
Abstract
Cancer arises from a multitude of disorders resulting in loss of differentiation and a stem cell-like phenotype characterized by uncontrolled growth. Polycomb Group (PcG) proteins are members of multiprotein complexes that are highly conserved throughout evolution. Historically, they have been described as essential for maintaining epigenetic cellular memory by locking homeotic genes in a transcriptionally repressed state. What was initially thought to be a function restricted to a few target genes, subsequently turned out to be of much broader relevance, since the main role of PcG complexes is to ensure a dynamically choregraphed spatio-temporal regulation of their numerous target genes during development. Their ability to modify chromatin landscapes and refine the expression of master genes controlling major switches in cellular decisions under physiological conditions is often misregulated in tumors. Surprisingly, their functional implication in the initiation and progression of cancer may be either dependent on Polycomb complexes, or specific for a subunit that acts independently of other PcG members. In this review, we describe how misregulated Polycomb proteins play a pleiotropic role in cancer by altering a broad spectrum of biological processes such as the proliferation-differentiation balance, metabolism and the immune response, all of which are crucial in tumor progression. We also illustrate how interfering with PcG functions can provide a powerful strategy to counter tumor progression.
Similar content being viewed by others

Introduction
Polycomb Group (PcG) proteins have first been described as main players in cellular memory known to maintain embryonic chromatin landscapes in a repressed transcriptional state throughout development. Counterintuitively, PcG proteins then appeared to be able to regulate the transcription of developmental genes involved in a wide range of highly dynamic biological processes such as differentiation, stem cell plasticity or cell cycle progression.1,2 In addition, mutations or dysregulations of PcG proteins have been extensively described in cancer.3 Knowing the importance of PcG proteins in transcriptional regulation, it was not surprising to find a correlation between modification of PcG activities and tumorigenesis. However, an early demonstration of a causal link between the ability of PcG complexes to promote or inhibit the transcription of oncogenes or tumor suppressor genes, respectively, has paved the way for work aimed at studying the different mechanisms by which PcG complexes are involved in the generation and the evolution of cancer cells.
Here, we first describe the molecular mechanisms underlying the recruitment and function of PcG proteins in gene regulation during normal development. We then review the involvement of Polycomb complexes in cancer, highlighting PcG-dependent disturbances of epigenetic processes in tumorigenesis. Next, we focus on the description of the latest discovered mechanisms linking Polycomb to cancer. PcG proteins have been extensively studied in hormone-dependent cancers where hormone-receptors interact directly with PcG proteins, modifying the transcriptional landscape of the affected cells. Furthermore, PcG proteins have been described as capable of modulating the metabolism and the immune response of the tumor microenvironment, both being hallmarks of cancer. Next, we focus on a new area of research involving mutated histones, also known as oncohistones, and discuss how these mutations can impact PcG behaviour in a tumoral context. Finally, we explain how PcG proteins are able to confer a non-genetic drug-resistance underlying the importance of epigenetics in cancer.
PcG Proteins
