
Abstract
Climate relicts hold considerable importance because they have resulted from numerous historical changes. However, there are major interspecific variations among the ways by which they survived climate changes. Therefore, investigating the factors and timing that affected population demographics can expand our understanding of how climate relicts responded to historical environmental changes. Here, we examined herbaceous hydrangeas of genus Deinanthe in East Asia, which show limited distributions and a remarkable disjunction between Japan and central China. Chloroplast genome and restriction site-associated DNA sequencing revealed that speciation event occurred in the late Miocene (ca. 7–9 Mya) in response to global climate change. Two lineages apparently remained not branched until the middle Quaternary, and afterwards started to diverge to regional population groups. The narrow endemic species in central China showed lower genetic diversity (He = 0.082), as its population size rapidly decreased during the Holocene due to isolation in montane refugia. Insular populations in the three Japanese islands (He = 0.137–0.160) showed a genetic structure that was inconsistent with sea barriers, indicating that it was shaped in the glacial period when its range retreated to coastal refugia on the exposed sea floor. Demographic modelling by stairway-plot analysis reconstructed variable responses of Japanese populations: some experienced glacial bottlenecks in refugial isolation, while post-glacial range expansion seemingly exerted founder effects on other populations. Overall, this study demonstrated the involvement of not just one, but multiple factors, such as the interplay between climate changes, geography, and other population-specific factors, that determine the demographics of climate relicts.
Similar content being viewed by others

Introduction
The modern temperate flora of the Northern Hemisphere has been shaped through geographic and climatic changes since the Tertiary period (65–2.6 million years ago [Mya]) (Manchester et al. 2009; Milne and Abbott 2002). Temperate forests were once distributed throughout high latitudes in large parts of Eurasia and North America during much of the Tertiary period (Wolfe 1975). As the global climate cooled and dried over the last 15 million years, the forests became restricted to limited regions having an equable climate (Donoghue and Smith 2004). Temperate plants that were adapted to the Tertiary climate were selectively extirpated in glacial stages of the Quaternary period and were later replaced by cold-adapted taxa (Momohara 2016; Zagwijn 1992) or survived as relicts in climate refugia (Milne and Abbott 2002; Tiffney 1985; Wen 1999).
Climate relicts are of considerable importance in evolutionary ecology because their presence resulted from numerous historical changes (Hampe and Jump 2011; Harrison and Noss 2017; Woolbright et al. 2014). These relicts can preserve ecological and evolutionary histories over millennia as well as reveal the population-level consequences of fragmentation and isolation (Woolbright et al. 2014). For example, the distributions of climate relicts are considered to reflect refugia that were stable in the past (Harrison and Noss 2017; Tang et al. 2018; Zhao et al. 2019), where they persisted under variations in warm and cold conditions due to relatively small altitudinal shifts (Hampe and Petit 2005; Tzedakis et al. 2002). Climate relicts are, therefore, valuable to understand their historical responses along geological epochs but also to explore the potential outcomes of future climate changes.
The Sino-Japanese Floristic Region of East Asia has harboured rich relict flora since the Tertiary period (Qiu et al. 2011; Wu and Wu 1996). In this region, relictual plants are concentrated in mountains in the humid warm-temperate and subtropical areas and found disjunctly in the subtropical/warm-temperate regions of mainland China and southern Japan (Tang et al. 2018). To investigate the species and population responses to historical climate changes, phylogeographic studies have been performed mainly for widespread woody taxa in East Asia. These studies have contributed to our understanding by reconstructing demographic histories in relation to environmental changes (Cao et al. 2020; Chou et al. 2011; Lu et al. 2020; Qi et al. 2012; Sakaguchi et al. 2012) and by detecting climate refugia that contributed to long-term persistence (Qi et al. 2014; Worth et al. 2013). However, our knowledge is still inadequate regarding whether the species’ life history could predict how climate relicts survived in East Asia through the climate deterioration in the Tertiary and glacial-interglacial cycles in the Quaternary (Harrison and Noss 2017), because most genetic studies in this region focused on widespread trees with high fecundity and dispersal abilities. It is now recognised that there is considerable variation among species in both the size of refugia and the duration during which species were confined to them and isolated from other populations (Bai et al. 2018; Stewart et al. 2010). Therefore, our understanding of the historical responses of climate relicts will benefit from investigating plants with diverse life-history traits, particularly the more narrowly distributed plants, which are considered to be more vulnerable to environmental changes (Cole 2003).
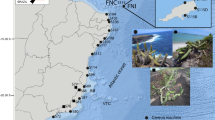
The oligotypic genus Deinanthe (section Deinanthe of Hydrangeaceae) (De Smet et al. 2015) includes two deciduous perennial herbs: Deinanthe bifida and Deinanthe caerulea. The flowers of Deinanthe are insect-pollinated, and small winged seeds (1–1.2 mm) are considered to be dispersed over a short distance by wind and gravity. The distribution of Deinanthe shows a remarkable disjunction between southern Japan and central China, as the species are separated by over 1800 km and interspersed by an unsuitable plain in East China (Fig. 1). D. bifida occurs discontinuously in the three main islands of Japan, inhabiting moist forest floor near streams (Kawanishi et al. 2006). Similarly, D. caerulea is narrowly endemic in moist valleys, particularly on limestone substrates at an elevation range of 700–1600 m in western Hubei, China (Wei and Bruce 2001) (Fig. 1). Although no fossils are available for Deinanthe, the ecology and biogeography of the two species indicate that they may well be climate relicts that long survived in separate regions. While the plant disjunctions between southern Japan and East China have been studied to test the role of land bridges during late Quaternary (Li et al. 2008; Qi et al. 2014; Qiu et al. 2009), the disjunctions between southern Japan and central/western China are rare (Wang 1989) and attained much less attentions despite their biogeographic importance.

A map showing the geographic distribution of Deinanthe species (a D. bifida in Japan, b D. caerulea in Hubei, China). These herbs show similarities in most morphological traits, but they qualitatively differ in calyx colour (white in D. bifida and blue in D. caerulea; a, b) and in how the two-lobed leaves are attached to the stem. The green and purple circles indicate the distribution records of D. bifida and D. caerulea, respectively. Small black dots represent the sampled populations. Areas with elevations exceeding 500 m above sea level and the continental shelf exposed during the last glacial maximum are shown by dark grey and pale blue colours, respectively.
By examining this system, we aimed to understand how relictual herbs with limited distributions survived climate changes since the Tertiary period. Specifically, we aimed to address the following questions: (1) How long were the two Deinanthe species isolated in insular and continental regions of East Asia? (2) Did the Deinanthe populations persist in situ through climatic oscillations during Quaternary or shift their distributions in response to climate changes? (3) How was the population demography of two species shaped through the warm and cold stages of the late Quaternary? To address these questions, we conducted phylogenetic analysis using chloroplast genome sequences and genome-wide single nucleotide polymorphisms (SNPs) as well as population genetic analysis accompanied by ecological niche modelling.
