
Abstract
Purpose
Radioulnar synostosis (RUS) can be syndromic or nonsyndromic. The genetic basis for several RUS syndromes have been reported. However, the genetic cause of nonsyndromic RUS (nsRUS) remains unknown.
Methods
We performed Giemsa (GTG) banding, Sanger sequencing, and exome sequencing on patients (n = 140) and families (n = 11) who suffered from RUS.
Results
GTG banding identified 10% RUS sporadic cases affected by sex chromosome aneuploidy. Sanger sequencing on candidate genes revealed noggin (NOG) rarely mutated in nsRUS. Exome sequencing identified 16 loss-of-function (LOF) and 6 missense variants (minor allele frequency [MAF] < 0.0001) in 22/117 nsRUS sporadic patients. Genetic association analysis found a significant association between SMAD6-LOF variants and nsRUS risk (odds ratio [OR] = 430, 95% confidence interval [CI]: 238–780, P < 0.000001). SMAD6 mutated in nsRUS was further confirmed by direct Sanger sequencing of SMAD6-coding regions on other unrelated cohorts of nsRUS cases or families. In summary, we detected 27 SMAD6 rare variants in nsRUS, most of which were LOF variants, 4 were de novo, and 3 were transmitted in families with autosomal dominant inheritance.
Conclusion
As an intracellular bone morphogenetic protein (BMP) antagonist gene, SMAD6 is frequently mutated in nsRUS.NOG, which encodes an extracellular BMP antagonist, is rarely mutated in nsRUS. This work is the first genetic study on nsRUS.
Similar content being viewed by others

INTRODUCTION
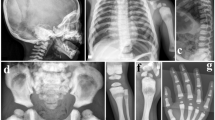
Radioulnar synostosis (RUS) is an abnormal connection between the radius and the ulna that limits the pronation and supination of the forearms.1,2 Post-traumatic or soft tissue–connected RUS is extremely rare.3,4 In the Chinese population, the overall RUS prevalence is 0.2‰ (ref. 5). In clinical settings, RUS generally refers to a congenital bony fusion of the proximal areas between the radius and the ulna (Fig. 1).1,2 Anatomically, two types of RUS have been described. Type I features a proximal fusion between the radius and the ulna. Type 2 is characterized by a fusion distal to the proximal radial epiphysis; it has a congenital dislocation of the radial head (Fig. 1).1 RUS can occur as a part of an underlying syndrome. To date, several RUS-related syndromes have been reported.6,7,8,9,10,11,12,13,14,15,16 Some of these syndromes have known genetic bases, including sex chromosome aneuploidy,6,7,8MECOM or HOXA11 mutated in amegakaryocytic thrombocytopenia with RUS,9B4GALT7 mutated in spondylodysplastic Ehlers–Danlos syndrome,10FGFR2 mutated in Crouzon or Pfeiffer syndrome,11TBX5 mutated in Holt–Oram syndrome,12 and BMPR1B disrupted in Pierre Robin syndrome.13 In 2012, an autosomal dominant family with multiple affected members suffered from Giuffrè–Tsukahara syndrome, which is a RUS-related syndrome.14,15 Since then, studies have focused on the genetic characteristics of human RUS. Nonsyndromic RUS (nsRUS) is a condition in which a patient suffers from ulnar–radius fusion with no other identifiable disease. This condition was initially described by Sandifort in 1793.17 Several studies have suggested that the inheritance of nsRUS is autosomal dominant.18,19,20 However, most RUS cases encountered in clinics are sporadic, and the genetic basis for nsRUS remains unexplored. Thus, the present study investigated 140 sporadic patients and 11 families with RUS of an unknown cause. Genetic analysis identified that 10% of patients with RUS are affected by sex chromosome aneuploidy, ~1% of patients with RUS are affected by noggin (NOG) pathogenic variants, 24 patients (~19%) have sporadic nsRUS, and 30% of families with nsRUS harbor SMAD6 rare variants. These findings have implications in the molecular diagnosis, genetic counseling, and prenatal diagnosis of human RUS.

Anatomical classification of radioulnar synostosis (RUS). X-ray images exhibit normal proximal radius and ulna of the right hand (a) and the RUS type II of the left hand (b) on a 7.5-year-old girl.c Image of RUS type I of the left hand on a 5.5-year-old boy.
