
Abstract
Until recently, mainly DNA sequencing has been used to identify variants within the gene coding for homogentisate dioxygenase (HGD, 3q13.33) that cause alkaptonuria (AKU), an autosomal recessive inborn error of metabolism of tyrosine. In order to identify possible larger genomic deletions we have developed a novel Multiplex Ligation-dependent Probe Amplification (MLPA) assay specific for this gene (HGD-MLPA) and tested it successfully in healthy controls and in patients carrying two known previously identified HGD deletions. Subsequently, we analysed 22 AKU patients in whom only one or none classical HGD variant was found by sequencing. Using HGD-MLPA and sequencing, we identified four larger deletions encompassing from 1 to 4 exons of this gene and we defined their exact breakpoints: deletion of exons 1–4 (c.1-8460_282 + 6727del), deletion of exons 5 and 6 (c.283-9199_434 + 1688del), deletion of exon 11 (c.775-1915_879 + 1293del), and deletion of exon 13 (c.1007-1709_1188 + 1121del). We suggest including MLPA in the DNA diagnostic protocols for AKU in cases where DNA sequencing does not lead to identification of both HGD variants.
Similar content being viewed by others


Results and discussion
Alkaptonuria (AKU) [OMIM 203500] is a classical rare autosomal recessive metabolic disorder characterised by ochronosis and ochronotic artropathy due to accumulation of dark melanin-like pigment in connective tissues [1,2,3]. AKU patients carry homozygous or compound heterozygous variants within the gene coding for homogentisate dioxygenase (HGD, 3q13.33) involved in metabolism of tyrosine (HGNC:4892) [4,5,6]. So far, mainly DNA sequencing has been used for identification of AKU-causing variants in the patients and all variants identified worldwide are summarised in the HGD gene mutation database (http://hgddatabase.cvtisr.sk/). In nine patients out of 550 listed in this database (December 2019), DNA sequencing was not able to identify any HGD variant possibly affecting protein function, moreover, in additional 27 AKU individuals, only one HGD change was found. Therefore, we have developed a novel HGD-MLPA assay in order to analyse such patients for possible larger genomic deletions within the gene. Unique synthetic MLPA probes for all 14 exons and intron 2 of the HGD gene were designed (Supplementary Table 1), following instructions from the synthetic probe design protocol (version 15, updated 09-09-2015, MRC Holland). Probes were synthetised by IDT (www.idtdna.com) and all Right Probe Oligonucleotides were 5′-phosphorylated. The HGD-MLPA probe mix was prepared by diluting and mixing of all 15 HGD gene probes, as indicated in the protocol. Human reference P200 (MRC Holland) was added into the mix and the analysis was performed according to the instructions of the producer. Genetic analyser ABI PRISM® 3130xl (Applied Biosystems) was used for sample separation and Coffalyser software (MRC Holland) for the peak analysis.
We tested the HGD-MLPA assay in five healthy controls and obtained a clear profile with no copy number changes for all tested HGD gene exons (Fig. 1A). Efficacy of our assay was further confirmed by successful identification of two known deletions: (i) the 765 bp deletion within the HGD intron 2 (c.87 + 8_88-31del) identified in our laboratory by sequencing [7] (Fig. 1B); and (ii) the previously reported deletion of the HGD exon 2 (c.16-272_87 + 305del) [8] (in homozygous and heterozygous state in Fig. 1C and 1D, respectively). The later deletion has so far been found also in our laboratory in ten families from Lebanon, Israel, Jordan and Italy (see the HGD mutation database).

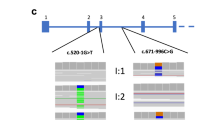
MLPA ratio charts of healthy control (A) and 7 AKU patients (B–H). HGD mutation database ID code is indicated for each patient (AKU_). HGD-MLPA identifies clearly a previously described deletion of intron 2 [7] (B, homozygous deletion), as well as deletion of exon 2 [8] shown in homozygous (C) and heterozygous form (D). We identified four novel deletions in heterozygous state: exon 13 (E), exons 1–4 (F), exons 5, 6 (G), and exon 11 (H). Patients are indicated by the specific allele/family code (AKU_xx_x), under which they can be identified in the HGD mutation database (http://hgddatabase.cvtisr.sk/).