Materials and methods
Reconstruction of chloroplast genome sequences
To obtain the chloroplast genome sequences of Deinanthe and the most related genus of Cardiandra (De Smet et al. 2015; Setoguchi et al. 2006), fresh leaf materials were collected from one individual each of D. bifida, D. caerulea, and Cardiandra amamioshimensis. After rinsing with deionised water, the leaf material was homogenised in a pre-chilled blender with 40 mL isolation buffer at 4 °C (Bookjans et al. 1984; Shi et al. 2012; Vieira et al. 2014) and used for chloroplast enrichment (Sakaguchi et al. 2017). The resultant pellet, including chloroplasts, was dissolved in 600 μL cetyl-trimethyl ammonium bromide (CTAB) buffer to extract DNA (Murray and Thompson 1980).
Purified DNA (50 ng) was used to construct DNA fragment libraries using the Ion Xpress Plus Fragment Library Kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, USA). An Ion PGM Hi-Q OT2 Kit (Thermo Fisher Scientific) was used to prepare template-positive ion sphere particles. DNA fragments were amplified by thermal cycling in microreactors using the Ion OneTouch 2 system (Thermo Fisher Scientific). Positive particles were isolated and purified using the Ion OneTouch ES system (Thermo Fisher Scientific). The particles were loaded onto Ion 318 chips and sequenced using an Ion PGM Sequencer (Thermo Fisher Scientific).
The raw reads were imported into CLC Genomics Workbench version 7.5.1 (CLC bio, Aarhus, Denmark) for adaptor and quality-based trimming. Low-quality bases were removed (quality limit = 0.03). Cleaned reads were assembled using MITObim version 1.8 (Hahn et al. 2013) guided by the complete chloroplast genome sequence of Hydrangea petiolaris (KY412466) obtained from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/).
Divergence time estimated based on chloroplast genome sequences
The chloroplast genome sequences of 28 species in Cornales (including the sequences of D. bifida, D. caerulea, and C. amamioshimensis obtained in this study [SRA accession numbers: DRX214874-DRX214876] and the data of 25 species [16 Hydrangeaceae species, each one species of Alangiaceae, Cornaceae, Curtisiaceae, Davidiaceae, Loasaceae, and each two species of Mastixiaceae and Nyssaceae] downloaded from GenBank) were aligned by the MAFFT programme (Katoh et al. 2017) and manually edited to delete incomplete sites, difficult-to-align regions and the sites with more than two alleles.
BEAST v1.10.4 (Drummond and Rambaut 2007) was used to estimate the divergence time of Deinanthe and related genera based on the chloroplast sequence alignment. Chloroplast genic regions comprising inverted repeat (IR), large single copy (LSC), and small single copy (SSC) regions were treated as different loci, and the best substitution models were separately selected per loci based on the Akaike information criterion (Akaike 1974) by jModelTest (Darriba et al. 2012). The selected models (TPM1uf + G for IR, GTR + I + G for LSC, and TVM + I for SSC) were set as substitution models, and an uncorrelated log-normal relaxed clock was assumed in the analysis (Drummond et al. 2002). To calibrate the phylogenetic tree, we employed the fossil age of Hironoia fusiformis in early Coniacian (Takahashi et al. 2002) for the crown of Cornales, which was set as a log-normal prior distribution with a mean of 1.0 Mya and standard deviation (SD) of 1.2 Mya, with an offset of 91.0 Mya. In addition, we applied a log-normal prior with a mean, SD, and offset of 1.2 Mya, 0.4 Mya and 71.5 Mya, respectively, for calibrating the crown of Davidia–Nyssa–Camptotheca, based on a fossil fruit of Davidia sp. from late Campanian (Manchester et al. 2015). Dating the diversification history within Hydrangeaceae has been a challenge; although the family are represented in the fossil record by several genera, many of these assignments are considered to be doubtful (Gandolfo et al. 1998). Also, previous phylogenetic studies reported variable estimates for the age of Hydrangeaceae, depending on molecular dataset, taxon sampling, and calibration methods (Fu et al. 2019; Xiang et al. 2011). Therefore, we assessed the effect of inclusion of variable secondary calibration points on divergence time estimate of Deinanthe–Cardiandra divergence, by applying two different estimates of Hydrangeaceae crown age [(i) unif(81.5, 89.1) Mya (old estimate) and (ii) unif(39.8, 78.8) Mya (recent estimate)] (Fu et al. 2019) together with an time estimate for Deutzia-Kirengeshoma crown [unif(21.1, 43.1)] (Kim et al. 2015). A birth-death process was specified as the tree prior. Markov chain Monte Carlo (MCMC) chains were run for 100 million generations with one tree sampled every 10,000 generations following a burn-in of the initial 20% of steps. We used the Tracer programme (Rambaut et al. 2018) to check the convergence of the chains to a stationary distribution. Two replicate runs were conducted to confirm the convergence and outcome. The maximum clade credibility tree with ages for each node was displayed and checked for more than 95% credibility in FigTree (Rambaut 2009).
DNA extraction and ddRAD sequencing of Deinanthe and related genera
Seventeen populations of D. bifida and four of D. caerulea (85 individuals in total; Table 1 and Table S1) covering the entire range of the species in South Japan and Central China were sampled. In addition, six related species of Hydrangeaceae were collected from East Asia (two samples of Cardiandra from Taiwan and Japan, Hydrangea macrophylla f. normalis [Central Honshu, Japan], H. petiolaris [Central Honshu, Japan], H. sikokiana [Shikoku, Japan], and Platycrater arguta [Shikoku, Japan]). The collected leaves were immediately dried with silica gel. The leaf samples (ca. 1.0 cm2) were kept at −80 °C and then pulverised using TissueLyser II (QIAGEN, Hilden, Germany). The leaf powder was then suspended in 2–4-(2-hydroxyethyl-1piperanzinyl) ethanesulfonic acid buffer (pH 8.0) and centrifuged (10,000 rpm 20 °C, 5 min) to remove polysaccharides. Total genomic DNA was extracted using a slightly modified CTAB method (Murray and Thompson 1980; Shi et al. 2012) and diluted in Tris-EDTA buffer.
Genomic DNA was used as an input for ddRAD sequencing (Peterson et al. 2012). The ddRAD library was prepared as described by Sakaguchi et al. (2018). Briefly, genomic DNA was digested with EcoRI and BglII (New England Biolabs, Ipswich, MA, USA), and adaptors were ligated at 37 °C overnight. The reaction solution was purified with AMPure XP (Beckman Coulter, Brea, CA, USA). Purified DNA was used for PCR amplification. Thermal cycling was initiated at 94 °C for 2 min, followed by 20 cycles of 98 °C for 10 s, 65 °C for 30 s, and 68 °C for 30 s. The PCR products were pooled, purified using AMPure XP, and then loaded onto a 2.0% agarose gel, and fragments were retrieved using E-Gel SizeSelect (Life Technologies, Carlsbad, CA, USA). Following quality assessment using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), the library was sequenced using an Illumina HiSeq 2000 (Illumina, San Diego, CA, USA). TRIMMOMATIC v.0.32 software was used for read trimming using the commands LEADING:19, TRAILING:19, SLIDING WINDOW:30:20, AVGQUAL:20, and MINLEN:51 (Bolger et al. 2014).
