
Abstract
Purifying selection tends to reduce nucleotide and haplotype diversity leading to increased linkage disequilibrium. However, detection of evidence for selection is difficult as the signature is confounded by wide variation in the recombination rate which has a complex relationship with selection. The effective bottleneck time (the ratio of the linkage disequilibrium map to the genetic map in Morgans) controls for variability in the recombination rate. Reduced effective bottleneck times indicate stronger residual linkage disequilibrium, consistent with increased selection. Using whole genome sequence data from one European and three Sub-Saharan African human populations we find, in the African samples, strong correlations between high gene densities and reduced effective bottleneck time for autosomal chromosomes. This suggests that gene-dense autosomes have been subject to increased purifying selection reducing effective bottleneck times compared to gene-poor autosomes. Although previous studies have shown unusually strong linkage disequilibrium for the sex chromosomes variation within the autosomes has not been recognised. The strongest relationship is between effective bottleneck time and the density of essential genes, which are likely targets of greater selective pressure (p = 0.006, for the 22 autosomes). The magnitude of the reduction in chromosome-specific effective bottleneck times from the least to the most gene-dense autosomes is ~17–21% for Sub-Saharan African populations. The effect size is greater in Sub-Saharan African populations, compared to a European sample, consistent with increased efficiency of selection in populations with larger effective population sizes which have not been subject to intense population bottlenecks as experienced by populations of European ancestry. The findings highlight the value of deeper analyses of selection within Sub-Saharan African populations.
Similar content being viewed by others


Introduction
An understanding of the impact of selection on genomes is important for the interpretation of population history, genome function and disease genomics. Patterns of linkage disequilibrium (LD) have been used extensively to find signatures of positive and purifying selection by recognition of reduced diversity (Voight et al. 2006; Huff et al. 2010). Tests include the identification of long-range haplotypes through, for example, extended haplotype homozygosity (EHH) which relies on the relationship between the frequency of an allele and the extent of LD surrounding it. In such regions a pattern of unusually long-range LD, given the allele’s population frequency, is found after correcting for the recombination rate (Sabeti et al. 2002).
Signatures of increased selection have also been demonstrated for whole chromosomes, specifically the sex chromosomes. The unique mode of inheritance of the X chromosome is likely to expose it to more intense selective pressure compared to the autosomal chromosomes. There is evidence that the X chromosome has been subject to strong selective sweeps (Dutheil et al. 2015) reducing nucleotide diversity around genes compared to autosomal chromosomes. Megabase-size regions spanning about one third of the X chromosome show a reduction in nucleotide diversity to less than 20% of the chromosome average (Nam et al. 2015). Several very strong selective sweeps are considered to have operated independently in these regions and testis-expressed ampliconic genes may have been the target: a mechanism to correct for distortions in the sex ratio is proposed (Dutheil et al. 2015). Gottipati et al. (2011) concluded that diversity on chromosome X has been impacted to a greater extent by selection at linked sites than the autosomes, probably through the increased exposure in males when damaging X-linked recessive variants are present in single copy. Veeramah et al. (2014) present evidence indicating that the X chromosome has been subject to elevated levels of both purifying and positive selection.
The Y chromosome also has unusually low levels of genetic diversity. Because most of the Y chromosome does not recombine all sites are effectively linked and selection operating on any individual site will affect all other sites. The Y chromosome is subject to purifying selection removing new deleterious mutations and reducing diversity at linked neutral sites (Wilson Sayres et al. 2014). Similarly, positive selection, operating on any beneficial mutations may decrease diversity at linked neutral sites. The Y chromosome is gene-poor and the number of sites which are subject to purifying selection exceeds the number of Y-linked coding sites (there are ~100,000 single copy coding sites). The highly repetitive ampliconic sequences span a much larger region (5.7 Mb) and genes in these regions are expressed exclusively in the testis and possibly subject to selection in relation to male fertility. The ampliconic region plays an important role in the restoration of mutation-free gene copies through intrachromosomal gene conversion (Bachtrog 2013).
The complex relationships between recombination and natural selection have received considerable attention over many years. It is known that selection impacts genetic diversity over greater physical distances in chromosome regions which have lower rates of recombination, through ‘hitch-hiking’ (Smith and Haigh 1974; Hudson and Kaplan 1995). However, recombination also improves the efficiency of selection whenever multiple linked loci are simultaneously under selective pressure (Hill and Robertson 1966; Castellano et al. 2016; Pengelly et al. 2019). If selection is more intense for genic sequences then genomic regions with a high density of coding sequences will contain more potential targets of selection compared to regions with low gene density. After controlling for the recombination rate, which varies widely along the length and between chromosomes, reductions in nucleotide (and hence haplotype) diversity in these regions suggest positive or purifying selection (Payseur and Nachman 2002; Cutter and Payseur 2013). Therefore it is expected that longer blocks of strong LD will surround substitutions at sites subject to selection. Autosomal chromosomes show very marked variations in gene density (for example, chromosome 19 has ~24 genes/Mb, compared to chromosome 13 which has only ~ 3 genes/Mb (Spataro et al. 2017)). Payseur and Nachman (2002) show that the number of genes in a genomic region is a reasonable predictor of selection intensity.