PcG proteins are highly conserved throughout metazoan evolution and are essential players in cellular identity. In Drosophila melanogaster, mutations in the Polycomb gene induce embryonic transformation of anterior segments into posterior segments by inducing ectopic expression of homeotic (Hox) genes.4,5 Subsequent work identified other mutations triggering derepression of Hox genes, leading to the identification of several genes that were defined as members of the Polycomb group. PcG proteins form two main epigenetic complexes, the Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2), which were later identified as transcriptional regulators targeting a large number of genes in genome-wide studies.6
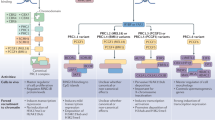
PRC2 is composed of the Embryonic Ectoderm Development (EED), Suppressor of Zeste 12 Homolog Protein (SUZ12) and Enhancer of Zeste Homolog 1/2 (EZH1/2) core constitutive subunits (Fig. 1a). EZH1/2 have a Su(var)3–9, Enhancer-of-zeste and Trithorax (SET) domain with a histone methyltransferase activity that mono-, di- or tri-methylates the lysine 27 of the histone H3 (H3K27me1/2/3).7 PRC2 can be divided into two sub-complexes, namely PRC2.1 and PRC2.2, characterized by the association with specific accessory proteins. PRC2.1 contains one of the three paralogous Polycomb-like (PCL) proteins PCL1/2/3, also known as PHF1, MTF2, PHF19 respectively, as well as PRC2-Associated LCOR Isoform 1/2 (PALI1/2) or Elongin B/C and PRC2-associated Protein (EPOP). In addition of the core subunits, PRC2.2 contains Jumonji and AT-Rich Interaction Domain containing 2 (JARID2) and Adipocyte Enhancer-Binding Protein 2 (AEBP2). Some PRC2 co-factors can have a negative impact on PRC2 methyltransferase activity. The Catalytic Antagonist of Polycomb (CATACOMB)-PRC2 variant presents a decrease in PRC2 enzymatic activity. Indeed, the CATACOMB (also known as EZHIP) gene is poorly expressed in physiological conditions, except in gonads,8 due to hypermethylation of its CpG islands (CGIs).9 While CATACOMB–PRC2 association does not impact PRC2 recruitment to chromatin, it lessens its ability to associate with sub-stochiometric co-factors that would otherwise enhance its enzymatic activity.8

a PRC2 can be sub-divided into PRC2.1 and PRC2.2. The core PRC2 with PCL1/2/3 and the PALI1/2 or EPOP subunits compose the PRC2.1 complex. The association between the core PRC2, JARID2 and AEBP2 constitute PRC2.2. b PRC1 complex can be sub-divided into two groups of complexes, namely cPRC1 and ncPRC1. cPRC1 is composed of RING1A/B associated with PCGF2/4 and CBX2/4/6–8. ncPRC1 is composed of either RYBP or YAF2 associated with one of the six PCGF proteins. The graphical representation of each complex is schematic and not aimed to represent the size, shape and relative position of the various subunits. AEBP2, Adipocyte enhancer-binding protein 2; AUTS2, Autism susceptibility candidate 2; BCOR, BCL6 corepressor; CBX2/4/6–8, Chromobox 2/4/6–8; CK2, Casein kinase 2; DCAF7, DDB1 and CUL4 associated factor 7; DP1, E2F dimerization partner 2; E2F6, E2F transcription factor 6; EED, Embryonic ectoderm development; EPOP, Elongin B/C and PRC2-associated protein; EZH1/2, Enhancer of zeste homolog 1/2; FBRS, Fibrosin; HDAC1/2, Histone deacetylase 1/2; HP1, Heterochromatin protein 1 gamma (here labeled as HP1, also named CBX3); JARID2, Jumonji AT rich interactive domain 2; KDM2B, Lysine demethylase 2B; L3MBTL2, Lethal(3) malignant brain tumor-like protein 2; MAX, Myc associated factor X; MGA, MAX gene associated protein; PALI1/2, PRC2 associated LCOR isoform 1/2; PCGF1–6, Polycomb group finger 1–6; PCL1–3, Polycomb like protein 1–3; PHC1–3, Polyhomeotic-like protein 1–3; RBBP4/7, Retinoblastoma binding protein 4/7; RING1A/B, Really interesting new gene 1B/A; RYBP, RING1 and YY1 binding protein; SCMH1/2, Sex comb on midleg homolog 1/2; SKP1, S-phase kinase associated protein 1; SUZ12, Suppressor of zeste 12 protein homolog; USP7, Ubiquitin specific peptidase 7; YAF2, YY1-associated factor 2; WDR5, WD repeat domain 5.