Intraspecific divergence time estimation
We used pyRAD (Eaton 2014) to process the ddRAD-seq reads of 15 samples (including nine Deinanthe samples and six samples of related genera) with the following parameter settings: minimum depth of coverage: 6, similarity threshold for sequence clustering within- and across-samples: 0.8, minimum taxon coverage: 6, and the minimum number of shared polymorphic sites in a locus: 6. A phylogenetic tree based on the dataset was estimated using the Bayesian approach as implemented in BEAST v1.8.2 (Drummond and Rambaut 2007). To calibrate the phylogenetic tree, the crown age for the Cardiandra and Deinanthe group was assumed to have a uniform prior distribution (11.1–25.7 Mya), based on our chloroplast phylogeny (see Results). The Hydrangea-Platycrater group was specified as an outgroup in this analysis. After specifying a birth-death process as the tree prior, GTR + G + I as the substitution model, and an uncorrelated log-normal relaxed clock (Drummond et al. 2002), Bayesian searches for tree topologies and node ages were conducted using BEAST v1.10.4 with the beagle library. MCMC chains were run for 100 million generations with one tree sampled every 10,000 generations following a burn-in of the initial 20% of steps. Parameter convergence was checked and the resultant tree was visualised as described above.
Population genetic analysis of Deinanthe
To investigate the genetic structure and diversity of Deinanthe species, we used pyRAD (Eaton 2014) to process the ddRAD-seq reads of 85 individuals (Table S1), with the following parameter settings: minimum depth of coverage: 6, similarity threshold for sequence clustering within- and across-samples: 0.8, minimum taxon coverage: 60, and the minimum number of shared polymorphic sites in a locus: 60. To filter potentially paralogous loci, SNP markers with observed excess heterozygosity (>0.5) were removed from the dataset (O’Leary et al. 2018). The SNPs were then analysed by BayeScan v2.0 software (Foll and Gaggiotti 2008), and the markers with a log-transformed posterior probability exceeding a threshold of 1.5 were considered to be candidate outliers of neutral evolution. The outliers were removed from the dataset to generate the SNP datasets comprising candidate neutral markers that are suitable for population genetics.
A maximum-likelihood tree for 85 individuals of two Deinanthe species was inferred using RAxML (Stamatakis 2014). Gaps were treated as missing data, and the GTRGAMMA model was specified as the substitution model for the dataset. Node support was assessed using 1000 bootstrap replicates. Principal component analysis (PCA) was applied to the filtered SNP dataset as a non-model-based ordination method to extract synthetic variables at which genetic variation between individuals is maximised. The R package ‘adegenet’ (Jombart 2008) was used to perform PCA analysis in R ver.3.4 (R Development Core Team 2017).
To infer the genetic structure within the samples, a model-based STRUCTURE analysis was performed for the ddRAD-seq data using STRUCTURE v.2.3 software (Falush et al. 2003; Pritchard et al. 2000). The population model was set to allow admixture, correlation of allele frequencies between clusters, and specification of the sampling location as prior information for clustering (Hubisz et al. 2009). Twenty independent simulations were run for each K (K = 1–10) with 100,000 burn-in steps followed by 100,000 MCMC steps. To explore the plausible number of gene pools, we monitored the changes in the average log-likelihood of the data, represented by LnP(D), of independent runs (Pritchard et al. 2000), and the second-order rate of change of LnP(D), designated as ΔK (Evanno et al. 2005).
To estimate the genetic diversity of the regional populations 21 populations were classified into five groups based on the genetic clustering at K = 5 in STRUCTURE (see “Results”). Genetic diversity indices (Ar: allelic richness, Ho: observed heterozygosity, He: expected heterozygosity, and FIS: inbreeding coefficient) of each group were calculated by the R ‘hierfstat’ package (Gouded 2005) using R ver.3.4. The R ‘diveRsity’ package was applied to detect any significant deviations from Hardy–Weinberg equilibrium. Multiple comparisons of each genetic diversity index were performed using Tukey’s method to detect any among-group differences following the analysis of variance using the R ‘multcomp’ package (Hothorn et al. 2012).
Furthermore, analysis of molecular variance (AMOVA) was applied to the ddRAD-seq dataset to partition genetic variances into different sources. Hierarchical models were considered for the dataset, including two species to quantify the relative contribution of among-species variance, and then for D. bifida to estimate among-region variance (four regions defined as above). Non-hierarchical AMOVA was performed for D. caerulea to estimate the among-population variance. The molecular variance was estimated and tests for the significance of each F-statistic based on the standard permutation method were conducted using GenAlEx 6.5 (Peakall and Smouse 2012).
Inference of population demographics using SNP frequency spectra
We used ipyRAD 0.9.42 (Eaton and Overcast 2016) for SNP discovery with the default parameter setting. To estimate the unfolded site frequency spectrum (SFS), we mapped sequences of each sample to FASTA files that were converted from the loci file in the ipyRAD output files. We extracted an individual sequence from this FASTA file for reference data. For Japanese populations (D. bifida in four regions of Chubu, Kinki, Chugoku, and Kyushu), one sample from China (SS145-1) was used as reference data. For the Chinese population (D. caerulea), we used one sample from the Japanese population (SS2-2) as reference data. Using the reference data of the other species allowed us to determine ancestral alleles for estimating the unfolded SFS of each population. We mapped read data with Burrows–Wheeler Aligner 0.7.17 (Li and Durbin 2009) and sorted and indexed mapped files using SAMtools 1.9 (Li et al. 2009). We estimated unfolded SFSs using the doSaf and realSFS functions in the ANGSD 0.931 programme (Korneliussen et al. 2014), while assuming Hardy-Weinberg equilibrium (HWE). Past population demographics using SFS in each population was inferred using stairway-plot v2 (Liu and Fu 2015) under a mutation rate of 7.5 × 10−9 (/site/year) (Ossowski et al. 2010) and a generation time of 5 years (the age at the first flowering; S. Sakaguchi, personal observation in a garden at Kyoto University), and the inferences were based on 500 iterations of the spectra.
Potential distribution shift from the last glacial maximum
An ecological niche model that can predict the changes in D. bifida distribution in Japan was constructed by coupling the current distribution records and bioclimatic variables. In total, 542 presence records of D. bifida were obtained from two databases, S-net (http://science-net.kahaku.go.jp/) and GBIF (https://www.gbif.org), and from our field surveys, from which duplicates were removed manually. Eight predictive variables included seven bioclimatic variables: (1) annual mean temperature, (2) maximum temperature of the warmest month, (3) mean temperature of the driest quarter, (4) mean temperature of the warmest quarter, (5) mean temperature of the coldest quarter, (6) precipitation of the wettest month, and (7) precipitation of the wettest quarter at 2.5 arc-min resolution, which were obtained from the Worldclim website (Hijmans et al. 2005) as well as the topographic slope calculated from ETOPO1 (Amante and Eakins 2009) using ArcGIS (ESRI, CA, USA). The seven bioclimatic variables were selected in this analysis, because they are considered to be ecologically important factor to limit the distribution of D. bifida and they did not show significant clamping effects when the model was projected onto the glacial conditions (see below). An ecological niche model was calibrated using the above data with the default parameter setting using the maximum entropy method implemented in MAXENT v3.2.1 (Phillips et al. 2006). To evaluate the constructed model, the accuracy of each model prediction was calculated using the receiver operating characteristic (ROC) curve and the area under the ROC curve (AUC) (Fawcett 2006).
