) showed a nearly complete genetic homogeneity, most likely because of large effective population sizes and dispersal abilities (Bailleul et al. 2018). Here, we report a similar atypical genetic homogeneity for the harbor porpoise in the Black Sea and adjacent waters, despite morphological evidence for heterogeneity between individuals from the Black Sea and Azov Sea. These examples raised the question of whether a single panmictic population exists or if this genetic homogeneity comes from a lack of analytical power, or from a genetic time-lag effect generating a “gray zone” of population differentiation. Deciphering among these hypotheses is rarely done in practice, but its implications for conservation and management of endangered species can be of paramount importance, as it is the case for harbor porpoises in the Black Sea (Birkun and Frantzis 2008).
Panmixia in harbor porpoises from the Black Sea and adjacent waters
The widespread genetic homogeneity observed in the Black Sea harbor porpoises is consistent with previous investigations that reported similar results between individuals from the Aegean and Black seas (Fontaine et al. 2012). Here, we report that this homogeneity further extends to the new zones surveyed in this study, including the Crimea peninsula, Kerch Strait, and Azov Sea. Genetic panmixia suggests that random mating occurs across the subspecies distribution or that population subdivision is too weak or too recent to have left a detectable signature on the genetic markers analyzed in this study. Such homogeneity is supported by the absence of clustering of the microsatellite genotypes in the STRUCTURE (Fig. 2c), PCA (Fig. 2d), and DAPC analyses (Supplementary Fig. S3), and the absence of significant differences in genetic diversity (Table 1) and allelic frequencies (Table 2). The POWSIM power analysis showed that this homogeneity does not result from a lack of power of the microsatellite data to reject panmixia, since simulated data sets with the same number of markers and comparable genetic diversity would be able to detect significant FST values as low as 0.008. Although sample sizes were small, mtDNA data also supported such homogeneity, with no differentiation between porpoises from the Black and Azov seas.
Previous studies (Viaud-Martinez et al. 2007; Tonay et al. 2017) reported significant differences in mtDNA-CR haplotype frequencies between porpoises from the Marmara Sea and those from the neighboring areas. This led Tonay et al. (2017) to suggest that a genetically differentiated population may exist in the Marmara Sea. In this study, we questioned this idea since the three samples analyzed here are the same as in Viaud-Martinez et al. (2007) and were also included in Tonay et al. (2017). All three individuals shared the same mitochondrial haplotype (see Appendix 1 in Viaud-Martinez et al. 2007). Their relatedness estimated with the microsatellite data (r = 0.55, Supplementary Fig. S1 and Table S3) indicated that these individuals are from the same family (parent–offspring or full sibling relationship). Such samplings of related individuals should be avoided in population genetic analyses, because this can produce spurious signals of population structure (Anderson and Dunham 2008; Rodriguez Ramilio and Wang 2012). Given the low sample size (n = 3 in the present study and in Viaud-Martinez et al. 2007; and n = 5 in Tonay et al. 2017) and its biased composition, no reliable conclusion can be drawn at this point. Unrelated samples from the Marmara Sea are required to resolve the status of the porpoise in that area.
