
Abstract
Two novel depsipeptides xenematides F and G (1, 2), were isolated from entomopathogenic Xenorhabdus budapestensis SN84 along with a known compound xenematide B. The structures of the two new molecules were elucidated using NMR, MS and Marfey’s method. The xenematide G (2) contains α-aminoheptanoic acid, a non-protein amino acid that is rarely found in secondary metabolites from entomopathogenic bacteria. Xenematides F and G were tested for antibacterial activity. Xenematide G (2) exhibited moderate antibacterial activity.
Similar content being viewed by others
Introduction
Members of the genus Xenorhabdus are symbionts of Steinernema nematodes and are important pathogens of insects [1]. After nematodes invade the host insect, entomopathogenic Photorhabdus spp. and Xenorhabdus spp. are released into the haemolymph of the insect host. Next, structurally diverse compounds produced by these bacteria are released into the insect to interfere with its metabolic processes [2]. Photorhabdus and Xenorhabdus play key roles in regulating bacteria–insect–nematode interactions, inducing the nematode infection process and protecting dead insects from fungal invasion [3, 4]. Several peptides, including the highly polar PAX (for peptide antimicrobial from Xenorhabdus) peptides [5], GameXPeptides [6], szentiamide [7], xentrivalpeptides [8] and xenematide [9, 10] have been identified and isolated from Xenorhabdus spp. In a previous study, five members of the PAX family showed significant activity against plants and human fungal pathogens, and moderate activity against several bacteria and yeast [5]. Two novel antimicrobial peptides isolated from Xenorhabdus budapestensis NMC-10 designated as GP-19 and EP-20 was active against Verticillium dahlia and Phytophthora capsici, respectively [11]. Rhabdopeptides/xenortide-like peptides were recently identified and show remarkable activity against trypanosoma and plasmodium [12, 13]. A derivative of the cyclic depsipeptide, xenematide A, exhibits antibacterial activity against both Gram-negative and Gram-positive bacteria [9].
Natural product research has strongly benefited in recent years from progress in sequencing technology and analytical chemistry [14, 15]. Many gene clusters encoding enzymes involved in natural product biosynthesis have been identified. Because the isolation and identification of new natural products using traditional methods demands a great deal of time, effort, and resources, the use of PCR to target the genomes of strains of interest has been increasingly used as alternative approach to discover new natural products [16,17,18]. In some studies, the biosynthesis genes have been exploited as highly relevant indicators of natural product production [19, 20]. Meanwhile, the use of real-time PCR using primers with specific melting temperatures corresponding to specific sequences had been shown to be a specific and highly efficient method to screen strains with identical natural product profiles [21].
The abundant production and significant activity of peptides prompted us to identify and isolate new peptides from Xenorhabdus. During our search for novel bioactive natural products in entomopathogenic nematodes [22, 23], a previously identified molecule, xenematide C, was isolated from X. budapestensis SN19 together with new xenematide E, and xenematide C exhibited moderate activity (EC50 = 22.71 μg ml−1) against Botrytis cinerea [24]. Considering the great effort expended in discovering the xenematide C-producing strain X. budapestensis SN19, as well as the limited yield and derivation of xenematides from strain SN19, in this study, we improved a method by real-time PCR to screen isolates of Xenorhabdus spp. for their ability to produce bioactive peptides. We demonstrated the utility and effectiveness of our method for identifying new xenematides from target producers. We have also discovered new cyclic depsipeptides xenematides F and G, the fermentation, isolation, structural elucidation and antimicrobial activity are described.
Materials and methods
General experimental procedures
UV spectra were recorded on a Thermo ND-1000 spectrophotometer, and IR spectra on a Bruker IFS-55. Preparation of a genomic DNA (gDNA) library, real-time PCR, DNA sequencing, phylogenetic analysis, as well as production of xenematide C is described in the Supplementary Methods.
Fermentation and extraction
A two-staged fermentation was performed as previously described [25]. For both stages, M medium (6.13 g glucose, 21.29 g peptone, 1.50 g MgSO4·7H2O, 2.46 g (NH4)2SO4, 0.86 g KH2PO4, 1.11 g K2HPO4, and 1.72 g NaSO4 in a final volume of 1 l H2O, pH 7.0) was used. A 250 ml flask containing 50 ml of M medium was inoculated with 100 μl of a bacterial solution and incubated with shaking (200 rpm) at 28 °C for 18 h to prepare the seed culture. Sixty 2-l flasks, each containing 400 ml of M medium, were subsequently inoculated with 20 ml of seed culture each and were allowed to ferment for 120 h under identical conditions.