The environment layers of seven bioclimatic variables for the last glacial maximum (LGM; ~21 thousand years ago [kya]) and the Mid-Holocene period (~6 kya) that were generated from two global circulation models of Community Climate System Model 4 (CCSM4) and Model for Interdisciplinary Research on Climate 3.2 (MIROC3.2) were downloaded from the Paleoclimate Modelling Intercomparison Project 2 (PMIP2) website (https://pmip2.lsce.ipsl.fr/). Based on these palaeoclimate and topographic slope layers, potential past distributions of D. bifida were predicted by the ecological niche model, which was calibrated as described above.
Results
Divergence time of Deinanthe species
The final alignment of the chloroplast genomes contained 17,204 total nucleotides, of which 1,838 sites were variable. The alignments were partitioned into IR (3 genes 1,455 bp), LSC (30 genes 14,680 bp), and SSC (3 genes 1,069 bp) regions and then used for Bayesian phylogenetic inference. In the resultant phylogenetic tree, seven Hydrangeaceae samples comprised a monophyletic clade with full support (Fig. 2a). Within this clade, herbaceous groups of Cardiandra and Deinanthe were clustered in a subclade (PP = 1.00), and their crown age was estimated to be 18.4 Mya (95% highest posterior density [HPD]: 11.1–25.7). Note that the time estimates of Cardiandra–Deinanthe crown varied depending on the phylogenetic trees, for which different calibration ages for Hydrangeaceae crown were taken into account [18.4 (13.7–28.7) −21.0 (10.9–24.9) Mya, Fig. S1]. Two Deinanthe species were estimated to have diverged at 8.9 Mya (95% HPD: 4.0–14.0) in the late Miocene.

a Bayesian phylogenetic tree of Hydrangeaceae and related families inferred based on chloroplast genome sequence data. This tree was calibrated by fossil ages of Cornales and Davidia–Nyssa–Camptotheca crowns and a secondary calibration age for Deutzia–Kirengeshoma crown. b Genome-wide SNP phylogeny of two Deinanthe species and related Hydrangeaceae species. The divergent time estimate from the dated chloroplast phytogenic tree was used for secondary calibration for the Deinanthe–Cardiandra node. Population codes correspond to those shown in Table S1. The bars and node supports represent 95% HPD and Bayesian posterior probability, respectively.
After read assembly using pyRAD, a concatenated ddRAD sequence matrix was obtained, comprising 143,015 nucleotides with 13,153 variable sites. The dated Bayesian tree based on the ddRAD-seq data recovered the two Deinanthe species as monophyletic (PP = 1.00) and as a sister taxon to Cardiandra (Fig. 2b). The estimated divergence time of Deinanthe species was inferred to be 7.1 Mya (95% HPD: 3.6–11.7), which was similar to the value obtained from the chloroplast phylogenetic tree. Intraspecific divergence was dated as beginning during the middle Quaternary at ca. 1.1 Mya for D. caerulea in China and 1.4 Mya for D. bifida in Japan.
Genetic structure and diversity within Deinanthe species
The pyRAD assembly and subsequent filtering processes yielded an SNP matrix consisting of 5,478 markers and 85 samples. An individual-based maximum-likelihood (ML) tree placed the two species distantly (Fig. 3a), and the individuals were clustered according to the sampling localities in most cases. The phylogeographic structure was apparent within D. bifida from Japan (Fig. 3a); moreover, the populations from the Chubu, Kinki, and Chugoku districts of Honshu and Kyushu Islands were clustered separately in the ML tree (see region names in Fig. 3b). The populations on Shikoku Island were an exception, as the individuals from the eastern and western parts were assigned to different groups (Fig. 3). This tendency was consistent in the PCA representation of the genetic structure of D. bifida (Fig. S2), which clustered eastern Shikoku individuals with those in Kinki and western Shikoku individuals with those in the Chugoku district.

a Maximum-likelihood phylogenetic tree of Deinanthe individuals inferred based on genome-wide SNP data. Differently coloured tip circles correspond to the geographic regions. Node support values are shown only for those greater than 99. b Geographic distributions of nuclear gene pools estimated by STRUCTURE analysis (Pritchard et al. 2000) based on genome-wide SNP data for Deinanthe species (K = 5). A neighbour‐joining tree showing the relationships of each gene pool are superimposed on the maps. Four regional populations in D. bifida are defined here as Chubu, Kinki, Chugoku, and Kyushu.
In the model-based clustering of STRUCTURE analysis, ΔK showed a sharp peak at K = 2 (Figure S3b), where the inferred two clusters perfectly corresponded to the two species (data not shown). Thus, K = 2 was considered to be the number of clusters that could capture the uppermost structure within the genetic data. A further increase in K increased the log-likelihood of the data monotonically, reaching a plateau at K = 5 (Fig. S3a). The clustering from K = 3 to K = 5 separated the regional populations in D. bifida; therefore, we considered that the clustering could be biologically interpretable up to K = 5. At K = 3, two clusters in D. bifida dominated the eastern and western parts of its distribution (indicated by the red and pale blue clusters, respectively), with a phylogeographic break located at ca. 133° E (Fig. S4). The two clusters in D. bifida were further divided into four regional clusters at K = 5 (Fig. 3b). In line with the non-model-based clustering (Fig. 3a and Fig. S2), the Shikoku populations were split into two groups (cluster III and IV in Fig. 3b), which were clustered with the populations outside the island.
Five regional populations (Table S1) were defined based on the clustering at K = 5 (Fig. 3b), for which a series of genetic summary statistics were calculated (Table 1). In D. bifida, the regional populations revealed similar genetic diversity, with Ar ranging between 1.14 and 1.16 and He between 0.137 and 0.160. The observed heterozygosity was higeher than the expected value among all population groups. Therefore, mean values of inbreeding coefficients (FIS) were consistently positive, but they did not significantly deviate from Hardy–Weinberg equilibrium due to relatively large variances (P > 0.01). Compared to the Japanese species, D. caelurea showed lower diversity (Ar = 1.08 and He = 0.082) and significantly positive FIS (P < 0.01).
Molecular variance was analysed for three datasets comprising (1) both Deinanthe species, (2) D. bifida with four regional populations, and (3) D. caerulea (Table 2). Hierarchical modelling showed that 55% of the variance could be significantly partitioned into the species difference (D. bifida vs. D. caerulea) with FRT = 0.552. Within D. bifida in Japan, the largest variance was estimated among individuals (57%, P < 0.01), while variances among regional populations (3%, FRT = 0.029) and among populations (20%, FST = 0.203) were relatively small. The population variance in D. caerulea was estimated to be 19% with FST = 0.190.