Genetic homogeneity is expected given the large dispersal abilities of porpoises
Large-scale genetic panmixia is expected and frequently reported in highly mobile marine species living in an environment where geographical barriers to dispersal are scarce (Quintela et al. 2014; Bailleul et al. 2018). In the case of the Black Sea porpoises, such homogeneity is expected given the large oceanographic connectivity among the adjacent seas (ex. Aydoğdu et al. 2018), the large dispersal abilities and habitat occupation of the species reported in other areas. For example, Nielsen et al. (2018) showed that the total habitat occupation of 72 porpoises tagged in the Danish waters of the North Sea could reach up to ~600,000 km2. Even more striking, 30 porpoises from Western Greenland displayed large-scale offshore movements and occupied a total habitat of 4,144,749 km2. Daily traveling rates can range between 20 and 50 km in a single day (Nielsen et al. 2018). Thus, the dispersal abilities of harbor porpoises are comparable to, or can exceed the total surface of the Black Sea (436,000 km2), Azov Sea (39,000 km2), Aegean Sea (214,000 km2), and Marmara Sea (11,350 km2). Furthermore, the continental climate prevailing in the northern Black Sea and Azov Sea can lead to rapid ice formation, forcing porpoises to leave the Azov Sea during winter when it becomes completely frozen (Matishov et al. 2014). Massive porpoise mortalities due to ice entrapment have been reported in the past (Kleinenberg 1956; Birkun 2002). Therefore, the absence of barriers to gene flow, the large dispersal abilities of the species, the small geographic scale, the frequent movements reported between the different seas (Kleinenberg 1956; Vishnyakova et al. 2013), and the unavailability of some habitats for part of the year, all point towards highly connected demes of porpoises in the Black Sea and adjacent waters. Our simulations (Fig. 3 and Supplementary Fig. S5) showed that moderate levels of connectivity (Ne.m = 10 migrants per generation) could lead to weak, but still rapidly detectable differences in allelic frequencies in <20 generations. Such a result is conservative, since the simulations assumed an effective number of reproducing individuals (Ne) of 1000 in each hypothetical diverging group. Previous Ne estimates for the Black Sea harbor porpoises ranged between 360 (Fontaine et al. 2010) and 700 individuals (Fontaine et al. 2012). Therefore, smaller Ne than those used in our simulations would lead to even faster genetic drift of the allele frequencies, and thus to a faster ability to detect high and significant FST values.
A short “gray zone” of population differentiation is expected in cetacean species
Genetic homogeneity does not necessarily imply demographic homogeneity. After a population split, a certain number of generations is required for genetic drift to change the allele frequencies in the diverging populations and reach a migration-drift equilibrium (Epps and Keyghobadi 2015). The time-lag before which genetic variation becomes a good proxy of demographic subdivision, the “gray zone” of population differentiation, is dependent on Ne and thus on the life-history traits of the species (Waples 1998; Gagnaire et al. 2015; Bailleul et al. 2018). In species exhibiting high fecundity and large population sizes (i.e., Ne between 104 and 107)—for example in fish such as herring, anchovy, salmon, blue shark—genetic drift can be ineffective and genetic differentiation very weak or even absent (Waples 1998; Gagnaire et al. 2015; Bailleul et al. 2018). Bailleul et al. (2018) showed that with a Ne value of 104 reproducing individuals, an average of 200 generations was required to obtain a detection capacity of significant FST in 95% of cases. However, this “gray zone” increased to 1000 generations with a Ne of 105 even if no gene flow occurred between the diverging populations. In those species, a lack of genetic differentiation can result from a range of situations spanning from nearly complete demographic independence among large-sized populations to the existence of a unique panmictic population (Palumbi 2003; Gagnaire et al. 2015).
In contrast, species with much lower fecundity and Ne (i.e., 102 to 104), such as cetacean species (Hoelzel 1998; Read 1999), should display a much shorter population “gray zone”. Our simulations confirmed this expectation (Fig. 3 and Supplementary Fig. S5). Assuming a current effective population size (Ne) of 1000 reproducing individuals, which is a conservative estimate for the porpoises in the Black Sea (Fontaine et al. 2010; 2012), rejection of panmixia was quickly achieved with good power even for moderate levels of gene flow between the diverging populations (Ne.m = 10 or 1% of the total population size). Indeed, panmixia was rejected in 95% of the cases in <20 generations (Fig. 3 and Supplementary Fig. S5). This “gray zone” would be even shorter with lower connectivity (seven generations with a Ne.m = 1 or 0.1%, Fig. 3 and Supplementary Fig. S5). In the case of harbor porpoises, a time-lag effect ranging between 7 and 20 generations would correspond to 70 to 200 years, assuming a conservative generation time of 10 years (Read 1999). This means that, for populations that are isolated long enough, genetic differentiation would be detectable with good power, unless the split occurred within the last 200 years. If Ne was smaller, which is likely the case for the Black Sea porpoises, genetic drift would be more efficient and population differentiation could be detected even more rapidly. It is thus unlikely that the genetic panmixia observed in harbor porpoises from the Black Sea and adjacent waters is the result from a population “gray zone” effect. When connectivity among demes increases and reaches 10% of the total population size (Ne.m = 100 migrants per generation), our simulations (Fig. 3 and Supplementary Fig. S5) showed that migration rates no longer allow demographic units to be independent. This result is consistent with previous studies (e.g., Palsbøll et al. 2007) and showed that populations can no longer be differentiated from a genetic and demographic perspective. Therefore, even if the effective population size is low for harbor porpoises in the Black Sea and adjacent waters, level of gene flow among demes is high enough to maintain genetic panmixia.
