
Abstract
Our goal in developing the MultiWaver software series was to be able to infer population admixture history under various complex scenarios. The earlier version of MultiWaver considered only discrete admixture models. Here, we report a newly developed version, MultiWaver 2.0, that implements a more flexible framework and is capable of inferring multiple-wave admixture histories under both discrete and continuous admixture models. MultiWaver 2.0 can automatically select an optimal admixture model based on the length distribution of ancestral tracks of chromosomes, and the program can estimate the corresponding parameters under the selected model. Specifically, for discrete admixture models, we used a likelihood ratio test (LRT) to determine the optimal discrete model and an expectation–maximization algorithm to estimate the parameters. In addition, according to the principles of the Bayesian Information Criterion (BIC), we compared the optimal discrete model with several continuous admixture models. In MultiWaver 2.0, we also applied a bootstrapping technique to provide levels of support for the chosen model and the confidence interval (CI) of the estimations of admixture time. Simulation studies validated the reliability and effectiveness of our method. Finally, the program performed well when applied to real datasets of typical admixed populations, such as African Americans, Uyghurs, and Hazaras.
Similar content being viewed by others


Introduction
Admixture history inference is a fundamental problem for studies on admixed populations [1]. Several methods have been developed to analyze the problem based on various kinds of population admixture information, such as break points of recombination [2], admixture linkage disequilibrium [3,4,5,6], and ancestral tracks [7,8,9,10,11,12]. The length distribution of ancestral tracts provides direct information concerning the decay of the ancestral segment length, which is closely related to the admixture history. Therefore, many methods have been developed based on this type of information [7,8,9,10,11, 13, 14]. The history of several classical admixed populations (African Americans, Mexicans, and Uyghur) have been well studied using these methods [15,16,17,18,19,20,21]. However, there are two shortcomings involved in these methods. First, before estimating the parameters of admixture history, a prior admixture model was required. Second, the prior admixture model was often an overly simplified scenario, such as a hybrid-isolation (HI), gradual admixture (GA), or continuous gene flow (CGF) models. Knowledge of admixture history is often lacking when real data are analyzed, especially for complex admixed populations [2, 15, 22,23,24]. Therefore, results can be unreliable in cases where the selected prior model deviates from the real admixture history.
In our previous work [13], we proposed some principles of parameter estimation and model selection under a general model, but our previous method could handle only three typical two-way admixture models. To solve this problem, we developed the MultiWaver program [25], which could select the optimal admixture model and estimate the corresponding parameters under a general discrete model. Our method could deal with complex admixture scenarios involving multiple ancestral populations with multiple admixture events. In principle, our model could also be used to analyze a population with continuous admixture, since the model can deal with admixture events at any generation. However, if the true admixture model is continuous, the number of parameters could be very large (each wave has two parameters, one admixture time and one admixture proportion); consequently, the model could become very complex. In MultiWaver, we applied a likelihood ratio test (LRT) to select the best-fit model. Using this method, more parameters means greater penalties. Thus, the method tends to select an optimal multiple-wave (discrete) model rather than a continuous model, and the continuous model is often neglected.
In this work, we extend the MultiWaver software to MultiWaver 2.0, which can handle both discrete and continuous models. In the new method, we consider four different models (the HI, GA, CGF, and multiple-wave models) (Fig. 1). Our new method can automatically select the optimal model among those candidate models, and the confidence interval (CI) of admixture time and supporting rate for each candidate model can be obtained via a bootstrapping procedure. We conducted simulation studies to demonstrate the effectiveness of our method. Finally, we applied our method to African Americans from the HapMap project phase III dataset [26] and to Uyghurs and Hazaras from the Human Genome Diversity Project (HGDP) dataset [27].