Demographic inference
The stairway-plot analysis of the SFSs of regional Deinanthe populations resulted in variable demographics in the last 0.3 million years (Fig. 4). The demographic histories earlier than 0.3 Mya are not presented here, as they showed spurious patterns that could not be interpreted according to Liu and Fu (2015). The Chubu and Kyushu populations of D. bifida in Japan showed a bottleneck where populations declined during LGM and then recovered during the Holocene climatic amelioration (Fig. 4a, d). The effective population sizes of the other two regional populations in Japan decreased at ca. 4 kya, a period roughly corresponding to the Holocene climate optimum (Fig. 4b, c). The Chinese species of D. caerulea were inferred to have experienced a significant population decline in the Holocene, collapsing to about 4000 individuals (an approximate 20-fold decline) at the most recent time.

a Chubu, b Kinki-east Shikoku, c Chugoku-west Shikoku, d Kyushu and e China. The effective population size is plotted from present time to 30 kya. Thick black lines indicate the median, and the light and dark grey areas represent the 2.5–97.5% and 12.5–87.5% percentile intervals, respectively. The inferences are based on 500 bootstrap samples of the unfolded site frequency spectra.
Ecological niche modelling of D. bifida
The Maxent model of D. bifida showed an AUC value of 0.841 ± 0.105 (mean ± SD), which indicates good model prediction. The predicted present distribution of D. bifida (Fig. 5a) mostly overlapped with the observed distributions (Fig. 1), except for the prediction of the northern Pacific coast of Honshu Island, where the species is actually absent. At 6 kya (Fig. 5b), the potential range of D. bifida was almost the same as the present distribution, although the logistic values were relatively low. Under the LGM climates, areas suitable to D. bifida contracted to several Pacific capes of ‘Japanese Island’, in which the three major islands of the present day were merged due to sea level depression (Fig. 5c, d). Coasts along the Sea of Japan did not seem to harbour LGM distributions for this species, indicating that post-glacial dispersals may have occurred from the Pacific sides of the palaeo-island.

a at present (0 kya), b at the Holocene Climatic Optimum (6 kya, CCSM4.0), at the last glacial maximum (LGM 21 kya; c CCSM4.0 and d MIROC3.2). Logistic values over a threshold of maximum sensitivity plus specificity are represented using heat colours. The distribution records of D. bifida at present are superimposed as grey points in the LGM maps. Palaeo-rivers during the LGM are illustrated by pale-blue lines.
Discussion
Lineage isolation since the tertiary and subsequent evolutionary stasis
Phylogenetic analysis based on chloroplast and nuclear genome markers consistently showed that the two Deinanthe species diverged in the late Miocene (8.9 and 7.1 Mya for chloroplast genomes and nuclear SNPs, respectively). The estimated divergence times safely excluded the scenario that the two species may be descendants of the common ancestor that migrated between Japan and the continent via land bridge(s) that emerged in the late Quaternary (Fig. 1) (Kimura 1996). Rather, the results suggested that Deinanthe had been isolated in each region for a longer geological time since the Tertiary. Following the Mid-Miocene Climatic Optimum (ca. 15 Mya), the global climate started to become cooler and drier (Holbourn et al. 2018; Zachos et al. 2001), leading to drastic shifts in the regional flora of East Asia from thermophilic species to more cool-temperate components (Pavlyutkin et al. 2016). Previous phylogenetic studies showed that climate changes in the late Miocene induced the allopatric lineage divergence of warm-temperate plants in the Sino-Japanese Floristic Region, such as Euptelea (Cao et al. 2020), Cardiocrinum (Lu et al. 2020), Asarum (Takahashi and Setoguchi 2018), and sect. Brachycalyx (Yoichi et al. 2017). Similarly, climate change is considered to be the driver of allopatric speciation in genus Deinanthe, which would have been adapted to the warmer and wetter Tertiary climate.
The dated phylogenetic trees (Fig. 2) revealed that, after the species divergence, both Deinanthe lineages (corresponding to the later species of D. bifida and D. caerulea) remained not branched for ca. 6 million years until the middle Quaternary. The long isolation history of these lineages is reflected by complete genetic sorting in species-specific clusters in STRUCTURE analysis (Fig. 3) and considerable genetic variance partitioned among species in AMOVA (Table 2). Although fossils are not available for this herbaceous genus, the only two Deinanthe lineages that can be seen today is considered as resulting from local extinctions of intraspecific lineages following speciation. In particular, the sporadic occurrence of D. caerulea in western Hubei (Wei and Bruce 2001) indicates that the species once attained wider distributions, but only a single lineage with lower genetic diversity (Table 1) currently survives in the stable montane refugia of central China (Lopez-Pujol et al. 2011; Tang et al. 2018). The surrounding regions currently lacking the species include East China (30–40° N), which is topographically flat and was once covered by dry steppe vegetation in the LGM (Harrison et al. 2001). Therefore, the current distribution of D. caerulea likely reflects the history that many habitats in East China became unsuitable during the Quaternary climate changes, which led to local extinctions. Successful recolonisation from climate refugia did not occur for this humidity-dependent species with limited dispersal ability.
Inconsistency between the genetic structure in D. bifida and insular geography
The geographic features of insular systems hinder the movement of land plants with low dispersal abilities, leading to increased genetic differentiation among islands. Thus, insular plant populations often show a pattern in which the genetic structure coincides spatially with the studied islands (Dias et al. 2016; Yoichi et al. 2017). However, contrary to this conventional expectation, the genetic structure of D. bifida spread across the three islands of Japan was not associated with the current insular geography; that is, two genetic groups (III and IV clusters) were commonly distributed in western Honshu and Shikoku islands regardless of sea barriers (Fig. 3). This genetic and geographic inconsistency indicates that the genetic structure was formed when populations were isolated under environmental conditions that differ from those of today. During the Middle to Late Quaternary, cold stages of climate cycles gradually extirpated Tertiary elements in Japan, such as Metasequoia (Momohara 1994), and warm-temperate forests are considered to have retreated to the Pacific coasts of ‘Japanese Island’ (Aoki et al. 2019; Tsukada 1983), in which the present-day islands were merged (Fig. 1). The ecological niche model of D. bifida reconstructed their potential LGM refugia in these southern areas (Fig. 5c, d), which may be analogous to former glacial distributions. Therefore, it seems likely that the Quaternary glacial climate was the isolating factor of D. bifida populations, and post-glacial expansions through lowland corridors along palaeo-rivers (Fig. 5) would have led to the current disjunct occurrence of genetic clusters (III and IV) on different islands (Fig. 3).
Contrasting demographic responses of narrow- and wider-ranging species to climate changes
Theory predicts that genetic drift exerts stronger effects on small populations, causing random loss of alleles (Kimura and Crow 1964; Wright 1931). Hence, species with restricted distributions tend to have smaller effective population sizes or less genetic diversity within populations (Cole 2003). In the case of Deinanthe, the narrow endemic D. caerulea showed the lowest levels of genetic diversity (Table 1), and its effective population size was inferred to continuously decrease during the Holocene (Fig. 4e). The inferred Holocene decline of D. caerulea can be interpreted by considering that the species experienced demographic bottlenecks during in situ altitudinal migrations in response to climate change and the subsequent isolation as small populations. Palynological records revealed that the temperate montane forests of western Hubei shifted to lower elevations on south-facing slopes during the last glacial period (Xiao et al. 2018), which induced glacial expansions of temperate plant populations (Lu et al. 2020). However, the increased temperature in the Holocene caused the lowland temperate forests to be replaced by subtropical evergreen forests. Consequently, the warm-temperate species of D. caerulea would have formed fragmented and small populations at higher elevations, thus gradually losing genetic diversity. In addition, the genetic signature of recent population decline does not exclude a possibility that human disturbance of natural forests over the last millennia may have acted in synergy with climate change on the warm-temperate forest including D. caelurea.
