
Abstract
The need for new antibacterial drugs to treat the increasing global prevalence of drug-resistant bacterial infections has clearly attracted global attention, with a range of existing and upcoming funding, policy, and legislative initiatives designed to revive antibacterial R&D. It is essential to assess whether these programs are having any real-world impact and this review continues our systematic analyses that began in 2011. Direct-acting antibacterials (47), non-traditional small molecule antibacterials (5), and β-lactam/β-lactamase inhibitor combinations (10) under clinical development as of December 2022 are described, as are the three antibacterial drugs launched since 2020. Encouragingly, the increased number of early-stage clinical candidates observed in the 2019 review increased in 2022, although the number of first-time drug approvals from 2020 to 2022 was disappointingly low. It will be critical to monitor how many Phase-I and -II candidates move into Phase-III and beyond in the next few years. There was also an enhanced presence of novel antibacterial pharmacophores in early-stage trials, and at least 18 of the 26 phase-I candidates were targeted to treat Gram-negative bacteria infections. Despite the promising early-stage antibacterial pipeline, it is essential to maintain funding for antibacterial R&D and to ensure that plans to address late-stage pipeline issues succeed.
Similar content being viewed by others

Introduction
Antibiotics are the foundation of modern medicine but are becoming increasingly ineffective due to growing levels of antimicrobial resistance, threatening global health. The adverse impact of drug-resistant infections is highlighted by a seminal analysis of the global burden of bacterial antimicrobial resistance in 2019, with 1.27 million deaths directly attributed to, and 4.9 million deaths associated with, resistant bacteria [1]. The development of new antibiotics, particularly new chemotypes or classes that can overcome existing resistance mechanisms, has been hindered by a failure of the healthcare system marketplace to adequately recognize and compensate for these products [2,3,4]. In addition to improved generic antibiotic sales, branded antibiotic prices have fallen since 2001 [5], aggravating the economic challenges. Recognition of the antibiotic crisis has led to the establishment of targeted funding initiatives for antibiotic development such as the Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) [6], INCATE [7], REPAIR Impact Fund [8], and the AMR Action Fund [9, 10], testing of new incentives to reimburse pharmaceutical companies such as a subscription ‘Netflix’ model [11,12,13,14], and legislative initiatives such as the PASTEUR (The Pioneering Antimicrobial Subscriptions To End Up surging Resistance) Act in the United States [15, 16]. There has also been an increase in the number of “non-traditional” antibacterials [17,18,19,20,21] being actively evaluated in clinical trials [21, 22]. Non-traditional antibacterials can be small molecules, monoclonal antibodies (mAbs), proteins or live biotherapeutics such as bacteria and bacteriophages that primarily affect bacteria growth or virulence indirectly with varying mechanisms such as toxin binding, cell adherence reduction, inhibition of antivirulence targets and drug resistance modification [21].
To assess whether these activities are improving the status quo, we have monitored antibacterial drug development since 2011 with reviews published in 2019 [23], 2015 [24], 2013 [25] and 2011 [26]. Complementary reviews with different approaches and analyses (but often with few or no chemical structures) are available. The Pew Trusts developed an online pipeline tracker that allows the visualization of changes in the pipeline from 2014–2020 [27], but their antibiotic resistance project was discontinued in December 2021 [28]. In 2022, the WHO published a report on antibacterial agents in both preclinical and clinical development in 2021 [22] and a journal article in 2022 [21]. The WHO also recently reviewed the preclinical and clinical antibacterial vaccine pipeline [29]. A 2021 review critically analyzed why compounds with Gram-negative (G-ve) activity have fallen out of the pipeline over the past decade [30], while two 2020 reviews covered both the clinical [31] and preclinical [32] antibacterial pipelines, with a third providing an overview of ‘novel’ antibacterial agents in various stages of development [33]. Reviews of patents from 2010–2021 focusing on compounds with activity against multi-drug resistant (MDR) G-ve bacteria [34], antibacterial combinations [35], and discovery strategies [36] have also been published.
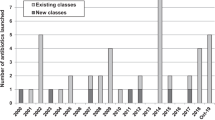
This review catalogs the small molecule antibacterial drugs launched since January 2012 in Table 1 and the yearly number of first-time antibacterial drugs launched by year since 2000 (Fig. 1, Table S1). International Nonproprietary Names (INN) are used for compound names when available. For completeness, Table 2 lists the non-traditional antibacterial drugs launched during this period. The new antibacterial drugs approved since the previous 2019 review [23], levonadifloxacin (1) and its prodrug alalevonadifloxacin (2), and contezolid (4) (Fig. 2), are analyzed. Consistent with previous reviews in this series [23,24,25,26], small molecule antibacterials (both traditional and non-traditional) and β-lactamase/β-lactam inhibitor (BL/BLI) combinations that are being evaluated in phase-I, -II, or -III clinical trials and under pre-approval regulatory evaluation as of 31 December 2022 are summarized (Tables 3–6, Figs. 3–13), along with their development status, mode of action (MoA), spectra of activity, historical discovery, and lead compound origin (natural product (NP), synthetic (S) or protein/mammalian peptides (P)). In the previous 2019 review [23], one antibody drug conjugate (ADC), DSTA4637S, was discussed, but its development has since been halted (Table 7). The clinical trial study codes, which are predominantly from ClinicalTrials.gov (NCT), are listed in parentheses for each trial, while non-registered trials are referenced at least in a Press Release or peer-reviewed publication. An overview of the drug development and approval process, on-line clinical trial databases antibiotic clinical trial categories and abbreviations can be found in the Supplementary Information. Prodrugs are grouped together with their active metabolites, while ongoing trials of antibacterial drugs already approved anywhere in the world are presented in Table S2. Compounds where no development activity has been identified since 2018 are listed in Table 7. The antibacterials in clinical development have been further analyzed by phase and source derivation (Fig. 14) and also compared with data reported in our 2011 [26], 2013 [25], 2015 [24] and 2019 [23] reviews (Fig. 15). An analysis of new antibacterial pharmacophores (Table 8, Figs. 16 and 17) and administration routes (Figs. S1 and S2) is also included. The administration routes in this review are as follows: po (oral), IV/po (intravenous oral switch); IV (intravenous), IV/topical (IV and topical), po topical (orally administered for Clostridioides difficile (formally Clostridium [37]) infections (CDI)), oral, topical and inhalation. The ‘po topical’ term distinguishes between oral administration to treat C. difficile infections and the gut microbiome compared to topical administration via creams, sprays, and eyedrops.

New small molecule antibacterial drugs and BL/BLI combinations launched from January 2000 to December 2022 with new classes highlighted

Structures of the recently lauched antibacterial drugs

Structure of the antibacterial in the NDA and MAA development stage (Table 3)

Structures of compounds in phase-III clinical trials (Table 3)

Structures of NP-derived compounds in phase-II clinical trials (Table 4)

Structures of synthetic compounds in phase-II clinical trials (Table 4)

Structures of small molecule non-traditional antibacterials in phase-II clinical trials (Table 4)

Structures of NP and peptide-derived compounds in phase-I clinical trials (Table 5)

Structures of synthetic-derived compounds in phase-I clinical trials (Table 5)

Structures of publicly disclosed small molecule non-traditional antibacterials in phase-I clinical trials (Table 5)

Structures of BLI and associated β-lactam antibacterial in NDA/MAA filing (Table 6)

Structures of BLIs in phase-III clinical trials (Table 6)

Structures of BLIs and associated β-lactam antibiotics in phase-I clinical trials (Table 6)

Compounds under clinical evaluation divided into development phases and their lead derivation source: natural product (NP) (NP-derived and NP-BLI), protein/mammalian peptide (P-derived) and synthetic (S) (S-derived and S-BLI)


Antibacterial compounds [natural product (NP), synthetic (S), protein/mammalian peptide (P)] and β-lactamase inhibitors (BLI)] with new antibacterial pharmacophores divided into development phases and their lead derivation source