Genetic homogeneity in the face of morphological heterogeneity
Significant morphological differences were previously reported between porpoises from the Black Sea and Azov Sea (Gol’din 2004; Gol’din and Vishnyakova 2015; 2016). These authors hypothesized that such phenotypic differences could reflect demographically, ecologically and genetically differentiated groups. However, our genetic results currently do not support this hypothesis. All the analyses and simulations conducted in this study pointed to genetic panmixia, which does not result from a lack of power of the genetic data set or from a population “gray zone” effect. Such a discrepancy between genetics and morphology has been widely reported (Rheindt et al. 2011). A first plausible explanation could be that the observed morphological variation between Azov and Black Sea porpoises is related to phenotypic plasticity. Adaptation to distinct ecological conditions can trigger differences in gene expression, leading to morphological variation without DNA modification (Duncan et al. 2014). If porpoises are adapted to distinct local environmental conditions, morphological differences could result from such phenotypic plasticity, without being underpinned by genetic variation. A second plausible hypothesis is that the few selectively neutral loci used in this study may not reveal genetic differentiation occurring in other places of the genome that are involved in ecological adaptation (Gagnaire et al. 2015). Markers evolving under divergent selection can form localized islands of differentiation, meaning regions of high-genetic differentiation along the genome. These are good evidence that divergent adaptive processes are ongoing (Turner and Hahn 2010). Examples of such genomic islands of differentiation have been reported in high fecundity, large population sizes, and highly dispersive species such as sticklebacks (Ravinet et al. 2018), cichlid fishes (Malinsky et al. 2015), and Anopheles mosquitoes (Turner and Hahn 2010). They are characteristic of incipient ecological differentiation in the presence of heterogeneous gene-flow along the genome. In these systems, portions of the genome involved in ecological adaptations would remain differentiated while the neutrally evolving portions of the genome would freely recombined and homogenize (Gagnaire et al. 2015). Such a pattern in harbor porpoises, if it exists, could be of paramount importance since it would suggest that locally adapted stocks occur in the Black Sea and could be the basis to define distinctive conservation units (Funk et al. 2012; Gagnaire et al. 2015). Testing such hypotheses will require genome-scale analyses and should be a perspective for future studies.
Conclusions
Deciphering among the various hypotheses to explain genetic panmixia in a species can be of paramount importance, especially when the species faces conservation issues. Here, we showed how empirical population genetic analyses and power analyses can be nicely complemented with a simulation-based framework to generate theoretical expectations to interpret patterns of weak genetic structure in highly mobile marine species with few or no barriers to dispersal. We illustrated this through the example of the harbor porpoises from the Black Sea and adjacent waters. Understanding the population structure of this endangered cetacean subspecies endemic to the Black Sea is crucially needed in order to assess the impact of various threats to its survival and inform management decisions. Despite previous evidence of phenotypic heterogeneity between porpoises from the Azov and Black seas, the present study did not reveal any departure from the panmixia hypothesis, suggesting that dispersal and gene flow are large enough to maintain genetic homogeneity at the scale of the Black Sea and adjacent waters. We showed that this result was not simply due to underpowered analyses. Using simulations, we also showed that given the small effective population observed in the Black Sea harbor porpoise, which is typical of many cetacean species facing conservation issues (i.e., 102 to 103 individuals), it is unlikely that the observed genetic panmixia is the result from a time-lag effect between demographic and genetic subdivision creating a “gray zone” of population differentiation. With small Ne estimates, simulations showed that the population “gray zone” is expected to be short, typically lower than 20 generations in presence of moderate gene flow (Ne.m = 10 migrants per generation). In the case of the Black Sea harbor porpoises, unless population subdivision is recent (≤200 years), the data set used in this study would have been able to detect a significant differentiation if it exists. We cannot rule out that other portions of the genome under natural selection could show significant differentiation among diverging groups adapting to distinct ecological conditions. Morphological differentiations between porpoises from the Azov Sea and the Black Sea could be consistent with this hypothesis, but could also reflect phenotypic plasticity. Disentangling these hypotheses will require whole genome analyses.