We note that the timescale in our demographic analysis was estimated by adopting a moderate mutation rate of 7.5 × 10−9 (/site/year) and a generation time of 5 years. As accurate information on the mutation and generation time for Deinanthe is lacking, the timing of demographic change needs to be interpreted with caution. For example, the generation time of Deinanthe can be longer than the assumed value, as the plants are perennial to reproduce over multiple times after first flowering. If this is true, our time estimate of demographic decline may be underestimated. Nevertheless, even if we assume a longer generation time of 10 years, the timing of population decline of D. caerulea still fall within Holocene period. Furthermore, the extent of population decline should be carefully considered, as the observed intraspecific genetic structure (Fig. 3a) and a significant deviation from HWE in D. caerulea could have played a role in generating false bottleneck signals (Chikhi et al. 2010). In contrast to D. caerulea, the potential distribution range of D. bifida during the LGM was considerably smaller and more fragmented than those under warmer climates (Fig. 5), suggesting that post-glacial expansion from southern refugia occurred. The dynamic distribution shift is consistent with the ‘expansion–contraction model’ of Quaternary biogeography (Hewitt 2004), which proposed an alternation between the contraction of species distribution during the glacial period and subsequent rapid expansions during interglacial periods. The genetic consequence of such a range shift usually involves a genetic diversity cline and differentiation among northern and southern populations (Petit et al. 2003; Sakaguchi et al. 2011; Sugahara et al. 2011). However, for the regional populations of D. bifida, more complicated and idiosyncratic demographic responses were detected by demographic inference (Fig. 4), which have not been reported for the forest trees in Japan. Stairway-plot analysis inferred that two populations (a: Chubu and d: Kyushu) experienced bottlenecks following the LGM, whereas the other two populations (b: Kinki-east Shikoku and c: Chugoku-west Shikoku) declined moderately during the Holocene. Such variable demographic patterns likely resulted from the relative extents to which the glacial contraction and post-glacial expansion negatively affected the population size. First, glacial range contractions as predicted by the ecological niche model could have caused genetic bottlenecks for the former population group, of which small populations persisted in refugia. Afterwards, population growth likely occurred along with climatic amelioration, which would have recovered the population size probably due to increased population density (Magri 2008). In the Japanese Archipelago, pollen fossils showed that the warm-temperate trees expanded their ranges from glacial refugia along coastal areas to inland and higher elevation areas (Gotanda and Yasuda 2008). Such post-glacial demographic changes have been genetically detected in a forest tree of Castanopsis (Aoki et al. 2019), and the associated temperate biota (Aoki et al. 2008; Kawamoto et al. 2007). Second, rapid range expansion can have adverse effects on genetic diversity by exerting founder effects in leading-edge populations (Waters et al. 2013). Founding events often result in the loss of genetic diversity (Le Corre and Kremer 1998) with rare long-distance dispersal events accelerating these founder effects (Austerlitz et al. 2000). This seems to have more strongly affected the latter Deinanthe population groups (Fig. 4b, c), which expanded to northerly ranges distant from glacial refugia (Fig. 5).
Conclusion
Our genetic assessment of the climate relict Deinanthe revealed that the two species are descendants of a few lineages that survived through Tertiary environmental changes and Quaternary climate cycles. Substantial extinctions, especially in continental regions, were speculated from genetic genealogies and current limited distributions of the narrow endemic species. Generally, climate relicts are assumed to be distributed in long-term refugial areas. This notion may apply to D. caerulea, which appears to have experienced altitudinal migrations in the montane refugia of central China. In contrast, the ranges of Japanese D. bifida populations were inferred to dynamically shift in response to the climate cycles of the late Quaternary. Furthermore, the demographic changes varied among the Japanese populations, as some suffered glacial bottlenecks and others were affected by interglacial declines despite being found in similar latitudinal ranges. This finding implies the involvement of not just one, but many factors, as the interplay between climate changes, geography, and other population-specific factors would have been involved in shaping the genetic demographics of relict species that are vulnerable to environmental changes.
Data availability
Raw short read data files are deposited in GenBank under accession numbers DRX214874-DRX214876 (accession numbers are shown in Table S2).
References
Akaike H (1974) New look at statistical-model identification. IEEE Trans Autom Control 19(6):716–723
Amante C, Eakins BW (2009) ETOPO1 Global Relief Model converted to PanMap layer format. NOAA-National Geophysical Data Center, PANGAEA. https://doi.org/10.1594/PANGAEA.769615
Aoki K, Kato M, Murakami N (2008) Glacial bottleneck and postglacial recolonization of a seed parasitic weevil, Curculio hilgendorfi, inferred from mitochondrial DNA variation. Mol Ecol 17(14):3276–3289
Aoki K, Tamaki I, Nakao K, Ueno S, Kamijo T, Setoguchi H et al. (2019) Approximate Bayesian computation analysis of EST-associated microsatellites indicates that the broadleaved evergreen tree Castanopsis sieboldii survived the Last Glacial Maximum in multiple refugia in Japan. Heredity 122(3):326–340
Austerlitz F, Mariette S, Machon N, Gouyon PH, Godelle B (2000) Effects of colonization processes on genetic diversity: Differences between annual plants and tree species. Genetics 154(3):1309–1321
Bai WN, Yan PC, Zhang BW, Woeste KE, Lin K, Zhang DY (2018) Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole-genome sequences. New Phytol 217(4):1726–1736
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Bookjans G, Stummann B, Henningsen K (1984) Preparation of chloroplast DNA from pea plastids isolated in a medium of high ionic strength. Anal Biochem 141(1):244–247
Cao Y-N, Zhu S-S, Chen J, Comes HP, Wang IJ, Chen L-Y et al. (2020) Genomic insights into historical population dynamics, local adaptation, and climate change vulnerability of the East Asian Tertiary relict Euptelea (Eupteleaceae). Evol Appl 13(8):2038–2055
Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA (2010) The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics 186(3):983–U347.