Data in this review were obtained by analyzing the scientific literature and internet sources such as company and funding organization websites, clinical trial registers, The Pew Charitable Trusts (Philadelphia, PA, USA) [28] and World Health Organization (WHO) (Geneva, Switzerland) pipeline analyses [21, 22] and biotechnology newsletters. Every effort has been made to ensure the accuracy of this data; however, it is possible that compounds in the early stages of clinical development have been overlooked as there is limited information available in the public domain.
Antibacterial drugs launched from January 2013 to December 2022
In the last 10 years, 19 new small molecule antibacterial drugs (eight NP-derived and 11 synthetic-derived) and four new BL/BLI combinations have been approved (Table 1 and S1, Figs. 1 and 2). Among these 19 antibacterial drugs, none was first-in-class, with the last being bedaquiline in 2012 (diarylquinoline class), which also was the first new tuberculosis (TB) drug class since 1963 [38]. Although the semi-synthetic pleuromutilin derivative lefamulin was approved in 2019 for systemic use for community-acquired bacterial pneumonia (CABP) infection, a topically administered pleuromutilin, retapamulin, was approved in 2007. While new classes of G-ve antibacterial drugs have been approved, new exemplars within existing classes, especially BL/BLI combinations, also show improved activity profiles against resistant G-ve bacteria.
Since the 2019 review [24] in this series, two new small molecule antibacterials (Table 1, Figs. 1 and 2), levonadifloxacin (1) (as its prodrug alalevonadifloxacin (2)) and contezolid (4) were first approved in India and China respectively.
Levonadifloxacin (1) (Emrok, WCK-771; IV), which is the arginine salt of the fluoroquinolone S-(–)-nadifloxacin, and its alanine prodrug alalevonadifloxacin (2) (Emrok O, WCK-2349; po) [39,40,41] were developed by Wockhardt (Mumbai, Republic of India). Both the IV and oral formulations were approved in January 2020 by the Indian Central Drugs Standard Control Organization (CDSCO) for the treatment of acute bacterial skin and skin structure infections (ABSSSI), including diabetic foot infections and concurrent bacteremia [42, 43]. Levonadifloxacin (1) has activity against G+ve bacteria including MRSA, as well as some G-ve bacteria [41], and a prescription-event monitoring study was recently published [44]. Racemic nadifloxacin was first approved in 1993 to topically treat acne and MRSA infections [45].
Contezolid (4) (Youxitai, MRX-1; IV) is an oxazolidinone developed by MicuRx Pharmaceuticals (Hayward, CA, USA and Shanghai, People’s Republic of China). It was approved by the Chinese National Medical Products Administration (NMPA) in June 2021 for the treatment of complicated skin and soft tissue infections (cSSTI), including, but not limited to, MSSA, MRSA, Streptococcus pyogenes and Streptococcus agalactiae [46,47,48]. The development pathway from contezolid (4) [49] to contezolid acefosamil (3) (MRX-4) was recently published [50]. The prodrug 3 provides dramatic improvements in solubility over the parent antibiotic (from 0.2 mg ml−1 to >200 mg ml−1), leading to exposure of contezolid (4) in rats after IV administration of contezolid acefosamil (3) like, or higher than, that from direct IV administration of 4. A phase-III trial (NCT05369052) evaluating contezolid acefosamil (3) (po)/contezolid (4) (IV) for diabetic foot infections compared to linezolid began in May 2022.
Three non-traditional antibacterial drugs (two mAbs and one biotherapeutic) were launched between 2013 and 2022 to treat bacterial infections (Table 2), compared to 19 traditional antibacterial drugs launched during this period (Table 1).
Obiltoxaximab [51, 52] is a mAb that neutralizes harmful toxins produced by Bacillus anthracis that was approved using the US FDA Animal Rule based on their efficacy in relevant animal models and safety in phase-I studies. Another mAb that also neutralizes B. anthracis toxins, raxibacumab [52, 53], was similarly approved in 2012. The mAb bezlotoxumab, which binds to toxin B produced by C. difficile [54, 55], was approved in 2016 to help prevent the recurrence of CDI after successfully completing two phase-III trials [56, 57].
In November 2022, a live biotherapeutic product, RBX2660 (Rebyota), was approved by the US FDA [58] to help prevent CDI following antibiotic treatment, based on phase-III trial data [59]. RBX2660 is a liquid suspension donor fecal microbiota that has been screened for bacterial, viral and parasitic pathogens [60, 61] that was developed by Rebiotix Inc (Roseville, MN, USA), which is part of Ferring Pharmaceuticals (Saint-Prex, Switzerland). There is also another phase-III trial (NCT03931941) in progress.
Although outside the cut-off period, another non-traditional antibacterial product, Vowst (SER-109), developed by Seres Therapeutics Inc (Cambridge, MA, USA) and Nestlé Health Science (Hoboken, NJ, USA) was approved by the US FDA on 26 April 2023.Footnote 1
Compounds undergoing clinical evaluation
Direct acting small molecules, mammalian-derived peptides and polymeric compounds currently undergoing clinical trials or under regulatory evaluation for the treatment of bacterial infections on 31 December 2022 are detailed in the following tables and figures: NDA and phase-III in Table 3 and 6 with structures in Figs. 3, 4, 11, and 12, phase-II in Table 4 with structures in Figs. 5–7, and phase-I in Tables 5 and 6 with structures in Figs. 8–10 and 13. Non-traditional antibacterial candidates that are not small molecules such as biotherapeutic microbiome modulation, phage therapy, and antibodies have not been included in this review.
Compounds in NDA/MAA filing (Table 3, Fig. 3)
Solithromycin (5) (T-4288, CEM-101; IV/po) is a semi-synthetic 2-fluoroketolide [62] that is being developed by FUJIFILM Toyama Chemical Co., Ltd. (Tokyo, Japan). In April 2019, an NDA was submitted to the Japanese Pharmaceuticals and Medical Devices Agency (PDMA) for use of 5 as a treatment for otorhinolaryngological bacterial infections. Although there have been no subsequent updates, 5 is still listed on their November 2022 pipeline as ‘NDA filing’ for otorhinolaryngology and as phase-III for respiratory infectious disease [63]. Solithromycin (5) was previously being developed in the USA and Europe for CABP but development was halted in 2016 and 2017 respectively [64].
Compounds in phase-III trials (Table 3, Fig. 4)
Sulopenem (6) (CP-70,429), which is a synthetic thiopenem BL first developed by Pfizer (New York, NY, USA) in the 1990s [65,66,67,68], and its prodrug sulopenem etzadroxil (7) (PF-03709270; po) are being developed as treatments for G-ve infections by Iterum Therapeutics (Dublin, Ireland). To date, three phase-III trials have been completed and have reported results: complicated intra-abdominal infections (cIAI) (NCT03358576), cUTI (NCT03357614) [69] and uUTI (NCT03354598) [70]. In November 2020, Interim filed an NDA for uUTIs with the FDA [71] for orally administered sulopenem etzadroxil (7) in combination with probenecid (8) [72]. Probenecid (8) is a marketed drug for gout and hyperuricemia that increases uric acid production, which inhibits BL tubular renal secretion that leads to a longer antibiotic half-life and higher serum concentrations [73]. However, the FDA issued a Complete Response Letter (CRL) in July 2021 that indicated that the NDA was not approvable in its present form [74]. In response to this CRL, Iterum initiated another phase-III trial (NCT05584657) in October 2022 to investigate sulopenem etzadroxil (7) + probenecid (8) compared to amoxicillin + clavulanic acid for uUTI, which is scheduled to finish in March 2024.
Nafithromycin (9) (WCK 4873; po) is an orally bioavailable ketolide being developed by Wockhardt Limited (Mumbai, Republic of India) that is being evaluated in a phase-III trial (CTRI/2019/11/021964) in India as an oral treatment for CABP. Nafithromycin (9) has broad spectrum antibacterial activity against G+ves such as S. pneumoniae and S. aureus and G-ves such as Haemophilus influenzae, Moraxella catarrhalis, Legionella pneumophila, Mycoplasma pneumoniae and Chlamydophila pneumoniae [75,76,77,78].
Gepotidacin (10) (GSK-2140944; po) is a new chemotype bacterial Type II topoisomerase inhibitor [79] (new triazaacenaphthylene class) being developed by Glaxo-SmithKline (GSK) (London, UK) for uUTI and gonorrhea. In November 2022, GSK announced that two phase-III trials (NCT04020341 and NCT04010539) for cUTI were stopped early for efficacy (positive news!), with an NDA planned for the first half of 2023 [80]. Gepotidacin (10) is also being evaluated in another cUTI phase-III trial with Japanese participants (NCT05630833), as well as a phase-III trial against uncomplicated urogenital gonorrhea caused by Neisseria gonorrhoeae (NCT04010539). Gepotidacin (10) has activity against a range of both G+ve and G-ve pathogens [81,82,83], including Mycobacteria [84], Stenotrophomonas maltophilia [85], Mycoplasma and Ureaplasma [86].
Zoliflodacin (11) (ETX0914, AZD0914; po) is another new chemotype topoisomerase inhibitor [87] (new spiropyrimidinetrione class) being developed by Entasis Therapeutics (Waltham, MA, USA), which was recently acquired by Innoviva (Burlingame, CA, USA) [88]. Zoliflodacin (11) is being evaluated in a phase-III trial (NCT03959527) as an oral treatment for uncomplicated gonorrhea [89,90,91] in partnership with the Global Antibiotics Research and Development Partnership (GARDP) (Geneva, Switzerland). GARDP has the right to register and commercialize 11 in low- and middle-income countries [92]. Zoliflodacin (11) also has activity against Mycoplasma genitalium, which could broaden its effectiveness as a treatment for sexually transmitted infections [93].
Benapenem (12) (IV) is a carbapenem that completed a phase-II/III trial in May 2020 (NCT04505683) as an intravenous treatment for cUTI, including pyelonephritis, by Sihuan Pharmaceutical (Beijing, People’s Republic of China). Benapenem (12) is structurally related to ertapenem and has a similar extended human half-life of 7 h, which supports once-daily IV dosing like ertapenem, an advantage over other carbapenems that require multiple daily dosing due to shorter half-lives [94, 95].
Epetraborole (13) (GSK2251052, AN3365, and BRII-658; po) is a benzoxaborole leucyl-tRNA synthetase (LeuRS) inhibitor [96], which is a new antibacterial target, being evaluated by AN2 Therapeutics (Menlo Park, CA, USA) in a phase-II/III (NCT05327803) against treatment-refractory Mycobacterium avium complex (MAC) lung disease. MAC accounts for up to 85% of non-tuberculosis mycobacteria (NTM) related lung disease [97]. Epetraborole (13) has also been reported to have in vivo activity against Mycobacterium abscessus, another NTM involved in lung infections [98, 99]. Epetraborole (13) was originally developed as a treatment for G-ve infections in phase-II trials for cUTI (NCT01381549) and cIAI (NCT01381562) but these studies were halted due to resistance developing in patients during the cUTI trial [100]. Brii Biosciences (Durham, NC, USA and Shanghai, People’s Republic of China) have licensed 13 for development in the Greater China region [101].
Traditional antibacterial compounds in phase-II trials (Table 4, Figs. 5 and 6)
Sanfetrinem cilexetil (14) (GV-104326; po) is a 1-(cyclohexyloxycarbonyloxy)ethyl ester prodrug of the trinem (tricyclic carbapenem) sanfetrinem (15) first developed in the 1990s by Glaxo Wellcome, which is now part of GSK (London, UK). Sanfetrinem (15) is active against a range of G+ve (e.g., S. aureus, S. pneumoniae and H. influenzae) and G-ve bacteria (e.g., E. coli, M. catarrhalis) [102,103,104]. Although sanfetrinem cilexetil (14) successfully completed a phase-II trial for respiratory infections in 1999, no further development work was undertaken until GSK started a phase-II trial (NCT05388448) in May 2022, which is evaluating 14 against rifampicin-susceptible pulmonary TB [105]. There has been a recent surge in interest in investigating carbapenem-type antibacterials as TB treatments, as evidenced by TASK (Cape Town, South Africa) leading a study that showed meropenem (66) in combination with amoxicillin + clavulanic acid had efficacy in a phase-II TB trial (NCT02349841) [106], as well as a consortium of private and public organizations that screened approximately 8,900 carbapenems against Mycobacterium tuberculosis (Mtb) [107].
MGB-BP-3 (16) (po topical) is a DNA binding antibacterial being developed by MGB Biopharma (Glasgow, UK) that successfully completed a phase-II trial (NCT03824795) in May 2020 for the treatment of C. difficile-associated diarrhea (CDAD) [108]. MGB-BP-3 (16) was discovered at the University of Strathclyde (Glasgow, UK) and was inspired by the actinomycetes-derived minor groove binders, distamycin, netropsin and thiazotropsin [109, 110]. In addition to activity against C. difficile, 16 has activity against a range of G+ve bacteria including S. aureus and Enterococcus faecalis but is not active against G-ve bacteria due to a lack of intracellular accumulation [111]. It was shown that two molecules of 16 bound to the minor groove of dsDNA, which then interfered with transcription, the supercoiling action of gyrase, and the relaxation and decatenation by topoisomerase IV enzymes in vitro [111]. This is mechanistically distinct from fluoroquinolones that cause an increase in double strand breaks, as well as induce recA and lexA SOS responses. A preprint has reported that 16 also binds to and inhibits multiple essential promoters on the S. aureus chromosome [112]. Furthermore, 16 is equally effective against ciprofloxacin-resistant and ciprofloxacin-susceptible strains [113].
Exeporfinium chloride (17) (XF-73; topical) is a photosensitizing porphyrin derivative with broad-spectrum G+ve activity [114,115,116] and a low propensity for developing resistance [117] being developed by Destiny Pharma (Brighton, UK). Exeporfinium chloride (17) successfully completed a phase-II trial (NCT03915470) in March 2021 that investigated its activity against nasal S. aureus in patients at risk of post-operative infections. Destiny Pharma plans to start two phase-III nasal decolonization trials in 2024 after securing a partnering deal [118].
Synthetic cannabidiol (18) (CBD, BTX 1801; topical) has been evaluated in a phase-II trial (ACTRN12620000456954) by Botanix Pharmaceuticals (Perth, Australia) for the clearance of nasally colonized S. aureus, as well as in phase-II trials in acne (BTX 1503, NCT03573518) and atopic dermatitis (BTX 1204, NCT03824405). Cannabidiol (18) is the major non-psychoactive component of cannabis (Cannabis sativa and C. indica) and its G+ve antibacterial activity, along with that of the major psychoactive compound Δ9-tetrahydrocannabinol, was reported as having potential as a topical antibacterial in 1976 [119]. Anti-MRSA activity of 18 was later confirmed in 2008 [120] and 2020 [121] studies, along with other analogs. In 2021, an in-depth study showed that 18 was active against drug resistant strains of S. aureus, S. pneumoniae, E. faecalis, Cutibacterium acnes and C. difficile, less active against S. pyogenes and S. agalactiae, weakly active against Mycobacterium smegmatis and barely active against Mtb [122]. While cannabidiol (18) was inactive against E. coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii, it also displayed activity against four G-ve bacteria: N. gonorrhoeae, Neisseria meningitidis, M. catarrhalis and L. pneumophila [122]. It was also demonstrated that 18 was active against MSSA and MRSA biofilms, was active in topical in vivo models (though highly formulation-dependent) and that its MoA involved cytoplasmic membrane disruption [122]. It was recently shown that 18 could also act as an adjuvant with bacitracin, a cell wall inhibitor, via inhibition of undecaprenyl pyrophosphate dephosphorylation [123]. Genomic analysis demonstrated that less susceptible S. aureus strains contained mutations in the transporter farE/farR efflux pump system [123]. Additionally, screening of the Nebraska Transposon Mutant Library identified that strains with insertions involved in menaquinone biosynthesis had increased susceptibility to 18 that could be reversed by the addition of menaquinone [123]. The menaquinone biosynthesis pathway has been shown to be a promising drug target for S. aureus [124, 125].
TNP-2092 (19) (CBR 2092; IV) is being developed by TenNor Therapeutics (Suzhou, People’s Republic of China) and completed a phase-II trial (NCT03964493) for the treatment of G+ve ABSSSI infections using IV dosing in September 2020. TenNor have also evaluated capsule administration of 19 for hyperammonemia/hepatic encephalopathy in a phase-II trial with patients with liver cirrhosis [126, 127], while a phase-I trial (NCT04294862) for Prosthetic Joint Infection (PJI) employed IV administration [128]. TNP-2092 (19) is a rifamycin-quinolizinone (lead ABT-719) hybrid G+ve antibacterial discovered by Cumbre Pharmaceuticals [126, 129, 130] and its MoA is via inhibition of the targets of both antibacterial components: RNA polymerase (rifamycin) and DNA gyrase and topoisomerase IV (quinolone/quinolizinone) [131].
TNP-2198 (20) (IV) is another hybrid being developed by TenNor Therapeutics (Suzhou, People’s Republic of China); in this case, a rifamycin-metronidazole hybrid [132] for microaerophilic and anaerobic infections, which include gastrointestinal diseases associated with Helicobacter pylori, bacterial vaginosis and CDAD [133]. An H. pylori phase-II trial (CTR20220625 [134]) of capsules of 20 in combination with rabeprazole tablets (used to treat peptic ulcer disease) and amoxicillin capsules was completed in September 2022. An X-ray crystal structure was recently published that showed 20 bound to the rifamycin binding site on RNA polymerase with the nitroimidazole portion interacting directly with the DNA template-strand in the RNA polymerase active-center cleft, forming a hydrogen bond with a base of the DNA template strand [132]. This is supportive of RNA polymerase inhibition being involved in the MoA of 20.
Afabicin (21) (Debio 1450, AFN 1720) [135,136,137] is a phosphate prodrug of afabicin desphosphono (22) (Debio 1452, AFN-1252; IV/po) being developed by Debiopharm Group (Lausanne, Switzerland). The lead compound was originally discovered by GSK (London, UK) and licensed to Affinium Pharmaceuticals, who were acquired by Debiopharm in February 2014. Afabicin (21) is being evaluated in a phase-II trial (NCT03723551) using an IV/oral switch strategy for the treatment of S. aureus bone or joint infections [138]. In an earlier phase-II trial (NCT02426918), 21 was shown to be clinically non-inferior to vancomycin/linezolid against staphylococcal ABSSSI [139]. Afabicin (21) specifically inhibits staphylococcal FabI [140,141,142], which is an essential enzyme in the final step of the fatty acid elongation cycle [143].
Peceleganan (23) (PL-5, V681; topical) is a 26-mer α-helical cationic hybrid peptide of cecropin A and melittin B [144, 145] being developed by Jiangsu ProteLight Pharmaceutical and Biotechnology (Jiangyin, People’s Republic of China). Peceleganan (23) is administered by spray and has successfully completed a phase-II trial in China (ChiCTR2000033334) for the treatment of bacterial wound infections [146]. No levels of 23 were detected in the patients’ blood. This indicated that there was minimal or no systemic exposure [146], a significant consideration since some cationic peptides have a history of causing nephrotoxicity. Peceleganan (23) has activity against both G+ve and G-ve bacteria [144, 145] and there are plans to start a phase-III trial in 2023.
Recce-327 (R327; topical and IV) is an acrolein polymer with a molecular weight range of 1–1.5 kDa [147] being evaluated by Recce Pharmaceuticals (Perth, Australia) in a phase-I/II (ACTRN12621000412831) for the treatment of G+ve and G-ve burn wound infections. A phase-I trial (ACTRN12621001313820) using IV administration of Reece-327 is being conducted with the goal of developing the polymer for serious bacterial infections such as sepsis in the future. It has been reported that the polymer disrupts bacterial cellular bioenergetics via membrane potential and/or ATP synthesis [148].
Pravibismane (24) (MBN-101, bismuth ethanedithiol, BisEDT; topical) is a broad spectrum antibacterial with anti-biofilm activity [149] that is being developed by Microbion Corporation (Bozeman, MT, USA). A phase-II trial (NCT05174806) evaluating 24 as a topical treatment for diabetic foot infections started in June 2022, while a phase-II trial (NCT02436876) using intraoperative administration in patients diagnosed with an orthopedic infection was completed in July 2018. This clinical work is supported by the Cystic Fibrosis Foundation (Bethesda, MD, USA) and CARB-X (Boston, MA, USA). It has been reported that 24 can cause bacterial membrane depolarization, which disrupts cellular bioenergetics [150]. Bismuth has intrinsic antibacterial activity and is a component of Pepto Bismol® (bismuth subsalicylate) [151] and Xeroform® (bismuth tribromophenate) [152], and is used in combination with antibiotics and a proton pump inhibitor to treat H. pylori infections [153]. There has recently been a resurgence in interest in the antibacterial activity of metal complexes [154, 155].
DNV-3837 (25) (MCB-3837; IV) is a phosphate prodrug of the oxazolidinone-quinolone hybrid DNV-3681 (26) (MCB-3681) being developed by Deinove (Montpellier, France). It is currently being evaluated in a phase-II CDI trial (NCT03988855) with IV administration [156]. Unfortunately, Deinove entered receivership proceedings in November 2022 [157]. The IV administration contrasts with most other antibacterials being developed for CDI, including non-traditionals [17, 21], that are almost exclusively delivered orally with little or no systemic distribution (po topical). DNV-3837 (25) also showed G+ve activity against MRSA, Francisella tularensis and B. anthracis [158,159,160].
Ibezapolstat (27) (ACX-362E; po topical) is a bis-substituted guanine derivative that is a bacterial DNA polymerase IIIC inhibitor [161,162,163,164] that is being evaluated in a phase-II CDI trial (NCT04247542) [165] by Acurx Pharmaceuticals (White Plains, NY, USA). DNA polymerase IIIC is a new target for clinical development and is an essential enzyme in bacteria with low guanine and cytosine content, such as Bacillus, Clostridioides, Enterococcus, Mycoplasma, Lactobacillus, Listeria, Pneumococcus, Staphylococcus and Streptococcus [163].
CRS3123 (28) (REP3123; po topical) is a methionyl tRNA synthetase (MetRS) inhibitor (new diaryldiamine class) being developed by Crestone (Boulder, CO, USA) that selectively acts on S. aureus and C. difficile MetRS with little effect on G-ve and mammalian orthologs [166, 167]. CRS3123 (28) prevents C. difficile sporulation, which leads to the inhibition of toxin production, and spares most normal gut flora [168]. CRS3123 (28) has completed two phase-I trials (NCT02106338 and NCT01551004) [169, 170] and is currently being evaluated in a CDI phase-II trial (NCT04781387) versus a vancomycin comparator. In the previous pipeline review [23], 28 was listed as having its development halted or discontinued. This a reminder that relatively long delays can occur in antibacterial drug development, which have been exacerbated by the COVID-19 pandemic due to disruptions to clinical trial enrollments and day-to-day operations of many organizations [171].
Anti-mycobacterial compounds in phase-II trials (Table 4, Fig. 6)