Four different types of admixture model. a Hybrid isolation (HI) model; b Gradual admixture (GA) model; c Continuous gene flow (CGF) model. POP1: the reference population 1; POP2: the reference population 2; m is the proportion of population 1 and α = 1 − m1/T. d The multiple-wave model, where POPki is the ancestral population of the ith admixture, αi is the admixture proportion of the ith admixture, and ti is the admixture time of the ith admixture
Materials and methods
Model selection and parameter estimation
In order to infer the admixture history, we need to select an optimal model from the four different models listed above. For discrete admixture models (the HI model and multiple-wave models), we apply the LRT method to select the optimal discrete model. The results are the same as those obtained using the MultiWaver software [25]. Next, we compare the optimal discrete model with continuous admixture models (GA and CGF). However, when we include the GA and CGF models in the analysis, we find that any pairs of GA, CGF, and discrete model are all not nested, which means that no model is a special case of another. The LRT method is unavailable in this case. Therefore, we apply another method, Bayesian Information Criterion (BIC) [28, 29], to select the optimal model. The value of the BIC can be calculated by the formula:
where k is the number of parameters, n is the sample size, and Lmax is the maximized value of the likelihood function. Details of the model selection procedure are illustrated in Fig. 2.

Flow chart of the algorithm for model selection. Lmax (GA), Lmax (CGFR), Lmax (CGFD), and Lmax (multi-wave) are the maximized values of the likelihood function under GA, CGFR, CGFD, and multiple-wave models, respectively. Best M is the optimal model, where M is the set of GA, CGFR, CGFD, and multiple-wave models
Whether one uses the LRT or BIC method, it is necessary to calculate Lmax and to estimate the parameters of the admixture models. In our previous study [13], we employed the length distributions of ancestral tracks under the HI, GA, and CGF models. These models involve only two parameters, the admixture proportion m and the admixture time T (Fig. 1a−c). Thus, we can easily calculate Lmax and the estimates of m and T via a binary search algorithm. For multiple-wave models, the length distribution of the ancestral tracks can be written as a mixed exponential distribution [25]. We can then use the expectation–maximization algorithm [30] to calculate Lmax and to estimate the admixture time (ti) and proportion (αi) (Fig. 1d); this produces the same estimates as the MultiWaver method [25]. After obtaining Lmax for the four models, we then select an optimal model via the LRT and BIC methods. The optimal model and the corresponding estimators of admixture time and proportion can then be used to describe the inferred admixture history. The details concerning the LRT are described in Supplementary Text 1.
Bootstrapping procedures
To assess the uncertainty in optimal model selection and estimated parameter values, we also apply the bootstrapping technique in MultiWaver 2.0 to obtain a degree of support for the chosen model and the CI of the admixture time. We conduct the bootstrapping by resampling the same number of segments with replacement and use these resampled segments to infer the admixture model and its corresponding admixture time. Details of the bootstrapping procedure are described in Supplementary Text 1. MultiWaver 2.0 can be downloaded at http://www.picb.ac.cn/PGG/resource.php.
Simulation studies
We conducted simulations to evaluate the performance of MultiWaver 2.0. The simulation data were generated by the forward-time simulator AdmixSim [31]. AdmixSim can be downloaded at http://www.picb.ac.cn/PGG/resource.php. The population size of the admixed population was arbitrarily set to 5000 and remained constant in our simulations, and the length of the simulated chromosome was 3.0 Morgans, which approximates the length of chromosome 1 of the human genome. At the end of the simulation, 100 “individuals” (pairs of chromosomes) were sampled, and the ancestral tracks were recorded.
For the symmetric admixture models (HI and GA) (Fig. 1a, b), the proportions of admixture varied from 20 to 50% in steps of 10%. For the asymmetric admixture model (CGF) (Fig. 1c), we divided the analysis into two sub-models. If population 1 was a gene flow recipient, we denoted it as a CGFR model; otherwise we denoted it as a CGFD model. We set the proportions of admixture in CGF model from 20 to 90% in steps of 10%, and we set the admixture time to 20, 40, 60, 80, and 100 generations. For the multiple-wave model (Fig. 1d), we considered a scenario of two ancestral populations with two-wave admixture. For simplicity, we assumed that in each wave of admixture the proportions (αi,1 ≤ i ≤ n) were equal. We used four values of admixture proportions: 0.2, 0.3, 0.4, and 0.5. The admixture time were set as five cases: (a) t2 = 10, T = 20, (b) t2 = 20, T = 40, (c) t2 = 40, T = 60, (d) t2 = 60, T = 80, and (e) t2 = 80, T = 100. Each simulation setting was repeated ten times for a total of 1400 simulations across the four admixture models. MultiWaver 2.0 was applied to the simulated data with the default settings and the results were recorded and summarized.