Chou Y-W, Thomas PI, Ge X-J, LePage BA, Wang C-N (2011) Refugia and phylogeography of Taiwania in East Asia. J Biogeogr 38(10):1992–2005
Cole CT (2003) Genetic variation in rare and common plants. Annu Rev Ecol Evolution Syst 34(1):213–237
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9(8):772
De Smet Y, Granados Mendoza C, Wanke S, Goetghebeur P, Samain M-S (2015) Molecular phylogenetics and new (infra) generic classification to alleviate polyphyly in tribe Hydrangeeae (Cornales: Hydrangeaceae). Taxon 64(4):741–753
Dias EF, Moura M, Schaefer H, Silva L (2016) Geographical distance and barriers explain population genetic patterns in an endangered island perennial. AoB PLANTS 8:plw072. https://doi.org/10.1093/aobpla/plw072
Donoghue MJ, Smith SA (2004) Patterns in the assembly of temperate forests around the Northern Hemisphere. Philos Trans R Soc Lond Ser B: Biol Sci 359(1450):1633–1644
Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W (2002) Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161(3):1307–1320
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Eaton DA, Overcast I (2016) ipyrad: interactive assembly and analysis of RADseq datasets. Bioinformatics 36(8):2592–2594
Eaton DAR (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30(13):1844–1849
Evanno G, Reganut E, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587
Fawcett T (2006) An introduction to ROC analysis. Pattern Recog Lett 27(8):861–874
Foll M, Gaggiotti OE (2008) A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Fu C-N, Mo Z-Q, Yang J-B, Ge X-J, Li D-Z, Xiang Q-Y et al. (2019) Plastid phylogenomics and biogeographic analysis support a trans-Tethyan origin and rapid early radiation of Cornales in the Mid-Cretaceous. Mol Phylogen Evol 140:106601
Gandolfo M, Nixon K, Crepet W (1998) Tylerianthus crossmanensis gen. et sp. nov. (aff. Hydrangeaceae) from the Upper Cretaceous of New Jersey. Am J Bot 85(3):376
Gotanda K, Yasuda Y (2008) Spatial biome change in southwestern Japan since the Last Glacial Maximum. Quat Int 184:84–93
Gouded J (2005) hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Resour 5(1):184–186
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res 41(13):e129. https://doi.org/10.1093/nar/gkt371
Hampe A, Jump AS (2011) Climate relicts: past, present, future. Annu Rev Ecol Evolu Syst 42(1):313–333
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8(5):461–467
Harrison S, Noss R (2017) Endemism hotspots are linked to stable climatic refugia. Ann Bot 119(2):207–214
Harrison SP, Yu G, Takahara H, Prentice IC (2001) Palaeovegetation—diversity of temperate plants in east Asia. Nature 413(6852):129–130
Hewitt GM (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond Ser B-Biol Sci 359(1442):183–195
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25(15):1965–1978
Holbourn AE, Kuhnt W, Clemens SC, Kochhann KGD, Jöhnck J, Lübbers J et al. (2018) Late Miocene climate cooling and intensification of southeast Asian winter monsoon. Nat Commun 9(1):1584
Hothorn T, Bretz F, Westfal P (2012) multcomp: simultaneous Inference in General Parametric Models. R package version 1:2–14
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9(5):1322–1332
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405
Katoh K, Rozewicki J, Yamada KD (2017) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20(4):1160–1166
Kawamoto Y, Shotake T, Nozawa K, Kawamoto S, Tomari K-I, Kawai S et al. (2007) Postglacial population expansion of Japanese macaques (Macaca fuscata) inferred from mitochondrial DNA phylogeography. Primates 48(1):27–40
Kawanishi M, Sakio H, Kubo M, Shimano K, Ohno K (2006) Effect of micro-landforms on forest vegetation differentiation and life-form diversity in the Chichibu Mountains, Kanto District, Japan. Vegetation Sci 23(1):13–24
Kim C, Deng T, Wen J, Nie Z-L, Sun H (2015) Systematics, biogeography, and character evolution of Deutzia (Hydrangeaceae) inferred from nuclear and chloroplast DNA sequences. Mol Phylogen Evol 87:91–104
Kimura M (1996) Quaternary paleogeography of the Ryukyu arc. J Geogr 105(3):259–285
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49(4):725–738
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinform 15(1):356
Le Corre V, Kremer A (1998) Cumulative effects of founding events during colonisation on genetic diversity and differentiation in an island and stepping-stone model. J Evol Biol 11(4):495–512
Li EX, Yi S, Qiu YX, Guo JT, Comes HP, Fu CX (2008) Phylogeography of two East Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol Phylogen Evol 49(3):702–714
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079
Liu X, Fu Y-X (2015) Exploring population size changes using SNP frequency spectra. Nat Genet 47(5):555–559
Lopez-Pujol J, Zhang F, Sun H, Ying T, Ge S (2011) Centres of plant endemism in China: places for survival or for speciation? J Biogeogr 38(7):1267–1280
Lu R-S, Chen Y, Tamaki I, Sakaguchi S, Ding Y-Q, Takahashi D et al. (2020) Pre-quaternary diversification and glacial demographic expansions of Cardiocrinum (Liliaceae) in temperate forest biomes of Sino-Japanese Floristic Region. Mol Phylogen Evol 143:106693
Magri D (2008) Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J Biogeogr 35(3):450–463
Manchester SR, Chen ZD, Lu AM, Uemura K (2009) Eastern Asian endemic seed plant genera and their paleogeographic history throughout the Northern Hemisphere. J Syst Evol 47(1):1–42
Manchester SR, Grímsson F, Zetter R (2015) Assessing the fossil record of asterids in the context of our current phylogenetic framework. Ann Mo Bot Gard 100(4):329–363
Milne RI, Abbott RJ (2002) The origin and evolution of Tertiary relict floras. Adv Bot Res 38:281–314
Momohara A (1994) Floral and paleoenvironmental history from the late Pliocene to middle Pleistocene in and around Central Japan. Palaeogeogr Palaeoclimatol Palaeoecol 108(3-4):281–293
Momohara A (2016) Stages of major floral change in Japan based on macrofossil evidence and their connection to climate and geomorphological changes since the Pliocene. Quat Int 397:93–105
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4325.