The production culture was centrifuged at 4723×g and 4 °C for 45 min to remove bacterial cells. The remaining broth was extracted with 4% XAD-16 (Sigma) resin for 4 h at room temperature with agitation. The resin was subsequently harvested by centrifugation and eluted four times with MeOH. The combined eluates were then concentrated under reduced pressure to obtain a dried extract.
Isolation of metabolites
The dried extract was re-dissolved in 50% MeOH (600 ml). The solution was extracted four times with an equal volume of CH2Cl2, after which the CH2Cl2 extract was collected and concentrated on a rotary evaporator under vacuum at 35 °C to yield 16.827 g of solid brown residue.
The CH2Cl2 extract was subjected to silica gel chromatography that was eluted stepwise with CH2Cl2:MeOH (100:0, 50:1, 20:1, 10:1, and 0:100, 3 l each) as the mobile phase to produce five fractions, F1–F5. Fraction F2 (1.3476 g) was subjected to gel chromatography on Sephadex LH-20 and was eluted with MeOH to yield xenematide C (426.5 mg) and a mixture of compounds. The mixture was subsequently purified by reverse-phase semi-preparative HPLC by applying a MeOH–H2O gradient (with 0.1% HCOOH added to both solvents) from 20% to 80% using a flow rate of 3.6 ml min−1 for 35 min. UV detection was performed at 210 nm. Xenematide B (36 mg), xenematides F (1, 11 mg), and G (2, 7.2 mg) eluted at 16.7, 20.5, and 27.9 min, respectively.
NMR spectra, HPLC and LC–MS analysis
Optical rotations were measured with an ATAGO AP-300 polarimeter. NMR spectra were recorded on a 600 MHz Bruker NMR spectrometer at room temperature. Carbon signals and the residual proton signals of DMSO-d6 (δC 39.50 and δH 2.50, respectively) were used for calibration. HPLC analysis was performed on an Agilent 1260 Infinity LC system coupled with a C18 column (Agilent ZORBAX Eclipse XDB, 4.60 × 250 mm, 5 μm). Marfey’s method was performed on a Shimadzu LC-2010A HT HPLC system coupled with a C18 column (Agilent ZORBAX Eclipse XDB, 4.60 × 150 mm, 5 μm). Semi-preparative HPLC was performed on an Agilent 1260 series system coupled with a C18 column (Agilent ZORBAX Eclipse XDB, 9.4 × 250 mm, 5 μm). High-resolution mass spectra were acquired using an Agilent 6500 series Q-TOF spectrometer. Column chromatography was performed using silica gel (100–200 mesh) and Sephadex LH-20 (GE Healthcare). The HPLC analysis of crude extracts containing xenematide C was performed using a 25 min solvent gradient (1 ml min−1) from 10% to 75% CH3CN in H2O, and xenematide C eluted at a retention time of 16.0 min. The peak areas at 210 and 280 nm were used to quantify xenematide C production on the basis of calibration curves with authentic xenematide C standards.
LC–MS was carried out on an Agilent 1260 Infinity LC coupled to a 6230 TOF equipped with an Agilent Extend C18 column (50 × 2.1 mm, 1.7 μm). Liquid chromatography was performed using a 17.0 min solvent gradient (0.2 ml min−1) at 50% CH3CN in H2O containing 0.1% formic acid. Xenematide C eluted at a retention time of 1.5 min. The extracted ion (m/z 624.2824) for the [xenematide C+H]+ ion verified xenematide C production.
Xenematide F (1): Colourless solid; UV (MeOH) λmax (log ε) 226 (3.51), 283 (2.84) nm; IR νC=O 1735 cm−1, νC–O–C 1168, 1096 cm−1, νC=O 1636(amide I) cm−1, δNH 1538(amide II) cm−1; \({[{\rm{\alpha}}]^{20}_{\mathrm{D}}}\) + 34.00 (c 1.00, MeOH); for 1H and 13C NMR data (see Table 1); 1H and 13C NMR, COSY, HSQC, and HMBC spectra are available as Supporting Information (Figs. S12–S16); HRESIMS m/z 590.2998 [M+H]+ (calculated for C32H40N5O6: 590.2978).