Application to analysis of real datasets
Several real admixed populations histories were analyzed by our method. First, we applied our method to African Americans. The data for African Americans and reference populations CEU and YRI were obtained from HapMap Project Phase III [26]. Next, we applied our method to reconstruct the population history of Uyghurs and Hazaras. We used the Han and French populations as the proxies for Eastern ancestry and Western ancestry, respectively [4]. Data used in this analysis were obtained from the HGDP dataset. Haplotype phasing was performed by SHAPEIT 2 [32]. Local ancestry was inferred by HAPMIX [33]. MultiWaver 2.0 was used to select the optimal model and to estimate the admixture time and proportion using tracks longer than 1 cm.
Results
MultiWaver 2.0 performed well in parameter estimation and model selection
With the extensively simulated data, we could systematically evaluate the performance of our method in regard to parameter estimation and model selection. The model was correctly selected in 88% of the simulations. For the simulations using the HI and GA scenarios, our method was able to distinguish the correct model in nearly all simulations; for the CGFR, CGFD, and multiple-wave models, our method identified the correct model with an accuracy of 82.0% (Table 1). We found that the simulations in which our method failed were often those including very recent admixture time and small admixture proportion.
We also evaluated the performance of our method for time estimation. Our method was able to estimate admixture time with high accuracy (Fig. 3). Figure 3 shows one set of simulations and the corresponding bootstrap results for CGFR, CGFD, GA, HI, and multiple-wave models. For the HI, CGF, and GA models, the results were highly consistent with the time simulated, while there was a slight overestimation for the multiple-wave model. We conclude that regardless of model selection or parameters estimated, our method performed well.

Admixture time estimated under four different types of admixture models. a CGFR model; b CGFD model; c GA model; d HI model; and e multiple-wave model. The x-coordinate is the admixture time in generations ago, with 0 being the present time. The y-coordinate is the density of admixture time estimated from 100 bootstrap-resampling datasets. There are five subgraphs for each model. Each subgraph represents the result from one simulation assuming a certain admixture time. The red dashed lines represent the given admixture time for the simulation. The admixture proportion was 0.3
Real data analysis
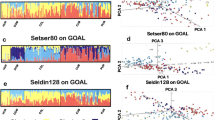
For African Americans, the program inferred the GA admixture model and the admixture time to be 12 generations (Fig. 4a, Table S1). In our previous study [13, 25], the African American population was inferred as a GA scenario with AdmixInfer and as a two-wave admixture model with MultiWaver. While both results are supported by various historical records, a best-fit model is desirable. The MultiWaver 2.0 program was able to solve this problem using a decision-making framework. We compared the likelihood of the two methods with the BIC and found that the GA model was the most likely scenario. In other words, the GA model appears to be superior to multiple-wave admixture models for African Americans.

Inferred admixture history based on analysis of real datasets. Inferred admixture history of a African Americans (ASW), where CEU and YRI were taken as the representative ancestral source populations of ASW; b Uyghurs, and c Hazaras. Han is Han population representing Asian ancestry, Fre is French population representing European ancestry
In addition, we applied our method to reconstruct the admixture histories of Uyghurs and Hazaras. These two admixed populations were inferred as GA types by AdmixInfer [13] and inferred as multiple-wave types by MultiWaver [25]. The results of MultiWaver 2.0 confirmed that the admixture pattern of Uyghurs and Hazaras followed a multiple-wave admixture model, rather than a GA or a CGF model (Fig. 4b, c, Table S1).
