
Abstract
Natural and artificial selection have led to substantial variation in the phenotypic traits of different populations. Therefore, there is a need to develop methods that are based on cross-population comparisons to discover loci related to specific traits. Here, we suggested a strategy to detect the genome selection signatures between populations based on the partial least squares (PLS) theory. Using the binary population indicator as the response variable in the PLS analysis, alleles under selection between populations were identified from the first PLS component. We explored the theory behind the PLS analysis to reveal its usefulness in detecting the loci under selection. Through the simulation study, the results showed that the PLS method had a better performance than the FST and EigenGWAS methods. In addition, by using the real data hapmap3, we found that rs11150606 in PRSS53 gene and rs1800414 in OCA2 gene were under selection between East Asian populations and three other populations, including African, American, and European populations. We concluded that this strategy was easily carried out and might supplement for the deficiency of the EigenGWAS method in some cases. To facilitate the application of this method, we developed an R script that is freely accessible at http://klab.sjtu.edu.cn/PLS/.
Similar content being viewed by others


Introduction
Identifying the genomic regions associated with phenotypic variation is a longstanding interest of biologists. An experimental design based on cross-population comparisons has been proven to be a useful method to discover loci related to specific traits (Kim et al. 2015). For example, by comparing the genome of the Yucatan Miniature pig breed to other large pig breeds, the signatures of the selection specific to body size were detected (Kim et al. 2015).
The principal component analysis (PCA) was recently used to detect the population structure. Based on the algorithm of the PCA analysis, some methods were developed to detect genome selection signatures (Chen et al. 2016; Duforet-Frebourg et al. 2016; Galinsky et al. 2016). Among these methods, Chen et al. (2016) postulated the EigenGWAS for finding loci under selection in structured populations by using the individual-level eigenvectors as phenotypes in a linear regression. This method detected the selection signatures that reflect the differences among samples in each eigenvector dimension. However, the previous cross-population comparison information was not considered in the PCA method.
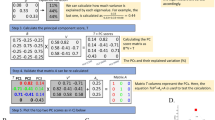
Therefore, the results calculated by PCA method may not accurately reflect the selection signatures between populations, especially when many sub-populations are considered. For example, assuming that there are two different base populations and each population is characterized with a distinct phenotype of tall and short individuals (Table 1), we should regard A and C as a group (tall) and B and D (short) as another group if our aim is to detect markers associated with the height variation. The PCA method does not consider this previous cross-population comparison, and if the analysis is implemented based on the PCA theory, the results obtained may not reflect the difference between population 1 and 2 (AC and BD). Hence, we aimed to apply the partial least squares (PLS) using the previous cross-population comparison information to optimize the PCA method.
The PLS is a supervised learning algorithm that uses the information about the response variables (Y) to construct the new components of the independent variables (X), and the extracted principle components obtained are highly associated with the response variables (Boulesteix and Strimmer 2007). Therefore, we used the binary population indicator into the PLS analysis as the response variable. In this study, we described the details of using the PLS method to explore the genome selection signatures between populations using both theoretical and real data. By using the real data, two scenarios described the PLS performance. Scenario 1 contains two sub-populations, and Scenario 2 contains more than two sub-populations. In addition, the EigenGWAS and FST (Weir and Cockerham 1984) methods were compared with the PLS method.
Materials and methods
Theory
We defined n as the total sample size and m as the number of genome makers. Let Xij (coded by 0, 1, or 2) be the genotype of sample i marker j. We defined the sample information from the different populations as a categorical variable (Y): one group was 0 and the other was 1. Thus, Y was a n × 1 matrix. First, we normalized Xij by subtracting the mean value and dividing the standard deviation of the marker j. The mean value of the marker j was \(\mu _j = \frac{{\mathop {\sum }\nolimits_{i = 1}^n X_{ij}}}{n}\). The standard deviation value of the marker j was \(s_j = \sqrt {\frac{{\mathop {\sum }\nolimits_{i = 1}^n (X_{ij} - \mu _j)^2}}{{n - 1}}}\). The normalized X matrix was denoted as X1. Similarly, we normalized Y as stated above. The normalized Y matrix was denoted as Y1.