Xenematide G (2): Colourless solid; UV (MeOH) λmax (log ε) 229 (3.72), 283 (3.25) nm; IR νC=O 1736 cm−1, νC–O–C 1169, 1096 cm−1, νC=O 1637(amide I) cm−1, δNH 1537(amide II) cm−1; \({[{\rm{\alpha}}]^{20}_{\mathrm{D}}}\) + 35.00 (c 1.00, MeOH); for 1H and 13C NMR data (see Table 1); 1H and 13C NMR, COSY, HSQC, and HMBC spectra are available as Supporting Information (Figs. S17–S21); HRESIMS m/z 604.3147 [M + H]+ (calculated for C33H42N5O6: 604.3135).
Determination of the absolute amino acid configurations
Compounds 1 and 2 (1 mg each) were hydrolysed with 6 M HCl (1 ml) at 120 °C for 3 h. The solutions were then evaporated to dryness, and the residues were re-dissolved in 1 ml of water and dried on a rotary evaporator. Next, 100 μl of 1 M sodium bicarbonate was added to each residue to form solutions. Amino acid standards were also individually dissolved in 100 μl of 1 M sodium bicarbonate at concentrations of 1 mg ml−1. The d, l-aminoheptanoic acid were purchased from Aladdin Biochemical Technology and J&K Scientific.
Thirty-five microlitres of Nα-(2,4-dinitro-5-fluorophenyl)-l-alaninamide (l-FDAA, Marfey’s Reagent, for threonine, leucine and tryptophan) [26] and 60 μl Nα-(2,4-dinitro-5-fluorophenyl)-d/l-leucinamide (d/l-FDLA, advanced Marfey’s reagent, for α-aminoheptanoic acid) [27] in acetone (10 mg ml−1) was added to each solution. The solutions were incubated in a water bath at 55 °C for 1 h. After cooling to room temperature, each solution was dissolved in MeCN for HPLC analysis. At 26 °C, MeCN/H2O containing 0.1% HCOOH was used as mobile phase using the following gradient: 0–5 min, 20% MeCN; 5–35 min, 20–60% MeCN; 35–40 min, 60–100% MeCN and 40–50 min, 100–20% MeCN. The flow rate was 1 ml min−1 with UV detection at 340 nm. The HPLC results for the amino acids were compared to the results of the standard samples [26].
Antifungal and antibacterial assays
Antifungal assays was performed with B. cinerea using previously described methods [28]. Minimum inhibitory concentrations (MICs) against bacteria were determined as previously reported [29]. The antibacterial activity of the two compounds was evaluated against Staphylococcus aureus ATCC 25923, Bacillus subtilis NCTC 2116, Bacillus cereus ATCC 10702, Pseudomonas aeruginosa A3, Shigella dysenteriae JS11910 and Salmonella enteritidis 1891. Streptomycin and Gentamycin were used as positive controls. The assays were performed in a 96-well microtiter plate format in duplicate, with two independent cultures for each strain. The two compounds were dissolved in DMSO (Sigma) and added to the cultures in wells to give a final concentration of DMSO of 2% that did not affect the growth of any of the tested strains. The effect of different dilutions of the compounds (up to 128 μg ml−1) on growth was assessed after 18 h incubation at 37 °C using a Thermo Multiskan GO plate reader at OD625. The MIC value was determined as the lowest concentration showing no growth compared to the control.
Results
Development of a strain prioritisation method by real-time PCR for cyclic depsipeptide discovery
Real-time PCR was performed to detect the fluorescence intensity of the target DNA. Melting curve analysis reveals the melting temperature (Tm) corresponds to the length, base composition, and nucleotide mismatch of PCR products and can be used as specificity indicator [30]. Without the need for gel electrophoresis, evaluating the Tm value of PCR products become a key factor in accelerating the discovery of natural products. We used SYBR Green I to perform real-time PCR with the genomic DNA (gDNA) samples of a Xenorhabdus genus strain collection for strain prioritisation, as it enabled us to perform a melting curve analysis of the PCR products. We isolated the xenematide producer X. budapestensis SN19 in our preliminary work, which produces a known xenematide (xenematide C). Because we expended a great deal of effort in isolating the xenematide, and the current repertoire of xenematide derivatives is currently limited, we screened our Xenorhabdus sp. strain collection at the Natural Product Laboratory of SYAU for newly identified xenematide C producers.