Data archiving
Data and scripts available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8w9ghx3h0
References
Allendorf FW, Luikart GH, Aitken SN (2012) Conservation and the genetics of populations. John Wiley & Sons (West Sussex, UK)
Anderson EC, Dunham KK (2008) The influence of family groups on inferences made with the program Structure. Mol Ecol Resour 8:1219–1229
Aydoğdu A, Pinardi N, Özsoy E, Danabasoglu G, Gürses Ö, Karspeck A (2018) Circulation of the Turkish Straits System under interannual atmospheric forcing. Ocean Sci 14:999–1019
Bailleul D, Mackenzie A, Sacchi O, Poisson F, Bierne N, Arnaud-Haond S (2018) Large-scale genetic panmixia in the blue shark (Prionace glauca): a single worldwide population, or a genetic lag-time effect of the ‘grey zone’ of differentiation? Evol Appl 11:1–42
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Begg GA, Waldman JR (1999) An holistic approach to fish stock identification. Fish Res 43:35–44
Birkun AA Jr. (2002) Cetacean habitat loss and degradation in the Black Sea. In: G. Notarbartolo di Sciara (ed.). Cetaceans of the Mediterranean and Black Seas: State of Knowledge and Conservation Strategies. A report to the ACCOBAMS Secrtetariat, (Monaco), February 2002. Section 8, pp 19
Birkun AA Jr, Frantzis A (2008) Phocoena phocoena ssp. relicta. The IUCN Red List of Threatened Species.: e.T17030A6737111. 10.2305–IUCN.UK.2008.RLTS.T17030A6737111.en. Downloaded on 04 April 2019.
Bowen WD (1997) Role of marine mammals in aquatic ecosystems. Mar Ecol Prog Ser 158:267–274
Csilléry K, Johnson T, Beraldi D, Clutton-Brock T, Coltman D, Hansson B et al. (2006) Performance of marker-based relatedness estimators in natural populations of outbred vertebrates. Genetics 173:2091–2101
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772
De Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Duncan EJ, Gluckman PD, Dearden PK (2014) Epigenetics, plasticity, and evolution: How do we link epigenetic change to phenotype? J Exp Zool B Mol Dev Evol 322:208–220
Earl DA, vonHoldt BM (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Epps CW, Keyghobadi N (2015) Landscape genetics in a changing world: disentangling historical and contemporary influences and inferring change. Mol Ecol 24:6021–6040
Excoffier L, Lischer HEL (2010) Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fontaine MC (2016) Harbour Porpoises, Phocoena phocoena, in the Mediterranean sea and adjacent regions: biogeographic relicts of the last glacial period. Adv Mar Biol 75:333–358
Fontaine MC, Baird SJE, Piry S, Ray N, Tolley KA, Duke S et al. (2007) Rise of oceanographic barriers in continuous populations of a cetacean: the genetic structure of harbour porpoises in Old World waters. BMC Biol 5:30
Fontaine MC, Galan M, Bouquegneau J-M, Michaux JR (2006) Efficiency of Fluorescent Multiplex Polymerase Chain Reactions (PCRs) for rapid genotyping of harbour porpoises (Phocoena phocoena) with 11 Microsatellite Loci. Aquat Mamm 32:301–304
Fontaine MC, Roland K, Calves I, Austerlitz F, Palstra FP, Tolley KA et al. (2014) Postglacial climate changes and rise of three ecotypes of harbour porpoises, Phocoena phocoena, in western Palearctic waters. Mol Ecol 23:3306–3321
Fontaine MC, Snirc A, Frantzis A, Koutrakis E, Öztürk B, Öztürk AA et al. (2012) History of expansion and anthropogenic collapse in a top marine predator of the Black Sea estimated from genetic data. Proc Natl Acad Sci USA 109:E2569–E2576
Fontaine MC, Tolley KA, Michaux JR, Birkun A, Ferreira M, Jauniaux T, Llavona Á, Öztürk B, Oztürk AA, Ridoux V, Rogan E, Sequeira M, Bouquegneau J-M, Baird SJE (2010) Genetic and historic evidence for climate-driven population fragmentation in a top cetacean predator: the harbour porpoises in European water. Proc R Soc B 277:2829–2837
Frankham R (2010) Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv 143:1919–1927
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496
Gagnaire PA, Broquet T, Aurelle D, Viard F, Souissi A, Bonhomme F et al. (2015) Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol Appl 8:769–786