Let T1 be the first principle component of X1, and let W1 be the first weight vector. Thus, T1 = X1W1, and W1TW1 = 1. The aim was to detect the W1 that maximized the correlation between T1 and Y1. The covariance between T1 and Y1 can be written as \({\mathrm{COV}}(T1,Y1) = \frac{1}{n}W1^TX1^TY1\), because X1 and Y1 are normalized (Boulesteix and Strimmer 2007). From the PLS algorithm, W1 is the first eigenvector of X1TY1Y1TX1 (Wold et al. 2001). It is difficult to carry out an eigendecomposition on X1TY1Y1TX1 which is a m × m matrix. Interestingly, when computing a singular vector decomposition on matrix Y1TX1, the right singular vector is the eigenvectors of (Y1TX1)TY1TX1 based on the singular vector decomposition algorithm (Kalman 2002). Thus, W1 can be obtained.
W1j shows the coefficient of each marker with respect to T1. Many methods have been studied to select the variables (Mehmood et al. 2012). To control the FDR (false discovery rate), we assumed that the coefficients were Gaussian with a zero mean (Duforet-Frebourg et al. 2016; Galinsky et al. 2016). Therefore, the variance of the W1j is \(V_{W1} = \frac{{{\sum} {\left( {W1_j - 0} \right)^2} }}{m}\), and VW1 is \(\frac{1}{m}\). Thus, \(\frac{{(W1_j - 0)^2}}{{V_{W1}}}\) follows a Chi-square distribution with one degree of freedom. Then, the statistic is m(W1j)2.
Real sequencing data
Hapmap3 data were downloaded from the http://www.hapmap.org (Altshuler et al. 2010). The SNPs with missing values were filtered. Finally, a total of 1184 samples and 363,251 SNPs (the minor allele frequency more than 0.05) were obtained. These samples came from 11 populations. The details of these samples are shown in Table S1. The pig data set was collected from the research of Wang et al. (2015). A total of 252 Taihu area pigs and 105,550 SNPs were obtained.
Simulation study
Two simulation studies were performed. In simulation 1, we used the Hapmap3 data. A total of 1184 human samples and 30,544 SNPs, sampled from the chromosome 1, were used. In simulation 2, we used the genotype data from the Taihu area pigs. A total of 252 pig samples and 9690 SNPs sampled from chromosome 1 were used. In each simulation study, we assigned genetic effects to 10 randomly selected markers. We defined the residual error of the phenotypes, which was drawn from a normal distribution. Then, the simulated phenotypes were obtained from the model y = Xkak + e, where the Xk vector (coded as zero, one and two for the three genotypes) stores the numerical codes of the genotypes for all the individuals of marker k. ak represents the effect of marker k. The residual error e of each individual, was randomly sampled from the normal distribution independently. The individual phenotype value that was more than the mean value was redefined as “1”. The individual phenotype value that was less than the mean value was redefined as “0”. Then, the response variable was obtained. We then performed EigenGWAS, FST and our approach. The PLS method was performed using the theory stated above. The EigenGWAS was performed by using the individual-level eigenvector, which has the maximal covariance with the response variable, as phenotypes in a linear regression. The individual-level eigenvectors were calculated by carrying out an eigendecomposition on an X1X1T matrix (Price et al. 2006). The FST method described by Weir and Cockerham (1984) was performed. Each simulation study was replicated 10 times. To compare the statistical power of the three methods, the threshold level was set as 1‰ and 1% of the SNPs with the most extreme values of the statistics. Then, the empirical statistical power was the ratio that was between the true SNP numbers detected under the threshold level and all the true SNP numbers.
Scenario one: two sub-populations
The aim of this scenario was to explore the selection signatures between the CEU (Utah residents with Northern and Western European ancestry from the CEPH collection) and TSI (Toscans in Italy) population. The sample size of the CEU and TSI populations were 165 and 88, respectively. Similar to the simulation study, the analysis was performed using the PLS, EigenGWAS, and FST methods. To ensure that there was no over-fitting, a 10-fold cross validation was performed. We randomly partitioned the samples into 10 parts of roughly equal size (9 parts had 25 samples and 1 part had 28 samples). We then used the 9 parts (X1Training) to predict the group information of the remaining portion of samples (X1Test). Using the training data (X1Training, Y1Training), the PLS component loading (W1Training) was obtained. Then, T1Training = X1TrainingW1Training and T1Test = X1TestW1Training. The estimate group value was obtained according to the mean distance between the test sample and the two different training groups in the T1 dimension. After all the parts were predicted, we estimated the time (et) when the prediction results of the test samples were accurately predicted. We randomly partitioned the samples into 10 parts of roughly equal size, and each partition was done10 times. Then, the predictability was defined as: \({\mathrm {Accuracy}} = \mathop {\sum}\nolimits_{t = 1}^{t = 10} {\frac{{e_t}}{{253}}}\).