The biosynthetic domain responsible for xenematide production in X. nematophila ATCC19061 was previously reported by Jason M. Crawford [10], and NRPS XNC1_2713 was indeed responsible for xenematide biosynthesis. The condensation (C) domain is considered to be an active site involved in xenematide biosynthesis. The gene sequences of active sites C1 and C2 were chosen to design the primer sets Xenf1/Xenr1, Xenf2/Xenr2 and Xenf6-JB/Xenr6-JB (Figs. S1and S2). Three primer sets were predicted to produce PCR amplicon sizes of 131, 90 and 135 bp. As the target fragments were relatively short, the specificity cut off was set at Tm ± 0.5 °C for each of the target amplicons. Four primers would specifically narrow the scope of our search among the correct hits and the relatively short target fragments could effectively save PCR time.
Strain prioritisation by real-time PCR for four conserved nucleotide sequences, affording two cyclic depsipeptide producers
The gDNA of the 47 strains was individually prepared and used as template for PCR with each of four sets of primers. Each amplification assay included one blank reaction (i.e., no template DNA) as a negative control and a reaction with strain SN19 gDNA as a positive control (Figure S6A–C).
As summarised in Figs. S6D, 2 of the 47 assayed strains (as well as strain SN19) were identified as putative hits using the primer sets Xenf1/Xenr1, Xenf2/Xenr2 and Xenf6-JB/Xenr6-JB. The primer sets Xenf1/Xenr1 and Xenf2/Xenr2 were designed based on highly conservative sequences to ensure high-precision matching with XNC1_2713-C2. The Xenf6-JB/Xenr6-JB primers set was designed to match XNC1_2713-C1. Sequencing of the resultant PCR amplicons revealed that strains SN18, SN19 and SN84 were likely correct hits.
Two identified positive strains produce xenematides and harbour xenematide biosynthetic sequences
To validate the production of xenematide C by these strains, all 10 strains which were preliminary or partly confirmed by three pairs of primers were evaluated in fermentation assays and assessed for xenematide C production. Two strains SN18 and SN84 were confirmed as xenematide producers (Fig. S3). DNA sequencing of the resultant PCR amplicons confirmed that each of the target genes was highly homologous (>85%) to sequences from known xenematide producers (Fig. S4). The hit strains were next subjected to polyphasic taxonomy studies. A polyphasic taxonomy study was performed using the 16S rRNA gene and ERIC-PCR on two hits. ERIC-PCR is a DNA polymorphism assay based on amplification of random DNA segments with single primers of arbitrary nucleotide sequence. DNA polymorphism analysis with ERIC-PCR can clearly distinguish microbial intraspecific differences [31]. While SN18 and SN84 were assigned as X. budapestensis by the phylogenetic tree based on the 16S rRNA gene (Fig. S5A), the intraspecific identification via ERIC-PCR revealed that SN18 was distinct from SN19 and SN84 (Fig. S5B). As determined by ERIC-PCR, the SN84 was similar to SN19, despite being isolated from different soil samples (Fig. S5B and Table S2).
Analysis and structural identification of three xenematides in strain SN84
Because of the richness of suspect xenematides we previously observed in strain SN84 and because of the experience we acquired in discovering xenematides in strain SN19, we chose strain SN84 as the strain to ferment and characterise xenematides from. An XAD extract was obtained from a liquid culture of strain SN84 (24 l). From the extract, two novel xenematides, named F and G (Fig. 1), and a known xenematide (xenematide B; Fig. S7) were isolated and purified using HPLC and gel chromatography. Xenematide B was readily identified based on its molecular mass and NMR spectroscopic data (Table S3). Xenematide B has been previously isolated from X. nematopila and was reported without NMR spectroscopic data [10]. The 1H and 13C NMR, COSY, HSQC, and HMBC spectra of xenematide B are available as Supporting Information (Figs. S7–S11).