PRC1 members form an even more diversified combination of variant complexes (Fig. 1b), which can be subdivided into canonical PRC1 (cPRC1) and non-canonical PRC1 (ncPRC1) complexes that all share a core PRC1 comprising one of six Polycomb Group Ring Finger 1–6 (PCGF1–6) proteins and RING1A/B, an E3 ubiquitin ligase catalyzing the mono-ubiquitination of lysine 119 of histone 2A (H2AK119ub in mammals or H2AK118ub in flies).10,11 cPRC1.2 and cPRC1.4 are respectively formed by PCGF2 or PCGF4 (also known as MEL-18 or BMI-1), RING1A/B and Sex Comb on Midleg Homolog 1/Like 2 (SCMH1/L2), and can be distinguished from ncPRC1s by the presence of one of Chromobox 2/4/6–8 (CBX2/4/6–8) proteins as well as one of the Polyhomeotic Homolog 1–3 (PHC1–3).6,12 In addition to RING1A/B and a PCGF1–6 protein, ncPRC1complexes assemble around RING1 and YY1-Binding Protein (RYBP) or YY1-Associated Factor 2 (YAF2) proteins, which are mutually exclusive homologous proteins able to bind to the same site on the C-terminal domain of RING1B.13,14 Moreover, the ncPRC1 complexes can be further classified by the identity of their PCGF subunit (PCGF1 for PRC1.1, PCGF2 for PRC1.2 and so on). Genome-wide analysis demonstrated that each PRC1 complex has its own chromatin targeting profile suggesting that the recruitment of cPRC1 and ncPRC1 depends on their differential compositions that in turn could contribute to pleiotropic functions.15
Molecular mechanisms modulating prc1 and prc2 recruitment
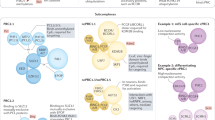
An important feature of the PRC1 and PRC2 core subunits is the absence of sequence-specific DNA-binding domains that would allow their direct recruitment to their target genes. PcG-mediated gene regulation therefore depends on components that direct their recruitment to specific chromatin domains. In a classical model, described in Drosophila melanogaster, PRC2 is first recruited on cis-regulatory sequences called Polycomb Response Elements (PREs) via consensus motifs for sequence-specific DNA-binding proteins that might interact with PRC2 subunits.16,17,18 PRC2, via its E(z) subunit, the Drosophila ortholog of EZH2/1, deposits H3K27me3. This H3K27me3 mark is then recognized by the cPRC1 PC subunit (ortholog of CBX).7,17,19,20 Subsequently, Sce — the ortholog of RING1A/B — ubiquitinates H2AK11810,11 (Fig. 2a). This model predicts co-occurrence of PRC1 and PRC2 at their target loci.

a First described in Drosophila melanogaster, the original pathway of PcG recruitment relies on two sequential steps. First, PRC2 is recruited to chromatin and deposits the repressive H3K27me3 mark via its EZH1/2 subunit. The repressive mark is then recognized by the CBX2/4/6–8 chromodomain, a subunit of cPRC1. Lastly, RING1A/B deposit the ubiquitination on H2AK119 (in Drosophila, H2AK118). While in Drosophila melanogaster PRC2 recruitment depends on specific transcription factors binding to PREs, in mammals PRC2 recruitment can occur at CGIs or depends on transcription factors or lncRNAs. More recent data suggest an alternative recruitment pathway, in which ncPRC1 complexes are recruited in a KDM2B-dependent manner which deposits the H2AK119 ubiquitination mark. In turn, this mark is recognized by the JARID2 subunit of PRC2.2. Furthermore, PRC2.1 binds the same targets via PCL1/2/3 proteins. Finally, cPRC1 is recruited via CBX2/4/6–8-mediated recognition of H3K27me3. Moreover, the new PcG proteins BAHD1 and BAHD2 have also been found to recognize the H3K27me3 repressive mark. Their interactions with HDACs generate a hypoacetylated chromatin state which participates in transcriptional silencing. b Chromatin compaction impairs the transcription of target genes. c PcG-mediated silencing depends on the inhibition of the transcriptional machinery while the repressive PRC2 and PRC1 marks are necessary to inhibit the deposition of active histone marks. d PRC1 participates in the higher-order 3D chromatin organization via its PHC subunits. The SAM domain of PHC-PRC1 is able to oligomerize which results in the maintenance of the transcriptionally repressed state. BAHD1, Bromo adjacent homology domain containing 1; CBP, CREB binding protein; HDAC, Histone deacetylase; RNA Pol II, RNA Polymerase II; SWI/SNF, Switch/sucrose non-fermentable.
However, ncPRC1 complexes do not possess CBX subunits that recognize H3K27me3 and only a small subset colocalizes with this PRC2-deposited mark.15 Moreover, mammalian ncPRC1s can act upstream of PRC2 by directly recognizing non-methylated DNA in CGIs leading to the ubiquitination of H2AK119 which is then recognized by PRC2-JARID2.21,22,23,24 These data suggest that ncPRC1 recruitment to a subset of their targets can act upstream of PRC2 recruitment (Fig. 2a).