Covariance between gene density and recombination rates can obscure the signatures of linked selection (Cutter and Payseur 2013). However, a useful measure is the effective bottleneck time (EBT) defined as the ratio of the LD unit (LDU) map length to the linkage map length in Morgans (Zhang et al. 2004). The EBT corresponds to the number of generations since an ‘effective’ bottleneck (a single bottleneck which reflects the combined impact of multiple bottlenecks on haplotype diversity over time). For example, Tapper et al. (2005) determined that the EBT for the X chromosome in the HapMap CEU population (representing Utah Residents with Northern and Western European ancestry) was substantially reduced compared to the autosomes consistent with increased purifying selection reducing diversity. Analysis of whole genome sequence (WGS) data from European and Nigerian population samples demonstrated a substantially higher resolution of LD structure compared to array-based panels (Pengelly et al. 2015). Using WGS data from individuals in four samples (representing European, Ethiopian, Zulu and Baganda populations) we describe patterns of variation in EBTs for autosomal chromosomes and relationship with gene density.
Methods
Processing of samples with European origin
Single nucleotide polymorphism (SNP) genotypes were obtained from WGS data from the Scripps Wellderly Genome Resource. Samples comprise 454 unrelated individuals with self-declared ethnically European origin (Erikson et al. 2016). Following Pengelly et al. (2015) SNPs with >5% missing genotypes and a significant Hardy–Weinberg deviation p-value (<0.001) were excluded (Wigginton et al. 2005). Rare SNPs are uninformative for LD and all SNPs with alternative minor allele frequencies (MAF) of < 0.01 were excluded from the maps. Completed LDU maps of chromosomes 1–22 contain 7,162,973 SNPs (Supplementary Table 1).
Processing of samples with African origin
WGS data from 320 healthy individuals from seven Sub-Saharan African (SSA) ethnolinguistic groups recruited to the African Genome Variation Project were considered (Gurdasani et al. 2015). To develop LD maps representing major ethnic populations, principal component analysis (PCA) was carried out using SNPRelate (Zheng et al. 2012). Individuals with >10% missing genotypes, SNPs with >10% missing genotypes and SNPs with significant Hardy-Weinberg deviation were removed. Only SNPs with MAF >5% were retained for PCA analysis. Identity by descent (IBD) was measured within each population and related individuals (IBD > 0.05) were removed. Squared correlation coefficients between allele counts were used to prune SNPs in strong LD (r2 > 0.5) with other SNPs. PCA analysis confirmed three major clusters mirroring distinct linguistic groupings across these populations (Supplementary Fig. 1). One Somali individual was excluded from further consideration due to apparent ethnic divergence from all the major groups. The Gumuz population formed a discrete cluster but comprises only 24 individuals so was not considered further. Samples from Amhara (n = 24), Oromo (n = 24), Somali (n = 23) and Wolayta (n = 24) sub-populations were considered together as a broader Ethiopian sub-population (n = 95). The two other populations with large sample size (n = 100 each), namely Zulu and Baganda, were considered independently.
Population-specific multivariant VCF files were generated for 295 individuals according to the PCA clustering pattern as described. Data from each population were pre-processed to exclude SNPs with >5% missing genotypes, SNPs with MAF < 0.01 in the respective sub-population and SNPs deviating from Hardy-Weinberg equilibrium at a significance threshold of p < 0.001.