Structures of compounds 1 and 2
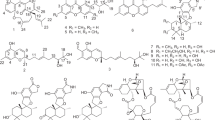
The molecular formula of 1 was determined to be C32H39N5O6 by HRESIMS analysis. The 1H and 13C NMR spectra of 1 were similar to that of xenematide C, showing identical signals for a β-alanine residue, a threonine residue, a tryptophan residue, and a phenylacetyl (PAA) group [10] (Table 1). However, further inspection of the 1H and 13C NMR spectra of 1 revealed signals from an isopropyl unit (δH 1.96, 0.88, and 0.87; δC 25.5, 22.3, and 22.3) which were not present in xenematide C. The isopropyl unit gave a clue to the presence of a leucine residue in 1. Furthermore, the phenylalanine residue signals observed in spectra of xenematide C were not present in 1, suggesting that the phenylalanine residue in xenematide C was substituted by a leucine residue in 1. This was further supported by key correlations observed in HMBC and COSY data (Fig. 2) and an accurate mass of m/z 590.2998 measured for the [M + H]+ ion. Based on these data, amino acid sequence of 1 was determined as PAA-Thr-Leu-Trp-β-Ala. The absolute configurations of the amino acids were determined using advanced Marfey’s method as l-threonine, l-leucine, and d-tryptophan. Compound 1 was given the name xenematide F.

Key COSY (bold) and HMBC (arrows) correlations supporting the structures of xenematides F and G (1, 2)
HRESIMS analysis indicated that compound 2 possessed the molecular formula C33H41N5O6. The 1H and 13C NMR spectra of 2 (Table 1) were similar to those of 1. Both 2 and 1 have a β-alanine residue, a threonine residue, a tryptophan residue, and a PAA group as building blocks. The primary difference between 1 and 2 is that the spectra of 2 showed signals for an n-butyl group (δH 1.48, 1.24, 1.24 and 0.85; δC 30.9, 25.0, 21.8 and 13.9) instead of the isopropyl group observed in spectra of 1. This suggested that the leucine residue in 1 was replaced by α-aminoheptanoic acid in 2. This was further supported by key correlations observed in HMBC and COSY experiments (Fig. 2). Absolute configurations of tryptophan and threonine were determined using Marfey’s method as d-tryptophan and l-threonine. Since 2 and 1 are both dextrorotatory and are co-occurring metabolites, it was assumed that they share the same biogenic origins. The stereochemistry of α-aminoheptanoic acid is determined by comparison with the control of d,l-aminoheptanoic acid using the advanced Marfey’s method. The absolute configuration of the α-aminoheptanoic acid in 2 was l-configuration (Fig. S24). Compound 2 was given the name xenematide G.
Antimicrobial susceptibility testing
Xenematide B, F and G were tested for antifungal activity against B. cinerea. The result show that three compounds demonstrated no notable activity. Xenematide has been reported to show antibacterial activities against a panel of pathogenic bacteria [9]. Therefore, the antimicrobial activities of xenematide B, F and G were tested against six pathogenic bacteria as shown in Table 2. The result suggest that xenematide B show insignificant bioactivity. Xenematide F show potent bioactivity against P. aeruginosa with MIC value of 32 μg ml−1. Its congener xenematide G has MIC value of 16 μg ml−1 against B. subtilis, which is similar to xenematide A [9].
Discussion
Peptides derived from NRPSs represent an important class of pharmaceutically relevant drugs. The prominent examples are the antibiotic daptomycin [32] or the immunosuppressant cyclosporine A [33]. In this study, two xenematide producers were identified, and two novel cyclic peptides were purified and structurally characterised. As a key depsipeptide node, the xenematide F (HRESIMS m/z 590.29) was predicted within subnetwork [18]. We isolated the pure compound and provided the NMR data and formula of xenematide F for the first time. As the first reported bioactivity xenematide, xenematide A has an MIC value of 10 µg ml−1 against Bacillus subtilis, Pseudomonas fluorescens, Pseudomonas syringae, Erwinia amylovora, and Ralstonia solanacearum [17]. In our study, the xenematide G has MIC value of 16 μg/ml against B. subtilis which is similar to xenematide A. In the mean time, the xenematide G contains α-aminoheptanoic acid, a non-protein amino acid that is rarely found in secondary metabolites from entomopathogenic bacteria. The special amino acid may provide a structural foundation for the novel bioactivity of xenematide G.