Delpazolid (29) (RMX2001, LCB01-0371; po) is an oxazolidinone developed by LegoChem Biosciences, Inc. (Daejeon, Republic of Korea), which has activity against G+ve bacteria [172], Mtb [173, 174] and NTMs [175, 176]. Delpazolid (29) is currently being evaluated in a phase-II TB trial (NCT04550832) in combination with standard-dose bedaquiline, delamanid and moxifloxacin, compared to standard-dose bedaquiline, delamanid and moxifloxacin alone. In addition, a combination of 25 and vancomycin is being evaluated against vancomycin alone for hospitalized adults with MRSA bacteremia in a phase-IIa trial (NCT05225558). An early bactericidal activity (EBA) [177] phase-II trial (NCT02836483) showed that 29 monotherapy reduced the log-CFU of Mtb in sputum by approximately 25%, and had fewer side effects than other oxazolidinones [178].
Sutezolid (30) (PF-2341272, PNU-100480; po) [179] is an oxazolidinone originally developed by Upjohn & Co (later was incorporated into Pfizer (New York, NY, USA)) with activity against TB [174, 180,181,182] and NTMs [176]. Sequella (Rockville, MD, USA) licensed 30 from Pfizer and completed a phase-II trial (NCT01225640) in December 2011 in naive patients with drug-sensitive pulmonary TB [183]. The European and Developing Countries Clinical Trials Partnership (EDCTP; The Hague, Netherlands) is leading a phase-II trial (NCT03959566) in partnership with Sequella evaluating a combination of 30 with bedaquiline, delamanid and moxifloxacin, compared against bedaquiline, delamanid and moxifloxacin alone. The TB Alliance (New York, NY, USA) and partners [184] will also evaluate sutezolid (30) in a phase-II (NCT05807399) and in combination with bedaquiline and pretomanid in a phase-II/III trial (NCT05686356) later in 2023.
Telacebec (31) (Q203; po) is an imidazo[1,2-a]pyridine amide [185,186,187] being developed by Qurient Co., Ltd. (Seongnam-si, Republic of Korea) that completed an EBA TB phase-II trial (NCT03563599) in September 2019 [188, 189]. The imidazo[1,2-a]pyridine amide pharmacophore was identified during phenotypic high-content assays in infected macrophages and 31 inhibits TB growth via targeting QcrB, which is a subunit of the menaquinol cytochrome c oxidoreductase (bc1 complex) [185, 190, 191]. Telacebec (31) also has promise as a treatment for Buruli ulcer (Mycobacterium ulcerans) [192, 193].
Fobrepodacin (32) (SPR720, pVXc-486; po) is a DNA gyrase inhibitor phosphate prodrug being investigated by Spero Therapeutics (Cambridge, MA, USA) in a phase-II trial (NCT05496374) with patients with MAC pulmonary disease. The active metabolite SPR719 (33) has activity against various Mycobacteria [194,195,196] and results from a phase-I trial (NCT03796910) suggested that predicted therapeutic exposures could be attained with once-daily oral administration [197]. Fobrepodacin (32) and SPR719 (33) were originally discovered by Vertex Pharmaceuticals (Boston, MA, USA) [198,199,200] and inhibit DNA synthesis via bacterial gyrase (GyrB) and topoisomerase IV ParE, which is a similar MoA to novobiocin [201].
BTZ-043 (34) (po) is the first member of a new benzothiazinone (BTZ) class of TB antibacterials that completed a phase-I/II trial (NCT04044001) in May 2022. This study evaluated the safety, tolerability and EBA of 34, and was led by the EDCTP (The Hague, Netherlands). BTZ-043 (34) inhibits the essential mycobacterial cell wall biosynthesis enzyme decaprenylphosphoryl‐β‐D‐ribose (DPR) 2′‐oxidase (DprE1) via in vivo reduction of the nitro group, generating a reactive nitroso intermediate that forms a covalent semi-mercaptal adduct with cysteine-387 [202,203,204,205,206]. It has been shown that BTZs can be de-aromatized in vivo through the formation of a Meisenheimer complex, which could also reduce their in vivo half-lives [207, 208]. A BTZ analog, macozinone (53, Fig. 9) is being evaluated in a phase-I trial.
Quabodepistat (35) (OPC-167832; po) is an antitubercular 3,4-dihydrocarbostyril derivative [209] being developed by Otsuka Pharmaceutical (Tokyo, Japan) that started a phase-II trial (NCT05221502) in April 2022 in combination with delamanid and bedaquiline, compared to a combination of rifampin, isoniazid, ethambutol, and pyrazinamide. Quabodepistat (35), which completed a phase-I/II trial in February 2022 (NCT03678688), exerts its anti-mycobacterial activity through inhibition of the cell wall synthesis enzyme DprE1 [210], which is the same target as BTZ-043 (34), macozinone (53) and TBA-7371 (38).
GSK3036656 (36) (GSK656; po) is a boron containing leucyl t-RNA synthetase inhibitor (new MoA) [211, 212] that GSK (London, UK) are currently investigating in a phase-II trial (NCT05382312) in combination with either delamanid, bedaquiline, both delamanid and bedaquiline or standard of care for 14 days in participants with newly diagnosed sputum smear positive drug-sensitive pulmonary TB. A phase-II EBA TB trial (NCT03557281) for 36 was completed in December 2021. A dechloro analog, epetraborole (13, Fig. 4), is currently in a phase-II/III trial (NCT05327803) against treatment-refractory MAC lung disease.
TBA-7371 (37) (po) is a substituted 1,4-azaindole that is being developed as a new TB treatment by the Global Alliance for TB Drug Development (New York, NY, USA), the Foundation for Neglected Disease Research (Bangalore, Republic of India) and the Bill & Melinda Gates Medical Research Institute (Cambridge, MA, USA). TBA-7371 (37) is currently being evaluated in a phase-II EBA and pharmacokinetic (PK) trial (NCT04176250) in patients with rifampicin-sensitive TB. TBA-7371 (37) is a non-covalent DprE1 inhibitor discovered by scaffold hopping from telacebec (31), which has a different mechanism [213,214,215].
Sudapyridine (38) (WX-081; po) is a bedaquiline analog with a chlorophenyl-methoxypyridyl group replacing the bedaquiline bromo-2-methoxy-3-quinolyl substituent [216] being developed by Shanghai Jiatan Biotech (Shanghai, People’s Republic of China). Sudapyridine (38) is being evaluated in a phase-II EBA trial (NCT04608955) in patients with susceptible and drug-resistant TB. Sudapyridine (38) has a similar in vitro and in vivo activity profile to bedaquiline, but had no adverse effects on blood pressure, heart rate, or qualitative ECG parameters during non-clinical toxicology studies [217]. Sudapyridine (38) also has in vitro activity against most NTM species [218].
Pyrifazimine (39) (TBI-166; po) is a clofazimine analog [219] (riminophenazine class) that completed a phase-II EBA TB trial (NCT04670120) in June 2021 run by the Institute of Materia Medica (Shanghai, People’s Republic of China), Chinese Academy of Medical Sciences (Beijing, People’s Republic of China) and Peking Union Medical College (Beijing, People’s Republic of China). Although clofazimine has been used to treat leprosy (Mycobacterium leprae infections) since 1962 and was recently incorporated into some short-course MDR-TB regimens [220, 221], its tissue accumulation can cause skin discoloration that can take months to clear. Pyrifazimine (39) was designed to maintain activity against TB, have improved PK/pharmacodynamics (PD) properties, and cause less skin discoloration [222,223,224,225].
Non-traditional antibacterial compounds in phase-II trials (Table 4, Fig. 7)
Fluorothiazinon (40) (ftortiazinon, fluorothyazinon, C-55; po) is an orally administered inhibitor of the bacterial type III secretion system (T3SS), which is a highly conserved G-ve anti-virulence target [226] Fluorothiazinon (40) was developed by the Gamaleya Research Institute of Epidemiology and Microbiology (Moscow, Russia) [227,228,229,230], and has been evaluated in a phase-II trial (NCT03638830) in combination with the cephalosporin cefepime (41) as a potential treatment for patients with cUTI caused by P. aeruginosa.
Dovramilast (42) (CC-11050, AMG-634; po) is an isoindole phosphodiesterase type 4 (PDE4) inhibitor being developed for TB [231, 232] and leprosy type 2 reactions by Medicines Development for Global Health (Melbourne, Australia), which licensed 42 from Amgen (Thousand Oaks, CA, USA) in December 2020 [233, 234]. Dovramilast (42) is being evaluated in a phase-II trial (NCT03807362) at The Leprosy Mission Nepal (Katmandu, Nepal) for patients with erythema nodosum leprosum (ENL), which is an inflammatory disorder triggered by leprosy. Another phase-II trial (NCT02968927) run by The Aurum Institute NPC (Johannesburg, South Africa) has been completed [235, 236]. PDE4 inhibitors are an adjunctive host-directed therapy designed to modulate the inflammatory response to Mtb infection by reducing, but not fully blocking, TNF-α production by the host cells. The NCT02968927 trial used 42 in combination with 2HRZE/4HR therapy, which is 2 months of isoniazid (H), rifampicin (R), pyrazinamide (Z) and ethambutol (E), followed by a continuation phase of 4 months of isoniazid and rifampicin, while the NCT03807362 trial examines the safety and efficacy of CC-11050 as a monotherapy.
Traditional antibacterial compounds in phase-I trials (Table 5, Figs. 8 and 9)
SPR206 (43) (IV) is a polymyxin analog being developed by Spero Therapeutics (Cambridge, MA, USA) with activity against MDR G-ve bacteria [237] and reduced nephrotoxicity compared to polymyxin. SPR206 (43) has completed three phase-I trials (NCT03792308, NCT04868292, and NCT04865393), with a phase-II trial planned for Q4 2023 [238]. Everest Medicines (Shanghai, People’s Republic of China) had licensed the rights for 43 in China, South Korea and several Southeast Asian countries [239], while Pfizer (New York, NY, USA) has the remaining rights outside of the USA [238].
MRX-8 (IV) is another polymyxin analog being developed by MicuRx (Hayward, CA, USA and Shanghai, People’s Republic of China) against G-ve bacteria [240,241,242] that completed a phase-I trial in 2021 (NCT04649541), while another phase-I is ongoing in China [243]. Although MRX-8’s structure has not been publicly disclosed, it is a polymyxin B analog with a fatty acid tail linked via a polar ester group to form a ‘soft’ prodrug [241, 244].
QPX-9003 (44) (F365, BRII-693; IV) is also a polymyxin derivative being developed by Qpex Biopharma (San Diego, CA, USA). It is a potential treatment for P. aeruginosa and A. baumannii infections and completed a phase-I trial in July 2022 (NCT04808414) [245]. QPX-9003 (44) was reported by researchers at Monash University (Melbourne, Australia) and Qpex to have reduced nephrotoxicity, acute toxicity and in vitro lung surfactant inactivation compared to other polymyxins [246]. Brii Biosciences (Durham, NC, USA and Shanghai, People’s Republic of China) have licensed QPX-9003 (44) for development in the Greater China region [101].
RG6319 (administration route not disclosed) is an inhibitor of LepB, which is an E. coli Type I signal peptidase (SPase), listed on Roche’s (Basel, Switzerland) pipeline as being evaluated in a phase-I clinical trial for cUTI [247]. SPases are enzymes that hydrolyze N-terminal signal peptides from proteins that are secreted across the cytoplasmic membrane and have a critical role in the viability and virulence of bacteria [248]. Although the structure of RG6319 has not been disclosed, Genetech (San Francisco, CA, USA) and The Scripps Research Institute (La Jolla, CA, USA) have been evaluating derivatives of the arylomycins, which are Streptomyces-derived SPase inhibitors, such as G0775 [249, 250].
Zifanocycline (45) (KBP-7072; IV/po) is a tetracycline derivative (aminomethylcycline) being developed by KBP BioSciences (Princeton, NJ, USA) that has completed three phase-I trials (NCT02454361, NCT02654626, and NCT04532957) and is currently being evaluated in another phase-I trial (NCT05507463). Zifanocycline (45) has broad spectrum antibacterial activity [251,252,253] and a preprint has disclosed an X-ray structure of 45 bound to the Thermus thermophilus 30 S ribosomal subunit [254]. As with CRS3123 (28), zifanocycline (45) was listed as discontinued or halted in the previous review [23].
Apramycin (46) (EBL-1003; IV) is an aminoglycoside being developed by Juvabis AG (Zurich, Switzerland) that completed a phase-I trial (NCT04105205) in October 2020. A new phase-I trial (NCT05590728) was recently started by the National Institute of Allergy and Infectious Diseases (NIAID; Rockville, MD, USA). Apramycin (46) has activity against carbapenem- and aminoglycoside-resistant Enterobacteriaceae, A. baumannii and P. aeruginosa [255, 256]. Apramycin (46) has been widely used as a veterinary antibiotic to treat E. coli and other G-ve infections [257], with European approval to treat colibacillosis and salmonellosis in calves, bacterial enteritis in pigs, colibacillosis in lambs and E. coli septicemia in poultry [258]. It was discovered in the 1960s at Eli Lilly & Co (Indianapolis, IN, USA) as a NP produced by Streptomyces tenebrarius [259, 260].
PLG0206 (47) (WLBU2; topical and IV) is a 24 residue membrane disrupting cationic peptide [261, 262] being evaluated by Peptilogics (Pittsburgh, PA, USA) in a phase-I trial (NCT05137314) for its potential to treat PJI in conjunction with the DAIR (debridement, antibiotics, and implant retention) surgical procedure after total knee arthroplasty. PLG0206 (47) has also successfully completed a phase-I trial with IV administration [263]. PLG0206 (47) has broad spectrum activity against G+ve and G-ve bacteria, including biofilms [261, 264, 265].
PL-18 (48) (HPRP-A1; topical) is a 15-mer α-helical cationic peptide derived from the N-terminus of the H. pylori ribosomal protein L1 (RpL1) that is being developed by Jiangsu ProteLight Pharmaceutical and Biotechnology (Jiangyin, People’s Republic of China). In August 2022, 48 started a phase-I trial (NCT05340790) in Australia for bacterial vaginosis using suppository administration. PL-18 (48) has activity against G-ve and G+ve bacteria [144, 145, 266, 267] and fungi [266], as well as induction of HeLa cell apoptosis [268] and hemolytic activity [266, 267]. These off-target activities suggest why topical administration is required for 48.
Murepavadin (49) (POL7080, RG7929; inhalation) is a synthetic 14-mer cyclic peptide derived from protegrin I being developed by Spexis (Basel, Switzerland), which was formed through a merger of EnBiotix and Polyphor in December 2021. Murepavadin (49) has potent and selective activity against P. aeruginosa via binding to the N-terminal of the β-barrel protein LptD (Imp/OstA), a novel MoA [269,270,271]. Murepavadin (49) is reported to be in a phase-I trial for cystic fibrosis using inhaled administration [272], and was previously investigated in two phase-III trials for the treatment of Pseudomonas nosocomial pneumonia (NCT03582007) and VAP infections (NCT03409679). However, these trials were halted due to adverse events — an increase in serum creatinine and acute kidney injury in the nosocomial pneumonia trial in 2019 [273].
TXA709 (50) (po) is an anti-MRSA prodrug of TXA707 (51) that has been evaluated in a phase-I trial conducted by TAXIS Pharmaceuticals (Monmouth Junction, NJ, USA) [274]. TXA707 (50) is an inhibitor of the new antibacterial target FtsZ, which is the bacterial homolog of tubulin that plays a critical role in bacterial cell wall division in both G+ve and G-ve bacteria [275, 276]. Prolysis Ltd (Oxford, UK) originally identified PC190723 [277,278,279] and replacement of its Cl substituent with a CF3 group in TXA707 (51) enhanced metabolic stability, PK properties and in vivo efficacy against S. aureus [280, 281].
RG6006 (RO7223280, Abx MCP; IV) is being developed by Roche (Basel, Switzerland) and a phase-I trial (NCT05614895) was started in December 2022 in critically ill participants with bacterial infections using IV administration. RG6006 will be developed as a treatment for A. baumannii infections [247] and is a tethered macrocyclic peptide [282, 283]; however, the structure and MoA have not been publicly disclosed.
BWC0977 (52) (IV/po) is an oxazolidinone containing ‘novel bacterial topoisomerase inhibitor’ (NBTI) [284] with similar activity against DNA gyrase GyrA and topoisomerase IV [284,285,286] being developed by Bugworks Research Inc (Bangalore, Republic of India). BWC 0977 (52) is being evaluated in a phase-1 trial (NCT05088421) using IV administration for treating critical care G-ve infections [287, 288] with later oral step-down administration.
Anti-mycobacterial compounds in phase-I trials (Table 5, Fig. 9)
Macozinone (53) (PBTZ169; po) is a benzothiazinone (BTZ) derivative [289] that was evaluated in a phase-II EBA TB trial (NCT03334734) by Nearmedic Plus LLC (Moscow, Russia), but the trial was discontinued in February 2018 due to slow enrollment. The Innovative Medicines for Tuberculosis (iM4TB) Foundation (Lausanne, Switzerland) is leading the development of 53 in the rest of the world and completed a Phase-I trial (NCT03776500) in March 2020. Macozinone (53) is a second generation analog of BTZ043 (34, Fig. 6) with the same MoA (inhibition of the mycobacterial cell wall biosynthesis enzyme DprE1) with superior physicochemical properties [289]; however, efforts have been undertaken to improve its PK and PD properties [290].
TBI-223 (54) (po) is an oxazolidinone [291] being developed by the TB Alliance (New York, NY, USA) and the Institute of Materia Medica (Shanghai, People’s Republic of China) that has completed two phase-I trials (NCT03758612 and NCT04865536). TBI-223 (54) was recently found to be active against S. aureus in MRSA mouse models [292].
TBAJ-876 (55) (po) is a bedaquiline analog (diarylquinolines class) with activity against Mtb [293] and M. abscessus [294], and minimal hERG channel inhibition [295, 296] that was discovered at the University of Auckland (Auckland, New Zealand). TBAJ-876 (55) is now being developed by the TB Alliance (New York, NY, USA) and completed a phase-I trial (NCT04493671) in November 2022, which focused on safety, tolerability, and PK. In September 2022, another phase-I trial (NCT05526911) was initiated that also evaluates its effects on CYP3A4 and P-glycoprotein. Like bedaquiline, 55 is an inhibitor of mycobacterial F-ATP synthase [297] but does not retain bedaquiline’s protonophore activity [298]. Cryogenic electron microscopy (cryo-EM) was recently used to show the binding of 55 to the Fo domain in M. smegmatis F1Fo-ATP synthase [299].
TBAJ-587 (56) (po) is another bedaquiline analog [295] with variations in the substituents on one pyridyl ring that lead to more potent in vitro and in vivo activity against Mtb [300]. TBAJ-587 (56) is currently in a phase-1 trial (NCT04890535) to evaluate its safety, tolerability, and PK.
GSK2556286 (57) (GSK-286; po) is a substituted uracil derivative being evaluated by GSK (London, UK) in a phase-I trial (NCT04472897) as a potential TB treatment [301]. GSK2556286 (57) was discovered by screening against Mtb that resides within human (THP-1) macrophage-like differentiated monocytes and had an IC50 of 0.07 µM [302]. In addition, 57 required cholesterol to show activity in an axenic culture and resistance mutations were mapped to Mtb adenylyl cyclase (cya) Rv1625c [302,303,304], which has been implicated in cholesterol utilization [305]. This is a new MoA.
Non-traditional antibacterial compounds in phase-I trials (Table 5, Fig. 10)
BVL-GSK098 (58) [306] (po) is the first member of a new non-traditional, anti-TB antibacterial class (spiroisoxazoline) being developed by BioVersys (Basel, Switzerland) and GSK (London, UK). BVL-GSK098 (58) completed a phase-I trial (NCT04654143) in May 2022. BVL-GSK098 works through inactivation of a Mtb TetR-like repressor, EthR2, which reverses ethionamide (59)-acquired resistance and increased basal sensitivity to 59 [307, 308]. A phase-II EBA trial (NCT05473195) is scheduled to evaluate ethionamide (59) with or without BVL-GSK098 (58) in participants with rifampicin- and isoniazid-susceptible pulmonary TB.
GSK3882347 (po) is an E coli Type 1 fimbrin D-mannose specific adhesin (FimH) inhibitor being evaluated by GSK (London, UK) and Fimbrion Therapeutics (St. Louis, MO, USA) with support from CARB-X (Boston, MA, USA) [309]. GSK3882347 completed a phase-I trial (NCT04488770) in May 2021 and is currently being evaluated in a Phase-Ib trial (NCT05138822) in participants with acute uUTI. A majority of UTIs are caused by uropathogenic E. coli (UPEC) [310], which use their type 1 pili to adhere to the cell wall via FimH adhesin [311]. Targeting the mannose-binding lectin domain of FimH prevents UPEC from binding to the bladder wall and is a promising antivirulence approach for UTI and Crohn’s Disease [312,313,314]. Although the structure of GSK3882347 has not been publicly disclosed, it is likely to be a mannose-derived biphenyl derivative [315].
ALS4 (po) is an S. aureus anti-virulence antibacterial being developed by Aptorum Therapeutics Limited (Hong Kong, People’s Republic of China) that has completed one phase-II trial (NCT05274802). Staphyloxanthin is a golden colored carotenoid with antioxidant activity that helps to neutralize reactive oxygen species (ROS) secreted by neutrophils, which protects bacteria [316, 317]. ALS4 is an inhibitor of 4,4ʹ-diapophytoene desaturase (CrtN), which is an enzyme involved in the biosynthesis of staphyloxanthin; however, although the structure of ALS4 has not been publicly disclosed, it is likely to be related to NP16 [318, 319].
β-Lactam/β-lactamase Inhibitor (BL/BLI) Combinations Undergoing Clinical Evaluation
The discovery of the Streptomyces-derived BLI clavulanic acid was a significant breakthrough that rescued the use of many BL antibiotics by inactivating enzymes responsible for their destruction. There have been four new BL/BLI combinations approved since 2014 (Table 1): Zerbaxa in 2014 (contains a new cephalosporin, ceftolozane), Avycaz in 2015 (contains a new DBO-type BLI, avibactam), Vabomere in 2017 (contains a new boronate-type BLI, vaborbactam), and Recarbrio in 2019 (contains a new DBO-type BLI, relebactam), but no new combinations were approved from 2019–2022. In this section, ten new BL/BLI combinations are currently being evaluated in clinical trials or under an NDA/MAA filing are discussed (Table 6, Figs. 11–13). It should be noted that BL/BLI combinations usually move straight from phase-I into phase-III trials.
BL/BLI combinations in NDA/MAA filing (Table 6, Fig. 11)
Durlobactam (60) (ETX2514) + sulbactam (61) (combination: SUL-DUR, ETX2514SUL; IV) is being developed by Entasis Therapeutics (a subsidiary of Innoviva, Burlingame, CA, USA) and completed a phase-III trial (NCT03894046) for treatment of infections caused by A. baumannii-calcoaceticus (ABC) complex [320,321,322] in June 2021. In this trial, SUL-DUR demonstrated statistical non-inferiority versus colistin for the primary end point of 28-day all-cause mortality in patients with carbapenem-resistant ABC infections and a significant difference in clinical cure rates, as well as a statistically significant reduction in nephrotoxicity [323]. On 17 April 2023, the US FDA Antimicrobial Drugs Advisory Committee voted 12-0 in favor of SUL-DUR for the treatment of adults with HABP/VABP caused by susceptible ABC strains.Footnote 2 Durlobactam (60) is a DBO-type BLI [324,325,326], while sulbactam (61) is a clavulanic acid-type BLI first launched in 1986 that also has direct-acting antibacterial activity against Acinetobacter spp., but requires co-administration of another BLI to restore its activity against MDR strains.
BL/BLI combinations in phase-III trials (Table 6, Fig. 12)