MATERIALS AND METHODS
Study subjects
The study protocol was approved by the Academic Committee of Hunan Children’s Hospital (approval number: HCHLL58, Changsha City, Hunan Province, China). Participants with RUS were ascertained from either the Child Healthcare Center or the Department of Orthopedics of Hunan Children’s Hospital from July 2010 to June 2018. All participants and their parents, as well as their family members (if available), provided written informed consent to take part in this study. Inclusion criteria involved the diagnosis of unilateral or bilateral RUS in the absence of identifiable syndromes examined by an orthopedic surgeon and a doctor of genetics. For the in-house controls used in data analysis, exome data were obtained from our in-house database, which included the exome data of 75 cases without RUS or any identifiable joint fusion disease. For BMP2 (rs1884302) frequency analysis, the genomic DNA of 27 healthy controls (24 males and 3 females) was obtained from individuals who underwent routine health examinations in our hospital.
Cytogenetic analysis
RUS cases and families were referred to our laboratory for cytogenetic analysis. The peripheral venous blood of the patients and their family members was collected in a vacutainer sodium heparin vial. Slides were prepared from phytohemagglutinin-stimulated peripheral lymphocyte cultures by using standard cytogenetic methods. Giemsa (GTG) banding at a 400-band level to a 550-band level was performed in accordance with the standard laboratory protocol. Two cultures corresponding to two series of slides from each sample were separately prepared and analyzed. At least 40 metaphases were analyzed for each individual.
Sanger sequencing
For each subject, genomic DNA was extracted from peripheral blood by using a DNA isolation kit (Cat# D3392–02; Omega Bio-Tek, Inc., Norcross, GA, USA) in accordance with the manufacturer’s instructions. Sanger sequencing was performed on the coding regions of NOG (NM_005450), GDF5 (NM_000557), SMAD6 (NM_005585), and a ~250 bp interval that included the BMP2-rs1884302 locus. Polymerase chain reaction (PCR) amplification was performed using genomic DNA as a template in a Goldstar® PCR kit (Cat# CW0655M; Jiangsu Kangwei Century Biotechnology Co., Ltd., Beijing, China). Sanger sequencing was conducted with a BigDye® Terminator v3.1 cycle sequencing kit (Applied Biosystems, Thermo Fisher Scientific, Inc., Waltham, MA, USA) in accordance with the manufacturer’s protocol. The amplified PCR products were purified with 70% ethanol (analytically pure) and then run on an Applied Biosystems™ 3500 Series Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Details about the primers and the PCR conditions in the current study are provided in Supplementary Table 1.
Exome sequencing
Exome sequencing was carried out for 117 sporadic nsRUS patients (Supplementary Table 2). Genomic DNA was evaluated through agarose gel electrophoresis and initially quantified with Qubit. Then, >0.6 µg of DNA for each patient was fragmented into 180–280 bp segments by a Covaris Bath Sonicator. A library was prepared and captured using an Agilent SureSelect Human All Exon V6 kit in accordance with the manufacturer’s protocol. The captured library was initially quantified using Qubit 2.0, and the insert size was detected with Agilent 2100. Then, the effective concentration of each sample was precisely quantified through qualitative PCR to ensure that the captured library has high quality. The quality-passed library was subsequently sequenced on an Illumina HiSeq X Ten Sequencing system (Illumina Inc., San Diego, CA, USA) to generate 2 × 150 bp paired-end reads. After a raw BCL file was converted to a raw FASTQ file, 11.9 G bases were obtained for each sample, and the average yield was ~15.8 Gb with an error rate of <0.1%. Furthermore, >80% bases had a Phred quality score of ≥30 (Q30).
Bioinformatics analysis
With raw data from exome sequencing, the adapter-contaminated and low-quality reads (“N” content >10% or >75% bases with a Phred quality score of <5) of each DNA sample were removed to obtain clean reads. Such reads were then mapped to the reference human genome (hg19; genome.ucsc.edu) by using the Burrows–Wheeler Aligner (version 0.7.8-r455, bio-bwa.sourceforge.net) to generate the alignment files (.bam). SAMtools (version 1.0, samtools.sourceforge.net) and Picard (version 1.111, broadinstitute.github.io/picard/) were then used to sort and remove duplicate reads by setting the default parameters. The mapping and coverage rates were evaluated on the basis of alignment files. In all of our sequenced samples, >99.8% reads were mapped to the reference genome hg19, and >99.6% of the target region was covered. SNVs and indels were detected by SAMtools (version 1.0) and then annotated by ANNOVAR (version 20160201, annovar.openbioinformatics.org/en/latest/), InterVar (version 20180118), and Phenolyzer (http://phenolyzer.usc.edu) for functional effects, allele frequencies, and phenotype correlations, respectively.