O’Leary SJ, Puritz JB, Willis SC, Hollenbeck CM, Portnoy DS (2018) These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol Ecol 27(16):3193–3206
Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG et al. (2010) The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. science 327(5961):92–94
Pavlyutkin BI, Yabe A, Golozubov V, Simanenko LF (2016) Miocene floral changes in the circum-Japan sea areas—their implications in the climatic changes and the time of japan sea opening. Mem Natl Sci Mus Tokyo 51:109–123
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics 28(19):2537–2539
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for De Novo SNP discovery and genotyping in model and non-model species. PLos ONE 7(5):e37135
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al. (2003) Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 300(5625):1563–1565
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190(3-4):231–259
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Qi X-S, Chen C, Comes HP, Sakaguchi S, Liu Y-H, Tanaka N et al. (2012) Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytol 196(2):617–630
Qi X-S, Yuan N, Comes H, Sakaguchi S, Qiu Y-X (2014) A strong ‘filter’ effect of the East China Sea land bridge for East Asia’s temperate plant species: inferences from molecular phylogeography and ecological niche modelling of Platycrater arguta (Hydrangeaceae). BMC Evol Biol 14(1):41
Qiu YX, Fu CX, Comes HP (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogen Evol 59(1):225–244
Qiu YX, Sun Y, Zhang XP, Lee J, Fu CX, Comes HP (2009) Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol 183(2):480–495
R Development Core Team (2017) R version 3.4.0: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Rambaut A (2009) FigTree ver. 1.3.1. http://tree.bio.ed.ac.uk/software/figtree/
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67(5):901
Sakaguchi S, Horie K, Kimura T, Nagano AJ, Isagi Y, Ito M (2018) Phylogeographic testing of alternative histories of single-origin versus parallel evolution of early flowering serpentine populations of Picris hieracioides L. (Asteraceae) in Japan. Ecol Res 33(3):537–547
Sakaguchi S, Qiu Y-X, Liu Y-H, Qi X-S, Kim S-H, Han J et al. (2012) Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol Ecol 21(15):3823–3838
Sakaguchi S, Takeuchi Y, Yamasaki M, Sakurai S, Isagi Y (2011) Lineage admixture during postglacial range expansion is responsible for the increased gene diversity of Kalopanax septemlobus in a recently colonised territory. Heredity 107(4):338–348
Sakaguchi S, Ueno S, Tsumura Y, Setoguchi H, Ito M, Hattori C et al. (2017) Application of a simplified method of chloroplast enrichment to small amounts of tissue for chloroplast genome sequencing. Appl Plant Sci 5(5):1700002
Setoguchi H, Yukawa T, Tokuoka T, Momohara A, Sogo A, Takaso T et al. (2006) Phylogeography of the genus Cardiandra based on genetic variation in cpDNA sequences. J Plant Res 119(4):401–405
Shi C, Hu N, Huang H, Gao J, Zhao Y-J, Gao L-Z (2012) An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 7(2):e31468
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Stewart JR, Lister AM, Barnes I, Dalen L (2010) Refugia revisited: individualistic responses of species in space and time. Proc R Soc B-Biol Sci 277(1682):661–671
Sugahara K, Kaneko Y, Ito S, Yamanaka K, Sakio H, Hoshizaki K et al. (2011) Phylogeography of Japanese horse chestnut (Aesculus turbinata) in the Japanese Archipelago based on chloroplast DNA haplotypes. J Plant Res 124(1):75–83
Takahashi D, Setoguchi H (2018) Molecular phylogeny and taxonomic implications of Asarum (Aristolochiaceae) based on ITS and matK sequences. Plant Species Biol 33(1):28–41
Takahashi M, Crane PR, Manchester SR (2002) Hironoia fusiformis gen. et sp. nov.; a cornalean fruit from the Kamikitaba locality (Upper Cretaceous, Lower Coniacian) in northeastern Japan. J Plant Res 115(6):463–473
Tang CQ, Matsui T, Ohashi H, Dong Y-F, Momohara A, Herrando-Moraira S et al. (2018) Identifying long-term stable refugia for relict plant species in East Asia. Nat Commun 9(1):4488
Tiffney BH (1985) Perspectives on the origin of the floristic similarity between eastern Asia and eastern North America. J Arnold Arbor 66(1):73–94
Tsukada M (1983) Vegetation and climate during the last glacial maximum in Japan. Quatern Res 19(2):212–235
Tzedakis PC, Lawson IT, Frogley MR, Hewitt GM, Preece RC (2002) Buffered tree population changes in a quaternary refugium: evolutionary implications. Science 297(5589):2044–2047
Vieira LdN, Faoro H, Fraga HPdF, Rogalski M, de Souza EM, de Oliveira Pedrosa F et al. (2014) An improved protocol for intact chloroplasts and cpDNA isolation in conifers. PLoS ONE 9(1):e84792
Wang WT (1989) Notes on disjunction in the flora of China. Bull Bot Res 9(1):1–16
Waters JM, Fraser CI, Hewitt GM (2013) Founder takes all: density-dependent processes structure biodiversity. Trends Ecol Evol 28(2):78–85
Wei Z, Bruce B (2001) Deinanthe, Vol 8. Science Press, Beijing, and Missouri Botanical Garden Press, St. Louis
Wen J (1999) Evolution of eastern Asian and eastern North American disjunct distributions in flowering plants. Annu Rev Ecol Syst 30(1):421–455
Wolfe JA (1975) Some aspects of plant geography of the northern hemisphere during the late Cretaceous and Tertiary. Ann Mo Botanical Gard 62(2):264–279
Woolbright SA, Whitham TG, Gehring CA, Allan GJ, Bailey JK (2014) Climate relicts and their associated communities as natural ecology and evolution laboratories. Trends Ecol Evol 29(7):406–416
Worth JRP, Sakaguchi S, Tanaka N, Yamasaki M, Isagi Y (2013) Northern richness and southern poverty: contrasting genetic footprints of glacial refugia in the relictual tree Sciadopitys verticillata (Coniferales: Sciadopityaceae). Biol J Linn Soc 108(2):263–277
Wright SJ (1931) Evolution in mendelian populations. Genetics 16(2):97–159
Wu Z, Wu S (1996) A proposal for a new floristic kingdom (realm)—the E. Asiatic kingdom, its delineation and characteristics. China Higher Education Press. Springer, Beijing
Xiang Q-Y, Thomas DT, Xiang QP (2011) Resolving and dating the phylogeny of Cornales—effects of taxon sampling, data partitions, and fossil calibrations. Mol Phylogen Evol 59(1):123–138
Xiao J, Shang Z, Shu Q, Yin J, Wu X (2018) The vegetation feature and palaeoenvironment significance in the mountainous interior of southern China from the Last Glacial Maximum. Sci China Earth Sci 61(1):71–81
Yoichi W, Jin XF, Peng CI, Tamaki I, Tomaru N (2017) Contrasting diversification history between insular and continental species of three‐leaved azaleas (Rhododendron sect. Brachycalyx) in East Asia. J Biogeogr 44(5):1065–1076
Zachos JC, Shackleton NJ, Revenaugh JS, Pälike H, Flower BP (2001) Climate response to orbital forcing across the Oligocene-Miocene boundary. Science 292(5515):274–278
Zagwijn WH (1992) The beginning of the ice age in Europe and its major subdivisions. Quat Sci Rev 11(5):583–591
Zhao Y-P, Fan G, Yin P-P, Sun S, Li N, Hong X et al. (2019) Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat Commun 10(1):4201
Acknowledgements
The authors are grateful to D. Fujiki, I. Tamaki, T. Teramine, N. Ishikawa, Y. Azuma, S. Mori, Y. Inoue, S. Kurata, and M. Yamamoto for their help in sample collection. This work was supported by the Japanese Society for the Promotion of Science (the Bilateral Program “The spatial and temporal dimensions and underlying mechanisms of lineage divergence and plant speciation of keystone species in Sino-Japanese Forest subkingdom”), the SICORP Program of the Japan Science and Technology Agency (grant no. 4-1403), and the Environment Research and Technology Development Fund (4-1605 and 4-1902) of the Ministry of the Environment, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Thomas Meagher
Supplementary information
Rights and permissions
About this article

Cite this article
Sakaguchi, S., Asaoka, Y., Takahashi, D. et al. Inferring historical survivals of climate relicts: the effects of climate changes, geography, and population-specific factors on herbaceous hydrangeas. Heredity 126, 615–629 (2021). https://doi.org/10.1038/s41437-020-00396-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00396-4
This article is cited by
-
Demographic history and conservation aspects of two relict shrubs (Daphnimorpha spp., Thymelaeaceae) showing disjunct and restricted distribution in the southern part of Japan
Tree Genetics & Genomes (2023)
-
Genome-wide assessment of population genetic and demographic history in Magnolia odoratissima based on SLAF-seq
Conservation Genetics (2023)