Scenario two: 11 sub-populations
The aim of this experiment was to explore the selection signatures between East Asians and other populations, including African, American, and European populations (Table S1). We pooled together the CHB (Han Chinese in Beijing, China), CHD (Chinese in Metropolitan Denver, Colorado), and JPT (Japanese in Tokyo, Japan) populations as a group, and the other eight populations were another group. Similar to the simulation study, the analysis was performed using the PLS, EigenGWAS, and FST methods.
Results
Simulation study
We found that the statistical power of the PLS method was higher than the FST and EigenGWAS methods in the two simulation studies whether the threshold was the top 1‰ or 1% (Table 2). The statistical power of the PLS method was slightly higher than the FST. The statistical power of the PLS and FST methods were much higher than the EigenGWAS method. In addition, the statistical powers of the PLS and FST methods in the human simulation study were much higher than in the pig simulation study.
Scenario one
For the PLS analysis, the TSI and CEU populations were clearly distinguished in the first PLS component dimensionality (Fig. 1a). The absolute value of the Pearson correlations (|r|) between the first PLS component and the prior class information was 0.994 (Table 3). A total of 455 SNPs having a p-value < 0.001 were detected (Fig. 2a). The most significant SNPs were located in chromosome 2 and were associated with the MCM6, DARS, and LCT genes. These three genes were located across a 68-kb area. Apart from chromosome 2, another significant SNP (rs916977, P = 2.13E-06, Rank = 43) was also located in the HERC2 gene in chromosome 15.

The population distribution in the PLS component. a The projected first PLS component for the CEU and TSI populations. b The projected first PLS component for the 11 human populations. The sampled populations were the following: East Asian (CHB, CHD, JPT); European (CEU, TSI); American (African ancestry in Southwest USA, ASW; Gujarati Indians in Houston, GIH; Mexican ancestry in Los Angeles, MEX); and African (Luhya in Webuye, LWK; Maasai in Kinyawa, MKK; Yoruba in Ibadan, YRI)

The Manhattan plot of the PLS results.The x-axis represents the SNPs, and the y-axis is the −log10(p). The red line in the middle is the genome-wide significant level at a p value of 0.001. a The SNP results of the PLS method in Scenario 1. b The SNP results of the PLS method in Scenario 2
To perform the EigenGWAS analysis, the |r| between the individual-level eigenvectors and the response variable were analyzed (Figure S1a). The |r| between the first eigenvector and the prior class information was 0.807, which was maximal (Table 3). Therefore, the EigenGWAS was performed using the first eigenvector as the phenotype information. The most significant areas detected in the EigenGWAS method were also located in chromosome 2 (Figure S2a). The genetic signals detected by the FST method are shown in Figure S2b. Considering all of the test SNPs, the Spearman rank correlation between the FST and the PLS was 0.978, and the correlation between the FST and the EigenGWAS was 0.541 (Table 3). In the cross-validation analysis, the mean accuracy was 100%. The estimated group values were all correct. One result of the cross-validation analysis is shown in Figure S3.
Scenario two
In the PLS analysis, the East Asians were clearly distinguished from the other populations in the first PLS component dimensionality (Fig. 2b). The |r| between the first PLS component and the prior class information was 0.901 (Table 3). A total of 244 SNPs were explored (p-value < 0.001) (Fig. 2b). The most significant SNPs were located in chromosomes X, 16 and 15. In chromosome 16, the most significant SNP was rs11150606 (ranked as top 2), which was located in the PRSS53 gene. In chromosome 15, a significant SNP rs1800414 (ranked as top 8), located in the OCA2 gene, was identified.
The |r| between the individual-level eigenvectors and the prior class information of samples are shown in Figure S1b. The |r| between the second eigenvector and the prior class information was maximal, which was 0.790 (Table 3). Therefore, the EigenGWAS was performed using the second eigenvector as the phenotypes. In the EigenGWAS analysis (Figure S4a), the rank orders, from small to large p-value, of rs11150606 and rs1800414 were 46 and 62, respectively. In the FST analysis (Figure S4b), the rank orders, from the large to small FST statistic, of rs11150606 and rs1800414 were 11 and 68, respectively. By considering the entire test SNPs, the Spearman rank correlation between the FST and the PLS was 0.975, and the Spearman rank correlation between the FST and the EigenGWAS was 0.269 (Table 3).