Discussion
MultiWaver 2.0 is an improved version of MultiWaver in that it can consider both discrete and continuous admixture models simultaneously. In MultiWaver 2.0, we apply the principles of the BIC to select the optimal model. Simulation studies suggest that our method is precise and efficient in model selection and parameter estimation.
Admixture history of a real population is often very complex. Previous methods have always required some strong pre-knowledge of the admixture pattern. If the admixture pattern is wrongly selected, the inferred admixture history may deviate from the actual history. Here, we provide a general framework to try to deal with this problem. MultiWaver 2.0 can automatically select the best-fit model from the candidates. Indeed, when the true admixture histories deviate from any of the given candidate models, our method might not have a good inference. However, the models we provided cover most admixture cases in the real data analysis and the framework of our method is much more flexible. In the future, if a new representative model is proposed, it can be easily introduced into this framework.
However, some problems remain. First, we found that the penalty for the number of parameters in the BIC method was not sufficient for our method of model selection; thus, the simulations under the CGFR and CGFD models were often wrongly determined as multiple-wave models. This was especially true for population with recent admixture time and small admixture proportion. Second, overestimation occurred for the admixture time when inferring admixture history under the multiple-wave model. This problem also occurred in MultiWaver. However, the overestimation was related to the admixture time and the admixture proportion of each wave. In our previous study, we used this relationship to adjust the estimation of admixture time [25].
Similar to other ancestral tracts information-based methods, our method is sensitive to the accuracy of local ancestry inference (LAI). For existing LAI methods, short ancestral tracts are very difficult to detect. To remove the influence of short ancestral segments, we suggest using only the ancestral tracks longer than a certain threshold C in our software. Besides the small tracts effect, the admixture model used by the LAI method is also a strong priori assumption. The history inference results might tend to be similar to those of the LAI model. To overcome this problem, joint inference of ancestral tracts and admixed history may be implemented in the future.
References
Yuan K, Zhou Y, Ni X, Wang Y, Liu C, Xu S. Models, methods and tools for ancestry inference and admixture analysis. Quant Biol. 2017;5:236–50.
Xu S, Huang W, Qian J, Jin L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am J Hum Genet. 2008;82:883–94.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y et al. Ancient admixture in human history. Genetics. 2012; 192:1065–93.
Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D et al. Inferring admixture histories of human populations using linkage disequilibrium. Genetics. 2013;193:1233–54.
Pickrell JK, Patterson N, Loh PR, Lipson M, Berger B, Stoneking M et al. Ancient west Eurasian ancestry in southern and eastern Africa. Proc Natl Acad Sci U S A. 2014;111:2632–7.
Zhou Y, Yuan K, Yu Y, Ni X, Xie P, Xing E et al. Inference of multiple-wave population admixture by modeling decay of linkage disequilibrium with polynomial functions. Heredity. 2017;118:503–10.
Pugach I, Matveyev R, Wollstein A, Kayser M, Stoneking M. Dating the age of admixture via wavelet transform analysis of genome-wide data. Genome Biol. 2011;12:R19.
Jin W, Li R, Zhou Y, Xu S. Distribution of ancestral chromosomal segments in admixed genomes and its implications for inferring population history and admixture mapping. Eur J Hum Genet. 2014;22:930–7.
Jin W, Wang S, Wang H, Jin L, Xu S. Exploring population admixture dynamics via empirical and simulated genome-wide distribution of ancestral chromosomal segments. Am J Hum Genet. 2012;91:849–62.
Gravel S. Population genetics models of local ancestry. Genetics 2012;191:607–19.
Pool JE, Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181:711–9.
Hellenthal G, Busby GB, Band G, Wilson JF, Capelli C, Falush D. et al. A genetic atlas of human admixture history. Science. 2014;343:747–51.
Ni X, Yang X, Guo W, Yuan K, Zhou Y, Ma Z et al. Length Distribution of Ancestral Tracks under a General Admixture Model and Its Applications in Population History Inference. Sci Rep. 2016;6:20048.