Galatius A, Gol’din PE (2011) Geographic variation of skeletal ontogeny and skull shape in the harbor porpoise (Phocoena phocoena). Can J Zool 89:869–879
Gol’din PE (2004) Growth and Body Size of the Harbour Porpoise, Phocoena phocoena (Cetacea, Phocoenidae), in the Sea of Azov and the Black Sea. Vestn Zoologii 38:59–73
Gol’din PE, Vishnyakova K (2016) Habitat shapes skull profile of small cetaceans: evidence from geographical variation in Black Sea harbour porpoises (Phocoena phocoena relicta). Zoomorphology 135:387–393
Gol’din PE, Vishnyakova KA (2015) Differences in skull size of harbour porpoises, phocoena phocoena (cetacea), in the Sea of Azov and the Black Sea: evidence for different morphotypes and populations. Vestn Zoologii 49:171–180
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Goudet J, Raymond M, de Meeus T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361
Hedgecock D, Barber PH, Edmands S (2007) Genetic approaches to measuring connectivity. Oceanography 20:70–79
Hoelzel AR (1998) Genetic structure of cetacean populations in sympatry, parapatry, and mixed assemblages: implications for conservation policy. J Hered 89:451–458
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Hudson RR (2000) A new statistic for detecting genetic differentiation. Genetics 155:2011–2014
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jombart T, Pontier D, Dufour A-B (2009) Genetic markers in the playground of multivariate analysis. Heredity 102:330–341
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Kleinenberg SE (1956) Mammals of the Black Sea and the Sea of Azov: an experience of biological and fisheries research. USSR Acad. Science Publ, Moscow
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Larsson LC, Charlier J, Laikre L, Ryman N (2008) Statistical power for detecting genetic divergence—organelle versus nuclear markers. Cons Genet 10:1255–1264
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity? Mol Ecol 19:3038–3051
Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP et al. (2015) Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 350:1493–1498
Matishov GG, Chikin AL, Dashkevich LV, Kulygin VV, Chikina LG (2014) The ice regime of the Sea of Azov and climate in the early 21st century. Dokl Earth Sci 457:1020–1024
Moura AE, Natoli A, Rogan E, Hoelzel AR (2013) Atypical panmixia in a European dolphin species (Delphinus delphis): implications for the evolution of diversity across oceanic boundaries. J Evol Biol 26:63–75
Nei M, Chesser RK (1983) Estimation of fixation indices and gene diversities. Ann Hum Genet 47:253–259
Nielsen NH, Teilmann J, Sveegaard S, Hansen RG, Sinding M, Dietz R et al. (2018) Oceanic movements, site fidelity and deep diving in harbour porpoises from Greenland show limited similarities to animals from the North Sea. Mar Ecol Prog Ser 597:259–272
Palsbøll PJ, Berube M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Palumbi SR (2003) Population genetics, demographic connectivity, and the design of marine reserves. Ecol Appl 13:146–158
Payne JL, Bush AM, Heim NA, Knope ML, McCauley DJ (2016) Ecological selectivity of the emerging mass extinction in the oceans. Science 353:1284–1286
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Peng B, Amos CI (2008) Forward-time simulations of non-random mating populations using simuPOP. Bioinformatics 24:1408–1409
Pew J, Muir PH, Wang J, Frasier TR (2015) related: an R package for analysing pairwise relatedness from codominant molecular markers. Mol Ecol Resour 15:557–561
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quintela M, Skaug HJ, Oien N, Haug T, Seliussen BB, Solvang HK et al. (2014) Investigating population genetic structure in a highly mobile marine organism: the minke whale Balaenoptera acutorostrata acutorostrata in the North East Atlantic. PLoS ONE 9:e108640
Rambaut A, Drummond AJ (2012) FigTree version 1.4.3. available at: tree.bio.ed.ac.uk/software/figtree.