LD map construction
For each of the four populations we constructed LD maps in LDUs for chromosomes 1–22 using the LDMAP programme (Lau et al. 2007; Pengelly et al. 2015). The programme constructs LD maps according to the Malécot–Morton model:
where \(\hat \rho\) is the association between a pair of SNPs, the asymptote L is ‘background’ association which is not due to linkage, M reflects association at zero distance with values ~1 consistent with monophyletic haplotypes and <1 with polyphyletic inheritance, ϵ is the rate of LD decline, and d is the physical distance in kilobases between SNPs. Parameters ϵ, L and M are estimated iteratively for each adjacent SNP–SNP interval. Map distances in LDUs describe the rate of decline of LD across each SNP interval computed as the product ϵd with cumulative map distances similar to the centimorgan scale. One LDU corresponds to the (highly variable) physical distance along the DNA sequence over which LD declines to background levels. LDUs plotted against chromosome location reveal ‘steps’ which reflect recombination hotspots and plateaus aligning with blocks of low haplotype diversity. The LDU map is constructed iteratively with the fit of the pairwise SNP association data to the kilobase map established through composite likelihood. Previous studies have shown LDU maps are largely insensitive to variation in the SNP marker density used for their construction, even when using much lower density SNP array data (Ke et al. 2004). Similarly, LDU map lengths have been shown to be largely stable and representative of population LD structure even for small population samples (Pengelly et al. 2015).
Computation of EBT
The EBT was defined for LDU maps by Zhang et al. (2004). Considering the ith interval between SNPs map distances in the corresponding linkage map can be expressed as wi Morgans (typically given as 100wi cM). Map distances along an LDU map correspond to wit, where t is the number of generations since LD began to decline from an effective hypothetical bottleneck, reflecting the cumulative impact of multiple bottlenecks. For populations these multiple bottlenecks drive reduced effective population size through mortality, migration, selective sweeps, or other factors. Differences between chromosomes suggest variation in intensity of purifying selection impacting the strength of LD after correcting for variable recombination rates. The LDU/w ratio describes the EBT in generations: evidence that some chromosomes within a population show relatively reduced EBTs suggests they have been subject to elevated selective pressure.
Alternative genetic recombination maps
The Kong et al. (2002) linkage map was constructed using 5136 microsatellite markers genotyped in 146 Icelandic families (869 individuals) and includes 1257 meiotic events spanning an autosomal chromosome length of 3436 cM (Supplementary Tables 2 and 3). Bhérer et al. (2017) used recombination data from six different sources and four datasets representing European populations to construct a ‘refined’ European linkage map spanning 3351 cM and representing 97,723 meioses. The majority of the samples used were from Icelandic families (Kong et al. 2014). Hinch et al. (2011) constructed a recombination map from African Americans (individuals with ~80% West African and 20% European ancestry) from 29,589 apparently unrelated African Americans genotyped on SNP arrays for genome-wide association studies. However, the map is constructed from population data and not families and has therefore been normalised by total map length. However, any systematic differences in relative chromosome lengths between a (largely) African population compared to European map should be evident.
Gene density
We considered gene densities for human chromosomes presented by Mayer et al. (2005) in their Fig. 1. We also computed gene densities using the total gene counts in Spataro et al. (2017) and the chromosome physical lengths in megabases from the human genome hg19 assembly (Supplementary Tables 2 and 4). The Spataro et al. (2017) study provides gene groups categorised according to essentiality and disease relationships. We computed gene densities within these gene groups (Supplementary Table 4).

LDU maps of chromosome 22 for three populations. The cumulative LDU scale is plotted against the physical location in kilobases for three populations. Map contours show close alignment across populations between steeper areas of rapid LD breakdown and flatter regions of strong LD. Map lengths reflect differences in effective bottleneck times between populations with a longer time to an effective bottleneck for the African Baganda and Ethiopian populations compared to the Wellderly sample of European origin
Chromosome regional analysis
To establish relationships between EBTs and gene densities in chromosome sub-regions the genome was firstly segmented into contiguous 10 Mb regions. Regions containing <30,000 SNP markers in the LD maps were excluded from further analysis, as these represent small regions truncated by the end of chromosomes or are regions containing extensive centromeric/heterochromatic sequences or other unsequenceable regions. Regional EBTs were calculated from the Bhérer et al. (2017) linkage map and the Zulu LDU map (Gurdasani et al. 2015). The regional densities of genes were computed counting a particular gene as within a region if the region contained at least half of the gene span.
Chromosome simulations
To independently validate real-data relationships between EBTs and gene densities we simulated chromosome populations with known parameters using the SLiM V3.3 software (Haller and Messer 2019). SLiM V3.3 is a highly flexible simulation software for modelling the chromosomes of individuals in a population. The length of the chromosomes simulated was 10 Mb. The simulations began from a population of 10,000 and ran for 5000 generations. The overall mutation rate was given as 10−8 per site per generation and the recombination rate was set at 10−8 per adjacent bases per generation. The model simulated two types of mutation: neutral mutations which did not affect fitness, and deleterious mutations, with a dominance coefficient of 0.5 and a fixed fitness effect of −0.03. Chromosomes contained genic and non-genic regions. Genes were fixed at 10,000 bp in length, and arranged evenly across the region, with rates of between five and 20 gene per Mb. Mutations arising in the non-genic regions were always neutral. Mutations arising within genes were designated neutral or deleterious at a ratio of 2:8. The final output in VCF format was sub-sampled with 100 chromosomes taken randomly from each simulation. VCF files were converted into .tped format using PLINK v1.90 (Purcell et al. 2007). SNPs with a MAF of <0.01 and markers deviating from Hardy–Weinberg Equilibrium at P < 0.001 significance were excluded. The simulated datasets were then passed to the LDMAP programme. Given the fixed recombination rate of 10−8 per site (i.e. 0.01 recombination events per MB per generation) and 10 Mb chromosomes, a linkage length on 0.1w was used for EBT calculations.