Taniborbactam (62) (VNRX-5133; IV) [327] + cefepime (41) is being developed by VenatoRx Pharmaceuticals (Malvern, PA, USA) and completed a phase-III trial (NCT03840148) in December 2021 for cUTI, including acute pyelonephritis. VenatoRx have revealed that cefepime-taniborbactam had a superior primary efficacy endpoint to the carbapenem meropenem (66) in this trial with a similar safety profile [328], and plan to submit an NDA to the US FDA in 2023 [329]. The taniborbactam (62) + cefepime (41) combination has activity against E. coli, K pneumoniae, carbapenemase-producing Enterobacterales and P. aeruginosa [330,331,332]. Taniborbactam (62) is a bicyclic boronate BLI [333] (new class) that is effective against both serine- and metallo-β-lactamases, including extended-spectrum β-lactamase (ESBL), OXA, KPC, NDM and VIM enzymes, but not IMP [327, 334], while cefepime (41) is a fourth-generation cephalosporin first approved in 1994.
Enmetazobactam (63) (AAI 101; IV) is a clavulanic acid-type BLI with a structure closely related to tazobactam with a methyl substituent on the tazobactam triazole ring. It has activity against ESBLs and some class A and D carbapenemases [335,336,337], and is being developed by Allecra Therapeutics (Weil am Rhein, Germany and Saint Louis, France). A combination of 63 and the cephalosporin cefepime (41) completed a phase-III trial (NCT03687255) in February 2020 for cUTI using IV administration, and successfully met criteria for non-inferiority, as well as superiority to piperacillin-tazobactam with respect to the primary efficacy outcome of clinical cure and microbiological eradication [338]. Allecra Therapeutics is planning to submit an MAA in Europe, followed by an NDA in the USA.
Zidebactam (64) (WCK 5107; IV) is a DBO-type BLI being developed by Wockhardt Limited (Mumbai, Republic of India) that inhibits PBPs and several β-lactamases, while enhancing BL activity [339] against A. baumannii, P. aeruginosa and CRE [340,341,342]. A combination of 63 and cefepime (41) (combination WCK 5222, FEP-ZID) started a phase-III trial (NCT04979806) in August 2022 as an IV administered treatment for cUTI and acute pyelonephritis. A phase-I trial (NCT05645757) of 63 in combination with the carbapenem ertapenem (combination WCK 6777) should commence soon, with this combination showing potent in vitro activity against many carbapenemases and β-lactamases [343].
BL/BLI combinations in phase-I trials (Table 6, Fig. 13)
Nacubactam (65) (OP0595, FPI-1459, RG6080, RO7079901; IV) is a DBO-type BLI [344,345,346], which was developed by Meiji Seika Pharma (Tokyo, Japan). Meiji Seika and Fedora Pharmaceuticals (Edmonton, AB, Canada) had previously partnered with Roche (Basel, Switzerland) [347, 348] and several phase-I trials have been completed (Meiji Seika: NCT02134834; Roche: NCT02975388, NCT03182504), as well as two phase-I trials in combination with meropenem (66) (Roche: NCT02972255, NCT03174795). Nacubactam (65) is still listed as OP0595 on Meiji Seika’s latest pipeline [349], while Fedora’s website indicates that the combination is available for licensing [350].
Xeruborbactam (67) (QPX7728; IV) is a bicyclic boronate BLI [333] (new class) being developed by Qpex Biopharma (San Diego, CA, USA) that displays broad spectrum β-lactamase inhibition, including against class B and class D enzymes [351,352,353], as well as some intrinsic G-ve antibacterial activity [354]. An IV administered combination of 67 and an undisclosed BL (QPX2014) has completed two phase-I trials (NCT04380207 and NCT05072444) with an aim to treat serious drug resistant Acinetobacter, Pseudomonas and Enterobacterales infections. An orally administered xeruborbactam prodrug, QPX7831 (68) [355] (po), completed a phase-I trial (NCT04578873) in August 2022 and there are plans to use 68 in combination with an undisclosed oral BL (QPX2015) to treat ESBLs and carbapenem-resistant Enterobacterales (CRE) infections.
A combination of the DBO-type BLI ETX0282 (69) (po) and the cephalosporin cefpodoxime proxetil (70), collectively called ETX0282CPDP, was evaluated in a phase-I trial that finished in September 2019 (NCT03491748) by Entasis Therapeutics (Waltham, MA, USA), who are now a wholly owned subsidiary of Innoviva (Burlingame, CA, USA). Both ETX0282 (69) and cefpodoxime proxetil (70) are esterase-cleavable prodrugs, of ETX1317 (71) and cefpodoxime (72) respectively, and the combination is being developed to treat multidrug resistant and CRE infections [356, 357]. ETX1317 (71) has an (R)-2-(N-oxy)-2-fluoroacetic acid unit in place of the N-oxy-sulfonic acid group present in other DBOs and displays some innate G-ve activity, in addition to BLI activity [356, 357].
A ledaborbactam etzadroxil (73) (VNRX-7145) + ceftibuten (74) combination (po) [358] is being developed by VenatoRx Pharmaceuticals (Malvern, PA, USA). This combination is currently being evaluated in two phase-I trials (NCT05527834 and NCT05488678) and has previously completed two other phase-I trials (NCT04243863 and NCT04877379). Ledaborbactam etzadroxil (73) is an esterase-cleavable prodrug of the bicyclic boronate-type BLI (new class [333]) of ledaborbactam (75) (VNRX-5236) [358], while 74 is a third-generation cephalosporin first approved in 1995. The ledaborbactam etzadroxil (73) + ceftibuten (74) combination is active against clinically-derived Enterobacterales that express ESBLs and serine carbapenemases [359,360,361].
A ternary combination therapy combining funobactam (76) (XNW-4107) + imipenem (77) + cilastatin (78) (IV) is being developed by Suzhou Sinovent Pharmaceuticals (Sinovent) (Suzhou, People’s Republic of China). Funobactam (76) is a DBO-type BLI [362], while imipenem (77) is a carbapenem-type BL that was approved in combination with cilastatin (78) in 1985, as well as in combination with the DBO relebactam and 78 in 2019 [363]. Cilastin (78) is a renal dehydropeptidase inhibitor that reduces the rate of 77 metabolism. Funobactam (76) has completed two phase-I trials (NCT04482569, NCT04802863) and two phase-I trials are ongoing (NCT04801043, NCT04787562). Two phase-III trials have been announced that will evaluate the funobactam (76) + imipenem (77) + cilastatin (78) combination against cUTI (NCT05204368) and HABP/VABP (NCT05204563).
CTB + AVP (PF-07612577; po) is a combination of the cephalosporin ceftibuten (74) (PF-06264006) and the DBO-type BLI avibactam (80) prodrug, AVP (79) (PF-07338233, ARX-006, ARX-1796), under development by Pfizer (New York, NY, USA). CTB + AVP is being evaluated in a phase-I trial (NCT05554237), which started in October 2022. Avibactam (80) in combination with ceftazidime (Avycaz) was first approved in 2015 by the US FDA and is used to treat cIAI and cUTI [364]. AVP (79) was first developed by Arixa Pharmaceuticals (Palo Alto, CA, USA) [365], who were acquired by Pfizer in October 2020 [366], and a prior phase-I trial (NCT03931876) had already been completed.
Compounds discontinued from clinical development
Analysis of compounds undergoing clinical trials
Numbers of compounds undergoing clinical evaluation and their source derivation
There were 62 antibacterial clinical candidates under clinical investigation (Figs. 14 and 15) on 31 December 2022 — ten BL/BLI inhibitor combinations and 52 small molecules, mammalian-derived peptides, and a direct acting polymer. Five of the 62 are non-traditional antibacterials that target virulence (fluorothyazinone (40), GSK3882347 and ALS4), resistance (BVL-GSK098 (58)) and host inflammation (dovramilast (42)) (Tables 4 and 5, Figs. 7 and 10). Of the ten BL/BLI combinations, one is in NDA/MAA (Table 6, Fig. 11), three are in phase-III (Table 6, Fig. 12) and six are in phase-I (Table 6, Fig. 13). Of the remaining 52 compounds, one is in NDA/MAA (Table 3, Fig. 3), six are in phase-III (Table 3, Fig. 4), 25 are in phase-II (Table 4, Figs. 5–7) and 20 are in phase-I (Table 5, Figs. 8–10). The source derivation of the 62 compounds was divided into 41 that were synthetically derived (S), 17 that were NP derived (NP), and four that were protein/mammalian peptide derived (P) (Fig. 14).
While there was a similar number of compounds in the different development phases in 2011, 2013 and 2015 analyses (except for a reduced number in phase-III trials in 2011 (6) compared to 2013 (16) and 2015 (15)), the number in phase-I trials increased to 22 in 2019 [23] from an average of 12 compounds in 2011-2015 [24,25,26], and this was even higher at 26 in 2022 (Fig. 15). The number of compounds in phase-II also increased (from 18 in 2019 to 25 in 2022), reflecting the successful progression of several of the 2019 phase-I candidates and the entry of new antibacterials. At least 18 of the 26 phase-I compounds target G-ve bacteria (11 traditional compounds, one anti-virulence and six BLI combinations), with four of these also possessing G+ve activity, while there are an additional eight with G+ve only activity (six against TB and two against MRSA). While the overall numbers are still low compared to other therapeutic disease indications, the clinical pipeline is now starting to resemble the more traditional progression of attrition, rather than the flat or inverse progressions seen in 2011 [26], 2013 [25] and 2015 [24], and this likely reflects the success of push incentives driving innovative antibiotic discovery [6,7,8,9,10].
New antibacterial pharmacophore analysis
A pharmacophore is the common subunit of active molecules that interact with biological targets. It is crucial to develop new antibacterial drugs with new MoA and/or pharmacophores to slow down drug resistance and to potentially allow the identification of new combination therapies. This is also why there is considerable excitement around the potential of non-traditional antibacterials, along with the yet-to-be-proven hypothesis that some modalities, such as antivirulence strategies, will not lead to resistance since bacterial survival is not directly targeted [17, 18, 20, 21].
In this review, new pharmacophores not previously found in human antibacterial drugs have been analyzed as a measure of antibacterial structure innovation (Table 8). In Table 8, compounds with new MoA not previously found in previously approved antibacterial drugs are underlined. The MoA of most traditional small molecule antibacterial drugs can be categorized into four major ‘macro’ level classes: cell wall, protein synthesis, DNA synthesis, and RNA synthesis inhibitors [367]. There are 34 different compounds — 15 in phase-I, 15 in phase-II and 4 in phase-III/NDA (Fig. 16) — and this total is significantly higher than identified in previous reviews: 11 in 2011 [26], 17 in 2013 [25], 15 in 2015 [24] and 19 in 2019 [23] (Fig. 17). Twenty-six of these compounds target the well-established ‘macro’ targets: cell wall (17), DNA (6) and protein synthesis inhibition (3). There are no novel RNA synthesis inhibitors in clinical development. Since the 2019 review [23], the boronate BLI class has expanded with the bicyclic boronates class, which includes taniborbactam (60), xeruborbactam (65) and ledaborbactam etzadroxil (71), now considered to be a new class [21, 22].
Existing antibacterial classes that inhibit the bacterial cell envelope include the BL, glycopeptide, polymyxin, daptomycin (lipopeptide), fosfomycin, and cycloserine classes. The new cell envelope acting antibacterials inhibit several different targets (LptD: murepavadin (49), FabI: afabicin (21), 3 × DprE: BTZ-043 (34) and macozinone (53), quabodepistat (35) and TBA-7371 (37), and FtsZ: TXA709 (62)) and six perturb bacterial membranes through less defined mechanisms (exeporfinium (17), cannabidiol (18), Recce-347, and the three cationic peptides, peceleganan (23), PLG0206 (47) and PL-18 (48)) (Table 8). Although the structure of RG6319 has not been disclosed, it is likely to be an arylomycin derivative that inhibits E. coli Type 1 signal peptidase, which is a key enzyme in transporting enzymes across the cytoplasmic membrane to the outer cell wall [248].
The (fluoro)quinolone class are DNA synthesis inhibitors (DNA gyrase GyrA and topoisomerase IV parC [201]) that are routinely used in clinical practice, while novobiocin, which is a DNA gyrase GyrB and topoisomerase IV ParE inhibitor, was briefly used as an antibacterial over 50 years ago [201, 368]. BWC0977 (52) belongs to a new antibacterial class and equally inhibits both DNA gyrase GyrA and topoisomerase IV. Fobrepodacin (32) is an ‘ethyl urea benzimidazole’ that also binds to both GyrB and ParE, gepotidacin (10) inhibits GyrA at a different binding site to the quinolones, and zoliflodacin (11) inhibits GyrB. Ibezapolstat (27) is the first member of the dichlorobenzyl guanine class that inhibits DNA polymerase IIIC, while MGB-BP-3 (16) is a DNA minor groove binder.
Bacterial protein synthesis inhibition can be caused by several compound classes including macrolides, aminoglycosides, tetracyclines, lincosamides, chloramphenicol, oxazolidinones, pleuromutilins and fusidic acid. There are two oxaborole-type leucine tRNA synthetase (LeuRS) inhibitors, epetraborole (13) and GSK3036656 (52), and one methionyl-tRNA synthetase inhibitor, CRS3123 (28), in clinical trials. The only marketed inhibitor of a tRNA synthetase is mupirocin, which targets isoleucyl-tRNA synthetase.
There are two direct-acting traditional and five non-traditional antibacterial compounds with new mechanisms. Telacebec (31) is an inhibitor of the mycobacterial respiratory cytochrome bc1 complex [185, 186, 369]. Inhibition of bacterial respiratory systems is an emerging MoA [369, 370] with three bedaquiline analogs, sudapyridine (38), TBAJ-876 (55) and TBAJ-587 (56) that are also in clinical development. GSK2556286 (57) was recently disclosed to be an adenylyl cyclase Rv1625c agonist, which interferes with cholesterol catabolism and reduces the levels of this critical carbon source [304]. RG6006 is a new antibacterial class but there is only limited public information available about the structure and MoA. There are three antivirulence compounds that employ totally different anti-virulence mechanisms: fluorothyazinone (40) inhibits the G-ve type III secretion system, GSK3882347 inhibits the binding of E. coli to host cell walls via FimH and ALS4 inhibits 4,4ʹ-diapophytoene desaturase (CrtN), which is an enzyme involved in the biosynthesis of staphyloxanthin. Finally, BVL-GSK098 (58) inactivates the TetR-like repressor, which reduces resistance to the TB drug ethionamide (59) and rescues its activity, which is conceptually similar to how BLIs restore the activity of BL antibiotics.
Administration analysis
The administration routes (po, oral; IV/po, intravenous oral switch; IV, intravenous; IV/topical, IV and topical; po topical, CDI oral; topical; inhalation; n/d, not disclosed) of the small molecule antibacterial compounds under clinical development were analyzed by development phase (Fig. S1) and lead source (Fig. S1). Oral administration predominates and 19 of the 30 (~63%) are being developed against mycobacteria, which is pivotal as anti-TB drug combinations are taken for multiple months and are often administered in countries with limited capacity to deliver IV treatments. The second highest category is IV administration with 15, while there are four candidates that can be used both IV and po and two for both IV and topical. This IV/oral switch strategy is a competitive advantage as it can be implemented when patients move from hospital-based IV administration to oral administration in wards or at home. Four candidates are being trialed using the po topical administration route, which is used to treat gastrointestinal infections, such as C. difficile and H. pylori. For these infections, drugs are usually orally administered, but most are not significantly systemically absorbed, which reduces the potential for toxicity; however, one of the CDI clinical candidates, DNV-3837 (25) is being investigated using IV administration. There are four topically-only administered candidates, while murepavadin (49) is being trialed with inhaled administration to treat P. aeruginosa infections in the lungs of cystic fibrosis patients. This is being undertaken to more efficiently deliver 49 into the lungs, but it may also ameliorate kidney toxicity that was observed in a prior nosocomial pneumonia trial [273].
Conclusion and outlook
The shape of the antibacterial pipeline has changed since our first analysis in 2011 [26]. At the front-end of the pipeline, there are now more than double the number of phase-I candidates (26) compared to 11 in 2015 [24] (Fig. 15). Funding initiatives have also helped to boost the number of phase-II (25) compounds since 2019 (18) (Fig. 15). Encouragingly, 16/26 (62%) of the compounds in phase-I and 14/25 (56%) in phase-II contain new pharmacophores (Figs. 16 and 17), with some also having new MoA (Table 8). Small molecule non-traditional antibacterial candidates are also starting to move through the pipeline with five in active development: fluorothyazinone (40), dovramilast (42), BVL-GSK098 (58), GSK3882347 and ALS4. Due to the increasing number of compounds with novel pharmacophores and targets in the pipeline (Table 8, Figs. 16 and 17), it is more likely that novel antibacterial drug classes will enter the clinic in the next few years, which is preferable to just expanding the pool of ‘me-too’ antibiotics. However, despite these early stage improvements, it is sobering to note that the overall antibacterial pipeline is still sparse compared to other therapeutic indications such as oncology (2,335 clinical trials in 2021 [371]) and even COVID-19 vaccines (180 in the pipeline in February 2023 [372]).
In contrast to the early-stage pipeline, the late-stage pipeline is still experiencing issues. There were only two new small molecule antibacterial drugs first approved between 2020 and 2022 (Table 1, Fig. 2): the fluoroquinolone levonadifloxacin (1) and its prodrug 2 in India in 2020 and the oxazolidinone contezolid (4) in China in 2021. There was also one ‘non-traditional’ live biotherapeutic product, Rebyota, approved in the USA in 2022 (Table 2). The last first-in-class small molecule approval was the anti-TB diarylquinoline bedaquiline in 2012. However, this could change, if the current phase-III candidates, gepotidacin (10), zoliflodacin (11) and epetraborole (13) (Table 3, Fig. 4), all of which have new pharmacophores, were granted approval to treat gonorrhea and G-ve bacteria, gonorrhea, and M. avium complex (MAC) infections respectively. A future approval of the durlobactam (60) + sulbactam (61) combination for the treatment of A. baumannii-calcoaceticus (ABC) complex infections would also be a welcome addition to the antibacterial armamentarium. There has also been a steady but small decline in the number of phase-III candidates from 2013 to 2022 (Fig. 15). It will be critical to monitor how many of the phase-I and -II candidates, especially the compounds with new pharmacophores, move into Phase-III and beyond in the next few years.
In addition to the difficulty in identifying novel lead compounds suitable for antibacterial drug development, the ability to secure funding for phase-III trials and NDA/MAAs, as well as the capacity to generate adequate revenue to get positive net returns on investment for marketed antibacterial drugs [2,3,4,5], have been major obstacles to antibacterial drug development. Hopefully funding from organizations such as the AMR Action Fund [9, 10] will help to ameliorate some of these funding issues, while the successful implementation of pull initiatives should help to improve financial returns [11,12,13,14,15,16]. Another welcome addition has been the US FDA’s Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) pathway that provides the potential for smaller, shorter, or fewer clinical trials (at least two phase-III trials are usually required) if the antibacterial drug candidate is “intended to treat a serious or life-threatening infection in a limited population of patients with unmet needs” [373]. However, the approved drug then carries a label restricting its use, which could limit future sales.
At least 19/26 (73%) phase-I compounds target G-ve bacteria (12 traditional compounds, one anti-virulence and six BL/BLI combinations), with four of these also possessing anti-G+ve activity. The high percentage of G-ve candidates being developed mirrors the clinical need and the recent focus of most funding schemes; however, the addition of G+ve activity to the 2022/23 CARB-X funding calls reflects the high mortality observed for global G+ve resistant infections in 2019 [1]. Only six of these 17 traditional G-ve antibacterial candidates are administered orally, with four of these being BL/BLI prodrugs. Although NPs have traditionally been the main source of novel antibacterials, 23 of the 34 (68%) of the compounds with new antibacterial pharmacophores were synthetically derived. There are also a substantial number of antitubercular drugs (TB and NTM) in the pipeline (19/62 (31%); one in phase-III, 12 in phase-II, and six in phase-I), showing the success of targeted funding for neglected diseases through organizations such as the TB Alliance (New York, NY, USA) and the Bill & Melinda Gates Foundation (Seattle, WA, USA).
In conclusion, despite the encouraging trends in phase-I and -II antibacterial drug candidates and plans to address issues with the late-stage pipeline, it is not the time to relax the urgency to continue to stimulate further antibacterial drug discovery and development.
Change history
07 November 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41429-023-00671-6
Notes
References
Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399:629–55. https://doi.org/10.1016/S0140-6736(21)02724-0.
Shlaes DM. The economic conundrum for antibacterial drugs. Antimicrob Agents Chemother. 2019;64:e02057–19. https://doi.org/10.1128/AAC.02057-19.
McKenna M. The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature. 2020;584:338–41. https://doi.org/10.1038/d41586-020-02418-x.
Outterson K. Estimating the appropriate size of global pull incentives for antibacterial medicines. Health Aff. 2021;40:1758–65. https://doi.org/10.1377/hlthaff.2021.00688.
Madden J, Outterson K. Trends in the global antibiotics market. Nat Rev Drug Disco. 2023;22:174. https://doi.org/10.1038/d41573-023-00029-5.
Alm RA, Gallant K. Innovation in antimicrobial resistance: the CARB-X perspective. ACS Infect Dis. 2020;6:1317–22. https://doi.org/10.1021/acsinfecdis.0c00026.
Alt S, Haggstrom D, Kessmann H, Kloss F, Schneider CE, Jäger T, et al. INCATE: a partnership to boost the antibiotic pipeline. Nat Rev Drug Disco. 2022;21:621–2. https://doi.org/10.1038/d41573-022-00138-7.
Engel A. Fostering antibiotic development through impact funding. ACS Infect Dis. 2020;6:1311–2. https://doi.org/10.1021/acsinfecdis.0c00069.
McCall B. New fund stimulates the ailing antibiotic pipeline. Lancet Infect Dis. 2020;20:1017. https://doi.org/10.1016/S1473-3099(20)30629-0.
Mullard A. Pharmaceutical firms commit US$1 billion to antibiotic development. Nat Rev Drug Disco. 2020;19:575–6. https://doi.org/10.1038/d41573-020-00143-8.
Mullard A. Pull incentives for antibiotics get push from the UK. Nat Rev Drug Disco. 2022;21:406. https://doi.org/10.1038/d41573-022-00088-0.
Glover RE, Singer AC, Roberts AP, Kirchhelle C. The antibiotic subscription model: fostering innovation or repackaging old drugs. Lancet Microbe. 2023;4:e2–e3. https://doi.org/10.1016/S2666-5247(22)00235-X.
Leonard C, Crabb N, Glover D, Cooper S, Bouvy J, Wobbe M et al. Can the UK ‘Netflix’ Payment Model Boost The Antibacterial Pipeline? Appl Health Econ Health Policy. 2023;21. https://doi.org/10.1007/s40258-022-00786-1.