Ravinet M, Yoshida K, Shigenobu S, Toyoda A, Fujiyama A, Kitano J (2018) The genomic landscape at a late stage of stickleback speciation: High genomic divergence interspersed by small localized regions of introgression. PLoS Genet 14:e1007358
Read AJ (1999) Harbour porpoise Phocoena phocoena (Linnaeus, 1758). In: Ridgway S, Harrison R (eds) Handbook of marine mammals, Academic Press San Diego, London, pp. 323–350
Rheindt FE, Székely T, Edwards SV, Lee PLM, Burke T, Kennerley PR et al. (2011) Conflict between genetic and phenotypic differentiation: the evolutionary history of a ‘lost and rediscovered’ shorebird. PLoS ONE 6:e26995
Rodriguez Ramilio ST, Wang J (2012) The effect of close relatives on unsupervised Bayesian clustering algorithms in population genetic structure analysis. Mol Ecol Resour 12:873–884
Rosel PE, Frantzis A, Lockyer C, Komnenou A (2003) Source of Aegean Sea harbour porpoises. Mar Ecol Prog Ser 247:257–261
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Ryman N, Palm S (2006) POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Molec Ecol Notes 6:600–602
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
R Core Team (2019) R: A language and environment for statistical computing. Vienna, Austria. https://www.R-project.org.
Tonay AM, Yazıcı Ö, Dede A, Bilgin S, Danyer E, Aytemiz I et al. (2017) Is there a distinct harbor porpoise subpopulation in the Marmara Sea? Mitochondrial DNA A DNA Mapp Seq Anal 28:558–564
Turner TL, Hahn MW (2010) Genomic islands of speciation or genomic islands and speciation? Mol Ecol 19:848–850
Veríssimo A, McDowell JR, Graves JE (2011) Population structure of a deep-water squaloid shark, the Portuguese dogfish (Centroscymus coelolepis). ICES J Mar Sci 68:555–563
Viaud-Martinez KA, Martinez Vergara M, Gol’din PE, Ridoux V, Öztürk AA, Öztürk B et al. (2007) Morphological and genetic differentiation of the Black Sea harbour porpoise Phocoena phocoena. Mar Ecol Prog Ser 338:281–294
Vishnyakova KA (2017) The harbor porpoise (Phocoena phocoena) in the Sea of Azov and the north-eastern Black Sea: population morphology and demography. Ph.D Thesis. Schmalhausen Institute of Zoology, National Academy of Sciences of Ukraine, Kiev
Vishnyakova K, Gol’din P (2015) Seasonality of strandings and bycatch of harbour porpoises in the Sea of Azov: the effects of fisheries, weather conditions and life history. ICES J Mar Sci 72:981–991
Vishnyakova KA, Savenko OV, Oleynikov EP, Gladilina EV, Gorokhova VR, Gol’din PE (2013) Shifting of the spring migration period of the porpoises (Phocoena phocoena relicta) in the Kerch Strait and in the north-eastern Black Sea in 2011–2012. Tr YugNIRO 51:32–35
Wang J (2002) An estimator for pairwise relatedness using molecular markers. Genetics 160:1203–1215
Waples RS (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89:438–450
Ward RD, Woodwark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44:213–232
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358
Winter DJ (2012) MMOD: an R library for the calculation of population differentiation statistics. Mol Ecol Resour 12:1158–1160
Acknowledgements
We thank Diane Bailleul for sharing the scripts to perform the simulations and Jan Veldsing for his assistance in the laboratory. We are also grateful to Jeanine Olsen, three anonymous reviewers, and the editor for their constructive feedbacks which greatly improved the manuscript. We thank Heïdi Lançon (Amphidae) for her professional English editing services. We would like also to thank the Center for Information Technology of the University of Groningen, and in particular Bob Dröge and Fokke Dijkstra, for their continuous support and for providing access to the Peregrine high-performance computing cluster. This study benefitted from the funding of the University of Groningen (The Netherlands). YBC was supported by a PhD fellowship from the University of Groningen.
Author information
Authors and Affiliations
Contributions
MCF and YBC designed the study; MCF supervised the study; KV and PG collected the samples in the field; JT performed the laboratory work and collected the data with help from MCF; YBC and MCF analyzed the data; YBC and MCF interpreted the results and wrote the manuscript with input and final approval from all the co-authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ben Chehida, Y., Thumloup, J., Vishnyakova, K. et al. Genetic homogeneity in the face of morphological heterogeneity in the harbor porpoise from the Black Sea and adjacent waters (Phocoena phocoena relicta). Heredity 124, 469–484 (2020). https://doi.org/10.1038/s41437-019-0284-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0284-1