Short read alignments at specific variant positions were visualized using Integrative Genomics Viewer (software.broadinstitute.org/software/igv/) as necessary.
Segregation analysis
The fragments that harbored SMAD6 and NOG variants of the probands were subjected to Sanger sequencing in their available family members to check whether or not the RUS phenotype was segregated with the candidate gene variants.
Statistical analysis
In the genetic association analysis, two control groups were used, namely, the in-house control group and the EAS_gnomAD control group. Logistic regression analysis was used to estimate the association between variant and nsRUS and to explore the association between the TC genotype of BMP2-rs1884302 and nsRUS. Statistical significance was considered when p < 0.05. Statistical analyses were conducted using IBM SPSS 20.0 (IBM SPSS, Inc., Chicago, IL).
RESULTS
GTG banding identified 14/140 sporadic RUS patients and 0/11 RUS families affected by sex chromosome aneuploidy
After excluding patients with clinically identifiable syndromes, such as amegakaryocytic thrombocytopenia with RUS;9 spondylodysplastic Ehlers–Danlos syndrome;10 Apert, Crouzon, or Pfeiffer syndrome; Holt–Oram syndrome;11,12 Pierre Robin syndrome;13 Giuffrè–Tsukahara syndrome;14,15 and RUS with polydactyly or severe mental retardation, we obtained 140 RUS sporadic cases and 11 RUS families. We successfully performed GTG banding on these 140 sporadic RUS cases, their available parents, and 11 RUS families. We detected that 14/140 sporadic RUS cases suffered from sex chromosome aneuploidy consisting of 47,XXY (six patients), 48,XXYY (four patients), 49,XXXXY (one patient), 45,X[8]/46,XY[134] (one patient), 47,XXY[10]/46,XY[10] (one patient), and 47,XXY[4]/46,XY[96] (one patient) (Supplementary Table 2 and Supplementary Data 1). None of them carried autosomal chromosomal abnormalities. Among the 14 RUS cases with sex chromosome aneuploidy, all of which were males, 12 were affected by bilateral RUS, and two were affected by right RUS (unilateral). The ratio of RUS type I:type II was 18:8. No chromosome abnormality was detected on all patients with RUS in 11 families. The genetic basis for the remaining 126 sporadic RUS cases and 11 RUS families (Fig. 2) was unknown. We defined all sporadic RUS or hereditary RUS as nsRUS without cause.

Clinical and genetic information of 11 hereditary families with nonsyndromic radioulnar synostosis (nsRUS).a Families with unknown genetic causes. b Families withSMAD6 rare variants.c Families with NOG rare variants. L:I type I RUS on the left upper limb, L:II type II RUS on the left upper limb, MT mutant type, NA DNA unavailable, R:I type I RUS on the right upper limb,R:II type II RUS on the right upper limb, WT wild type.
General information on 126 sporadic nsRUS patients and 11 nsRUS families
Supplementary Table 2 shows the basic data of 126 patients with sporadic nsRUS, as follows: (1) age of diagnosis, ranging from 11 days to 15 years; (2) males were more frequent than females, that is, 97 males and 29 females (77% male, 23% female); (3) unilateral RUS was more frequent than bilateral RUS, that is, 58 bilateral and 68 unilateral (left: 41, right: 27); (4) type I RUS (108) was more frequent than type II RUS (76); (5) types I and II RUS could occur on the same patients. Among the 58 cases with bilateral RUS, 15 had one hand affected by type I RUS and the opposite hand affected by type II RUS, implying that both types were not separate entities.
For the 11 nsRUS families (Fig. 2), although male-to-male, male-to-female, and female-to-male direct transmission occurred, inheritance was consistent with autosomal dominance.18,19,20 We did not observe female-to-female transmission, and the number of males (19) was more than that of females (5). Therefore, we modified the inheritance of family nsRUS to autosomal dominant, and males exhibited greater susceptibility than females. In a single RUS family, type I and II RUS could occur, further implying that both types were not separate entities.