Silverman R, Towse A. An ambitious USG advanced commitment for subscription-based purchasing of novel antimicrobials and its expected return on investment Washington DC, USA: Center for Global Development; 2021 [Accessed 24 January 2023]. Available from: https://www.cgdev.org/publication/ambitious-usg-advanced-commitment-subscription-based-purchasing-novel-antimicrobials.
The Pioneering Antimicrobial Subscriptions To End Up surging Resistance Act of 2021 or the PASTEUR Act of 2021; 2021 [Accessed 24 January 2023]. Available from: https://www.congress.gov/bill/117th-congress/house-bill/3932.
Dall C. For PASTEUR Act advocates, the finish line is in sight for antibiotic development aid 2022 [Accessed 24 January 2023]. Available from: https://www.cidrap.umn.edu/antimicrobial-stewardship/pasteur-act-advocates-finish-line-sight-antibiotic-development-aid.
Theuretzbacher U, Piddock LJV. Non-traditional antibacterial therapeutic options and challenges. Cell Host Microbe. 2019;26:61–72. https://doi.org/10.1016/j.chom.2019.06.004.
Shlaes DM. Innovation, nontraditional antibacterial drugs, and clinical utility. ACS Infect Dis. 2021;7:2027–8. https://doi.org/10.1021/acsinfecdis.1c00227.
Rex JH, Fernandez Lynch H, Cohen IG, Darrow JJ, Outterson K. Designing development programs for non-traditional antibacterial agents. Nat Commun. 2019;10:3416. https://doi.org/10.1038/s41467-019-11303-9.
Duffy EM, Buurman ET, Chiang SL, Cohen NR, Uria-Nickelsen M, Alm RA. The CARB-X portfolio of nontraditional antibacterial products. ACS Infect Dis. 2021;7:2043–9. https://doi.org/10.1021/acsinfecdis.1c00331.
Butler MS, Gigante V, Sati H, Paulin S, Al-Sulaiman L, Rex JH, et al. Analysis of the clinical pipeline of treatments for drug-resistant bacterial infections: despite progress, more action is needed. Antimicrob Agents Chemother. 2022;66:e01991–21. https://doi.org/10.1128/aac.01991-21.
World Health Organization. 2021 Antibacterial agents in clinical and preclinical development: an overview and analysis. World Health Organization: Geneva, Switzerland, 2022. Available from: https://www.who.int/publications/i/item/9789240047655.
Butler MS, Paterson DL. Antibiotics in the clinical pipeline in October 2019. J Antibiot. 2020;73:329–64. https://doi.org/10.1038/s41429-020-0291-8.
Butler MS, Blaskovich MAT, Cooper MA. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot. 2017;70:3–24. https://doi.org/10.1038/ja.2016.72.
Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J Antibiot. 2013;66:571–91. https://doi.org/10.1038/ja.2013.86.
Butler MS, Cooper MA. Antibiotics in the clinical pipeline in 2011. J Antibiot. 2011;64:413–25. https://doi.org/10.1038/ja.2011.44.
Pew Trusts. Analysis shows continued deficiencies in antibiotic development since 2014. Visualization tool lets users track changes in the pipeline from 2014 [Accessed 24 January 2023]. Available from: https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2019/five-year-analysis-shows-continued-deficiencies-in-antibiotic-development.
Pew Trusts. Tracking the global pipeline of antibiotics in development, March 2021 [Accessed 24 January 2023]. Available from: https://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2021/03/tracking-the-global-pipeline-of-antibiotics-in-development.
Frost I, Sati H, Garcia-Vello P, Hasso-Agopsowicz M, Lienhardt C, Gigante V, et al. The role of bacterial vaccines in the fight against antimicrobial resistance: an analysis of the preclinical and clinical development pipeline. Lancet Microbe. 2023;4:e113–e25. https://doi.org/10.1016/S2666-5247(22)00303-2.
Prasad Neha K, Seiple Ian B, Cirz Ryan T, Rosenberg Oren S. Leaks in the pipeline: a failure analysis of Gram-negative antibiotic development from 2010 to 2020. Antimicrob Agents Chemother. 2022;66:e00054–22. https://doi.org/10.1128/aac.00054-22.
Theuretzbacher U, Bush K, Harbarth S, Paul M, Rex JH, Tacconelli E, et al. Critical analysis of antibacterial agents in clinical development. Nat Rev Microbiol. 2020;18:286–98. https://doi.org/10.1038/s41579-020-0340-0.
Theuretzbacher U, Outterson K, Engel A, Karlén A. The global preclinical antibacterial pipeline. Nat Rev Microbiol. 2020;18:275–85. https://doi.org/10.1038/s41579-019-0288-0.
Vila J, Moreno-Morales J, Ballesté-Delpierre C. Current landscape in the discovery of novel antibacterial agents. Clin Microbiol Infect. 2020;26:596–603. https://doi.org/10.1016/j.cmi.2019.09.015.
Jiménez MC, Kowalski L, Souto RB, Alves IA, Viana MD, Aragón DM. New drugs against multidrug-resistant Gram-negative bacteria: a systematic review of patents. Future Microbiol. 2022;17:1393–408. https://doi.org/10.2217/fmb-2022-0104.
Si Z, Pethe K, Chan-Park MB. Chemical basis of combination therapy to combat antibiotic resistance. JACS Au. 2023;3:276–92. https://doi.org/10.1021/jacsau.2c00532.
Walesch S, Birkelbach J, Jézéquel G, Haeckl FPJ, Hegemann JD, Hesterkamp T, et al. Fighting antibiotic resistance—strategies and (pre)clinical developments to find new antibacterials. EMBO Rep. 2023;24:e56033. https://doi.org/10.15252/embr.202256033.
Editorial. C difficile−a rose by any other name. Lancet Infect Dis. 2019;19:449. https://doi.org/10.1016/S1473-3099(19)30177-X.
Médecins Sans Frontières. Bedaquiline: First new tuberculosis drug in 50 years (Press release 31 December 2012). 2012 [Accessed 27 January 2023]. Available from: https://www.msf.org/bedaquiline-first-new-tuberculosis-drug-50-years.
de Souza NJ, Gupte SV, Deshpande PK, Desai VN, Bhawsar SB, Yeole RD, et al. A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus. J Med Chem. 2005;48:5232–42. https://doi.org/10.1021/jm050035f.
Veeraraghavan B, Bakthavatchalam YD, Manesh A, Lal B, Swaminathan S, Ansari A, et al. India-discovered levonadifloxacin & alalevonadifloxacin: A review on susceptibility testing methods, CLSI quality control and breakpoints along with a brief account of their emerging therapeutic profile as a novel standard-of-care. Indian J Med Microbiol. 2023;41:71–80. https://doi.org/10.1016/j.ijmmb.2022.11.005.
Bhagwat S, Nandanwar M, Kansagara A, Patel A, Takalkar S, Chavan R, et al. Levonadifloxacin, a novel broad-spectrum anti-MRSA benzoquinolizine quinolone agent: review of current evidence. Drug Des Devel Ther. 2019;13:4351–65. https://doi.org/10.2147/DDDT.S229882.
India’s First New Discovery Antibiotics from Wockhardt Granted Indian Regulatory Approval (Press release 16 January 2020) [Accessed 16 December 2022]. Available from: http://www.wockhardt.com/pdfs/Press-Release-16-01-2020.pdf.
Bhatia A, Mastim M, Shah M, Gutte R, Joshi P, Kumbhar D, et al. Efficacy and safety of a novel broad-spectrum anti-MRSA agent levonadifloxacin compared with linezolid for acute bacterial skin and skin structure infections: a phase 3, openlabel, randomized study. J Assoc Physicians India. 2020;68:30–6.
Mehta Y, Sutar AR, Zirpe K, Kothari JN, Alapati C, Pathak M, et al. Prescription-event monitoring study on safety and efficacy of levonadifloxacin (oral and I.V.) in management of bacterial infections: findings of real-world observational study. Int J Appl Basic Med Res. 2022;12:30–6. https://doi.org/10.4103/ijabmr.ijabmr_602_21.
Jacobs MR, Appelbaum PC. Nadifloxacin: a quinolone for topical treatment of skin infections and potential for systemic use of its active isomer, WCK 771. Expert Opin Pharmacother. 2006;7:1957–66. https://doi.org/10.1517/14656566.7.14.1957.
China NMPA approves MicuRx’s contezolid for treatment of drug-resistant bacterial infection (Press release 2 June 2021) [Accessed 16 December 2022]. Available from: https://www.micurx.com/703.html.
Hoy SM. Contezolid: first approval. Drugs. 2021;81:1587–91. https://doi.org/10.1007/s40265-021-01576-0.
Zhao X, Huang H, Yuan H, Yuan Z, Zhang Y. A Phase III multicentre, randomized, double-blind trial to evaluate the efficacy and safety of oral contezolid versus linezolid in adults with complicated skin and soft tissue infections. J Antimicrob Chemother. 2022;77:1762–9. https://doi.org/10.1093/jac/dkac073.
Gordeev MF, Yuan ZYY. New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. J Med Chem. 2014;57:4487–97. https://doi.org/10.1021/jm401931e.
Liu J, Wang W, Wang C, Zhang L, Zhang X, Liu S, et al. Discovery of antibacterial contezolid acefosamil: innovative O-acyl phosphoramidate prodrug for IV and oral therapies. ACS Med Chem Lett. 2022;13:1030–5. https://doi.org/10.1021/acsmedchemlett.2c00191.
Greig SL. Obiltoxaximab: first global approval. Drugs. 2016;76:823–30. https://doi.org/10.1007/s40265-016-0577-0.
Xu W, Ohanjandian L, Sun J, Cui X, Suffredini D, Li Y, et al. A systematic review and meta-analysis of preclinical trials testing anti-toxin therapies for B. anthracis infection: a need for more robust study designs and results. PLoS One. 2017;12:e0182879. https://doi.org/10.1371/journal.pone.0182879.
Tsai C-W, Morris S. Approval of raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule”. Front Microbiol. 2015;6:1320. https://doi.org/10.3389/fmicb.2015.01320.
Kufel WD, Devanathan AS, Marx AH, Weber DJ, Daniels LM. Bezlotoxumab: a novel agent for the prevention of recurrent Clostridium difficile infection. Pharmacotherapy. 2017;37:1298–308. https://doi.org/10.1002/phar.1990.
Granata G, Schiavone F, Pipitone G. Bezlotoxumab in patients with a primary Clostridioides difficile infection: a literature review. Antibiotics. 2022;11:1495. https://doi.org/10.3390/antibiotics11111495.
Kelly CP, Poxton IR, Shen J, Wilcox MH, Gerding DN, Zhao X, et al. Effect of endogenous Clostridioides difficile toxin antibodies on recurrence of C. difficile infection. Clin Infect Dis. 2020;71:81–6. https://doi.org/10.1093/cid/ciz809.
Johnson S, Citron DM, Gerding DN, Wilcox MH, Goldstein EJC, Sambol SP, et al. Efficacy of bezlotoxumab in trial participants infected with Clostridioides difficile strain BI associated with poor outcomes. Clin Infect Dis. 2021;73:e2616–e24. https://doi.org/10.1093/cid/ciaa1035.
Ferring receives U.S. FDA approval for REBYOTA™ (fecal microbiota, live-jslm) – a novel first-in-class microbiota-based live biotherapeutic (Press release 30 November 2022) [Accessed 16 December 2022]. Available from: https://ferringusa.com/?press=ferring-receives-u-s-fda-approval-for-rebyota-fecal-microbiota-live-jslm-a-novel-first-in-class-microbiota-based-live-biotherapeutic.
Khanna S, Assi M, Lee C, Yoho D, Louie T, Knapple W, et al. Efficacy and safety of RBX2660 in PUNCH CD3, a Phase III, randomized, double-blind, placebo-controlled trial with a bayesian primary analysis for the prevention of recurrent Clostridioides difficile infection. Drugs. 2022;82:1527–38. https://doi.org/10.1007/s40265-022-01797-x.
Ray A, Jones C. Does the donor matter? Donor vs patient effects in the outcome of a next-generation microbiota-based drug trial for recurrent Clostridium difficile infection. Future Microbiol. 2016;11:611–6. https://doi.org/10.2217/fmb.16.10.
Orenstein R, Dubberke E, Hardi R, Ray A, Mullane K, Pardi DS, et al. Safety and durability of RBX2660 (microbiota suspension) for recurrent Clostridium difficile infection: Results of the PUNCH CD study. Clin Infect Dis. 2015;62:596–602. https://doi.org/10.1093/cid/civ938.
Donald B, Surani S, Deol H, Mbadugha U, Udeani G. Spotlight on solithromycin in the treatment of community-acquired bacterial pneumonia: design, development, and potential place in therapy. Drug Des Devel Ther. 2017;11:3559–66. https://doi.org/10.2147/DDDT.S119545.
FUJIFILM Toyama Chemical Co., Ltd. Pipeline (as of Nov. 2022) [Accessed 16 December 2022]. Available from: https://www.fujifilm.com/fftc/en/about/rd/pipeline.
Fierce Biotech. Cempra pulls EU antibiotic filing following EMA questioning (28 March 2017) [Accessed 16 December 2022]. Available from: https://www.fiercebiotech.com/biotech/cempra-pulls-eu-antibiotic-filing-following-ema-questioning.
Minamimura M, Taniyama Y, Inoue E, Mitsuhashi S. In vitro antibacterial activity and β-lactamase stability of CP-70,429 a new penem antibiotic. Antimicrob Agents Chemother. 1993;37:1547–51. https://doi.org/10.1128/AAC.37.7.1547.
Foulds G, Knirsch AK, Lazar JD, Tensfelt TG, Gerber N. Pharmacokinetics of the penem CP-65,207 and its separate stereoisomers in humans. Antimicrob Agents Chemother. 1991;35:665–71. https://doi.org/10.1128/AAC.35.4.665.
Brenek SJ, Caron S, Chisowa E, Delude MP, Drexler MT, Ewing MD, et al. Development of a practical and convergent process for the preparation of sulopenem. Org Process Res Dev. 2012;16:1348–59. https://doi.org/10.1021/op300131e.
Brenek SJ, Caron S, Chisowa E, Colon-Cruz R, Delude MP, Drexler MT, et al. Development of a second-generation process to antibacterial candidate sulopenem. Org Process Res Dev. 2012;16:1338–47. https://doi.org/10.1021/op300130p.
Dunne MW, Aronin SI, Das AF, Akinapelli K, Breen J, Zelasky MT, et al. Sulopenem for the treatment of complicated urinary tract infections including pyelonephritis: a phase 3, randomized trial. Clin Infect Dis. 2023;76:78–88. https://doi.org/10.1093/cid/ciac704.
Dunne MW, Aronin SI, Das AF, Akinapelli K, Zelasky MT, Puttagunta S, et al. Sulopenem or ciprofloxacin for the treatment of uncomplicated urinary tract infections in women: a phase 3, randomized trial. Clin Infect Dis. 2023;76:66–77. https://doi.org/10.1093/cid/ciac738.
Iterum Therapeutics announces U.S. FDA filing acceptance of New Drug Application for oral sulopenem (Press release 25 January 2021) [Accessed 16 December 2022]. Available from: https://www.iterumtx.com/news/press-releases/detail/58/iterum-therapeutics-announces-u-s-fda-filing-acceptance-of.
Dunne M, Dunzo E, Puttagunta S. A phase 1 study to assess the pharmacokinetics of sulopenem etzadroxil (PF-03709270). Open Forum Infect Dis. 2017;4:S525–S6. https://doi.org/10.1093/ofid/ofx163.1369.
Cox VC, Zed PJ. Once-daily cefazolin and probenecid for skin and soft tissue infections. Ann Pharmacother. 2004;38:458–63. https://doi.org/10.1345/aph.1D2.
Iterum Therapeutics receives Complete Response Letter from U.S. Food and Drug Administration for oral sulopenem (Press release 26 July 2021) [Accessed 16 December 2022]. Available from: https://www.iterumtx.com/news/press-releases/detail/73/iterum-therapeutics-receives-complete-response-letter-from.
Rodvold KA, Gotfried MH, Chugh R, Gupta M, Friedland HD, Bhatia A. Comparison of plasma and intrapulmonary concentrations of nafithromycin (WCK 4873) in healthy adult subjects. Antimicrob Agents Chemother. 2017;61:e01096–17. https://doi.org/10.1128/aac.01096-17.
Hackel MA, Karlowsky JA, Dressel D, Sahm DF. Determination of disk diffusion and MIC quality control ranges for nafithromycin (WCK 4873), a new lactone-ketolide. J Clin Microbiol. 2017;55:3021–7. https://doi.org/10.1128/JCM.00972-17.
Zhou M, Wu L, Kang W, Li Y, Zhang G, Zhang J, et al. In vitro activity of lactone ketolide nafithromycin (WCK 4873) against Streptococcus pneumoniae isolates enriched with macrolide-resistance phenotype collected from mainland China. JAC Antimicrob Resist 2022;4:dlac103. https://doi.org/10.1093/jacamr/dlac103.
Kohlhoff S, Hammerschlag MR. In vitro activity of nafithromycin (WCK 4873) against Chlamydia pneumoniae. Antimicrob Agents Chemother. 2021;65:e00585–21. https://doi.org/10.1128/AAC.00585-21.
Gibson EG, Bax B, Chan PF, Osheroff N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect Dis. 2019;5:570–81. https://doi.org/10.1021/acsinfecdis.8b00315.
EAGLE-2 and EAGLE-3 phase III trials for gepotidacin stopped early for efficacy following pre-planned interim analysis by Independent Data Monitoring Committee (Press release 3 November 2022) [Accessed 17 December 2022]. Available from: https://www.gsk.com/en-gb/media/press-releases/gsk-announces-phase-iii-trials-for-gepotidacin/.
Flamm RK, Farrell DJ, Rhomberg PR, Scangarella-Oman NE, Sader HS. Gepotidacin (GSK2140944) in vitro activity against Gram-positive and Gram-negative bacteria. Antimicrob Agents Chemother. 2017;61:e00468–17. https://doi.org/10.1128/aac.00468-17.
Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, et al. In vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob Agents Chemother. 2016;60:1918–23. https://doi.org/10.1128/AAC.02820-15.
Hackel MA, Karlowsky JA, Canino MA, Sahm DF, Scangarella-Oman NE. In vitro activity of gepotidacin against Gram-negative and Gram-positive anaerobes. Antimicrob Agents Chemother. 2022;66:e02165–21. https://doi.org/10.1128/aac.02165-21.
Ahmad MN, Garg T, Singh S, Shukla R, Malik P, Krishnamurthy RV, et al. In vitro and in vivo activity of gepotidacin against drug-resistant mycobacterial infections. Antimicrob Agents Chemother. 2022;66:e00564–22. https://doi.org/10.1128/aac.00564-22.
Sanders MI, Ali E, Buer J, Steinmann J, Rath P-M, Verhasselt HL, et al. Antibacterial activity of the novel drug gepotidacin against Stenotrophomonas maltophilia − an in vitro and in vivo study. Antibiotics. 2022;11:192. https://doi.org/10.3390/antibiotics11020192.
Waites KB, Crabb DM, Xiao L, Duffy LB. In vitro activities of gepotidacin (GSK2140944) and other antimicrobial agents against Human mycoplasmas and ureaplasmas. Antimicrob Agents Chemother. 2017;61:e01064–17. https://doi.org/10.1128/aac.01064-17.
Kern G, Palmer T, Ehmann DE, Shapiro AB, Andrews B, Basarab GS, et al. Inhibition of Neisseria gonorrhoeae type II topoisomerases by the novel spiropyrimidinetrione AZD0914. J Biol Chem. 2015;290:20984–94. https://doi.org/10.1074/jbc.M115.663534.
Innoviva Completes Acquisition of Entasis Therapeutics (Press release 11 July 2022) [Accessed 17 December 2022]. Available from: https://investor.inva.com/news-releases/news-release-details/innoviva-completes-acquisition-entasis-therapeutics.
Jacobsson S, Golparian D, Alm RA, Huband M, Mueller J, Jensen JS, et al. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob Agents Chemother. 2014;58:5585–8. https://doi.org/10.1128/AAC.03090-14.
Unemo M, Ahlstrand J, Sánchez-Busó L, Day M, Aanensen D, Golparian D, et al. High susceptibility to zoliflodacin and conserved target (GyrB) for zoliflodacin among 1209 consecutive clinical Neisseria gonorrhoeae isolates from 25 European countries, 2018. J Antimicrob Chemother. 2021;76:1221–8. https://doi.org/10.1093/jac/dkab024.
Taylor SN, Marrazzo J, Batteiger BE, Hook EW, Seña AC, Long J, et al. Single-dose zoliflodacin (ETX0914) for treatment of urogenital gonorrhea. N Engl J Med. 2018;379:1835–45. https://doi.org/10.1056/NEJMoa1706988.
GARDP website. Sexually Transmitted Infections, Zoliflodacin drug development project [Accessed 17 December 2022]. Available from: https://gardp.org/sexually-transmitted-infections/.
Damião Gouveia AC, Unemo M, Jensen JS. In vitro activity of zoliflodacin (ETX0914) against macrolide-resistant, fluoroquinolone-resistant and antimicrobial-susceptible Mycoplasma genitalium strains. J Antimicrob Chemother. 2018;73:1291–4. https://doi.org/10.1128/aac.00564-22.
Zhao C-Y, Lv Y, Zhu Y, Wei M-J, Liu M-Y, Ji X-W, et al. A first-in-human safety, tolerability, and pharmacokinetics study of benapenem in healthy Chinese volunteers. Antimicrob Agents Chemother. 2019;63:e02188–18. https://doi.org/10.1128/aac.02188-18.
Yang H, Zhang M, Chen Y, Ren H, Zhang H, Yu C, et al. Pharmacokinetics of benapenem for injection in subjects with mild to moderate renal impairment. Eur J Clin Pharm. 2022;78:1079–86. https://doi.org/10.1007/s00228-022-03317-y.
Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, et al. Discovery of a novel class of boron-based antibacterials with activity against Gram-negative bacteria. Antimicrob Agents Chemother. 2013;57:1394–403. https://doi.org/10.1128/AAC.02058-12.
van Ingen J, Obradovic M, Hassan M, Lesher B, Hart E, Chatterjee A, et al. Nontuberculous mycobacterial lung disease caused by Mycobacterium avium complex - disease burden, unmet needs, and advances in treatment developments. Expert Rev Respir Med. 2021;15:1387–401. https://doi.org/10.1080/17476348.2021.1987891.
Ganapathy US, Gengenbacher M, Dick T. Epetraborole is active against Mycobacterium abscessus. Antimicrob Agents Chemother. 2021;65:e01156–21. https://doi.org/10.1128/AAC.01156-21.
Sullivan JR, Lupien A, Kalthoff E, Hamela C, Taylor L, Munro KA, et al. Efficacy of epetraborole against Mycobacterium abscessus is increased with norvaline. PLoS Pathog. 2021;17:e1009965. https://doi.org/10.1371/journal.ppat.1009965.
O’Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, et al. Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob Agents Chemother. 2015;59:289–98. https://doi.org/10.1128/AAC.03774-14.
Brii Biosciences Pipeline [Accessed 2 January 2023]. Available from: https://www.briibio.com/en/science/pipeline/.
Tamura S, Miyazaki S, Tateda K, Ohno A, Ishii Y, Matsumoto T, et al. In vivo antibacterial activities of sanfetrinem cilexetil, a new oral tricyclic antibiotic. Antimicrob Agents Chemother. 1998;42:1858–61. https://doi.org/10.1128/AAC.42.7.1858.
Doern GV, Pierce G, Brueggemann AB. In vitro activity of sanfetrinem (GV104326), a new trinem antimicrobial agent, versus Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. Diagn Microbiol Infect Dis. 1996;26:39–42. https://doi.org/10.1016/S0732-8893(96)00173-3.
Sader HS, Gales AC. Emerging strategies in infectious diseases. Drugs. 2001;61:553–64. https://doi.org/10.2165/00003495-200161050-00001.
Working Group on TB Drugs Pipeline - Sanfertrinem [Accessed 21 December 2022]. Available from: https://www.newtbdrugs.org/pipeline/compound/sanfetrinem.
Diacon AH, van der Merwe L, Barnard M, von Groote-Bidlingmaier F, Lange C, García-Basteiro AL, et al. β-Lactams against tuberculosis — new trick for an old dog. N Engl J Med. 2016;375:393–4. https://doi.org/10.1056/NEJMc1513236.
Gold B, Zhang J, Quezada LL, Roberts J, Ling Y, Wood M, et al. Identification of β-lactams active against Mycobacterium tuberculosis by a consortium of pharmaceutical companies and academic institutions. ACS Infect Dis. 2022;8:557–73. https://doi.org/10.1021/acsinfecdis.1c00570.
MGB Biopharma announces successful outcome from Phase II clinical study with MGB-BP-3 – a potential new gold standard, first-line treatment for Clostridium difficile infection (CDI) (Press release 19 May 2020) [Accessed 21 December 2022]. Available from: https://www.mgb-biopharma.com/mgb-biopharma-announces-successful-outcome-from-phase-ii-clinical-study-with-mgb-bp-3-a-potential-new-gold-standard-first-line-treatment-for-clostridium-difficile-infection-cdi/.
Anthony NG, Breen D, Clarke J, Donoghue G, Drummond AJ, Ellis EM, et al. Antimicrobial lexitropsins containing amide, amidine, and alkene linking groups. J Med Chem. 2007;50:6116–25. https://doi.org/10.1021/jm070831g.
Suckling CJ. The antibacterial drug MGB-BP3: from discovery to clinical trial. Chem Biol Interface. 2015;5:166–74.
Hind C, Clifford M, Woolley C, Harmer J, McGee LMC, Tyson-Hirst I, et al. Insights into the spectrum of activity and mechanism of action of MGB-BP-3. ACS Infect Dis. 2022;8:2552–63. https://doi.org/10.1021/acsinfecdis.2c00445.
Kerr L, Browning DF, Lemonidis K, Salih T, Hunter IS, Suckling CJ, et al. Novel antibiotic mode of action by repression of promoter isomerisation. bioRxiv. 2021. https://doi.org/10.1101/2020.12.31.424950.
Kaushik V, Tiwari M, Tiwari V. Interaction of RecA mediated SOS response with bacterial persistence, biofilm formation, and host response. Int J Biol Macromol. 2022;217:931–43. https://doi.org/10.1016/j.ijbiomac.2022.07.176.
Ooi N, Miller K, Hobbs J, Rhys-Williams W, Love W, Chopra I. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J Antimicrob Chemother. 2009;64:735–40. https://doi.org/10.1093/jac/dkp299.
Farrell DJ, Robbins M, Rhys-Williams W, Love WG. In vitro activity of XF-73, a novel antibacterial agent, against antibiotic-sensitive and -resistant Gram-positive and Gram-negative bacterial species. Int J Antimicrob Agents. 2010;35:531–6. https://doi.org/10.1016/j.ijantimicag.2010.02.008.