The existence of mammalian PREs is still controversial.25,26 The analysis of PRC2 genome binding identified the enrichment for CGIs characterized by low levels of DNA methylation, that could therefore act as PREs in mammals27,28,29 (Fig. 2a). Thanks to their Polycomb-like (PCL) extended domain, the PCL proteins PHF1, MTF2 or PHF19 preferentially bind unmethylated CpG-containing DNA sequences,30 promote PRC2 binding to CGIs31,32 and stabilize the dimerization of PRC2.33 The accessory subunits — JARID2 and AEBP2 — are also important for PRC2 recruitment, via recognition of the H2AK119ub mark, as well as for deposition of H3K27me3 at specific PcG targets34 (Fig. 2a).
Additional mechanisms involve a PRC1-independent transcriptional repression. Indeed, the proteins Bromo Adjacent Homology Domain Containing protein 1 (BAHD1) and BAH Domain And Coiled-Coil Containing 1 (BAHCC1/BAHD2) possess a C-terminal Bromo Adjacent Homology (BAH) domain which recognizes H3K27me3.35,36,37,38 Moreover, BAHD1 acts as a scaffold protein that recruits additional co-repressors such as Histone DeAcetylases (HDACs).35,39 Alternative recruiting mechanisms also involve long non-coding RNAs (lncRNAs) as well as specific transcription factors (reviewed respectively in40,41) (Fig. 2a).
Polycomb recruitment is also modulated by the chromatin landscape. Indeed, Trithorax Group (TrxG) proteins counteract Polycomb-mediated gene silencing by decorating chromatin with active histone marks such as H3K4me1/2/36 and a fine-tuned balance between these two complexes is critically important. SWI/SNF and COMPASS complex subunits are the main TrxG proteins, respectively involved in chromatin remodeling and H3K4 methylation.42 Interestingly, MLL2/COMPASS binds specific promoters and trimethylates H3K4 to promote MLL2-dependent gene transcription.43 Upon loss of MLL2, H3K27me3 decorates MLL2-dependent genes and represses them.43 However, in MLL2 depleting context, H3K27me3 spreading is prevented by DNA methylation at CpG islands.44 Dual deletion of MLL2 and DNA methylation increases the repressive mark spreading while diluting its level, which ultimately leads to transcription of the corresponding genes.43 Moreover, spreading of the PRC2 mark is also counteracted by H3K36me2, which is deposited by NSD1.45
In summary, the molecular mechanisms deployed by PcG complexes to specifically target the genome remain a major area of interest with important consequences for understanding how target genes are specified. Coordinating Polycomb action with key developmental orchestrators, including transcription factors, involves a wide spectrum of tissue- and time-specific players. Future studies should provide insight into this complex Polycomb recruitment network.
PcG protein function in gene silencing
PcG-mediated transcriptional regulation has been widely portrayed as gene silencing and suggested to be mediated by various mechanisms. First, PcG complexes can mediate chromatin compaction46,47,48 (Fig. 2b). In Drosophila melanogaster, mutations in cPRC1 genes were shown to induce decompaction of the Hox clusters, followed by ectopic Hox gene expression which began a few hours later.49 In Ring1b-knockout mouse embryonic stem cells (mESCs) chromatin decompaction and expression of Hox genes occur even though the H3K27me3 repressive mark is still present.50 Surprisingly, this phenotype is rescued by a catalytic mutant form of RING1B, suggesting that its E3-ubiquitin ligase is dispensable for PcG-mediated silencing.50 This latter result contrasts with research suggesting a role for the H2AK119ub mark in maintaining PcG-dependent repression.11,24,51,52
Second, a switch from a transcriptional repressive state to an active state can be induced by competition between BAF — an ATP-dependent chromatin remodeling complex part of SWI/SNF family — and Polycomb complexes.12 BAF-dependent eviction of PcG proteins opens chromatin architecture after H3K27me3 and H2AK119Ub depletion.53 Strikingly, in a dominant-negative BAF mutant background, accumulation of PRC1 and PRC2 on chromatin does not necessarily trigger changes in chromatin landscapes, suggesting that DNA-accessibility to BAF is PcG-independent.54