Discussion
PLS is a supervised method that is specifically established to address the problem of making good predictions in multivariate problems (Mehmood et al. 2012). The PLS framework selects the component that has the maximal covariance with the response variable. In our study, the response variable was the binary code for two groups, and the important variables were selected according to SNP coefficients. Through the simulation study, the results showed that the PLS method identified the genetic selection signatures and had a better performance than the FST and EigenGWAS methods. In addition, the statistical power of the FST was higher than the EigenGWAS. The previous cross-population comparison information was not considered in the PCA method. Thus, the results calculated by the PCA method may not actually reflect the selection signatures between the populations. This may be the reason why the statistical power of the EigenGWAS method was lower than the FST and PLS methods. Compared to the FST method, the PLS method combines both the theory of the PCA and a correlation analysis. For the large variable data in the genetic selection signatures analysis, the relationships between variables were not considered in the FST analysis, since it was a single locus statistic. Thus, this might be the reason why the statistical power of the FST method was lower than the PLS method in the simulation study. The cross-validation analysis was implemented considering the fact that the PLS method might suffer from the over-fitting problem for the large variables and the small sample data. In the cross-validation analysis, the estimate group values were all correct. This result might reflect the credibility of the PLS method in our study. Using this strategy, we detected selection signatures in a cross-population comparison design based on the real sequencing data.
In Scenario 1, the aim was to explore the selection signatures between the TSI and CEU populations. We retrieved well-known signals of adaptation in humans that were associated with lactase persistence (LCT) (Segurel and Bon 2017) and eye color (HERC2) (Sturm et al. 2008) (Fig. 1). In Scenario 2, we detected two significant SNPs, namely, rs11150606 and rs1800414. The two SNPs were missense variants. It is reported that rs11150606 influences hair shape, and the effect of this locus is demonstrated in a real molecular experiment (Adhikari et al. 2016). Therefore, considering the difference in the hair shape between East Asians and other populations, rs1800414 should be regarded as the true selection signature between the East Asians and other populations. It is reported that rs1800414 was associated with pigmentation in East Asians populations (Edwards et al. 2010). Interestingly, this SNP is not found at high frequencies of this polymorphism in any population outside of East Asia (Yuasa et al. 2011). These studies reflect that rs1800414 should be regarded as the true selection signature between the East Asians and other populations. Thus, from the results of Scenarios 1 and 2, we concluded that our strategy was feasible for exploring the genome selection signatures between populations.
Furthermore, the EigenGWAS and FST methods were compared with the PLS method. EigenGWAS regards the individual-lever eigenvector as the phenotype of the genome-wide association study. Based on the PCA theory, the eigenvector was extracted from the genotype information. Thus, we hypothesized that the individual-lever eigenvector may not actually reflect the information of the cross-population comparison and demonstrated this in the real study cases. Scenario 1 and Scenario 2 were a cross-population comparison design. We found that the correlation between the individual-level eigenvectors and the response variable was lower than the correlation between the PLS component and the response variable. In addition, the SNP rank correlation between EigenGWAS and FST was lower than the correlation between PLS and FST (Table 3). The FST method is the most wildly used for exploring selection signatures between populations (Vatsiou et al. 2016). Considering the robustness of the FST analysis, less bias might be identified in this method. Thus, the EigenGWAS method in Scenarios 1 and 2 might identify much bias results. From the results of the Spearman rank analysis in Scenarios 1 and 2, the PLS method was extremely similar to the FST method. The reason for this result might be that the prior information was also considered in the FST analysis as it was in the PLS method. Although the prior information was regarded as a categorical variable in the PLS analysis, a continuous axis (the PLS component) was obtained (Fig. 1) as in the PCA method. The continuous axis might provide useful information of the genetic variation within groups (Price et al. 2006). Therefore, in this respect, the EigenGWAS and PLS methods had a better performance in visualization in comparison with the FST method.
Another advantage of the PLS analysis in exploring the genetic selection signatures was the lower computing complexity. To carry out the EigenGWAS analysis, the individual-level eigenvectors need to be calculated first. Then, this is followed by the estimation of the SNP effect one after the other through a genome-wide association study. In addition, to carry out the FST analysis, the SNP effect needs to be calculated one by one. However, in the PLS analysis, all of the SNPs effects were generated at the same time from the SVD analysis on the matrix Y1TX1. A computer with the CPU of Intel(R) Core(TM) i5-4460 3.20 GHz and RAM of 8.00 GB was used to analyze the Scenario 1 data. We wrote the R script, and the analysis was performed in the R platform (x86_64-w64-mingw32/x64). The time it took generate the analysis from the input of the genotype data files to the output of the SNP p-value files was approximately 45 s. During the analysis, it took just 3 s for the execution of the SVD analysis. In addition, in order to facilitate the application of the PLS strategy, we developed a freely accessible R script and provided example data. It is freely accessible at http://klab.sjtu.edu.cn/PLS/.