Subsequently, we performed HGD-MLPA in 22 clinically confirmed AKU patients previously analysed by DNA sequencing [7, 9] (Supplementary Table 2), 13 of whom had a single heterozygous HGD pathogenic variant, and 4 none. The remaining five cases carried intronic variants that were shown to have effect on correct HGD splicing [7]. However, we analysed them in order to exclude possible other DNA changes.
Our new HGD-MLPA assay identified a heterozygous deletion of exon 13 in five cases from Italy (Fig. 1E), a heterozygous deletion of exons 1–4 in a patient from Germany (mother is of Peruvian origin) (Fig. 1F), and a heterozygous deletion of both exons 5 and 6 in one patient from Netherland (Fig. 1G) [7]. No copy number changes were found in five cases with confirmed splicing variant and in the remaining ten analysed cases (Supplementary Table 2).
Many AKU patients are reported based on sequencing results as homozygotes for some HGD variant (HGD mutation database). However, sequencing is not able to identify a possible hidden hemizygosity, the actual presence of only one allele carrying a certain variant, while the same region on the other allele is deleted. Therefore, we analysed a cohort of 72 patients, in whom sequencing indicated homozygosity, by our assay [7, 9, 10]. Indeed, in one Italian patient previously reported to be homozygous for c.1102 A > G variant in exon 13, p.(Met368Val), our HGD-MLPA assay uncovered heterozygous deletion of exon 13 (AKU_DB_176, Supplementary Table 2) [9].
In addition, in one patient reported to be homozygous for a splicing variant c.16-1 G > A in intron 1, p.(Tyr6_Gln29del) [7], a heterozygous deletion of exon 11 was found (Fig. 1H), indicating that this patient carries three independent HGD variants (AKU_TR2_0119, Supplementary table 2). Six more AKU patient who carry three likely pathogenic HGD variants are listed in the HGD mutation database (AKU_TR2_0129, AKU_DB_36, AKU_DB_93, AKU_DB_94, AKU_DB_33, AKU_DB_175, AKU_DB_287).
As indicated in Supplementary Table 2, 14 of the patients reported in this paper were included in the SONIA2 clinical study (DevelopAKUre project) and results of their MLPA analysis (presence or absence of the deletions) have been previously reported in Eur J Hum Genet [7]. Here, we define the exact breakpoints of all four novel deletions. In brief, we designed specific PCR primers within the regions flanking each deletion, and all HGD alleles carrying the deletion were amplified from corresponding patients´ DNA and subsequently sequenced. Results are summarised in Table 1 and Fig. 2, as well as updated in the HGD mutation database (http://hgddatabase.cvtisr.sk).

Details on deletion breakpoints of four novel HGD gene deletions, as defined by sequencing of the fusion DNA products obtained from the patients´ DNA. Deletions were identified by HGD-MLPA assay as deletion of: ex1–4 (A), ex5-6 (B), ex11 (C), and ex13 (D). Red underlined letters indicate a sequence flanking the deletion breakpoints from both sides, whereas black capital letters show the first and the last 25 nucleotides of the deleted region.
CNVs in general are caused by different mutational mechanisms, including DNA recombination-, replication- and repair associated processes (recently summarised in Pos et al. [11]). Recurrent rearrangements are usually caused by non-allelic homologous recombination and repeated sequences, including low-copy repeats and high-copy repeats (e.g. LINEs, SINEs-including Alu sequences), are typically enriched near breakpoints. We used Repeat Masker (http://www.repeatmasker.org/) in order to test for the presence of such sequences within the regions of 1000 bp both upstream and downstream from all identified deletion breakpoints (Supplementary material). Our results show that none of the breakpoints overlaps with some Alu sequence. However, AluSx was found 45 bp upstream of intron 6 breakpoint in the patient with exons 5 and 6 deletion, as well as 185 bp downstream of intron 10 breakpoint in the patient with exon 11 deletion. AluJb sequence was found 750 bp upstream of intron 4 deletion breakpoint and AluY sequence 158 bp downstream of intron 12 deletion breakpoint involved in the generation of exon 1–4 and exon13 deletions, respectively. Interestingly, recently Nakama et al. [12] pointed out that intronic antisense Alu elements may contribute to alternative splicing and transcriptomic diversity in some genes, especially when splice acceptor sites are suboptimal.