Multiple-synostosis gene NOG mutated in 1/126 sporadic patients and in 1/11 nsRUS families
Variants in NOG (MIM 602991) on 17q22 or GDF5 (MIM 601146) on 20q11.2 are two major genes responsible for human multiple synostosis.21,22 The NOG- and GDF5-coding regions of 126 patients with nsRUS and 11 families with nsRUS were subjected to Sanger sequencing. However, we detected two variants on NOG but none on GDF5 when the variants were filtered with the 1000G database (minor allele frequency [MAF] < 0.0001). The NOG missense variants were c.310C>A:p.L104M and c.248C>T:p.P83L (Fig. 2, Supplementary Data 2). The c.248C>T:p.P83L cosegregated with nsRUS in a family trio, the proband was affected by unilateral RUS, and his father was affected by bilateral RUS (Fig. 2, Supplementary Data 2). The c.310C>A:p.L104M occurred on a sporadic patient with left RUS, and his mother carried the variant with minor finger deformity but did not present RUS (Fig. 2, Supplementary Data 2). NOG is encoded as a secretory protein.21 Functional assay revealed that c.310C>A:p.L104M and c.248C>T:p.P83L mutants were less secretory than wild-type (WT) NOG (Supplementary Data 2).
Performance of exome sequencing on 117 nsRUS sporadic patients revealed 22/117 patients harbored SMAD6 rare variants
Exome sequencing was performed on 117 sporadic patients with nsRUS (Supplementary Table 2). The variants were detected on 24,738 genes (Fig. 3a). nsRUS is a rare and dominant disorder.5,19,20 Thus, it should be attributed to a mutated gene with rare heterozygous variant. The number of candidate genes was reduced to 8140, considering the heterozygous, not benign, MAF < 0.0001 (All_gnomAD, ExAC, 1000 Genomes and ESP6500), coding region variants (Fig. 3a). For these 8140 genes, we focused on genes with damaging variants, such as loss-of-function (LOF) and damaging missense (D-mis) variants; the gene number was reduced to 4042 (Fig. 3a). The gene number was reduced to 2804 after using Phenolyzer. We focused on genes whose damaging variants were detected in more than one nsRUS patient; the gene number was reduced to 742 (Fig. 3a). We subsequently performed a rare variant (MAF < 0.0001) burden analysis of the numbers of patient variants per gene versus the numbers found in the control cohort (75 in-house controls); 432 genes remained (p < 0.05). To further discriminate the major gene for nsRUS, we considered genes whose damaging variants accounted for >3 nsRUS cases; the gene number was reduced to 28 (Supplementary Table 3). Finally, we compared the frequency of rare variants (MAF < 0.0001) in these 28 genes in 117 nsRUS cases and 123,136 unrelated controls (All_gnomAD). Q–Q plots showed that damaging variants on SMAD6 were most significantly enriched in nsRUS patients (Fig. 3b, Supplementary Table 3). SMAD6 encodes an inhibitor of bone morphogenetic protein (BMP) signaling. Reports suggested that SMAD6 frequently mutates in a bone fusion disease, that is, midline craniosynostosis.23,24 Then, we rechecked the data of theSMAD6 gene for cases that received exome sequencing in this study. We found that the average coverage (depth >20) of the coding regions of SMAD6 was 100%, whereas the average coverage (>5×) of 5′UTR of SMAD6 was 20.9%, and the average coverage (>5×) of 3′UTR ofSMAD6 was 40.7%.

Analyzing strategies and preliminary results of 117 exome sequencing data.a Filtering strategies for searching of pathogenic genes of nonsyndromic radioulnar synostosis (nsRUS). LOF loss-of-function variants, D-mis = damaging missense variants, when two or more of four software programs (a–d) predict the single-nucleotide variant (SNV) is damaging. b Quantile–quantile (Q–Q) plots of odds ratio of observed rare damaging variants in 117 nsRUS cases vs. in 123,136 controls of the All_gnomAD database (LOF and D-mis, MAF < 0.0001) for the filtered/prioritized 28 genes.MAF minor allele frequency, SNP single-nucleotide polymorphism.
After Sanger sequencing validation, we identified 22 SMAD6 rare variants (MAF < 0.0001) that occurred on 22 nsRUS patients (Table 1, Supplementary Data 3). Results of logistic regression showed that the SMAD6-LOF variants were significantly associated with increased risk of nsRUS (odds ratio [OR] = 430, 95% confidence interval [CI]: 237.535–780.076, P < 0.000001), and the SMAD6-LOF + D-mis variants were significantly associated with nsRUS with an OR of 59.6 (95% CI: 37.168–95.419; P < 0.000001) (Supplementary Table 4).
Segregation analysis was used to test whether or not theseSMAD6 variants segregated with nsRUS. Parental genomic DNA was available for 11 probands with SMAD6 rare variants. Sanger sequencing validated that four variants were de novo (the paternity relationship for each trio was validated, Supplementary Table 5), six were inherited from the probands’ unaffected mother, and one was inherited from the probands’ unaffected father (Table 1 and Supplementary Data 3).