Results
LD maps
LD maps were constructed from the Wellderly sample (Erikson et al. 2016) and samples from three SSA populations: Ethiopia, Zulu and Baganda (Gurdasani et al. 2015). The latter were combined from smaller population groups defined by principal component analysis. Supplementary Fig. 1 shows the PCA plot made using SNP data from seven SSA sub-populations. Although PCA decomposition of genotypes demonstrates very close alignment between Zulu and Baganda populations we constructed independent LD maps for both populations. LD maps in LDUs constructed for the 22 autosomal chromosomes include ~7.2 million SNPs from the Wellderly sample and ~13.6–14.4 million SNPs for each of the SSA populations (Supplementary Table 1). Figure 1 shows the LDU contour for a representative chromosome (chromosome 22) for Wellderly, Ethiopia and Baganda populations demonstrating close alignment across populations between regions of LD breakdown (‘steps’) and flatter plateau regions of lower haplotype diversity (strong LD). The overall increased LDU map lengths for SSA populations compared to Wellderly, reflect, as expected for these populations, increased times since an effective population bottleneck. The Ethiopian map is intermediate consistent with increased admixture with populations of European and Middle Eastern origin perhaps through multiple events within the last 3000 years (Hodgson et al. 2014; Busby et al. 2016).
Map lengths for the autosomes total ~63K LDUs for Wellderly and 107–130K LDUs for the three SSA populations (Supplementary Table 2). The Wellderly map length exceeds a previous estimate by Tapper et al. (2005), using HapMap array genotype data from the CEU population, of 57,819 LDUs. However, although the Tapper et al. (2005) maps also included the X chromosome which was excluded here and SNPs were selected using a MAF cut-off of >0.05, unlike the MAF of >0.01 used here. Inclusion of a higher SNP density in the current analysis increases LDU map length through greater resolution of chromosome regions poorly represented in lower density maps (Pengelly et al. 2015).
Effective bottleneck times
Figure 2 and Supplementary Table 3 give EBTs by population and chromosome. EBTs were computed using three alternative linkage maps: the Kong et al. (2002) map, made from Icelandic pedigrees (‘Kong map’); the Bhérer et al. (2017) map which includes a larger number of meioses and is derived from a combined European data set which includes many Icelandic families (‘Bhérer map’), and the Hinch et al. (2011) map made using data from African–American samples (‘Hinch map’). The Hinch map is slightly longer than the Bhérer and Kong maps (at ~3,523 cM, Supplementary Table 2) and therefore contributes relatively reduced EBTs for all four populations although the impact is across all autosomes and not biased towards specific chromosomes. There is relatively limited difference in the pattern of EBTs defined using alternate linkage maps within a population (Fig. 2). Using the Bhérer map to define EBTs indicates 1876, 3073, 3801 and 3793 generations to an effective bottleneck for Wellderly, Ethiopia, Zulu and Baganda populations respectively. The observed pattern is consistent with the expectation from numerous other studies of reduced effective population sizes experienced by European populations. Figure 2 demonstrates the much increased variance in EBTs for different chromosomes for SSA populations relative to the Wellderly group. The Ethiopian population is intermediate with relatively reduced variance in EBTs compared to the more southerly located SSA populations but with increased variance compared to the Wellderly population. Populations in the Horn of Africa have experienced substantial gene-flow from Middle Eastern and European populations during the past 3000 years (Hodgson et al. 2014) and their genomes show evidence consistent with back-to-Africa migratory events (Pickrell et al. 2014; Busby et al. 2016). As a result, Eurasian admixture over a variety of timescales has generated relatively increased LD demonstrated by reduced LDU map lengths compared to the other SSA populations considered here.