Ooi N, Miller K, Randall C, Rhys-Williams W, Love W, Chopra I. XF-70 and XF-73, novel antibacterial agents active against slow-growing and non-dividing cultures of Staphylococcus aureus including biofilms. J Antimicrob Chemother. 2010;65:72–8. https://doi.org/10.1093/jac/dkp409.
MacLean RC. Assessing the potential for Staphylococcus aureus to evolve resistance to XF-73. Trends Microbiol. 2020;28:432–5. https://doi.org/10.1016/j.tim.2020.03.011.
XF-73 nasal final Phase 3 development plans (Press release 7 December 2022) [Accessed 21 December 2022]. Available from: https://otp.tools.investis.com/clients/uk/destiny_pharma_ltd/rns/regulatory-story.aspx?cid=2241&newsid=1650054.
van Klingeren B, ten Ham M. Antibacterial activity of Δ9-tetrahydrocannabinol and cannabidiol. Antonie van Leeuwenhoek. 1976;42:9–12. https://doi.org/10.1007/BF00399444.
Appendino G, Gibbons S, Giana A, Pagani A, Grassi G, Stavri M, et al. Antibacterial cannabinoids from Cannabis sativa: A structure−activity study. J Nat Prod. 2008;71:1427–30. https://doi.org/10.1021/np8002673.
Martinenghi LD, Jønsson R, Lund T, Jenssen H. Isolation, purification, and antimicrobial characterization of cannabidiolic acid and cannabidiol from Cannabis sativa L. Biomolecules. 2020;10:900. https://doi.org/10.3390/biom10060900.
Blaskovich MAT, Kavanagh AM, Elliott AG, Zhang B, Ramu S, Amado M, et al. The antimicrobial potential of cannabidiol. Commun Biol. 2021;4:7. https://doi.org/10.1038/s42003-020-01530-y.
Wassmann CS, Rolsted AP, Lyngsie MC, Torres-Puig S, Kronborg T, Vestergaard M, et al. The menaquinone pathway is important for susceptibility of Staphylococcus aureus to the antibiotic adjuvant, cannabidiol. Microbiol Res. 2022;257:126974. https://doi.org/10.1016/j.micres.2022.126974.
Choi S-R, Frandsen J, Narayanasamy P. Novel long-chain compounds with both immunomodulatory and MenA inhibitory activities against Staphylococcus aureus and its biofilm. Sci Rep. 2017;7:40077. https://doi.org/10.1038/srep40077.
Johnston JM, Bulloch EMM. Advances in menaquinone biosynthesis: sublocalisation and allosteric regulation. Curr Opin Struct Biol. 2020;65:33–41. https://doi.org/10.1016/j.sbi.2020.05.005.
Yuan Y, Wang X, Xu X, Liu Y, Li C, Yang M, et al. Evaluation of a dual-acting antibacterial agent, TNP-2092, on gut microbiota and potential application in the treatment of gastrointestinal and liver disorders. ACS Infect Dis. 2020;6:820–31. https://doi.org/10.1021/acsinfecdis.9b00374.
TenNor reports positive Phase II results for TNP-2092 capsule (Press release 23 November 2021) [Accessed 21 December 2022]. Available from: http://www.tennorx.com/en/h-nd-86.html?fromColId=148.
Fisher CR, Schmidt-Malan SM, Ma Z, Yuan Y, He S, Patel R. In vitro activity of TNP-2092 against periprosthetic joint infection–associated staphylococci. Diagn Microbiol Infect Dis. 2020;97:115040. https://doi.org/10.1016/j.diagmicrobio.2020.115040.
Ma Z, Lynch AS. Development of a dual-acting antibacterial agent (TNP-2092) for the treatment of persistent bacterial infections. J Med Chem. 2016;59:6645–57. https://doi.org/10.1021/acs.jmedchem.6b00485.
Robertson GT, Bonventre EJ, Doyle TB, Du Q, Duncan L, Morris TW, et al. In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: microbiology profiling studies with Staphylococci and Streptococci. Antimicrob Agents Chemother. 2008;52:2324–34. https://doi.org/10.1128/AAC.01651-07.
Robertson GT, Bonventre EJ, Doyle TB, Du Q, Duncan L, Morris TW, et al. In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: studies of the mode of action in Staphylococcus aureus. Antimicrob Agents Chemother. 2008;52:2313–23. https://doi.org/10.1128/AAC.01649-07.
Ma Z, He S, Yuan Y, Zhuang Z, Liu Y, Wang H, et al. Design, synthesis, and characterization of TNP-2198, a dual-targeted rifamycin-nitroimidazole conjugate with potent activity against microaerophilic and anaerobic bacterial pathogens. J Med Chem. 2022;65:4481–95. https://doi.org/10.1021/acs.jmedchem.1c02045.
TenNor discloses TNP-2198 for the treatment of H. pylori Infections in the Journal of Medicinal Chemistry (Press release 18 February 2022) [Accessed 21 December 2022]. Available from: http://www.tennorx.com/en/h-nd-95.html.
Guangzhou Yushi Pharmaceutical Technology Co. Website [Accessed 21 December 2022]. Available from: https://www.yscro.com/search.html?q=TNP-2198.
Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, et al. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob Agents Chemother. 2002;46:3118–24. https://doi.org/10.1128/AAC.46.10.3118-3124.2002.
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Disco. 2007;6:29–40. https://doi.org/10.1038/nrd2201.
Hafkin B, Kaplan N, Murphy B. Efficacy and safety of AFN-1252, the first Staphylococcus-specific antibacterial agent, in the treatment of acute bacterial skin and skin structure infections, including those in patients with significant comorbidities. Antimicrob Agents Chemother. 2015;60:1695–701. https://doi.org/10.1128/AAC.01741-15.
Menetrey A, Janin A, Pullman J, Overcash JS, Haouala A, Leylavergne F, et al. Bone and joint tissue penetration of the Staphylococcus-selective antibiotic afabicin in patients undergoing elective hip replacement surgery. Antimicrob Agents Chemother. 2019;63:e01669–18. https://doi.org/10.1128/aac.01669-18.
Wittke F, Vincent C, Chen J, Heller B, Kabler H, Overcash JS, et al. Afabicin, a first-in-class antistaphylococcal antibiotic, in the treatment of acute bacterial skin and skin structure infections: Clinical noninferiority to vancomycin/linezolid. Antimicrob Agents Chemother. 2020;64:e00250–20. https://doi.org/10.1128/AAC.00250-20.
Parsons JB, Kukula M, Jackson P, Pulse M, Simecka JW, Valtierra D, et al. Perturbation of Staphylococcus aureus gene expression by the enoyl-acyl carrier protein reductase inhibitor AFN-1252. Antimicrob Agents Chemother. 2013;57:2182–90. https://doi.org/10.1128/AAC.02307-12.
Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, et al. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother. 2012;56:5865–74. https://doi.org/10.1128/AAC.01411-12.
Dyon-Tafani V, Josse J, Dieppois G, Ferry T, Laurent F. Antimicrobial activity of the new FabI inhibitor afabicin desphosphono against intraosteoblastic Staphylococcus aureus. Int J Antimicrob Agents. 2021;57:106321. https://doi.org/10.1016/j.ijantimicag.2021.106321.
Rana P, Ghouse SM, Akunuri R, Madhavi YV, Chopra S, Nanduri S. FabI (enoyl acyl carrier protein reductase) - A potential broad spectrum therapeutic target and its inhibitors. Eur J Med Chem. 2020;208:112757. https://doi.org/10.1016/j.ejmech.2020.112757.
Feng Q, Huang Y, Chen M, Li G, Chen Y. Functional synergy of α-helical antimicrobial peptides and traditional antibiotics against Gram-negative and Gram-positive bacteria in vitro and in vivo. Eur J Clin Microbiol Infect Dis. 2015;34:197–204. https://doi.org/10.1007/s10096-014-2219-3.
Chen Y, Mant CT, Farmer SW, Hancock REW, Vasil ML, Hodges RS. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J Biol Chem. 2005;280:12316–29. https://doi.org/10.1074/jbc.M413406200.
Wei Y, Wu J, Chen Y, Fan K, Yu X, Li X, et al. Efficacy and safety of PL-5 (peceleganan) spray for wound infections: A phase IIb randomized clinical trial. Ann Surg. 2023;277:43–9. https://doi.org/10.1097/SLA.0000000000005508.
MST Access. Recce Pharmaceuticals (RCE.AX) Initiation of Coverage 2021 [Accessed 16 February 2023]. Available from: https://www.recce.com.au/PDF-Flip/Recce_iPoster.pdf.
Dilizia M, Tsunemoto H, Quach D, Sharp M, Prendergast J, Graham J. Elucidating the mechanism of action of novel polymer-based. Synthetic anti-infective compound RECCE® 327. 2021 [Accessed 16 February 2023]. Available from: https://www.recce.com.au/PDF-Flip/Recce_iPoster.pdf.
Folsom JP, Baker B, Stewart PS. In vitro efficacy of bismuth thiols against biofilms formed by bacteria isolated from human chronic wounds. J Appl Microbiol. 2011;111:989–96. https://doi.org/10.1111/j.1365-2672.2011.05110.x.
Baker B. 1289. Pravibismane is a potent, broad spectrum anti-infective small molecule that rapidly disrupts bacterial bioenergetics and halts bacterial growth. Open Forum Infect Dis. 2020;7:S659–S60. https://doi.org/10.1093/ofid/ofaa439.1472.
Bierer DW. Bismuth subsalicylate: history, chemistry, and safety. Clin Infect Dis. 1990;12:S3–S8. https://doi.org/10.1093/clinids/12.supplement_1.s3.
Barillo DJ, Barillo AR, Korn S, Lam K, Attar PS. The antimicrobial spectrum of Xeroform®. Burns. 2017;43:1189–94. https://doi.org/10.1016/j.burns.2016.10.023.
Alkim H, Koksal AR, Boga S, Sen I, Alkim C. Role of bismuth in the eradication of Helicobacter pylori. Am J Ther. 2017;24:e751–e7. https://doi.org/10.1136/gutjnl-2015-311019.
Frei A, Zuegg J, Elliott AG, Baker M, Braese S, Brown C, et al. Metal complexes as a promising source for new antibiotics. Chem Sci. 2020;11:2627–39. https://doi.org/10.1039/C9SC06460E.
Frei A, Verderosa A, Elliott AG, Zuegg J, Blaskovich MAT. Metals to combat antimicrobial resistance. Nat Rev Chem. 2023;7:202–24. https://doi.org/10.1038/s41570-023-00463-4.
DEINOVE is now ready to start Phase II clinical trial for its antibiotic compound DNV3837 (Press Release 16 May 2019) [Accessed 2 February 2023]. Available from: http://www.deinove.com/en/news/all-press-releases/deinove-now-ready-start-phase-ii-clinical-trial-its-antibiotic-compound-dnv3837.
DEINOVE announces the opening of receivership proceedings (Press release 7 November 2022) [Accessed 22 December 2022]. Available from: https://www.deinove.com/en/news/all-press-releases/deinove-announces-opening-receivership-proceedings.
Rashid M-U, Dalhoff A, Weintraub A, Nord CE. In vitro activity of MCB3681 against Clostridium difficile strains. Anaerobe. 2014;28:216–9. https://doi.org/10.1016/j.anaerobe.2014.07.001.
Rashid M-U, Dalhoff A, Bäckström T, Björkhem-Bergman L, Panagiotidis G, Weintraub A, et al. Ecological impact of MCB3837 on the normal human microbiota. Int J Antimicrob Agents. 2014;44:125–30. https://doi.org/10.1016/j.ijantimicag.2014.03.016.
Dalhoff A, Rashid MU, Kapsner T, Panagiotidis G, Weintraub A, Nord CE. Analysis of effects of MCB3681, the antibacterially active substance of prodrug MCB3837, on human resident microflora as proof of principle. Clin Microbiol Infect. 2015;21:767.e1–.e4. https://doi.org/10.1016/j.cmi.2015.05.025.
Wright GE, Brown NC, Xu W-C, Long Z-Y, Zhi C, Gambino JJ, et al. Active site directed inhibitors of replication-specific bacterial DNA polymerases. Bioorg Med Chem Lett. 2005;15:729–32. https://doi.org/10.1016/j.bmcl.2004.11.016.
Xu W-C, Wright GE, Brown NC, Long Z-Y, Zhi C-X, Dvoskin S, et al. 7-Alkyl-N2-substituted-3-deazaguanines. Synthesis, DNA polymerase III inhibition and antibacterial activity. Bioorg Med Chem Lett. 2011;21:4197–202. https://doi.org/10.1016/j.bmcl.2011.05.093.
Xu W-C, Silverman MH, Yu XY, Wright G, Brown N. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorg Med Chem. 2019;27:3209–17. https://doi.org/10.1016/j.bmc.2019.06.017.
Murray B, Wolfe C, Marra A, Pillar C, Shinabarger D. In vitro activity of the novel antibacterial agent ibezapolstat (ACX-362E) against Clostridioides difficile. J Antimicrob Chemother. 2020;75:2149–55. https://doi.org/10.1093/jac/dkaa134.
Garey KW, McPherson J, Dinh AQ, Hu C, Jo J, Wang W, et al. Efficacy, safety, pharmacokinetics, and microbiome changes of ibezapolstat in adults with Clostridioides difficile infection: a phase 2a multicenter clinical trial. Clin Infect Dis. 2022;75:1164–70. https://doi.org/10.1093/cid/ciac096.
Critchley IA, Green LS, Young CL, Bullard JM, Evans RJ, Price M, et al. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother. 2009;63:954–63. https://doi.org/10.1093/jac/dkp041.
Citron DM, Warren YA, Tyrrell KL, Merriam V, Goldstein EJC. Comparative in vitro activity of REP3123 against Clostridium difficile and other anaerobic intestinal bacteria. J Antimicrob Chemother. 2009;63:972–6. https://doi.org/10.1093/jac/dkp037.
Ochsner UA, Bell SJ, O’Leary AL, Hoang T, Stone KC, Young CL, et al. Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J Antimicrob Chemother. 2009;63:964–71. https://doi.org/10.1093/jac/dkp042.
Nayak SU, Griffiss JM, Blumer J, O’Riordan MA, Gray W, McKenzie R, et al. Safety, tolerability, systemic exposure, and metabolism of CRS3123, a methionyl-tRNA synthetase inhibitor developed for treatment of Clostridium difficile, in a Phase 1 study. Antimicrob Agents Chemother. 2017;61:e02760–16. https://doi.org/10.1128/AAC.02760-16.
Lomeli BK, Galbraith H, Schettler J, Saviolakis GA, El-Amin W, Osborn B, et al. Multiple-ascending-dose phase 1 clinical study of the safety, tolerability, and pharmacokinetics of CRS3123, a narrow-spectrum agent with minimal disruption of normal gut microbiota. Antimicrob Agents Chemother. 2019;64:e01395–19. https://doi.org/10.1128/AAC.01395-19.
Baedeker M, Ringel MS, Möller CC, Schulze U. 2022 FDA approval number dips: a COVID-19 hangover. Nat Rev Drug Disco. 2023;22:91. https://doi.org/10.1038/d41573-023-00007-x.
Jeong J-W, Jung S-J, Lee H-H, Kim Y-Z, Park T-K, Cho Y-L, et al. In vitro and in vivo activities of LCB01-0371, a new oxazolidinone. Antimicrob Agents Chemother. 2010;54:5359–62. https://doi.org/10.1128/AAC.00723-10.
Zong Z, Jing W, Shi J, Wen SA, Zhang T, Huo F, et al. Comparison of in vitro activity and MIC distributions between the novel oxazolidinone delpazolid and linezolid against multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis in China. Antimicrob Agents Chemother. 2018;62:e00165–18. https://doi.org/10.1128/AAC.00165-18.
Wang C, Wang G, Huo F, Xue Y, Jia J, Dong L et al. Novel oxazolidinones harbor potent in vitro activity against the clinical isolates of multidrug-resistant Mycobacterium tuberculosis in China. Front Med. 2022;9. https://doi.org/10.3389/fmed.2022.1067.
Kim TS, Choe JH, Kim YJ, Yang C-S, Kwon H-J, Jeong J, et al. Activity of LCB01-0371, a novel oxazolidinone, against Mycobacterium abscessus. Antimicrob Agents Chemother. 2017;61:e02752–16. https://doi.org/10.1128/AAC.02752-16.
Kim DH, Kim S-Y, Koh W-J, Jhun BW. In vitro activity of oxazolidinone against nontuberculous mycobacteria, including macrolide-resistant clinical isolates. Antimicrob Agents Chemother. 2021;65:e02306–20. https://doi.org/10.1128/AAC.02306-20.
Donald PR, Sirgel FA, Venter A, Parkin DP, Seifart HI, van de Wal BW, et al. Early bactericidal activity of antituberculosis agents. Expert Rev Anti Infect Ther. 2003;1:141–55. https://doi.org/10.1586/14787210.1.1.141.
Kim JS, Kim Y-h, Lee SH, Kim YH, Kim J-w, Kang JY, et al. Early bactericidal activity of delpazolid (LCB01-0371) in patients with pulmonary tuberculosis. Antimicrob Agents Chemother. 2022;66:e01684–21. https://doi.org/10.1128/aac.01684-21.
Barbachyn MR, Hutchinson DK, Brickner SJ, Cynamon MH, Kilburn JO, Klemens SP, et al. Identification of a novel oxazolidinone (U-100480) with potent antimycobacterial activity. J Med Chem. 1996;39:680–5. https://doi.org/10.1021/jm950956y.
Cynamon MH, Klemens SP, Sharpe CA, Chase S. Activities of several novel oxazolidinones against Mycobacterium tuberculosis in a murine model. Antimicrob Agents Chemother. 1999;43:1189–91. https://doi.org/10.1128/AAC.43.5.1189.
Alffenaar JWC, van der Laan T, Simons S, van der Werf TS, van de Kasteele PJ, de Neeling H, et al. Susceptibility of clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob Agents Chemother. 2011;55:1287–9. https://doi.org/10.1128/AAC.01297-10.
Williams KN, Stover CK, Zhu T, Tasneen R, Tyagi S, Grosset JH, et al. Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model. Antimicrob Agents Chemother. 2009;53:1314–9. https://doi.org/10.1128/AAC.01182-08.
Wallis RS, Dawson R, Friedrich SO, Venter A, Paige D, Zhu T, et al. Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. PLoS One. 2014;9:e94462. https://doi.org/10.1371/journal.pone.0094462.
TB Alliance. Our Pipeline. Sutezolid. 2023 [Accessed 14 April 2023]. Available from: https://www.tballiance.org/portfolio/trial/12018.
Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med. 2013;19:1157–60. https://doi.org/10.1038/nm.3262.
Kang S, Kim RY, Seo MJ, Lee S, Kim YM, Seo M, et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J Med Chem. 2014;57:5293–305. https://doi.org/10.1038/nm.3262.
Kim J, Choi J, Kang H, Ahn J, Hutchings J, Niekerk CV, et al. Safety, tolerability, pharmacokinetics, and metabolism of telacebec (Q203) for the treatment of tuberculosis: a randomized, placebo-controlled, multiple ascending dose phase 1B trial. Antimicrob Agents Chemother. 2023;67:e01123–22. https://doi.org/10.1128/aac.01123-22.
de Jager VR, Dawson R, van Niekerk C, Hutchings J, Kim J, Vanker N, et al. Telacebec (Q203), a new antituberculosis agent. N Engl J Med. 2020;382:1280–1. https://doi.org/10.1056/NEJMc1913327.
Lee BS, Pethe K. Telacebec: an investigational antibacterial for the treatment of tuberculosis (TB). Expert Opin Invest Drugs. 2022;31:139–44. https://doi.org/10.1080/13543784.2022.2030309.
Moraski GC, Seeger N, Miller PA, Oliver AG, Boshoff HI, Cho S, et al. Arrival of imidazo[2,1-b]thiazole-5-carboxamides: Potent anti-tuberculosis agents that target QcrB. ACS Infect Dis. 2016;2:393–8. https://doi.org/10.1021/acsinfecdis.5b00154.
Lee BS, Kalia NP, Jin XEF, Hasenoehrl EJ, Berney M, Pethe K. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. J Biol Chem. 2019;294:1936–43. https://doi.org/10.1074/jbc.RA118.005732.
Thomas SS, Kalia NP, Ruf M-T, Pluschke G, Pethe K. Toward a single-dose cure for Buruli ulcer. Antimicrob Agents Chemother. 2020;64:e00727–20. https://doi.org/10.1128/AAC.00727-20.
Komm O, Almeida DV, Converse PJ, Omansen TF, Nuermberger EL. Impact of dose, duration, and immune status on efficacy of ultrashort telacebec regimens in mouse models of Buruli ulcer. Antimicrob Agents Chemother. 2021;65:e01418–21. https://doi.org/10.1128/AAC.01418-21.
Brown-Elliott BA, Rubio A, Wallace RJ Jr. In vitro susceptibility testing of a novel benzimidazole, SPR719, against nontuberculous mycobacteria. Antimicrob Agents Chemother. 2018;62:e01503–18. https://doi.org/10.1128/aac.01503-18.
Pidot SJ, Porter JL, Lister T, Stinear TP. In vitro activity of SPR719 against Mycobacterium ulcerans, Mycobacterium marinum and Mycobacterium chimaera. PLoS Negl Trop Dis. 2021;15:e0009636. https://doi.org/10.1371/journal.pntd.0009636.
Aragaw WW, Cotroneo N, Stokes S, Pucci M, Critchley I, Gengenbacher M, et al. In vitro resistance against DNA gyrase inhibitor SPR719 in Mycobacterium avium and Mycobacterium abscessus. Microbiol Spectr. 2022;10:e01321–21. https://doi.org/10.1128/spectrum.01321-21.
Talley AK, Thurston A, Moore G, Gupta VK, Satterfield M, Manyak E, et al. First-in-human evaluation of the safety, tolerability, and pharmacokinetics of SPR720, a novel oral bacterial DNA gyrase (GyrB) inhibitor for mycobacterial infections. Antimicrob Agents Chemother. 2021;65:e01208–21. https://doi.org/10.1128/AAC.01208-21.
O’Dowd H, Shannon DE, Chandupatla KR, Dixit V, Engtrakul JJ, Ye Z, et al. Discovery and characterization of a water-soluble prodrug of a dual inhibitor of bacterial DNA gyrase and topoisomerase IV. ACS Med Chem Lett. 2015;6:822–6. https://doi.org/10.1021/acsmedchemlett.5b00196.
Locher CP, Jones SM, Hanzelka BL, Perola E, Shoen CM, Cynamon MH, et al. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob Agents Chemother. 2015;59:1455–65. https://doi.org/10.1128/AAC.04347-14.
Grillot A-L, Tiran AL, Shannon D, Krueger E, Liao Y, O’Dowd H, et al. Second-generation antibacterial benzimidazole ureas: Discovery of a preclinical candidate with reduced metabolic liability. J Med Chem. 2014;57:8792–816. https://doi.org/10.1021/jm500563g.
Durcik M, Tomašič T, Zidar N, Zega A, Kikelj D, Mašič LP, et al. ATP-competitive DNA gyrase and topoisomerase IV inhibitors as antibacterial agents. Expert Opin Ther Pat. 2019;29:171–80. https://doi.org/10.1080/13543776.2019.1575362.
Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–4. https://doi.org/10.1126/science.1171583.
Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, et al. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci USA. 2012;109:11354–9. https://doi.org/10.1073/pnas.1205735109.
Trefzer C, Škovierová H, Buroni S, Bobovská A, Nenci S, Molteni E, et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase DprE1. J Am Chem Soc. 2012;134:912–5. https://doi.org/10.1021/ja211042r.
Neres J, Pojer F, Molteni E, Chiarelli LR, Dhar N, Boy-Röttger S, et al. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci Transl Med. 2012;4:150ra21. https://doi.org/10.1126/scitranslmed.3004395.
Shi J, Lu J, Wen SA, Zong Z, Huo F, Luo J, et al. In vitro activity of PBTZ169 against multiple Mycobacterium species. Antimicrob Agents Chemother. 2018;62:e01314–18. https://doi.org/10.1128/aac.01314-18.
Kloss F, Krchnak V, Krchnakova A, Schieferdecker S, Dreisbach J, Krone V, et al. In vivo dearomatization of the potent antituberculosis agent BTZ043 via Meisenheimer complex formation. Angew Chem, Int Ed. 2017;56:2187–91. https://doi.org/10.1002/anie.201609737.
Liu R, Krchnak V, Brown SN, Miller MJ. Deuteration of BTZ043 extends the lifetime of Meisenheimer intermediates to the antituberculosis nitroso oxidation state. ACS Med Chem Lett. 2019;10:1462–6. https://doi.org/10.1021/acsmedchemlett.9b00308.
Hariguchi N, Chen X, Hayashi Y, Kawano Y, Fujiwara M, Matsuba M, et al. OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother. 2020;64:e02020–19. https://doi.org/10.1128/AAC.02020-19.
OPC-167832 (Working Group on New TB Drugs) [Accessed 4 January 2023]. Available from: https://www.newtbdrugs.org/pipeline/compound/opc-167832.
Li X, Hernandez V, Rock FL, Choi W, Mak YSL, Mohan M, et al. Discovery of a potent and specific M. tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J Med Chem. 2017;60:8011–26. https://doi.org/10.1021/acs.jmedchem.7b00631.
Tenero D, Derimanov G, Carlton A, Tonkyn J, Davies M, Cozens S, et al. First-time-in-human study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment. Antimicrob Agents Chemother. 2019;63:e00240–19. https://doi.org/10.1128/aac.00240-19.
Shirude PS, Shandil R, Sadler C, Naik M, Hosagrahara V, Hameed S, et al. Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem. 2013;56:9701–8. https://doi.org/10.1021/jm401382v.
Shirude PS, Shandil RK, Manjunatha MR, Sadler C, Panda M, Panduga V, et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J Med Chem. 2014;57:5728–37. https://doi.org/10.1021/jm500571f.
Chatterji M, Shandil R, Manjunatha MR, Solapure S, Ramachandran V, Kumar N, et al. 1,4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob Agents Chemother. 2014;58:5325–31. https://doi.org/10.1128/AAC.03233-14.
Huang Z, Luo W, Xu D, Guo F, Yang M, Zhu Y, et al. Discovery and preclinical profile of sudapyridine (WX-081), a novel anti-tuberculosis agent. Bioorg Med Chem Lett. 2022;71:128824 https://doi.org/10.1016/j.bmcl.2022.128824.
Yao R, Wang B, Fu L, Li L, You K, Li Y-G, et al. Sudapyridine (WX-081), a novel compound against Mycobacterium tuberculosis. Microbiol Spectr. 2022;10:e02477–21. https://doi.org/10.1128/spectrum.02477-21.
Zhu R, Shang Y, Chen S, Xiao H, Ren R, Wang F, et al. In vitro activity of the sudapyridine (WX-081) against non-tuberculous mycobacteria isolated in Beijing, China. Microbiol Spectr. 2022;10:e01372–22. https://doi.org/10.1128/spectrum.01372-22.
Zhang D, Lu Y, Liu K, Liu B, Wang J, Zhang G, et al. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J Med Chem. 2012;55:8409–17. https://doi.org/10.1021/jm300828h.
Dey T, Brigden G, Cox H, Shubber Z, Cooke G, Ford N. Outcomes of clofazimine for the treatment of drug-resistant tuberculosis: a systematic review and meta-analysis. J Antimicrob Chemother. 2012;68:284–93. https://doi.org/10.1093/jac/dks389.