Breakpoints in intron 4 and intron 6 leading to the deletion of exons 5–6 deletion overlapped with sequences of CR1 Mam (LINE/CR1) and MER4B-int (LTR/ERV1), respectively (Supplementary material), but none of them had a counterpart on the other side of the deletion breakpoint.
Therefore, we believe that here described deletions most likely represent non-recurrent rearrangements, which are known to emerge due to several mechanisms: some of which are non-replicative, including non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ); others are replicative, such as replication slippage, fork stalling and template switching (FoSTeS) or microhomology-mediated break-induced replication (MMBIR) [13]. Occurrence of replication errors, eventually leading to CNVs, may be favoured by the formation of non-B DNA structures (e.g. hairpins/cruciforms, Z-DNA, triplexes (H-DNA), tetraplexes, slipped DNA), which are configurations typical for the sequences rich in short repetitive sequence motifs (e.g. inverted, direct and mirror repeats) [14, 15].
Thus, we searched for microhomologies at each breakpoint by visual inspection of the original sequences and fusion product, as well as screened surrounding sequences for potential non-B DNA structures (Supplementary material). In the case of exon 11 deletion and exon 1–4 deletion, we observed a microhomology of 4 (ATGT) and 3 (TTC) homologous nucleotides, respectively. Microhomology at the breakpoints leading to exon 13 and exons 5–6 deletions was only 1nt (A). However, in the case of the later deletion, after one nucleotide mismatch that follows (A), there are 2 more homologous nucleotides (AGTT/AATT) (see Supplementary material).
Non-B DNA Motif Search Tool (https://nonb-abcc.ncifcrf.gov/apps/nBMST/default/) was used in order to search for possible non-B DNA Motifs within the region of 100 bp both upstream and downstream from the deletion breakpoint (Supplementary material). We also tested for the presence of the Palindromic sequences using EMBOSS tool (https://www.bioinformatics.nl/cgi-bin/emboss/palindrome) and DNA folding was analysed using mFOLD web server (http://www.unafold.org/DNA_form.php). Our analysis shows that no relevant non-B DNA motifs were present in the vicinity of the breakpoints, except of a simple repeat (TCCC)n that starts 5 bp downstream of intron 11 breakpoint involved in exon 11 deletion. However, we could see several palindromic sequences that may lead to the formation of hairpin structures. This finding has been confirmed by mFOLD analysis, which showed a high probability of single-stranded state for regions around deletion-breakpoint (Supplementary material).
In conclusion, we believe that especially in case of deletions of exons 1–4 and exon 11, FoSTES/MMBIR could be the most likely mechanism, while for deletions of exon 5–6 and exon 13, NHEJ or MMEJ are more probable, instead.
We can conclude that our MLPA assay has proven effective in detecting copy number changes within the HGD gene and it should be included in a variant detection protocol for AKU, since larger genomic deletions seems to be rather frequent in AKU. The HGD mutation database lists DNA sequencing results from 717 AKU patients worldwide (June 2021). There are 8 deletions out of 249 so far described variants. However, there are still 41 patient reported with only one or no HGD variant identified, including 10 cases from this study that have been here already analysed by MLPA and show no HGD gene exons copy number changes. It is possible that some of these patients carry some deep intronic variants affecting splicing. This could be verified by the analysis of patients’ cDNA, which is not available at the moment.