Finally, as a summary of our work and a prospect for a possible extension, multiple response variables were considered in exploring the genetic selection signatures. In this paper, we applied partial least squares to explore the genome selection signatures between the populations. Through the simulation study, the results showed that the PLS method had a better performance than the FST and EigenGWAS methods. Using real human genome data, we demonstrated that it was feasible and fast to explore the genome selection signatures using partial least squares in a cross-population comparison design. However, a single response variable was considered in the analysis. For some special study cases, i.e., if the aim of the study design is to detect genetic differences among multiple populations, multiple response variables should be prepared. We suggest that the response variable should be created as a matrix. Each column is a binary indicator vector representing the presence (“1”) or absence (“0”) of the population indicated. Then, the genetic difference among multiple populations might be detected.
References
Adhikari K, Fontanil T, Cal S, Mendoza-Revilla J, Fuentes-Guajardo M, Chacon-Duque JC et al. (2016) A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat Commun 7:10815
Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature 467:52–58
Boulesteix AL, Strimmer K (2007) Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinform 8:32–44
Chen GB, Lee SH, Zhu ZX, Benyamin B, Robinson MR (2016) EigenGWAS: finding loci under selection through genome-wide association studies of eigenvectors in structured populations. Heredity 117:51–61
Duforet-Frebourg N, Luu K, Laval G, Bazin E, Blum MG (2016) Detecting genomic signatures of natural selection with principal component analysis: application to the 1000 genomes data. Mol Biol Evol 33:1082–1093
Edwards M, Bigham A, Tan J, Li S, Gozdzik A, Ross K et al. (2010) Association of the OCA2 polymorphism His615Arg with melanin content in east Asian populations: further evidence of convergent evolution of skin pigmentation. PLoS Genet 6:e1000867
Galinsky KJ, Bhatia G, Loh PR, Georgiev S, Mukherjee S, Patterson NJ et al. (2016) Fast principal-component analysis reveals convergent evolution of ADH1B in Europe and East Asia. Am J Hum Genet 98:456–472
Kalman D (2002) A singularly valuable decomposition: the SVD of a matrix. The American University, Washington, p 5
Kim H, Song KD, Kim HJ, Park W, Kim J, Lee T et al. (2015) Exploring the genetic signature of body size in Yucatan miniature pig. PLoS One 10:e0121732
Mehmood T, Liland KH, Snipen L, Saebo S (2012) A review of variable selection methods in partial least squares regression. Chemom Intell Lab Syst 118:62–69
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Segurel L, Bon C (2017) On the evolution of lactase persistence in humans. Annu Rev Genom Hum Genet 18:297–319
Sturm RA, Duffy DL, Zhao ZZ, Leite FP, Stark MS, Hayward NK et al. (2008) A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am J Hum Genet 82:424–431
Vatsiou AI, Bazin E, Gaggiotti OE (2016) Detection of selective sweeps in structured populations: a comparison of recent methods. Mol Ecol 25:89–103
Wang Z, Chen Q, Yang Y, Liao R, Zhao J, Zhang Z et al. (2015) Genetic diversity and population structure of six Chinese indigenous pig breeds in the Taihu Lake region revealed by sequencing data. Anim Genet 46:697–701
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wold S, Sjostrom M, Eriksson L (2001) PLS-regression: a basic tool of chemometrics. Chemom Intell Lab Syst 58:109–130
Yuasa I, Harihara S, Jin F, Nishimukai H, Fujihara J, Fukumori Y et al. (2011) Distribution of OCA2 *481Thr and OCA2 *615Arg, associated with hypopigmentation, in several additional populations. Leg Med 13:215–217
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant nos. U1402266 and 31772552).
Author contributions
H.S., Y.P. and Q.W. conceived the study; Y.P. and Q.W. supervised the study. H.S., Z.Z., Z.X., Q.Z., P.M. and O.B.S. collected the data used in the study; and H.S. analyzed the data and wrote the manuscript. All the authors read and edited the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sun, H., Zhang, Z., Olasege, B.S. et al. Application of partial least squares in exploring the genome selection signatures between populations. Heredity 122, 288–293 (2019). https://doi.org/10.1038/s41437-018-0121-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0121-y

{kind=link}
{kind=link}
{kind=link}
{kind=link}