Box plot of effective bottleneck times of autosomes for four populations and alternative linkage maps. Effective bottleneck times computed using the Kong, Bhérer and Hinch linkage maps show small differences with relatively reduced effective bottleneck times when using the African–American Hinch map compared to the Bhérer linkage map from a European sample. The magnitude of variation in chromosome-specific effective bottleneck times follows the trend European < Ethiopian < Zulu/Baganda and is consistent whichever linkage map is used
EBT and gene density
The calculation of gene density for each chromosome depends in part on how the coding regions are defined. We considered alternative gene densities given by Mayer et al. (2005) and Spataro et al. (2017) (Supplementary Table 4). The latter provides gene counts in different gene groups: non-disease non-essential (NDNE) which are genes not known to be involved in disease phenotypes or classed as ‘essential’; complex non-Mendelian (CNM) genes known to contain common variation involved in complex but not currently implicated in Mendelian phenotypes; complex-Mendelian (CM) genes containing variants known to be involved in both complex and Mendelian phenotypes; Mendelian non-complex (MNC) genes known to contain Mendelian, but not complex trait variation and essential non-disease (END) essential genes not known to contain disease variation. END genes were defined as having a mouse ortholog showing pre-natal, peri-natal or post-natal lethality in mouse knockouts. Gene densities from both Mayer et al. and Spataro et al. indicate that chromosomes 13, 4 and 18 have the lowest density of genes (Mayer et al. 2005; Spataro et al. 2017). Mayer et al. (2005) has chromosomes 19, 17 and 22 as the most gene dense and Spataro et al. (2017) has 19, 17 and 11 as most gene dense. Table 1, supplementary Table 5 and Fig. 3 show relationships between gene densities and EBTs. Negative correlations between gene densities and EBTs are found for all four populations although only as a trend for the Wellderly population (Table 1). Correlations are stronger for the Mayer et al. gene densities than the Spataro et al. ‘all’ gene densities although the density of END genes shows the strongest correlation with EBTs. From the linear fit the reduction in EBT from chromosomes with the lowest density of END genes to the highest density for the SSA populations (Fig. 3) is ~17–21% corresponding to ~514–748 generations. The density of genes associated with only complex phenotypes (CNM, Table 1) shows non-significant correlations.

Effective bottleneck times (using Bhérer map) against the density of essential non-disease genes (END). Chromosome numbers corresponding to the points plotted are given in circles above the graph. A strong trend following reduced effective bottleneck times for chromosomes with increasing density of essential genes is shown for the SSA populations (Ethiopia, Zulu, Baganda) with a weaker and non-significant trend for the European (Wellderly) population. Linear trends indicate ~ 17%, 20% and 21% reduction in effective bottleneck times for Ethiopia, Baganda and Zulu populations respectively for chromosomes with lowest to highest density of essential genes. The difference is only ~5% for the European (Wellderly) population
Figure 4 shows the relationship between EBTs in sub-chromosomal regions across the genome (computed using the Bhérer linkage map and the Zulu LDU map, Supplementary Table 2) and the density of END genes in each region. The pattern is consistent with the relationship observed for whole chromosomes of reduced EBTs associated with higher gene densities (for the 269 10 Mb regions, correlation r = −0.328, P < 0.00001).

Effective bottleneck times in 10 Mb regions computed from the Bhérer map and the Zulu LDU map versus the regional density of essential non-disease genes (END). The trend towards reduced EBTs with an increasing regional density of essential genes is evident. The linear trend is consistent with a ~ 57% reduction in EBTs for regions with the lowest to highest density of essential genes
The data are supportive of the hypothesis that chromosomes and their sub-regions which have a high density of essential genes are subject to stronger purifying selection reflected in consistently reduced EBTs. The trend is weak for the Wellderly sample and strongest for both the Zulu and Baganda populations (Fig. 3). The impact of defining EBT using the alternative Kong and Hinch linkage maps has a variable impact on the significance of correlations but trends remain consistent (Supplementary Table 5).
Chromosome simulations using known input parameters were performed to investigate the relationship between gene density and EBT. Figure 5 shows LDU maps from simulated chromosomes where only gene densities are varied. All other parameters were fixed between populations, including recombination and mutation rates, however mutations falling in genic regions were assumed to have a higher chance of being deleterious with non-genic regions subject only to neutral mutation. The results demonstrate a relationship between increasing gene density and decreasing LDU map length. Given the fixed recombination rate per chromosome the LDU map lengths are readily converted into EBTs (Fig. 6) demonstrating a strong negative correlation with gene density (P < 0.001). The simulations provide support to the relationship shown here in real data between increased gene density and reduced EBTs reflecting purifying selection in gene-dense chromosomes.