Further Sanger sequencing confirmed rare SMAD6 variants in 2/8 sporadic nsRUS patients and 3/10 nsRUS families
We performed Sanger sequencing of SMAD6-coding regions for an unrelated cohort of nsRUS patients (10 transmitted families and 8 sporadic cases). We further detected five rare variants (MAF < 0.0001) for this cohort of nsRUS patients. Among them,SMAD6 variants segregating with nsRUS were detected in 3/10 families, and SMAD6 variants were detected in 2/8 sporadic cases (Fig. 2, Table 1 and Supplementary Data 3).
Genotype–phenotype correlation for SMAD6 mutated in 24 sporadic cases and three families with nsRUS
The human SMAD6 gene (NM_005585), which is located on 15q22.31, consists of four exons that encode a BMP-signaling inhibitor. This inhibitor consists of 496 amino acids with an amino-terminal region (N domain) and a conserved carboxy-terminal MH2 domain.25 We identified a total of 27 rare variants (19 LOF and 8 missense) on SMAD6 that occurred in 24/125 sporadic cases and 3/10 families with nsRUS. Among the 19 LOF variants, 14 (73.7%) occurred on exon 1 of SMAD6 (Fig. 4a) and 16/19 LOF variants are located on the N domain of the SMAD6 protein (Fig. 4b). Among the eight rare missense variants of SMAD6 (Fig. 4b, c), three are located on the MH2 domain (among them, c.1412G>A:p.G471D is on the D3 loop motif), and five are evenly distributed on the N domain section (from amino acids 154 to 267) of the protein.

Pattern diagram of 27 nonsyndromic radioulnar synostosis (nsRUS)SMAD6variants consisting of 19 loss-of-function (LOF) and eight missense variants.aSMAD6 exons and the distribution of the 19 LOF variants. b Pattern diagram of the SMAD6 protein showing N-terminal domain and MH2 domains and the positions of SMAD6 variants. L3 loop, PLDLS, and PY motifs are shown according to a study elsewhere.27 Variants above the protein diagram are rare LOF, whereas variants under the diagram are rare missense. c Multiple alignment of the SMAD protein families with eight SMAD6 missense variants is indicated by the arrowhead. cDNA complementary DNA.
We identified 30 nsRUS patients (sporadic and family patients) who harbored SMAD6 rare variants (Table 1). Among them, 25 were males and five were females (male:female ratio was 5:1). In addition, 17 were affected by bilateral RUS, and 13 were affected by unilateral RUS (left: 9, right: 4). Among the 47 limbs affected by RUS, 35 were type I and 12 were type II RUS.
DISCUSSION
Elbow joint is formed at the end of the humerus, the radius, and the ulna. The elbow is first identifiable 35 days after conception; at this stage, the cartilaginous anlagen of the humerus, the radius, and the ulna is continuous.1,2 Subsequently, programmed segmentation produces elbow joints, which include three articulations, namely, humeroulnar, humeroradial, and radioulnar articulations.2 RUS is an upper limb skeletal malformation characterized by bony fusion at the proximal aspect of the radius and the ulna.1,2 If these bones do not appropriately separate, then two articulations, namely, humeroradial and radioulnar, are affected, and the muscles that rotate the radius and the ulna are absent.26 Consequently, patients with RUS suffer from defects of supination and pronation of the forearms; some severely affected patients also suffer from the defects of adduction or abduction of elbow joints.26 Several RUS-related syndromes caused by gene variant or cytogenetic abnormalities have been described.6,7,8,9,10,11,12,13,14,15,16,17 However, the most common form of RUS in clinics is nsRUS, and its genetic characteristics are largely unexplored.
BMP signaling plays crucial roles in bone and joint morphogenesis. The intensity and duration of BMP signaling must be precisely regulated by two types of inhibitory factors, namely, extracellular and intracellular BMP antagonists.27 The importance of extracellular BMP antagonists, such as NOG and chordin, has been documented.28,29 For instance, the variant of NOG causes a variable spectrum of human joint synostosis or bone fusion diseases.21,29 Several cases with truncating NOG variants also exhibit cranial malformation.30,31 NOG-null mice exhibit enlarged growth plates and joint fusions.28 To test if NOG is mutated in RUS, we performed Sanger sequencing of NOG-coding regions on 146 cases with nsRUS, which included a combination of sporadic and family patients. However, only two missense variants were detected, as follows: one was from a sporadic patient and another was directly transmitted with RUS in a family trio. Functional assay indicated that both mutants could be secreted to the extracellular environment, but the amount of secreted mutants was less than those of the WT protein. Then, we suggested that these NOG variants were pathogenic but the effects were attenuated. These data imply that NOG pathogenic variants are a rare cause of human nsRUS.