Data availability
The datasets/sequences generated and analysed during the current study are available from the corresponding author on reasonable request. All patients and their variants identified during the current study were inserted in the HGD mutation database (http://hgddatabase.cvtisr.sk/) and can be identified based on the specific anonymous allele/family codes indicated in Supplementary Table 2.
References
Garrod AE. Croonian lectures on inborn errors of metabolism, lecture II: alkaptonuria. Lancet. 1908;2:73–9.
Zannoni VG, Lomtevas N, Goldfinger S. Oxidation of homogentisic acid to ochronotic pigment in connective tissue. Biochim Biophys Acta. 1969;177:94–105.
La Du BN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem. 1958;230:251–60.
Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:19–24.
Fernández-Cañón JM, Peñalva MA. Molecular characterization of a gene encoding a homogentisate dioxygenase from Aspergillus nidulans and identification of its human and plant homologues. J Biol Chem. 1995;270:21199–205.
Granadino B, Beltrán-Valero de Bernabé D, Fernández-Cañón JM, Peñalva MA, Rodríguez de Córdoba S. The human homogentisate 1,2-dioxygenase (HGO) gene. Genomics. 1997;43:115–22.
Ascher DB, Spiga O, Sekelska M, Pires DEV, Bernini A, Tiezzi M, et al. Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU. Eur J Hum Genet. 2019;27:888–902.
Zouheir Habbal M, Bou-Assi T, Zhu J, Owen R, Chehab FF. First report of a deletion encompassing an entire exon in the homogentisate 1,2-dioxygenase gene causing alkaptonuria. PLoS ONE. 2014;9:e106948.
Nemethova M, Radvanszky J, Kadasi L, Ascher DB, Pires DE, Blundell TL, et al. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: focus on ‘black bone disease’ in Italy. Eur J Hum Genet. 2016;24:66–72.
Zatkova A, Sedlackova T, Radvansky J, Polakova H, Nemethova M, Aquaron R, et al. Identification of 11 Novel Homogentisate 1,2 Dioxygenase Variants in Alkaptonuria Patients and Establishment of a Novel LOVD-Based HGD Mutation Database. JIMD Rep. 2012;4:55–65.
Pos O, Radvanszky J, Buglyo G, Pos Z, Rusnakova D, Nagy B, et al. DNA copy number variation: Main characteristics,evolutionary significance, and pathological aspects. Biomed J. 2021;44:548–59.
Nakama M, Otsuka H, Ago Y, Sasai H, Abdelkreem E, Aoyama Y, et al. Intronic antisense Alu elements have a negative splicing effect on the inclusion of adjacent downstream exons. Gene. 2018;664:84–9.
Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–64.
Chen L, Zhou W, Zhang L, Zhang F. Genome architecture and its roles in human copy number variation. Genomics Inf. 2014;12:136–44.
Zhao J, Bacolla A, Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2010;67:43–62.
Acknowledgements
We would like to thank Drs Berardino Porfirio (Florence, Italy), Robert Aquaron (Marsei, France), Caterina Aurizi (Rome, Italy) and Annalisa Santucci (Siena, Italy) for providing DNA samples of AKU patients.
Funding
DNA analysis of AKU samples was supported by the European Commission Seventh Framework Programme funding granted in 2012 (DevelopAKUre, project number: 304985) and Slovak grant agency VEGA (grant number 02/0040/20).
Author information
Authors and Affiliations
Contributions
MS and AS performed MLPA analysis, AS performed deletion breakpoint analysis, including DNA sequencing. AS and AZ performed breakpoint region sequence analysis. AZ initiated and supervised the project, analyzed sequences, and prepared manuscript, including table and figures. All authors contributed to finalize the manuscript and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
DNA samples analysed were collected during the DevelopAKUre project (was approved by Independent ethics committees at each study center), or during routine diagnostics. No additional ethical approval was required.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Soltysova, A., Sekelska, M. & Zatkova, A. Breakpoints characterisation of the genomic deletions identified by MLPA in alkaptonuria patients. Eur J Hum Genet 31, 485–489 (2023). https://doi.org/10.1038/s41431-022-01042-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01042-9