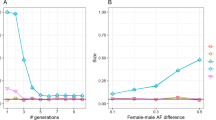
LDU map lengths of gene-dense and gene-sparse chromosomes simulated using SLiM software. LDU maps made from a sample of 100 individuals from a simulated population using SLiM V3.3. The same parameters were applied to all populations, including recombination rates, with the only variation being in gene density, ranging between five and twenty genes per Mb. Reduced LDU lengths (and correspondingly reduced EBTs in the gene-dense chromosome populations) is consistent with real-data findings in SSA populations

Relationship between EBT and gene density for chromosomes simulated using SLiM software. LDU maps made from a sample of 100 individuals from a simulated population using SLiM V3.3. Increased gene density is negatively correlated with EBT as predicted and demonstrated in the SSA data. This relationship is statistically significant, calculated using the default F-test in R
Discussion
Selection which acts to increase the frequency of beneficial mutations and eliminate detrimental mutations is more efficient in large populations (Cutter and Payseur 2013). Therefore the intensity of hitch-hiking and the impact of background selection is increased within populations with greater effective population size (Ne) (Charlesworth 2009). Given increased Ne in SSA populations, compared to populations with European ancestry, this expectation is consistent with differences in EBTs which indicate chromosome-specific patterns of selection. It is known that intense population bottlenecks, such as those experienced by European populations on leaving Africa, may erase historically-present signatures of selection at linked sites (Cutter and Payseur 2013). This might account for the much reduced evidence (trend only) for a relationship between chromosome EBTs and gene density in the Wellderly sample which has European ancestry.
Accurate computation of EBTs depends on availability of population-specific genetic linkage maps, unless the pattern of recombination is broadly conserved across populations. The Kong linkage map from Icelandic families and the Bhérer map, from a combined data set representing European populations, yield consistently similar EBTs. The lack of a recombination map from a pure SSA population presents difficulties. The Hinch map represents an African-American population, with a high proportion of SSA ancestry, although it is constructed from population rather than family data and has therefore been normalised for total map length. There is little indication that recombination rates differ by chromosome sufficiently to impact the pattern of chromosome-specific EBTs and there is good evidence that broad patterns of recombination are conserved across human populations (Jorgenson et al. 2005; Serre et al. 2005). The ratio of recombination rates for two populations has been shown to be constant along the chromosomes such that genetic linkage maps made from European populations are considered valid to make inferences in other populations (Serre et al. 2005). However, on finer scales there is evidence that more of the genome is recombinationally active in West Africans (Hinch et al. 2011), which have a larger number of recombination hotspots making crossovers more evenly distributed across chromosomes compared to Europeans. Only limited variation in EBTs is evident when defined using the three alternative genetic linkage maps, consistent with the suggestion that European-based linkage maps are adequate for making wider inferences in other populations.
Increased haplotype diversity, and therefore overall reduced LD, amongst SSA populations reflects their extended population history. Detection of selection events is enhanced by both the extended history of SSA populations, providing sufficient time for selective forces to create a detectable signal, and the reduced impact of population bottlenecks which may erase signatures of selection. Although some signatures of recent adaptation since the divergence of African and non-African populations have been identified (Hamblin and Di Rienzo 2000; Sabeti et al. 2002; Bersaglieri et al. 2004; Lamason et al. 2005; Pickrell et al. 2009) there is limited evidence for frequent hard selective sweeps since migration out of Africa (Teshima et al. 2006) and identification of such events is confounded by more prominent evolutionary forces such as population bottlenecks, drift and stratification (Coop et al. 2009; Berg et al. 2018).
Evidence established over a number of years demonstrates that selection has impacted whole chromosomes to different degrees. The sex chromosomes show strong signatures of increased selection relative to the autosomes as a whole. X chromosome hemizygosity in males results in accelerated adaptive evolution across X-linked genes (Vicoso and Charlesworth 2006). Recessive X-linked mutations in males are subject to selection and become readily fixed in the population even when their deleterious burden in females surmounts their advantageous utility in males (Nam et al. 2015). As a result, the X chromosome is enriched for sexually antagonistic alleles that are pertinent to male and female reproductive fitness (Sangrithi and Turner 2018). Although gene density on the X is generally low compared to autosomes there is evidence from diversity that the targets of selection include its coding regions. Hammer et al. (2010) considered the relationship between normalised nucleotide diversity and genetic distance from genes on both the X chromosome and autosomes. Considering nongenic regions in 0.1 cM bins, diversity was substantially lower within bins located close to genes compared to bins further away. Although the magnitude of the effect was more marked on the X chromosome compared to the autosomes a broadly similar relationship was observed for both.