Mirnejad R, Asadi A, Khoshnood S, Mirzaei H, Heidary M, Fattorini L, et al. Clofazimine: a useful antibiotic for drug-resistant tuberculosis. Biomed Pharmacother. 2018;105:1353–9. https://doi.org/10.1016/j.biopha.2018.06.023.
Xu J, Wang B, Fu L, Zhu H, Guo S, Huang H, et al. In vitro and in vivo activities of the riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63:e02155–18. https://doi.org/10.1128/aac.02155-18.
Zhang Y, Zhu H, Fu L, Wang B, Guo S, Chen X, et al. Identifying regimens containing TBI-166, a new drug candidate against Mycobacterium tuberculosis in vitro and in vivo. Antimicrob Agents Chemother. 2019;63:e02496–18. https://doi.org/10.1128/aac.02496-18.
Zhu H, Fu L, Wang B, Chen X, Zhao J, Huang H, et al. Activity of clofazimine and TBI-166 against Mycobacterium tuberculosis in different administration intervals in mouse tuberculosis models. Antimicrob Agents Chemother. 2021;65:e02164–20. https://doi.org/10.1128/AAC.02164-20.
Ding Y, Zhu H, Fu L, Zhang W, Wang B, Guo S, et al. Superior efficacy of a TBI-166, bedaquiline, and pyrazinamide combination regimen in a murine model of tuberculosis. Antimicrob Agents Chemother. 2022;66:e00658–22. https://doi.org/10.1128/aac.00658-22.
Lv C, Li Y, Wei Y, Wang J, Yu H, Gao F, et al. Research progress on small molecular inhibitors of the Type 3 secretion system. Molecules. 2022;27:8348. https://doi.org/10.3390/molecules27238348.
Nesterenko LN, Zigangirova NA, Zayakin ES, Luyksaar SI, Kobets NV, Balunets DV, et al. A small-molecule compound belonging to a class of 2,4-disubstituted 1,3,4-thiadiazine-5-ones suppresses Salmonella infection in vivo. J Antibiot. 2016;69:422–7. https://doi.org/10.1038/ja.2015.131.
Zigangirova NA, Zayakin ES, Kapotina LN, Kost EA, Didenko LV, Davydova DY, et al. Development of chlamydial type III secretion system inhibitors for suppression of acute and chronic forms of chlamydial infection. Acta Nat. 2012;4:87–97.
Sheremet AB, Zigangirova NA, Zayakin ES, Luyksaar SI, Kapotina LN, Nesterenko LN, et al. Small molecule inhibitor of type three secretion system belonging to a class 2,4-disubstituted-4H-[1,3,4]-thiadiazine-5-ones improves survival and decreases bacterial loads in an airway Pseudomonas aeruginosa infection in mice. Biomed Res Int. 2018:5810767. https://doi.org/10.1155/2018/5810767.
Zigangirova NA, Nesterenko LN, Sheremet AB, Soloveva AV, Luyksaar SI, Zayakin ES, et al. Fluorothiazinon, a small-molecular inhibitor of T3SS, suppresses Salmonella oral infection in mice. J Antibiot. 2021;74:244–54. https://doi.org/10.1038/s41429-020-00396-w.
Subbian S, Koo M-S, Tsenova L, Khetani V, Zeldis JB, Fallows D, et al. Pharmacologic inhibition of host phosphodiesterase-4 improves isoniazid-mediated clearance of Mycobacterium tuberculosis. Front Immunol. 2016;7:238. https://doi.org/10.3389/fimmu.2016.00238.
Subbian S, Tsenova L, Holloway J, Peixoto B, O’Brien P, Dartois V, et al. Adjunctive phosphodiesterase-4 inhibitor therapy improves antibiotic response to pulmonary tuberculosis in a rabbit model. eBioMedicine. 2016;4:104–14. https://doi.org/10.1016/j.ebiom.2016.01.015.
Amgen licenses AMG 634, an investigational treatment for tuberculosis and leprosy, to Medicines Development for Global Health (Press release 22 December 2020) [Accessed 4 January 2023]. Available from: https://www.medicinesdevelopment.com/news/mdgh-announces-in-licensing-of-investigational-treatment-for-tb-and-leprosy-from-amgen.
Working Group on New TB Drugs, CC-11050 [Accessed 4 January 2023]. Available from: https://www.newtbdrugs.org/pipeline/compound/cc-11050.
Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Edward VA, et al. Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med. 2021;9:897–908. https://doi.org/10.1016/S2213-2600(20)30448-3.
Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Sebe M, et al. Lung and blood early biomarkers for host-directed tuberculosis therapies: Secondary outcome measures from a randomized controlled trial. PLoS One. 2022;17:e0252097. https://doi.org/10.1371/journal.pone.0252097.
Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, et al. Evaluation of the in vitro activity of new polymyxin B analogue SPR206 against clinical MDR, colistin-resistant and tigecycline-resistant Gram-negative bacilli. J Antimicrob Chemother. 2020;75:2609–15. https://doi.org/10.1093/jac/dkaa217.
Spero Therapeutics announces third quarter 2022 operating results and provides business update (Press release 14 November 2022) [Accessed 2 January 2023]. Available from: https://s3.amazonaws.com/b2icontent.irpass.cc/2748/rl113755.pdf.
Spero Therapeutics signs license agreement with Everest Medicines to develop, manufacture and commercialize SPR206 in Asia, with option for SPR741 rights, and initiates SPR206 Phase 1 Clinical Trial (Press release 7 January 2019) [Accessed 2 February 2023]. Available from: https://www.globenewswire.com/news-release/2019/01/07/1681644/0/en/Spero-Therapeutics-Signs-License-Agreement-with-Everest-Medicines-to-Develop-Manufacture-and-Commercialize-SPR206-in-Asia-with-Option-for-SPR741-Rights-and-Initiates-SPR206-Phase-1.html.
Lepak AJ, Wang W, Andes DR. Pharmacodynamic evaluation of MRX-8, a novel polymyxin, in the neutropenic mouse thigh and lung infection models against Gram-negative pathogens. Antimicrob Agents Chemother. 2020;64:e01517–20. https://doi.org/10.1128/AAC.01517-20.
Duncan LR, Wang W, Sader HS. In vitro potency and spectrum of the novel polymyxin MRX-8 tested against clinical isolates of Gram-negative bacteria. Antimicrob Agents Chemother. 2022;66:e00139–22. https://doi.org/10.1128/aac.00139-22.
Wu S, Yin D, Zhi P, Guo Y, Yang Y, Zhu D, et al. In vitro activity of MRX-8 and comparators against clinical isolated Gram-negative bacilli in China. Front Cell Infect Microbiol. 2022;12:829592. https://doi.org/10.3389/fcimb.2022.829592.
MicuRx Pipeline [Accessed 2 January 2023]. Available from: https://www.micurx.com/pipeline.
Gordeev MF, Liu J, Wang X, Yuan Z. inventors; MicuRx Pharmaceuticals, assignee. Antimicrobial polymyxins for treatment of bacterial infections. USA patent 9771394. 2019.
Griffith D, Carmeli Y, Gehrke S, Morgan E, Dudley M, Loutit J. 217. A phase 1 study of the safety, tolerability, and pharmacokinetics of multiple doses of the lipopeptide QPX9003 in healthy adult subjects. Open Forum Infect Dis. 2022;9. https://doi.org/10.1093/ofid/ofac492.295.
Roberts KD, Zhu Y, Azad MAK, Han M-L, Wang J, Wang L, et al. A synthetic lipopeptide targeting top-priority multidrug-resistant Gram-negative pathogens. Nat Commun. 2022;13:1625. https://doi.org/10.1038/s41467-022-29234-3.
Roche Pipeline [Accessed 3 January 2023]. Available from: https://www.roche.com/solutions/pipeline.
Szałaj N, Benediktsdottir A, Rusin D, Karlén A, Mowbray SL, Więckowska A. Bacterial type I signal peptidase inhibitors - Optimized hits from nature. Eur J Med Chem. 2022;238:114490. https://doi.org/10.1016/j.ejmech.2022.114490.
Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature. 2018;561:189–94. https://doi.org/10.1038/s41586-018-0483-6.
Tan YX, Peters DS, Walsh SI, Holcomb M, Santos-Martins D, Forli S, et al. Initial analysis of the arylomycin D antibiotics. J Nat Prod. 2020;83:2112–21. https://doi.org/10.1021/acs.jnatprod.9b01174.
Pfaller MA, Li L, Liu Q, Zhang J, Huband MD, Lindley JM, et al. In vitro activity of a novel aminomethylcycline antibacterial (KBP-7072), a third-generation tetracycline, against clinical isolates with molecularly characterized tetracycline resistance mechanisms. JAC Antimicrob Resist. 2021;3:dlab177. https://doi.org/10.1093/jacamr/dlab177.
Huband MD, Thompson JD, Gurung ND, Liu Q, Li L, Zhang J, et al. Activity of the novel aminomethylcycline KBP-7072 and comparators against 1057 geographically diverse recent clinical isolates from the SENTRY Surveillance Program, 2019. Antimicrob Agents Chemother. 2022;66:e01397–21. https://doi.org/10.1128/AAC.01397-21.
Han R, Ding L, Yang Y, Guo Y, Yin D, Wu S, et al. In vitro activity of KBP-7072 against 536 Acinetobacter baumannii complex isolates collected in China. Microbiol Spectr. 2022;10:e01471–21. https://doi.org/10.1128/spectrum.01471-21.
Kaminishi T, Schedlbauer A, Ochoa-Lizarralde B, Astigarraga ED, Çapuni R, Yang F et al. The third-generation tetracycline KBP-7072 exploits and reveals a new potential of the primary tetracycline binding pocket. bioRxiv. 2018:508218. https://doi.org/10.1101/508218.
Juhas M, Widlake E, Teo J, Huseby DL, Tyrrell JM, Polikanov YS, et al. In vitro activity of apramycin against multidrug-, carbapenem- and aminoglycoside-resistant Enterobacteriaceae and Acinetobacter baumannii. J Antimicrob Chemother. 2019;74:944–52. https://doi.org/10.1093/jac/dky546.
Gysin M, Hon PY, Tan P, Sengduangphachanh A, Simmalavong M, Hinfonthong P, et al. Apramycin susceptibility of multidrug-resistant Gram-negative blood culture isolates in five countries in Southeast Asia. Int J Antimicrob Agents. 2022;60:106659. https://doi.org/10.1016/j.ijantimicag.2022.106659.
Tian E, Muhammad I, Hu W, Wu Z, Li R, Lu X, et al. Tentative epidemiologic cut-off value and resistant characteristic detection of apramycin against Escherichia coli from chickens. FEMS Microbiol Lett. 2019;366:fnz196. https://doi.org/10.1093/femsle/fnz196.
Committee for veterinary medicinal products. Apramycin. Summary Report (1) 1997 [Accessed 24 January 2023]. Available from: https://www.ema.europa.eu/en/documents/mrl-report/apramycin-summary-report-1-committee-veterinary-medicinal-products_en.pdf.
Thompson RQ, Presti EA. Nebramycin, a new broad-spectrum antibiotic complex. III. Isolation and chemical-physical properties. Antimicrob Agents Chemother. 1968:332–40.
O’Connor S, Lam LKT, Jones ND, Chaney MO. Apramycin, a unique aminocyclitol antibiotic. J Org Chem. 1976;41:2087–92. https://doi.org/10.1021/jo00874a003.
Huang DB, Brothers KM, Mandell JB, Taguchi M, Alexander PG, Parker DM, et al. Engineered peptide PLG0206 overcomes limitations of a challenging antimicrobial drug class. PLoS One. 2022;17:e0274815. https://doi.org/10.1371/journal.pone.0274815.
Heinrich F, Salyapongse A, Kumagai A, Dupuy FG, Shukla K, Penk A, et al. Synergistic biophysical techniques reveal structural mechanisms of engineered cationic antimicrobial peptides in lipid model membranes. Chem Eur J. 2020;26:6247–56. https://doi.org/10.1002/chem.202000212.
Huang D, Dobbins D, Ghahramani P, Friedland I, Steckbeck J. A phase 1 study of the safety, tolerability, and pharmacokinetics of single ascending doses of a first-in-human engineered cationic peptide, PLG0206, intravenously administered in healthy subjects. Antimicrob Agents Chemother. 2022;66:e01441–21. https://doi.org/10.1128/AAC.01441-21.
Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015;59:1329–33. https://doi.org/10.1128/AAC.03937-14.
Huang D, Pachuda N, Sauer JM, Dobbins D, Steckbeck J. The engineered antibiotic peptide PLG0206 eliminates biofilms and is a potential treatment for periprosthetic joint infections. Antibiotics. 2022;11:41. https://doi.org/10.3390/antibiotics11010041.
Zhao L, Huang Y, Gao S, Cui Y, He D, Wang L, et al. Comparison on effect of hydrophobicity on the antibacterial and antifungal activities of α-helical antimicrobial peptides. Sci China Chem. 2013;56:1307–14. https://doi.org/10.1007/s11426-013-4884-y.
Mai X-T, Huang J, Tan J, Huang Y, Chen Y. Effects and mechanisms of the secondary structure on the antimicrobial activity and specificity of antimicrobial peptides. J Pept Sci. 2015;21:561–8. https://doi.org/10.1002/psc.2767.
Hao X, Yan Q, Zhao J, Wang W, Huang Y, Chen Y. TAT modification of alpha-helical anticancer peptides to improve specificity and efficacy. PLoS One. 2015;10:e0138911. https://doi.org/10.1371/journal.pone.0138911.
Andolina G, Bencze L-C, Zerbe K, Müller M, Steinmann J, Kocherla H, et al. A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from Pseudomonas aeruginosa. ACS Chem Biol. 2018;13:666–75. https://doi.org/10.1021/acschembio.7b00822.
Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327:1010–3. https://doi.org/10.1126/science.1182749.
Botos I, Noinaj N, Buchanan SK. Insertion of proteins and lipopolysaccharide into the bacterial outer membrane. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160224. https://doi.org/10.1098/rstb.2016.0224.
Spexis Pipeline. Inhaled Murepavadin [Accessed 2 January 2023]. Available from: https://spexisbio.com/pol7080/.
Polyphor temporarily halts enrollment in the phase III studies of murepavadin for the treatment of patients with nosocomial pneumonia (Press release 9 May 2019) [Accessed 2 February 2023]. Available from: https://spexisbio.com/news/corporate-news-details/?newsid=1775911.
TAXIS Pharmaceuticals Pipeline [Accessed 2 January 2023]. Available from: https://www.taxispharma.com/research-development/our-pipeline/.
Carro L. Recent progress in the development of small-molecule FtsZ inhibitors as chemical tools for the development of novel antibiotics. Antibiotics. 2019;8:217 https://doi.org/10.3390/antibiotics8040217.
Elsen NL, Lu J, Parthasarathy G, Reid JC, Sharma S, Soisson SM, et al. Mechanism of action of the cell-division inhibitor PC190723: Modulation of FtsZ assembly cooperativity. J Am Chem Soc. 2012;134:12342–5. https://doi.org/10.1021/ja303564a.
Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science. 2008;321:1673–5. https://doi.org/10.1126/science.1159961.
Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, et al. Creating an antibacterial with in vivo efficacy: synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J Med Chem. 2010;53:3927–36. https://doi.org/10.1021/jm9016366.
Stokes NR, Baker N, Bennett JM, Berry J, Collins I, Czaplewski LG, et al. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob Agents Chemother. 2013;57:317–25. https://doi.org/10.1128/aac.01580-12.
Kaul M, Mark L, Zhang Y, Parhi AK, Lyu YL, Pawlak J, et al. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2015;59:4845–55. https://doi.org/10.1128/AAC.00708-15.
Lepak AJ, Parhi A, Madison M, Marchillo K, VanHecker J, Andes DR. In vivo pharmacodynamic evaluation of an FtsZ inhibitor, TXA-709, and its active metabolite, TXA-707, in a murine neutropenic thigh infection mode. Antimicrob Agents Chemother. 2015;59:6568–74. https://doi.org/10.1128/aac.01464-15.
Mattei P. Discovery of RG6006, a tethered macrocyclic peptide targeting Acinetobacter baumannii (15th Winter Conference on Medicinal & Bioorganic Chemistry, Steamboat Springs Resort, CO, USA) 2022 [Accessed 23 February 2023]. Available from: https://mbcfconference.com/wp-content/uploads/2022/04/Patrizio-Mattei-2023-MBCF-abstract.pdf.
Bleicher K, Cheang D, Giorgio PD, Hu T, Mattei P, Schmitz P, et al. inventors; Hoffmann La Roche Inc, assignee. Peptide macrocycles against Acinetobacter baumannii. USA patent 11505573. 2022.
Desai J, S S, Kumar S, Sharma R. Novel bacterial topoisomerase inhibitors (NBTIs)—a comprehensive review. Eur J Med Chem Rep. 2021;3:100017. https://doi.org/10.1016/j.ejmcr.2021.100017.
Mohamed SHP, Bharatham N, Katagihallimath N, Sharma S, Nandishaiah R, Ramachandran V et al. inventors; Bugworks Research Inc, assignee. Heterocyclic compounds useful as anti-bacterial agents and method for production thereof. USA patent 10912780. 2021.
Hameed S, Sharma S, Nandishaiah R, Katagihallimath N, Bharatham N, Shanbhag A, et al. BWC0977, a novel dual target topoisomerase inhibitor: antimicrobial potency, spectrum and mechanism of action (ECCMID 2019, Amsterdam, Netherlands, Poster #P1844). 2019 [Accessed 23 February 2023]. Available from: https://bugworksresearch.com/wp-content/uploads/2021/11/e-20190415_ECCMID_P1844.pdf.
Raveendran S, Thomas T, Sharma M, Rajagopal S, Prabhumurthy S, Nagaraj S, et al. In vitro activity of BWC0977 (a novel bacterial topoisomerase inhibitor) and comparators against recent clinical Enterobacteriaceae and non-fermenter Isolates from two hospitals in Bengaluru, India (ECCMID 2019, Amsterdam, Netherlands, Poster #P1845). 2019 [Accessed 23 February 2023]. Available from: https://bugworksresearch.com/wp-content/uploads/2021/11/d-20190415_ECCMID_P1845.pdf.
Wiederhold NP, McElmeel M, Patterson TF, Slayden RA, Cummings JE, Ramachandran V, et al. In vitro activity of BWC0977, a novel bacterial topoisomerase inhibitor, against molecularly characterized Enterobacteriaceae & non-fermenter isolates of the CDC collection and key biodefense pathogens (ECCMID 2019, Amsterdam, Netherlands, Poster #1845). 2019 [Accessed 23 February 2023]. Available from: https://bugworksresearch.com/wp-content/uploads/2021/11/c-20190415_ECCMID_P1846.pdf.
Makarov V, Mikušová K. Development of macozinone for TB treatment: an update. Appl Sci. 2020;10:2269. https://doi.org/10.3390/app10072269.
Koryakova A, Shcherbakova V, Riabova O, Kazaishvili Y, Bolgarin R, Makarov V. Antituberculosis macozinone extended-release tablets to enhance bioavailability: a pilot pharmacokinetic study in beagle dogs. Microbiol Spectr. 2023;11:e02327–22. https://doi.org/10.1128/spectrum.02327-22.
Working Group on New TB Drugs, TBI-223 [Accessed 2 January 2023]. Available from: https://www.newtbdrugs.org/pipeline/compound/tbi-223.
Gordon O, Dikeman DA, Ortines RV, Wang Y, Youn C, Mumtaz M, et al. The novel oxazolidinone TBI-223 is effective in three preclinical mouse models of methicillin-resistant Staphylococcus aureus infection. Microbiol Spectr. 2022;10:e02451–21. https://doi.org/10.1128/spectrum.02451-21.
Almeida D, Converse PJ, Li S-Y, Upton AM, Fotouhi N, Nuermberger EL. Comparative efficacy of the novel diarylquinoline TBAJ-876 and bedaquiline against a resistant Rv0678 mutant in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2021;65:e01412–21. https://doi.org/10.1128/AAC.01412-21.
Sarathy JP, Ganapathy US, Zimmerman MD, Dartois V, Gengenbacher M, Dick T. TBAJ-876, a 3,5-dialkoxypyridine analogue of bedaquiline, is active against Mycobacterium abscessus. Antimicrob Agents Chemother. 2020;64:e02404–19. https://doi.org/10.1128/AAC.02404-19.
Sutherland HS, Tong AST, Choi PJ, Blaser A, Conole D, Franzblau SG, et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg Med Chem. 2019;27:1292–307. https://doi.org/10.1016/j.bmc.2019.02.026.
Choi PJ, Conole D, Sutherland HS, Blaser A, Tong AST, Cooper CB, et al. Synthetic studies to help elucidate the metabolism of the preclinical candidate TBAJ-876—a less toxic and more potent analogue of bedaquiline. Molecules. 2020;25:1423. https://doi.org/10.3390/molecules25061423.
Sarathy JP, Ragunathan P, Shin J, Cooper CB, Upton AM, Grüber G, et al. TBAJ-876 retains bedaquiline’s activity against subunits c and ε of Mycobacterium tuberculosis F-ATP synthase. Antimicrob Agents Chemother. 2019;63:e01191–19. https://doi.org/10.1128/AAC.01191-19.
Sarathy JP, Ragunathan P, Cooper CB, Upton AM, Grüber G, Dick T. TBAJ-876 displays bedaquiline-like mycobactericidal potency without retaining the parental drug’s uncoupler activity. Antimicrob Agents Chemother. 2020;64:e01540–19. https://doi.org/10.1128/AAC.01540-19.
Krah A, Grüber G, Bond PJ. Binding properties of the anti-TB drugs bedaquiline and TBAJ-876 to a mycobacterial F-ATP synthase. Curr Res Struct Biol. 2022;4:278–84. https://doi.org/10.1016/j.crstbi.2022.09.001.
Xu J, Converse PJ, Upton AM, Mdluli K, Fotouhi N, Nuermberger EL. Comparative efficacy of the novel diarylquinoline TBAJ-587 and bedaquiline against a resistant Rv0678 mutant in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2021;65:e02418–20. https://doi.org/10.1128/AAC.02418-20.
Working Group on New TB Drugs, GSK-286 [Accessed 3 January 2023]. Available from: https://www.newtbdrugs.org/pipeline/compound/gsk-286.
Nuermberger EL, Martínez-Martínez MS, Sanz O, Urones B, Esquivias J, Soni H, et al. GSK2556286 is a novel antitubercular drug candidate effective in vivo with the potential to shorten tuberculosis treatment. Antimicrob Agents Chemother. 2022;66:e00132–22. https://doi.org/10.1128/aac.00132-22.
Mehta V, Khanppnavar B, Schuster D, Kantarci I, Vercellino I, Kosturanova A, et al. Structure of Mycobacterium tuberculosis Cya, an evolutionary ancestor of the mammalian membrane adenylyl cyclases. eLife. 2022;11:e77032. https://doi.org/10.7554/eLife.77032.
Brown KL, Wilburn KM, Montague CR, Grigg Jason C, Sanz O, Pérez-Herrán E, et al. Cyclic AMP-mediated inhibition of cholesterol catabolism in Mycobacterium tuberculosis by the novel drug candidate GSK2556286. Antimicrob Agents Chemother. 2023;67:e01294–22. https://doi.org/10.1128/aac.01294-22.
VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 2015;11:e1004679. https://doi.org/10.1371/journal.ppat.1004679.
Working Group on New TB Drugs, BVL-GSK098 [Accessed 4 January 2023]. Available from: https://www.newtbdrugs.org/pipeline/compound/bvl-gsk098.
Blondiaux N, Moune M, Desroses M, Frita R, Flipo M, Mathys V, et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science. 2017;355:1206–11. https://doi.org/10.1126/science.aag1006.
Guieu B, Jourdan J-P, Dreneau A, Willand N, Rochais C, Dallemagne P. Desirable drug–drug interactions or when a matter of concern becomes a renewed therapeutic strategy. Drug Disco Today. 2021;26:315–28. https://doi.org/10.1016/j.drudis.2020.11.026.
CARB-X funds GSK to develop a new drug for urinary tract infections (UTI) caused by Escherichia coli bacteria (Press release 22 September 2020) [Accessed 4 January 2023]. Available from: https://carb-x.org/carb-x-news/carb-x-announces-funding-for-gsk-to-develop-a-new-drug-for-urinary-tract-infections-uti-caused-by-escherichia-coli-bacteria/.
Ronald A. The etiology of urinary tract infection: traditional and emerging pathogens. Am J Med. 2002;113:14–9. https://doi.org/10.1016/S0002-9343(02)01055-0.
Foroogh N, Rezvan M, Ahmad K, Mahmood S. Structural and functional characterization of the FimH adhesin of uropathogenic Escherichia coli and its novel applications. Micro Pathog. 2021;161:105288. https://doi.org/10.1016/j.micpath.2021.105288.
Mydock-McGrane LK, Hannan TJ, Janetka JW. Rational design strategies for FimH antagonists: new drugs on the horizon for urinary tract infection and Crohn’s disease. Expert Opin Drug Disco. 2017;12:711–31. https://doi.org/10.1080/17460441.2017.1331216.
Kalas V, Hibbing ME, Maddirala AR, Chugani R, Pinkner JS, Mydock-McGrane LK, et al. Structure-based discovery of glycomimetic FmlH ligands as inhibitors of bacterial adhesion during urinary tract infection. Proc Natl Acad Sci USA. 2018;115:E2819–E28. https://doi.org/10.1073/pnas.1720140115.
Maddirala AR, Klein R, Pinkner JS, Kalas V, Hultgren SJ, Janetka JW. Biphenyl Gal and GalNAc FmlH lectin antagonists of uropathogenic E. coli (UPEC): Optimization through iterative rational drug design. J Med Chem. 2019;62:467–79. https://doi.org/10.1021/acs.jmedchem.8b01561.
Bishop MJ, Stewart EL, Widdowson KL, Janetka JW, McGrane LK. inventors; GlaxoSmithKline Intellectual Property Development Ltd, Fimbrion Therapeutics Inc, assignee. C-mannoside compounds useful for the treatment of urinary tract infections. USA patent 11111262. 2021.
Liu C-I, Liu GY, Song Y, Yin F, Hensler ME, Jeng W-Y, et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–4. https://doi.org/10.1126/science.1153018.
Elmesseri RA, Saleh SE, Elsherif HM, Yahia IS, Aboshanab KM. Staphyloxanthin as a potential novel target for deciphering promising anti-Staphylococcus aureus agents. Antibiotics. 2022;11:298. https://doi.org/10.3390/antibiotics11030298.
Kao YTR, Gao P, Li X, Liu M. inventors; Versitech Ltd, assignee. Compounds affecting pigment production and methods for treatment of bacterial diseases. USA patent 11052078. 2021.
Gao P, Davies J, Kao RYT. Dehydrosqualene desaturase as a novel target for anti-virulence therapy against Staphylococcus aureus. mBio. 2017;8:e01224–17. https://doi.org/10.1128/mBio.01224-17.