Pugach I, Matveev R, Spitsyn V, Makarov S, Novgorodov I, Osakovsky V et al. The Complex Admixture History and Recent Southern Origins of Siberian Populations. Mol Biol Evol. 2016;33:1777–95.
Feng QD, Lu Y, Ni XM, Yuan K, Yang YJ, Yang X et al. Genetic History of Xinjiang's Uyghurs Suggests Bronze Age Multiple-Way Contacts in Eurasia. Mol Biol Evol. 2017;34:2572–82.
Kidd JM, Gravel S, Byrnes J, Moreno-Estrada A, Musharoff S, Bryc K et al. Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation. Am J Hum Genet. 2012;91:660–71.
Baharian S, Barakatt M, Gignoux CR, Shringarpure S, Errington J, Blot WJ et al. The Great Migration and African-American Genomic Diversity. PLoS Genet. 2016;12:e1006059.
Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G et al. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 2011;7:e1001373.
Price AL, Patterson N, Yu F, Cox DR, Waliszewska A, McDonald GJ et al. A genomewide admixture map for Latino populations. Am J Hum Genet. 2007;80:1024–36.
Tian C, Hinds DA, Shigeta R, Adler SG, Lee A, Pahl MV et al. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet. 2007;80:1014–23.
Wang S, Ray N, Rojas W, Parra MV, Bedoya G, Gallo C et al. Geographic patterns of genome admixture in Latin American Mestizos. PLoS Genet. 2008;4:e1000037.
Xu S, Jin L. A genome-wide analysis of admixture in Uyghurs and a high-density admixture map for disease-gene discovery. Am J Hum Genet. 2008;83:322–36.
Lipson M, Loh PR, Patterson N, Moorjani P, Ko YC, Stoneking M et al. Reconstructing Austronesian population history in Island Southeast Asia. Nat Commun. 2014;5:4689.
Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am J Hum Genet. 2015;96:37–53.
Ni X, Yuan K, Yang X, Feng Q, Guo W, Ma Z et al. Inference of multiple-wave admixtures by length distribution of ancestral tracks. Heredity. 2018:1.
International HapMap C, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8.
Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–4.
Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6:461–4.
Wit E, van den Heuvel E, Romeijn JW. ‘All models are wrong…’: an introduction to model uncertainty. Stat Neerl. 2012;66:217–36.
Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc B.1977:39:1−38.
Yang X, Ni X, Zhou Y, Guo W, Yuan K, Xu S. AdmixSim: a forward-time simulator for various and complex scenarios of population admixture. bioRxiv. 2016:037135.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81.
Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519.
Acknowledgements
This work was supported by the Strategic Priority Research Program (XDB13040100) and Key Research Program of Frontier Sciences (QYZDJ-SSW-SYS009) of the Chinese Academy of Sciences (CAS), the Fundamental Research Funds for the Central Universities (2017JBM071, 2017YJS197), the National Natural Science Foundation of China (NSFC) (91731303, 31771388, 11426237, and 31711530221), the National Science Fund for Distinguished Young Scholars (31525014), the Program of Shanghai Academic Research Leader (16XD1404700), the National Key Research and Development Program (2016YFC0906403), and Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), the China Postdoctoral Science Foundation (2017M620595), the National Center for Mathematics and Interdisciplinary Sciences of CAS. SX also gratefully acknowledges the support of the National Program for Top-Notch Young Innovative Talents of the “Wanren Jihua” Project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ni, X., Yuan, K., Liu, C. et al. MultiWaver 2.0: modeling discrete and continuous gene flow to reconstruct complex population admixtures. Eur J Hum Genet 27, 133–139 (2019). https://doi.org/10.1038/s41431-018-0259-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0259-3
This article is cited by
-
Locked Tension Band Wiring: A Modified Technique for Olecranon Fractures—A Multicenter Study Comparing Clinical Outcomes and Complications with Conventional Methods
Indian Journal of Orthopaedics (2023)
-
AdmixSim 2: a forward-time simulator for modeling complex population admixture
BMC Bioinformatics (2021)
-
Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0
Nature Communications (2021)