The evidence here indicates a strong relationship between chromosome-specific EBTs and the density of essential genes. Classical examples of positive selection in the human genome have been demonstrated for individual genes and include non-essential genes such as SLC24A5 on chromosome 15 (Sabeti et al. 2002) and LCT (Bersaglieri et al. 2004) and EDAR (Sabeti et al. 2007) which are both on chromosome 2. However, evidence for a relationship between variation in chromosome-specific EBTs and the density of all genes (not just essential genes) is consistent with purifying selection impacting numerous targets across the genome. It is known that chromosomes and regions with high gene densities are subject to weaker positive selection due to Hill-Robertson interference (Hill and Robertson 1966) but are under increased purifying selection acting on functionally deleterious mutations (Castellano et al. 2016). Our findings show that the impact of these processes can be observed at the level of whole chromosomes, at least in SSA populations.
Only small differences in EBTs are evident between different autosomes in the European population studied here. Historical emphasis on studies in European populations may have made chromosome-specific signatures of selection difficult to detect. The greatly increased availability of samples derived from European compared to SSA populations has reduced the possibility of detecting chromosome-specific differences in selection intensity for different autosomes. The evidence presented here supports deeper analysis of signatures of selection within SSA populations and the expectation that studies in these populations offer greater power to detect chromosome (and gene) specific signatures of selection.
Conclusions
The increased lengths of LDU maps constructed from SSA human populations, compared to a map from a population of European ancestry are consistent with the expectation of increased EBTs in these populations. Previous focus on European populations has suggested that EBTs are relatively constant for all autosomal chromosomes, although reduced for the X chromosome in line with evidence that it has undergone unusually intense selective sweeps. However, analysis of SSA populations shows variability in EBTs across the autosomes with reduced EBTs for some chromosomes, such as chromosomes 17 and 19, which have high a density of essential genes. This is consistent with a pattern of strong purifying selection (and weaker positive selection due to Hill-Robertson interference) reducing the diversity of gene-dense chromosomes. The findings strongly support efforts to analyse patterns of selection in SSA populations since the power to detect signals is enhanced through their extended population history and reduced impact of intense population bottlenecks.
References
Bachtrog D (2013) Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet 14:113–124
Berg JJ, Harpak A, Sinnott-Armstrong N, Jørgensen AM, Mostafavi H, Field Y et al. (2018) Reduced signal for polygenic adaptation of height in UK Biobank. bioRxiv: 354951
Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA et al. (2004) Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet 74:1111–1120
Bhérer C, Campbell CL, Auton A (2017) Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nat Commun 8:14994
Busby GB, Band G, Si Le Q, Jallow M, Bougama E, Mangano VD (2016) Admixture into and within sub-Saharan Africa. Elife 5:e15266
Castellano D, Coronado-Zamora M, Campos JL, Barbadilla A, Eyre-Walker A (2016) Adaptive evolution is substantially impeded by Hill-Robertson interference in Drosophila. Mol Biol Evol 33:442–455
Charlesworth B (2009) Effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195–205
Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D et al. (2009) The role of geography in human adaptation. PLoS Genet 5:e1000500.
Cutter AD, Payseur BA (2013) Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet 14:262–274
Dutheil JY, Munch K, Nam K, Mailund T, Schierup MH (2015) Strong selective sweeps on the X chromosome in the human-chimpanzee ancestor explain its low divergence. PLOS Genet 11:e1005451
Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA et al. (2016) Whole-genome sequencing of a healthy aging cohort. Cell 165:1002–1011
Gottipati S, Arbiza L, Siepel A, Clark AG, Keinan A (2011) Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nat Genet 43:741–743
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K et al. (2015) The African Genome Variation Project shapes medical genetics in Africa. Nature 517:327–332
Haller BC, Messer PW (2019) SLiM 3: forward genetic simulations beyond the Wright–Fisher Model (R Hernandez, Ed.). Mol Biol Evol 36:632–637
Hamblin MT, Di Rienzo A (2000) Detection of the signature of natural selection in humans: evidence from the duffy blood group locus. Am J Hum Genet 66:1669–1679
Hammer MF, Woerner AE, Mendez FL, Watkins JC, Cox MP, Wall JD (2010) The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nat Genet 42:830–831
Hill WG, Robertson A (1966) The effect of linkage on limits to artificial selection. Genet Res 8:269–294
Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD et al. (2011) The landscape of recombination in African Americans. Nature 476:170–175
Hodgson JA, Mulligan CJ, Al-Meeri A, Raaum RL (2014) Early Back-to-Africa migration into the Horn of Africa. PLoS Genet 10:e1004393
Hudson RR, Kaplan NL (1995) Deleterious background selection with recombination. Genetics 141:1605–1617
Huff CD, Harpending HC, Rogers AR (2010) Detecting positive selection from genome scans of linkage disequilibrium. BMC Genomics 11:8
Jorgenson E, Tang H, Gadde M, Province M, Leppert M, Kardia S et al. (2005) Ethnicity and human genetic linkage maps. Am J Hum Genet 76:276–290
Ke X, Hunt S, Tapper W, Lawrence R, Stavrides G, Ghori J et al. (2004) The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum Mol Genet 13:577–588
Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B et al. (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247
Kong A, Thorleifsson G, Frigge ML, Masson G, Gudbjartsson DF, Villemoes R et al. (2014) Common and low-frequency variants associated with genome-wide recombination rate. Nat Genet 46:11–16
Lamason RL, Mohideen M-APK, Mest JR, Wong AC, Norton HL, Aros MC et al(2005) SLC24A5, a Putative Cation Exchanger, Affects Pigmentation in Zebrafish and Humans Science 310:1782–1786
Lau W, Kuo T-YTY, Tapper W, Cox S, Collins A (2007) Exploiting large scale computing to construct high resolution linkage disequilibrium maps of the human genome. Bioinformatics 23:517–519
Mayer R, Brero A, von Hase J, Schroeder T, Cremer T, Dietzel S (2005) Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol 6:44
Nam K, Munch K, Hobolth A, Dutheil JY, Veeramah KR, Woerner AE et al. (2015) Extreme selective sweeps independently targeted the X chromosomes of the great apes. Proc Natl Acad Sci USA 112:6413–6418
Payseur BA, Nachman MW (2002) Gene Density and Human Nucleotide Polymorphism. Mol Biol Evol 19:336–340
Pengelly RJ, Tapper W, Gibson J, Knut M, Tearle R, Collins A et al. (2015) Whole genome sequences are required to fully resolve the linkage disequilibrium structure of human populations. BMC Genomics 16:666
Pengelly RJ, Vergara-Lope A, Alyousfi D, Jabalameli MR, Collins A (2019) Understanding the disease genome: gene essentiality and the interplay of selection, recombination and mutation. Brief Bioinform 20:267–273
Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D et al. (2009) Signals of recent positive selection in a worldwide sample of human populations. Genome Res 19:826–837
Pickrell JK, Patterson N, Loh P-R, Lipson M, Berger B, Stoneking M et al. (2014) Ancient west Eurasian ancestry in southern and eastern Africa. Proc Natl Acad Sci 111:2632–2637
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MARR, Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF et al. (2002) Detecting recent positive selection in the human genome from haplotype structure. Nature 419:832–837
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C et al. (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449:913–918
Sangrithi MN, Turner JMA (2018) Mammalian X chromosome dosage compensation: perspectives from the germ line. BioEssays 40:1800024
Serre D, Nadon R, Hudson TJ (2005) Large-scale recombination rate patterns are conserved among human populations. Genome Res 15:1547–1552
Smith JM, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23:23–35
Spataro N, Rodríguez JA, Navarro A, Bosch E (2017) Properties of human disease genes and the role of genes linked to Mendelian disorders in complex disease aetiology. Hum Mol Genet 26:489–500
Tapper W, Collins A, Gibson J, Maniatis N, Ennis S, Morton NE (2005) A map of the human genome in linkage disequilibrium units. Proc Natl Acad Sci USA 102:11835–11839
Teshima KM, Coop G, Przeworski M (2006) How reliable are empirical genomic scans for selective sweeps? Genome Res 16:702–712
Veeramah KR, Gutenkunst RN, Woerner AE, Watkins JC, Hammer MF (2014) Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Mol Biol Evol 31:2267–2282
Vicoso B, Charlesworth B (2006) Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet 7:645–653
Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4:e72
Wigginton JE, Cutler DJ, Abecasis GRGR (2005) A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 76:887–893
Wilson Sayres MA, Lohmueller KE, Nielsen R (2014) Natural selection reduced diversity on human Y chromosomes. PLoS Genet 10:e1004064
Zhang W, Collins A, Gibson J, Tapper WJ, Hunt S, Deloukas P et al. (2004) Impact of population structure, effective bottleneck time, and allele frequency on linkage disequilibrium maps. Proc Natl Acad Sci 101:18075–18080
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28:3326–3328
Acknowledgements
The authors acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jabalameli, M.R., Horscroft, C., Vergara-Lope, A. et al. Gene-dense autosomal chromosomes show evidence for increased selection. Heredity 123, 774–783 (2019). https://doi.org/10.1038/s41437-019-0272-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0272-5
This article is cited by
-
Chromosomal instability in aneuploid acute lymphoblastic leukemia associates with disease progression
EMBO Molecular Medicine (2023)