Exome sequencing was performed to search for the disease gene of nsRUS. We identified 27 heterozygous SMAD6 variants in 24 sporadic patients and three families with nsRUS. Among these SMAD6 variants, four were de novo, three were cosegregated with RUS in families, eight were transmitted from unaffected parents, and the remaining variants had unknown origins.
SMAD6 is an intracellular BMP antagonist that specifically inhibits BMP signaling by at least three approaches as follows: (1) blocking BMP signaling by binding to activated BMP type I receptor, (2) aggregating type I receptor degradation by recruiting E3 ubiquitin ligases to type I receptor, and (3) binding directly to SMAD1, thereby interfering the SMAD1–SMAD4 complex formation.32,33,34 These signaling steps converge on shared nuclear targets that promote bone formation. SMAD6-null mice exhibit an enhanced activity of proliferative and hypertrophic chondrocytes, which are associated with increased collagen production in axial and appendicular skeletal development.27 In humans, SMAD6 variants have previously been associated with a variety of different phenotypes, namely, craniosynostosis,23,24 congenital heart disease,35,36 bicuspid aortic valve/thoracic aortic aneurysm,37,38,39 and intellectual disability.40
Timberlake et al.23,24 detected 18 SMAD6 rare variants from 384 patients with nonsyndromic midline craniosynostosis. Then, SMAD6 mutated in 4.7% patients with midline craniosynostosis.23,24 Craniosynostosis is a congenital bone fusion disease in which one or more cranial sutures close prematurely. Among the 18SMAD6 pathogenic variants detected in midline craniosynostosis, several variants are de novo or transmitted with a certain phenotype in families, and the remaining variants are sporadic.23,24 The penetrance of craniosynostosis associated with SMAD6 mutation is reduced. Timberlake et al. also implicated two locus inheritance for the entity as genotyping for the “C” risk allele of a candidate common variant located ~325 kb downstream of the BMP2 gene (BMP2-Chr20–7106289T-C, rs1884302) demonstrated that 14/17 cases (82%) with a SMAD6 rare variant and the risk “C” allele had craniosynostosis, whereas only 3/17 (18%) cases with a SMAD6 rare variant and no rs1884302 risk allele were affected.23
After excluding the index cases, we obtained ten SMAD6-LOF carriers (Supplementary Fig. 1). Among these ten carriers, only two have nsRUS. Therefore, the penetrance of RUS in SMAD6-LOF cases is reduced (~20%). This phenomenon is consistent with the data, suggesting that incomplete penetrance of midline craniosynostosis is observed in SMAD6-mutated cases.23,24 Then, we hypothesized that SMAD6 variants combined with BMP2-Chr20–7106289T-C cocontributed to the nsRUS phenotype. Two independent analyses were conducted using data from two control groups (in-house control group and the EAS_gnomAD control group) (Supplementary Table 6). However, no significant associations were observed between BMP2-Chr20–7106289T-C genotype and nsRUS in cases with SMAD6 rare variants (Supplementary Table 7). Meanwhile, the transmission disequilibrium test was performed but did not support the cocontribution of BMP2-Chr20–7106289T-C to nsRUS (Supplementary Fig. 1).
A gene variant other than BMP2-Chr20–7106289T-C might exist to modify SMAD6 variants given the incomplete penetrance of nsRUS in SMAD6-mutated cases, thereby causing human nsRUS. However, when considering nearly half of the SMAD6-mutated patients affected by unilateral RUS (Table 1), other factors may interact with SMAD6 rare variants, leading to nsRUS. Further studies are necessary to identify the modifier gene, screen the interacting factors in SMAD6 mutation carriers, and interpret the incomplete penetrance of SMAD6 mutations in this research.
We analyzed the SMAD6 variant spectra of five types of disorders (nsRUS, craniosynostosis, congenital heart disease,35,36 bicuspid aortic valve/thoracic aortic aneurysm,37,38,39 and intellectual disability40). As shown in Supplementary Fig. 2, three SMAD6-LOF variants of nsRUS have been reported in craniosynostosis (p.G88fs and p.P156fs) or bicuspid aortic valve/thoracic aortic aneurysm (p.Y279X), none of the eight SMAD6 D-mis variants have been reported in other disorders,35,36,37,38,39,40 and the SMAD6-LOF variants of nsRUS tend to be close to the C-terminal of SMAD6 protein.