Barnes MD, Kumar V, Bethel CR, Moussa SH, O’Donnell J, Rutter JD, et al. Targeting multidrug-resistant Acinetobacter spp.: sulbactam and the diazabicyclooctenone β-lactamase inhibitor ETX2514 as a novel therapeutic agent. mBio. 2019;10:e00159–19. https://doi.org/10.1128/mBio.00159-19.
McLeod SM, Shapiro AB, Moussa SH, Johnstone M, McLaughlin RE, de Jonge BLM, et al. Frequency and mechanism of spontaneous resistance to sulbactam combined with the novel β-lactamase inhibitor ETX2514 in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2018;62:e01576–17. https://doi.org/10.1128/aac.01576-17.
Granata G, Taglietti F, Schiavone F, Petrosillo N. Durlobactam in the treatment of multidrug-resistant Acinetobacter baumannii infections: a systematic review. J Clin Med. 2022;11:3258. https://doi.org/10.3390/jcm11123258.
Innoviva announces FDA acceptance and priority review of New Drug Application for Sulbactam-Durlobactam (SUL-DUR) (Press release 30 November 2022) [Accessed 7 January 2023]. Available from: https://investor.inva.com/news-releases/news-release-details/innoviva-announces-fda-acceptance-and-priority-review-new-drug.
Durand-Réville TF, Guler S, Comita-Prevoir J, Chen B, Bifulco N, Huynh H, et al. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat Microbiol. 2017;2:17104. https://doi.org/10.1038/nmicrobiol.2017.104.
Shapiro AB, Gao N, Jahić H, Carter NM, Chen A, Miller AA. Reversibility of covalent, broad-spectrum serine β-lactamase inhibition by the diazabicyclooctenone ETX2514. ACS Infect Dis. 2017;3:833–44. https://doi.org/10.1021/acsinfecdis.7b00113.
O’Donnell J, Preston RA, Mamikonyan G, Stone E, Isaacs R. Pharmacokinetics, safety, and tolerability of intravenous durlobactam and sulbactam in subjects with renal impairment and healthy matched control subjects. Antimicrob Agents Chemother. 2019;63:e00794–19. https://doi.org/10.1128/AAC.00794-19.
Liu B, Trout REL, Chu G-H, McGarry D, Jackson RW, Hamrick J, et al. Discovery of taniborbactam (VNRX-5133): A broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem. 2020;63:2789–801. https://doi.org/10.1021/acs.jmedchem.9b01518.
Venatorx Pharmaceuticals announces positive results for Phase 3 clinical trial (CERTAIN-1) of cefepime-taniborbactam for treatment of cUTI (Press release 10 March 2022) [Accessed 7 January 2023]. Available from: https://www.venatorx.com/press-releases/venatorx-pharmaceuticals-announces-positive-results-for-phase-3-clinical-trial-certain-1-of-cefepime-taniborbactam-for-treatment-of-cuti/.
Venatorx Pharmaceuticals presents data on investigational cefepime-taniborbactam at IDWeek 2022 (Press release 20 October 2022) [Accessed 7 January 2023]. Available from: https://www.venatorx.com/press-releases/venatorx-pharmaceuticals-presents-data-on-investigational-cefepime-taniborbactam-at-idweek-2022/.
Meletiadis J, Paranos P, Georgiou P-C, Vourli S, Antonopoulou S, Michelaki A, et al. In vitro comparative activity of the new beta-lactamase inhibitor taniborbactam with cefepime or meropenem against Klebsiella pneumoniae and cefepime against Pseudomonas aeruginosa metallo-beta-lactamase-producing clinical isolates. Int J Antimicrob Agents. 2021;58:106440 https://doi.org/10.1016/j.ijantimicag.2021.106440.
Roach EJ, Uehara T, Daigle DM, Six DA, Khursigara CM. The next-generation β-lactamase inhibitor taniborbactam restores the morphological effects of cefepime in KPC-producing Escherichia coli. Microbiol Spectr. 2021;9:e00918–21. https://doi.org/10.1128/Spectrum.00918-21.
Hernández-García M, García-Castillo M, Ruiz-Garbajosa P, Bou G, Siller-Ruiz M, Pitart C, et al. In vitro activity of cefepime-taniborbactam against carbapenemase-producing Enterobacterales and Pseudomonas aeruginosa isolates recovered in Spain. Antimicrob Agents Chemother. 2022;66:e02161–21. https://doi.org/10.1128/aac.02161-21.
Lence E, González-Bello C. Bicyclic boronate β-lactamase inhibitors: the present hope against deadly bacterial pathogens. Adv Ther. 2021;4:2000246. https://doi.org/10.1002/adtp.202000246.
Krajnc A, Brem J, Hinchliffe P, Calvopina K, Panduwawala T, Lang PA, et al. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J Med Chem. 2019;62:8544–56. https://doi.org/10.1021/acs.jmedchem.9b00911.
Mameli M, Vezzelli A, Verze’ S, Biondi S, Motta P, Greco A, et al. Liquid chromatography–tandem mass spectrometry for the simultaneous quantitation of enmetazobactam and cefepime in human plasma. J Pharm Biomed Anal. 2019;174:655–62. https://doi.org/10.1016/j.jpba.2019.06.041.
Crandon JL, Nicolau DP. In vivo activities of simulated human doses of cefepime and cefepime-AAI101 against multidrug-resistant Gram-negative Enterobacteriaceae. Antimicrob Agents Chemother. 2015;59:2688–94. https://doi.org/10.1128/AAC.00033-15.
Crandon JL, Nicolau DP. In vitro activity of cefepime/AAI101 and comparators against cefepime non-susceptible Enterobacteriaceae. Pathogens. 2015;4:620–5. https://doi.org/10.3390/pathogens4030620.
Kaye KS, Belley A, Barth P, Lahlou O, Knechtle P, Motta P, et al. Effect of cefepime/enmetazobactam vs piperacillin/tazobactam on clinical cure and microbiological eradication in patients with complicated urinary tract infection or acute pyelonephritis: A randomized clinical trial. JAMA. 2022;328:1304–14. https://doi.org/10.1001/jama.2022.17034.
Papp-Wallace KM, Nguyen NQ, Jacobs MR, Bethel CR, Barnes MD, Kumar V, et al. Strategic approaches to overcome resistance against Gram-negative pathogens using β-lactamase inhibitors and β-lactam enhancers: Activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J Med Chem. 2018;61:4067–86. https://doi.org/10.1021/acs.jmedchem.8b00091.
Moya B, Barcelo IM, Bhagwat S, Patel M, Bou G, Papp-Wallace KM, et al. Potent β-lactam enhancer activity of zidebactam and WCK 5153 against Acinetobacter baumannii, including carbapenemase-producing clinical isolates. Antimicrob Agents Chemother. 2017;61:e01238–17. https://doi.org/10.1128/aac.01238-17.
Sader HS, Mendes RE, Duncan LR, Carvalhaes CG, Castanheria M. Antimicrobial activity of cefepime/zidebactam (WCK 5222), a β-lactam/β-lactam enhancer combination, against clinical isolates of Gram-negative bacteria collected worldwide (2018–19). J Antimicrob Chemother. 2022;77:2642–9. https://doi.org/10.1093/jac/dkac233.
Guo Y, Han R, Jiang B, Ding L, Yang F, Zheng B, et al. In vitro activity of new β-lactam–β-lactamase inhibitor combinations and comparators against clinical isolates of Gram-negative bacilli: Results from the China Antimicrobial Surveillance Network (CHINET) in 2019. Microbiol Spectr. 2022;10:e01854–22. https://doi.org/10.1128/spectrum.01854-22.
Mushtaq S, Garello P, Vickers A, Woodford N, Livermore DM. Activity of ertapenem/zidebactam (WCK 6777) against problem Enterobacterales. J Antimicrob Chemother. 2022;77:2772–8. https://doi.org/10.1093/jac/dkac280.
Morinaka A, Tsutsumi Y, Yamada M, Suzuki K, Watanabe T, Abe T, et al. OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam ‘enhancer’. J Antimicrob Chemother. 2015;70:2779–86. https://doi.org/10.1093/jac/dkv166.
Asempa TE, Motos A, Abdelraouf K, Bissantz C, Zampaloni C, Nicolau DP. Efficacy of Human-simulated epithelial lining fluid exposure of meropenem-nacubactam combination against class a serine β-lactamase-producing Enterobacteriaceae in the neutropenic murine lung infection model. Antimicrob Agents Chemother. 2019;63:e02382–18. https://doi.org/10.1128/aac.02382-18.
Livermore DM, Warner M, Mushtaq S, Woodford N. Interactions of OP0595, a novel triple-action diazabicyclooctane, with β-lactams against OP0595-resistant Enterobacteriaceae mutants. Antimicrob Agents Chemother. 2016;60:554–60. https://doi.org/10.1128/AAC.02184-15.
Garde D. Roche inks a $750M antibiotics deal as it re-embraces the field (Fierce Biotech 13 Jan 2015) [Accessed 2 February 2023]. Available from: http://www.fiercebiotech.com/story/roche-inks-750m-antibiotics-deal-it-re-embraces-field/2015-01-13.
Garde D. Roche, Meiji and Fedora join forces to tackle increasing bacterial resistance to antibiotics [Accessed 2 February 2023]. Available from: https://www.fiercebiotech.com/biotech/roche-meiji-and-fedora-join-forces-to-tackle-increasing-bacterial-resistance-to-antibiotics.
Meiji Seika Pipeline, As of November 8, 2022 [Accessed 8 January 2023]. Available from: https://www.meiji.com/global/pharmaceuticals/pipeline.html.
Fedora Pharmaceuticals Website, Our Story [Accessed 8 Januray 2023]. Available from: https://www.fedorapharma.com/about-us.
Hecker SJ, Reddy KR, Lomovskaya O, Griffith DC, Rubio-Aparicio D, Nelson K, et al. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J Med Chem. 2020;63:7491–507. https://doi.org/10.1021/acs.jmedchem.9b01976.
Lomovskaya O, Tsivkovski R, Sun D, Reddy R, Totrov M, Hecker S, et al. QPX7728, an ultra-broad-spectrum β-lactamase inhibitor for intravenous and oral therapy: Overview of biochemical and microbiological characteristics. Front Microbiol. 2021;12. https://doi.org/10.3389/fmicb.2021.697180.
Lomovskaya O, Rubio-Aparicio D, Tsivkovski R, Loutit J, Dudley M. The ultrabroad-spectrum beta-lactamase inhibitor QPX7728 restores the potency of multiple oral beta-lactam antibiotics against beta-lactamase-producing strains of resistant Enterobacterales. Antimicrob Agents Chemother. 2022;66:e02168–21. https://doi.org/10.1128/aac.02168-21.
Sun D, Tsivkovski R, Pogliano J, Tsunemoto H, Nelson K, Rubio-Aparicio D, et al. Intrinsic antibacterial activity of xeruborbactam in vitro: assessing spectrum and mode of action. Antimicrob Agents Chemother. 2022;66:e00879–22. https://doi.org/10.1128/aac.00879-22.
Reddy KR, Parkinson J, Sabet M, Tarazi Z, Boyer SH, Lomovskaya O, et al. Selection of QPX7831, an orally bioavailable prodrug of boronic acid β-lactamase inhibitor QPX7728. J Med Chem. 2021;64:17523–9. https://doi.org/10.1021/acs.jmedchem.1c01722.
Durand-Réville TF, Comita-Prevoir J, Zhang J, Wu X, May-Dracka TL, Romero JAC, et al. Discovery of an orally available diazabicyclooctane inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J Med Chem. 2020;63:12511–25. https://doi.org/10.1021/acs.jmedchem.0c00579.
Miller AA, Shapiro AB, McLeod SM, Carter NM, Moussa SH, Tommasi R, et al. In Vitro characterization of ETX1317, a broad-spectrum β-lactamase inhibitor that restores and enhances β-lactam activity against multi-drug-resistant Enterobacteriales, including carbapenem-resistant strains. ACS Infect Dis. 2020;6:1389–97. https://doi.org/10.1021/acsinfecdis.0c00020.
Trout RE, Zulli A, Mesaros E, Jackson RW, Boyd S, Liu B, et al. Discovery of VNRX-7145 (VNRX-5236 etzadroxil): an orally bioavailable β-lactamase inhibitor for Enterobacterales expressing Ambler class A, C, and D enzymes. J Med Chem. 2021;64:10155–66. https://doi.org/10.1021/acs.jmedchem.1c00437.
Karlowsky JA, Hackel MA, Sahm DF. In vitro activity of ceftibuten/VNRX-5236 against urinary tract infection isolates of antimicrobial-resistant Enterobacterales. Antimicrob Agents Chemother. 2022;66:e01304–21. https://doi.org/10.1128/AAC.01304-21.
Chatwin CL, Hamrick JC, Trout REL, Myers CL, Cusick SM, Weiss WJ, et al. Microbiological characterization of VNRX-5236, a broad-spectrum β-lactamase inhibitor for rescue of the orally bioavailable cephalosporin ceftibuten as a carbapenem-sparing agent against strains of Enterobacterales expressing extended-spectrum β-lactamases and serine carbapenemases. Antimicrob Agents Chemother. 2021;65:e00552–21. https://doi.org/10.1128/AAC.00552-21.
Karlowsky JA, Wisek MG, Hackel MA, Pevear DC, Moeck G, Sahm DF. Ceftibuten-ledaborbactam activity against multidrug-resistant and extended-spectrum-β-lactamase-positive clinical isolates of Enterobacterales from a 2018–2020 global surveillance collection. Antimicrob Agents Chemother. 2022;66:e00934–22. https://doi.org/10.1128/aac.00934-22.
Wu Y, Huang S, Chen X, Hu Y, Liu X. inventors; Suzhou Sinovent Pharmaceuticals, assignee. β-Lactamase inhibitor and use thereof. USA patent 11078202. 2021.
Heo Y-A. Imipenem/cilastatin/relebactam: a review in Gram-negative bacterial infections. Drugs. 2021;81:377–88. https://doi.org/10.1007/s40265-021-01471-8.
Dietl B, Martínez LM, Calbo E, Garau J. Update on the role of ceftazidime-avibactam in the management of carbapenemase-producing Enterobacterales. Future Microbiol. 2020;15:473–84. https://doi.org/10.2217/fmb-2020-0012.
Gordon EM, Duncton MAJ, Gallop MA. Orally absorbed derivatives of the β-lactamase inhibitor avibactam. Design of novel prodrugs of sulfate containing drugs. J Med Chem. 2018;61:10340–4. https://doi.org/10.1021/acs.jmedchem.8b01389.
Biospace. Arixa Pharmaceuticals announces acquisition by Pfizer’s hospital business 2020 [Accessed 7 January 2023]. Available from: https://www.biospace.com/article/releases/arixa-pharmaceuticals-announces-acquisition-by-pfizer-s-hospital-business/.
Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010;8:423–35. https://doi.org/10.1038/nrmicro2333.
Bisacchi GS, Manchester JI. A new-class antibacterial—almost. Lessons in drug discovery and development: a critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infect Dis. 2015;1:4–41. https://doi.org/10.1021/id500013t.
Zhou S, Wang W, Zhou X, Zhang Y, Lai Y, Tang Y, et al. Structure of Mycobacterium tuberculosis cytochrome bcc in complex with Q203 and TB47, two anti-TB drug candidates. eLife. 2021;10:e69418. https://doi.org/10.7554/eLife.69418.
Hards K, Adolph C, Harold LK, McNeil MB, Cheung C-Y, Jinich A, et al. Two for the price of one: attacking the energetic-metabolic hub of mycobacteria to produce new chemotherapeutic agents. Prog Biophys Mol Biol. 2019;152:35–44. https://doi.org/10.1016/j.pbiomolbio.2019.11.003.
IQVIA Institute. Global Oncology Trends 2022 2022 [Accessed 28 February 2023]. Available from: https://www.iqvia.com/insights/the-iqvia-institute/reports/global-oncology-trends-2022.
World Health Organization. COVID-19 vaccine tracker and landscape 2023 [Accessed 28 February 2023]. Available from: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines.
USA FDA. Limited Population Pathway for Antibacterial and Antifungal Drugs – the LPAD Pathway 2020 [Accessed 23 February 2023]. Available from: https://www.fda.gov/drugs/development-resources/limited-population-pathway-antibacterial-and-antifungal-drugs-lpad-pathway.
Noviello S, Huang DB, Corey GR. Iclaprim: a differentiated option for the treatment of skin and skin structure infections. Expert Rev Anti Infect Ther. 2018;16:793–803. https://doi.org/10.1080/14787210.2018.1536545.
Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, et al. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother. 2005;56:968–74. https://doi.org/10.1093/jac/dki319.
Sacksteder KA, Protopopova M, Barry CE, Andries K, Nacy CA. Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012;7:823–37. https://doi.org/10.2217/fmb.12.56.
Infectex announces positive phase 2b-3 clinical trial results of SQ109 for the treatment of multidrug-resistant pulmonary tuberculosis (Press Release 21 March 2017) [Accessed 2 February 2023]. Available from: https://www.newtbdrugs.org/sites/default/files/downloads/Infectex%20Press%20Release.2017.0321.pdf.
Borisov SE, Bogorodskaya EM, Volchenkov GV, Kulchavenya EV, Maryandyshev AO, Skornyakov SN, et al. Efficiency and safety of chemotherapy regimen with SQ109 in those suffering from multiple drug resistant tuberculosis. Tuberc Lung Dis. 2018;96:6–18. https://doi.org/10.21292/2075-1230-2018-96-3-6-18.
Sattar A, Thommes P, Payne L, Warn P, Vickers RJ. SMT19969 for Clostridium difficile infection (CDI): In vivo efficacy compared with fidaxomicin and vancomycin in the hamster model of CDI. J Antimicrob Chemother. 2015;70:1757–62. https://doi.org/10.1093/jac/dkv005.
Bassères E, Endres BT, Khaleduzzaman M, Miraftabi F, Alam MJ, Vickers RJ, et al. Impact on toxin production and cell morphology in Clostridium difficile by ridinilazole (SMT19969), a novel treatment for C. difficile infection. J Antimicrob Chemother. 2016;71:1245–51. https://doi.org/10.1093/jac/dkv498.
Collins DA, Riley TV. Ridinilazole: a novel, narrow‐spectrum antimicrobial agent targeting Clostridium (Clostridioides) difficile. Lett Appl Microbiol. 2022;75:526–36. https://doi.org/10.1111/lam.13664.
Summit Therapeutics Presents Ri-CoDIFy Trial Results for Microbiome-Sparing Ridinilazole at IDWeek 2022 (Press release 20 October 2022) [Accessed 2 February 2023]. Available from: https://www.smmttx.com/app/uploads/2022/10/2022_PR_1020_ID-Week-2022-Results-_-FINAL.pdf.
Reck F, Bermingham A, Blais J, Capka V, Cariaga T, Casarez A, et al. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – identification of LYS228. Bioorg Med Chem Lett. 2018;28:748–55. https://doi.org/10.1016/j.bmcl.2018.01.006.
Blais J, Lopez S, Li C, Ruzin A, Ranjitkar S, Dean CR, et al. In vitro activity of LYS228, a novel monobactam antibiotic, against multidrug-resistant Enterobacteriaceae. Antimicrob Agents Chemother. 2018;62:e00552–18. https://doi.org/10.1128/AAC.00552-18.
Oka D, Changkwanyeun R, Yamaguchi T, Nakajima C, Suzuki Y, Matsumoto M. In vitro antibacterial activity of OPS-2071 against Gram-positive and Gram-negative enteropathogenic bacteria. J Antimicrob Chemother. 2022;77:3248–55. https://doi.org/10.1093/jac/dkac308.
Oka D, Yamaya N, Kuno T, Asakawa Y, Shiragiku T, Chen L, et al. In vitro and in vivo antibacterial activities of a novel quinolone compound, OPS-2071, against Clostridioides difficile. Antimicrob Agents Chemother. 2021;65:e01170–20. https://doi.org/10.1128/AAC.01170-20.
Falagas ME, Skalidis T, Vardakas KZ, Voulgaris GL, Papanikolaou G, Legakis N. Activity of TP-6076 against carbapenem-resistant Acinetobacter baumannii isolates collected from inpatients in Greek hospitals. Int J Antimicrob Agents. 2018;52:269–71. https://doi.org/10.1016/j.ijantimicag.2018.03.009.
Morgan CE, Zhang Z, Bonomo RA, Yu EW. An analysis of the novel fluorocycline TP-6076 bound to both the ribosome and multidrug efflux pump AdeJ from Acinetobacter baumannii. mBio. 2022;13:e03732–21. https://doi.org/10.1128/mbio.03732-21.
Grossman TH, Fyfe C, O’Brien W, Hackel M, Minyard MB, Waites KB, et al. Fluorocycline TP-271 is potent against complicated community-acquired bacterial pneumonia pathogens. mSphere. 2017;2:e00004–17. https://doi.org/10.1128/mSphere.00004-17.
Grossman TH, Anderson MS, Drabek L, Gooldy M, Heine HS, Henning LN, et al. The fluorocycline TP-271 is efficacious in models of aerosolized Bacillus anthracis infection in BALB/c mice and cynomolgus macaques. Antimicrob Agents Chemother. 2017;61:e01103–17. https://doi.org/10.1128/aac.01103-17.
Grossman TH, Anderson MS, Christ D, Gooldy M, Henning LN, Heine HS, et al. The fluorocycline TP-271 is efficacious in models of aerosolized Francisella tularensis SCHU S4 Infection in BALB/c mice and cynomolgus macaques. Antimicrob Agents Chemother. 2017;61:e00448–17. https://doi.org/10.1128/aac.00448-17.
Stainton SM, Abdelraouf K, Utley L, Pucci MJ, Lister T, Nicolau DP. Assessment of the in vivo activity of SPR741 in combination with azithromycin against multidrug-resistant Enterobacteriaceae isolates in the neutropenic murine thigh infection model. Antimicrob Agents Chemother. 2018;62:e00239–18. https://doi.org/10.1128/aac.00239-18.
Corbett D, Wise A, Langley T, Skinner K, Trimby E, Birchall S, et al. Potentiation of antibiotic activity by a novel cationic peptide: Potency and spectrum of activity of SPR741. Antimicrob Agents Chemother. 2017;61:e00200–17. https://doi.org/10.1128/aac.00200-17.
Halasohoris SA, Scarff JM, Pysz LM, Lembirik S, Lemmon MM, Biek D, et al. In vitro and in vivo activity of GT-1, a novel siderophore cephalosporin, and GT-055, a broad-spectrum β-lactamase inhibitor, against biothreat and ESKAPE pathogens. J Antibiot. 2021;74:884–92. https://doi.org/10.1038/s41429-021-00472-9.
Nguyen LP, Park CS, Pinto NA, Lee H, Seo HS, Vu TN et al. In vitro activity of a novel siderophore-cephalosporin LCB10-0200 (GT-1), and LCB10-0200/avibactam, against carbapenem-resistant Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa strains at a tertiary hospital in Korea. Pharmaceuticals. 2021;14. https://doi.org/10.3390/ph14040370.
Weiss A, Delavenne E, Matias C, Lagler H, Simon D, Li P, et al. Topical niclosamide (ATx201) reduces Staphylococcus aureus colonization and increases Shannon diversity of the skin microbiome in atopic dermatitis patients in a randomized, double-blind, placebo-controlled phase 2 trial. Clin Transl Med. 2022;12:e790. https://doi.org/10.1002/ctm2.790.
Peck M, Rothenberg ME, Deng R, Lewin-Koh N, She G, Kamath AV, et al. A phase 1, randomized, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of DSTA4637S, an anti-Staphylococcus aureus thiomab antibody-antibiotic conjugate, in healthy volunteers. Antimicrob Agents Chemother. 2019;63:e02588–18. https://doi.org/10.1128/aac.02588-18.
Deng R, Zhou C, Li D, Cai H, Sukumaran S, Carrasco-Triguero M, et al. Preclinical and translational pharmacokinetics of a novel THIOMAB™ antibody-antibiotic conjugate against Staphylococcus aureus. mAbs. 2019;11:1162–74. https://doi.org/10.1080/19420862.2019.1627152.
Acknowledgements
The authors would like to thank the researchers, organizations, and companies who responded to enquiries.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
MATB has conducted antibiotic research that was funded by Botanix Pharmaceuticals and has received funding from CARB-X. MATB is an inventor of patents describing novel antibiotics.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The authors of the above article have noticed that prodrug contezolid acefosamil (3) was incorrectly stated as being approved rather than contezolid (4). This has been corrected throughout the review. The changes are below: In the sentence in the third paragraph of the introduction beginning “The new antibacterial drugs approved since the previous 2019 review…”, “contezolid acefosamil (3)” should have been “contezolid (4)”. In Table 1, the drug name “contezolid acefosamil (3) (prodrug)” should have been “contezolid (4)”, without “(prodrug)”. In the sentence beginning “Since the 2019 review…”, in paragraph 2 of the section “Antibacterial drugs launched from January 2013 to December 2022”, “contezolid acefosamil (3)” should have been “contezolid (4)”. In the fourth paragraph of the section “Antibacterial drugs launched from January 2013 to December 2022”, several changes have been made. In the first sentence, “Contezolid acefosamil (3)” has been changed to “Contezolid (4)”, “(Youxitai, MRX-4, po)” has been changed to “(Youxitai, MRX-1, IV)”, and the word “prodrug” has been removed. In the third sentence, “(MRX-1)” has been removed, and “3” has been changed to “contezolid acefosamil (3)”. In the fourth sentence, “The prodrug” has been changed to “The prodrug 3”. In the “Conclusion and outlook” section, in the sentence beginning “There were only two new small molecule antibacterial drugs…”, “the oxazolidinone contezolid acefosamil (3)” should have read “the oxazolidinone contezolid (4)”. In addition, there was also an error in the “New antibacterial pharmacophore analysis” section where it was stated that gepotidacin (10) inhibited GyrB and not GyrA. The mode of action of gepotidacin (10) is correctly described elsewhere in the review. GyrB has been corrected to GyrA. The original article has been corrected.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Butler, M.S., Henderson, I.R., Capon, R.J. et al. Antibiotics in the clinical pipeline as of December 2022. J Antibiot 76, 431–473 (2023). https://doi.org/10.1038/s41429-023-00629-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-023-00629-8
This article is cited by
-
Endophytes: a uniquely tailored source of potential antibiotic adjuvants
Archives of Microbiology (2024)
-
Current trends, limitations and future research in the fungi?
Fungal Diversity (2024)