An improved real-time PCR method was used to prioritise the most promising strains from a microbial strain collection for xenematide production. The high-throughput and short extension time advantages of this method allowed the time of discovery and cost associated with traditional approaches to be decreased. Thus, the biosynthetic relationship of peptides and their producing NRPSs has become a focus of research [34]. Based on the modification of NRPSs, new NRPSs, peptides, peptide derivatives, functionalizable peptides and peptide-based compounds could potentially be created [35]. As strain banks expand with microbial isolates, efficient molecular biology techniques, such as real-time PCR should be introduced to accelerate natural product discovery [36]. The primers were designed to amplify relatively short target fragments (~100 bp) to effectively shorten the required assay time without sacrificing accuracy. Based on the principle of real-time PCR, relatively short target fragments can also avoid instances where the Ct value appears too late [30].
References
Richards GR, Goodrich-Blair H. Masters of conquest and pillage: Xenorhabdus nematophila global regulators control transitions from virulence to nutrient acquisition. Cell Microbiol. 2009;11:1025–33.
Crawford JM, Kontnik R, Clardy J. Regulating alternative lifestyles in entomopathogenic bacteria. Curr Biol. 2010;20:69–74.
Goodrich-Blair H, Clarke DJ. Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol Microbiol. 2007;64:260–8.
Shi YM, Bode HB. Chemical language and warfare of bacterial natural products in bacteria–nematode–insect interactions. Nat Prod Rep. 2018;35:309–35.
Gualtieri M, Aumelas A, Thaler J-O. Identification of a new antimicrobial lysine-rich cyclolipopeptide family from Xenorhabdus nematophila. J Antibiot. 2009;62:295–302.
Bode HB, Reimer D, Fuchs SW, Kirchner F, Dauth C, Kegler C, et al. Determination of the absolute configuration of peptide natural products by using stable isotope labeling and mass spectrometry. Chem Eur J. 2012;18:2342–8.
Ohlendorf B, Simon S, Wiese J, Imhoff JF. Szentiamide, an N-formylated cyclic depsipeptide from Xenorhabdus szentirmaii DSM 16338T. Nat Prod Commun. 2011;6:1247–50.
Zhou Q, Dowling A, Heide H, Wöhnert J, Brandt U, Baum J, et al. Xentrivalpeptides A–Q: depsipeptide diversification in Xenorhabdus. J Nat Prod. 2012;75:1717–22.
Lang G, Kalvelage T, Peters A, Wiese J, Imhoff JF. Linear and cyclic peptides from the entomopathogenic bacterium Xenorhabdus nematophilus. J Nat Prod. 2008;71:1074–7.
Crawford JM, Portmann C, Kontnik R, Walsh CT, Clardy J. NRPS substrate promiscuity diversifies the xenematides. Org Lett. 2011;13:5144–7.
Xiao Y, Meng FL, Qiu DW, Yang XF. Two novel antimicrobial peptides purified from the symbiotic bacteria Xenorhabdus budapestensis NMC-10. Peptides. 2012;35:253–60.
Reimer D, Cowles KN, Proschak A, Nollmann FI, Dowling AJ, Kaiser M, et al. Rhabdopeptides as insect-specific virulence factors from entomopathogenic bacteria. ChemBioChem. 2013;14:1991–7.
Zhao L, Kaiser M, Bode HB. Rhabdopeptide/Xenortide-like peptides from Xenorhabdus innexi with terminal amines showing potent antiprotozoal activity. Org Lett. 2018;20:5116–20.
Piel J. Approaches to capturing and designing biologically active small molecules produced by uncultured microbes. Annu Rev Microbiol. 2011;65:431–53.
Brachmann AO, Bode HB. Identification and bioanalysis of natural products from insect symbionts and pathogens. Adv Biochem Eng Biot. 2013;135:123–55.
Masanori F, Satoshi B, Toshio T, Masaaki K, Yasuo O, Masahiro T, et al. Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew Chem. 2013;125:11821–5.
Owen JG, Reddy BVB, Ternei MA, Charlop-Powers Z, Calle PY, Kim JH, et al. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc Natl Acad Sci USA. 2013;110:11797–802.
Tobias NJ, Wolff H, Djahanschiri B, Grundmann F, Kronenwerth M, Shi YM, et al. Natural product diversity associated with the nematode symbionts Photorhabdus and Xenorhabdus. Nat Microbiol. 2017;2:1676–85.
Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132:2469–93.
Bachmann BO, Van Lanen SG, Baltz RH. Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J Ind Microbiol Biotechnol. 2014;41:175–84.
Hindra Huang TT, Yang D, Rudolf JD, Xie PF, Xie G, et al. Strain prioritization for natural product discovery by a high-throughput real-time PCR method. J Nat Prod. 2014;77:2296–303.
Shi DS, An R, Zhang WB, Zhang GL, Yu ZG. Stilbene derivatives from Photorhabdus temperata SN259 and their antifungal activities against phytopathogenic fungi. J Agric Food Chem. 2017;65:60–65.
Bi YH, Gao CZ, Yu ZG. Rhabdopeptides from Xenorhabdus budapestensis SN84 and their nematicidal activities against Meloidogyne incognita. J Agric Food Chem. 2018;66:3833–9.
Lu XZ, Shi DS, Gao CZ, Tian XM, Bi YH, Yu ZG. Isolation and identification of secondary metabolites from Xenorhabdus budapestensis SN19. Nat Prod ResDev. 2016;6:828–32.
Yu ZG, Vodanovic-Jankovic S, Kron M, Shen B. New WS9326A Congeners from Streptomyces sp. 9078 inhibiting Brugia malayi asparaginyl-tRNA synthetase. Org Lett. 2012;14:4946–9.
Marfey P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-Difluoro-2,4-dinitrobenzene. Carls Res Commun. 1984;49:591–6.
Fujii K, Ikai Y, Oka H, Suzuki M, Harada K. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: combination of Marfey’s method with massspectrometry and its practical application. Anal Chem. 1997;69:5146–51.
Fang XL, Li ZZ, Wang YH, Zhang X. In vitro and in vivo antimicrobial activity of Xenorhabdus bovienii YL002 against Phytophthora capsici and Botrytis cinerea. J Appl Microbiol. 2011;111:145–54.
Kronvall G. Single-strain regression analysis for determination of interpretive breakpoints for cefoperazone disk diffusion susceptibility testing. J Clin Microbiol. 1983;17:975–80.
Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ. 2005;29:151–9.
Versalovic J, Koeuth T, Lupski R. Distribution of repetitive DNA sequences in eubacteria and application to finerpriting of bacterial enomes. Nucleic Acids Res. 1991;19:6823–31.
Baltz RH, Miao V, Wrigley SK. Natural products to drugs: daptomycin and related lipopeptide antibiotics. ChemInform. 2006;37:717–41.
Lawen A. Biosynthesis of cyclosporins and other natural peptidyl prolyl cis/trans isomerase inhibitors. BBA-Gen Subj. 2015;1850:2111–20.
Cai XF, Nowak S, Wesche F, Bischoff I, Kaiser M, Fürst R, et al. Entomopathogenic bacteria use multiple mechanisms for bioactive peptide library design. Nat Chem. 2016;9:379–86.
Bozhüyük KAJ, Fleischhacker F, Linck A, Wesche F, Tietze A, Niesert C-P, et al. De novo design and engineering of non-ribosomal peptide synthetases. Nat Chem. 2017;10:275–309.
Muangpat P, Yooyangket T, Fukruksa C, Suwannaroj M, Yimthin T, Sitthisak S, et al. Screening of the antimicrobial activity against drug resistant bacteria of Photorhabdus and Xenorhabdus associated with entomopathogenic nematodes from Mae Wong National Park, Thailand. Front Microbiol. 2017;8:1142.
Acknowledgements
This work are supported by Liaoning Province Fund for Nature (No. 01032017001), Shenyang Agricultural University Postdoctoral Fund (No. 770215012) and Shenyang Agricultural University Introducing Talent Fund (No. 880415016).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Xi, X., Lu, X., Zhang, X. et al. Two novel cyclic depsipeptides Xenematides F and G from the entomopathogenic bacterium Xenorhabdus budapestensis. J Antibiot 72, 736–743 (2019). https://doi.org/10.1038/s41429-019-0203-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0203-y
This article is cited by
-
Recombineering using RecET-like recombinases from Xenorhabdus and its application in mining of natural products
Applied Microbiology and Biotechnology (2022)