In this study, several other genes (aside from SMAD6) approached exome-wide significance in these analyses of dominant alleles (Supplementary Table 3). However, most of these genes are functionally unrelated to joint/bone fusion or genes, the variants of which are detected in GC-rich (or repeat) regions. Therefore, further validation assay and gene functional experiments are required.
Conclusion
The extracellular BMP antagonist gene NOG is rarely mutated, but the intracellular BMP antagonistSMAD6 is frequently mutated in human nsRUS. The first genetic study on nsRUS emphasizes the importance of SMAD6 in joint morphogenesis.
Change history
10 June 2019
The original version of this Article contained an error in the spelling of the author Yimin Zhu, which was incorrectly given as Yiming Zhu. This has now been corrected in both the PDF and HTML versions of the Article.
References
Bauer M, Jonsson K. Congenital radioulnar synostosis. Scand J Plast Reconst Surg. 1988;22:251–255.
Elliott AM, Kibria L, Reed MH. The developmental spectrum of proximal radioulnar synostosis. Skeletal Radiol. 2010;39:49–54.
Bergeron JSG, Desy NM, Bernstein M, Harvey EJ. Management of posttraumatic radioulnar msynostosis. Am Acad Orthop Surg. 2012;20:450–458.
Okrent DH, McFadden JC. Congenital radioulnar synostosis. Orthopedics. 1986;9:1452–1456.
Wang J. Congenital radioulnar synostosis. In: Ji S, Pan S, Wang J (eds), Pediatric Orthopaedics (In Chinese) pp. 92-94. Yingxiongshan Road 189, Jinan: Shandong Science and Technology Press; 1998.
Cho YG, Kim DS, Lee HS, Cho SC, Choi SI. A case of 49, XXXXX in which the extra X chromosomes. J Clin Pathol. 2004;57:1004–1006.
Tartaglia N, Davis S, Hench A, Nimishakavi S, Beauregard R, Reynolds A, et al. A new look at XXYY syndrome: medical and psychological features. Am J Med Genet A. 2008;146A:1509–1522.
De Smet L, Fryns JP. Unilateral radio-ulnar synostosis and idic-Y chromosome. Genet Couns. 2008;19:425–427.
Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, encoding ncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet. 2015;97:848–854.
Ritelli M, Dordoni C, Cinquina V, Venturini M, Calzavara-Pinton P, Colombi M. Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers-Danlos syndrome. Orphanet J Rare Dis. 2017;12:153.
Schaefer F, Anderson C, Can B, Say B. Novel mutation in the FGFR2 gene at the same codon as the Crouzon syndrome mutations in a severe Pfeiffer syndrome type 2 case. Am J Med Genet. 1998;75:252–255.
Wall LB, Piper SL, Habenicht R, Oishi SN, Ezaki M, Goldfarb CA. Defining features of the upper extremity in Holt-Oram syndrome. J Hand Surg Am. 2015;40:1764–1768.
Yang Y, Yuan J, Yao X, Zhang R, Yang H, Zhao R, et al. BMPR1B mutation causes Pierre Robin sequence. Oncotarget. 2017;8:25864–25871.
Dalal AB, Sarkar A, Priya TP, Nandineni MR. Giuffrè-Tsukahara syndrome: evidence for X-linked dominant inheritance and review. Am J Med Genet A. 2010;152A:2057–2060.
Zhu Y, Jin K, Mei H, Li L, Liu Z, Yang Y, et al. A family with radio-ulnar synostosis, scoliosis, and thick vermilion of lips: a novel syndrome or variant of Giuffrè-Tsukahara syndrome? Am J Med Genet A. 2012;158A:2036–2042.
Charvat KA, Hornstein L, Oestreich AE. Radio-ulnar synostosis in Williams syndrome. A frequently associated anomaly. Pediatr Radiol. 1991;21:508–510.
Simmons BP, Southmayd WW, Riseborough EJ. Congenital radioulnar synostosis. J Hand Surg Am. 1983;8:829–838.
Hansen OH, Andersen NO. Congenital radio-ulnar synostosis: report of 37 cases. Acta Orthop. Scand. 1970;41:225–230.
Rizzo R, Pavone V, Corsello G, Sorge G, Neri G, Opitz JM. Autosomal dominant and sporadic radio-ulnar synostosis. Am J Med Genet. 1997;68:127–134.
Spritz RA. Familial radioulnar synostosis. J Med Genet. 1978;15:160–162.
Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R, et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat. Genet. 1999;21:302–304.
Dawson K, Seeman P, Sebald E, King L, Edwards M, Williams J, et al. GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet. 2006;78:708–712.
Timberlake AT, Choi J, Zaidi S, Lu Q, Nelson-Williams C, Brooks ED, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. eLife. 2016;5:e20125.
Timberlake AT, Furey CG, Choi J, Nelson-Williams C, et al. De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. Proc Natl Acad Sci U S A. 2017;114:E7341–E7347.
Miyazawa K, Miyazono K, Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol. 2017;9:a022095.
Beals RK. The normal carrying angle of the elbow. A radiographic study of 422 patients. Clin Orthop Relat Res. 1976;119:194–196.
Estrada KD, Retting KN, Chin AM, Lyons KM. Smad6 is essential to limit BMP signaling during cartilage development. J Bone Miner Res. 2011;26:2498–2510.
LJ Brunet, McMahon JA, McMahon AP, et al. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457.
Potti TA, Petty EM, Lesperanceum MM. A comprehensive review of reported heritable noggin‐associated syndromes and proposed clinical utility of one broadly inclusive diagnostic term: NOG‐related‐symphalangism spectrum disorder (NOG‐SSD). Hum Mutat. 2011;32:1–10.
van den Ende JJ, Mattelaer P, Declau F, Vanhoenacker F, Claes J, Van Hul E, et al. The facio-audio-symphalangism syndrome in a four generation family with a nonsense mutation in the NOG-gene. Clin Dysmorphol. 2005;14:73–80.
Rudnik-Schöneborn S, Takahashi T, Busse S, Schmidt T, Senderek J, Eggermann T, et al. Facioaudiosymphalangism syndrome and growth acceleration associated with a heterozygous NOG mutation. Am J Med Genet A. 2010;152A:1540–1544.
Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626.
Inoue Y, Imamura T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107–2112.
Hata A, Lagna G, Massague J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197.
Tan HL, Glen E, Töpf A, Hall D, O’Sullivan JJ, Sneddon L, et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat. 2012;33:720–727.
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601.
Gillis E, Kumar AA, Luyckx I, Preuss C, Cannaerts E, van de Beek G, et al. Candidate gene resequencing in a large bicuspid aortic valve-associated thoracic aortic aneurysm cohort: SMAD6 as an important contributor. Front Physiol. 2017;8:730.
Park JE, Park JS, Jang SY, Park SH, Kim JW, Ki CS, et al. novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol Genet Genomic Med. 2019;7:e620.
Luyckx I, MacCarrick G, Kempers M, Meester J, Geryl C, Rombouts O, et al. Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur J Hum Genet. 2019. https://doi.org/10.1038/s41431-019-0363-z [Epub ahead of print].
Lelieveld SH, Reijnders MR, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–1196.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81271946 to Y. Zhu and 31501017 to Y.Y.) and Hunan Health Commission Research Fund (B2019019). The authors are grateful to the family members who participated in this study. We thank Rui Zhao (from Hunan Children’s Hospital, Changsha City, China) and Zhong Nanbert (from Institute for Basic Research in Developmental Disabilities, Staten Island, NY, United States) for their suggestions and help in this study. Special thanks to Xun Li, Zhenqing Luo, and Shiting Xiang (Hunan Children’s Hospital, Changsha City, China) for their statistical analysis and technique works.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yang, Y., Zheng, Y., Li, W. et al. SMAD6 is frequently mutated in nonsyndromic radioulnar synostosis. Genet Med 21, 2577–2585 (2019). https://doi.org/10.1038/s41436-019-0552-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0552-8
Keywords
This article is cited by
-
Clinical interest of molecular study in cases of isolated midline craniosynostosis
European Journal of Human Genetics (2023)
-
Novel Loss of Function (G15D) Mutation on RAC2 in a Family with Combined Immunodeficiency and Increased Levels of Immunoglobulin G, A, and E
Journal of Clinical Immunology (2023)
-
SMAD6-deficiency in human genetic disorders
npj Genomic Medicine (2022)
-
Case series of congenital pseudarthrosis of the tibia unfulfilling neurofibromatosis type 1 diagnosis: 21% with somatic NF1 haploinsufficiency in the periosteum
Human Genetics (2022)
-
Case report: two novel VPS13B mutations in a Chinese family with Cohen syndrome and hyperlinear palms
BMC Medical Genetics (2019)