Third, the maintenance of the repressed state of PcG target genes also depends on the PcG ability to block initiation and elongation of transcription (Fig. 2c). In particular, RING1-mediated ubiquitination maintains RNA polymerase in a poised state.52,55,56 In mESCs, PRC2 can methylate Elongin A to block transcription.57 The H2AK119ub and H3K27me3 repressive histone marks respectively repress deposition of the H3K4me2/3 and H3K27Ac active histone marks.58,59 Moreover, in flies, PRC1-PC binds to CBP and inhibits its H3K27 acetyltransferase activity, contributing to a repressive state60 while on the other hand TRX or TRX-related (TRR) association to CBP antagonizes PcG-mediated silencing.61 Interestingly, while the trimethylated form of H3K27 has been extensively studied, less is known about the importance of H3K27me2 in transcriptional repression. Remarkably, the dimethylated H3K27 mark represents 70% of total histone H3 against 4% for the trimethylated form.62 Their distribution is mutually exclusive, indeed, the methylated state of H3K27 correlates with different transcriptional states. It is suggested that H3K27me2 coats most of chromatin in order to protect chromatin changes mediated by Histone Acetyl Transferase (HAT).62
Finally, PcG proteins actively participate in the three-dimensional (3D) organization of the genome, adding a higher-order layer through which they contribute to gene regulation (Fig. 2d). PcG proteins can drive the formation of 3D-loops between regulatory elements such as promoters and enhancers.63,64 Loop formation involves the cPRC1-PH subunit, that oligomerizes via its SAM domain, but is independent of the cPRC1 catalytic activity.65,66,67 Consistent with a function for 3D architecture in gene regulation, PRC1 knockout in ESCs leads to loss of promoter–promoter contacts resulting in transcriptional upregulation of PRC1 target genes.68
PcG protein function in transcriptional activation
Interestingly, an involvement of PcG proteins in transcriptional activation has been suggested in pathological as well as in physiological contexts69 and is now better understood at the molecular level (reviewed in70). Morey et al. described that only 31% of the cPRC1 and ncPRC1 target genes overlap.71 While cPRC1 target genes are strongly repressed, ncPRC1 target genes are overall expressed and involved in dynamic processes such as metabolism and cell cycle progression.71 Thus, H3K27me3-independent PRC1 recruitment appears to be an important feature that favors transcriptionally active states by PcG proteins.72,73,74 For instance, PRC1.5 includes the component Autism Susceptibility candidate 2 (AUTS2) that recruits CK2 and p300 which, respectively, inhibits the E3-ubiquitin ligase activity by phosphorylating RING1B and deposits acetylation on histone tails to facilitate transcription.15,75 For PRC1.5-AUTS2 target genes, the concomitant enrichment for the H3K4me3 and H4K16Ac active marks, the presence of the RNA polymerase II and a reduction of the H3K27me3 repressive mark lead to transcriptional activation.75 The transcription factor NRF1 is involved in PRC1.5-AUTS2 recruitment to its target genes, providing an example for sequence-specific targeting of a PRC1 complex in mammals.76
To recapitulate, it is the specific composition of the PcG complexes, as well as the dynamics of their chromatin binding and replacement through cell lineages that determine their transcriptional impact.15,71,77 While canonical PcG proteins maintain cellular memory, such as in stem cells where they support self-renewal properties by repressing lineage-specific genes, ncPRC1s control differentiation in more subtle ways. By fine-tuning transcription, PcG proteins are master contributors of cell fate determination,1 in particular in the control of a balance between proliferation and differentiation. On the other hand, loss of this fine balance upon misregulation of PcG-dependent mechanisms can cause pathogenesis.
Polycomb in cancer
Altering the proper functions of PcG can affect cellular identity, therefore promoting tumorigenesis (Tables 1, 2).
PRC2 in cancer
Polycomb dysregulation in cancer has been the subject of extensive studies since Varambally et al. demonstrated that EZH2 overexpression is associated with advanced stage and poor prognosis in prostate cancer.78 Quantitative and qualitative EZH2 dysregulation has been frequently described in solid malignancies including lung, hepatocellular, breast, colorectal, pancreatic cancers as well as in several hematologic malignancies.79,80 EZH2 expression can be regulated by specific transcription factors, including the MLL-AF9 fusion protein, or by miRNAs that will induce EZH2 mRNA decay.81,82 Dysregulation of those specific regulators participates in the tumorigenesis onset. EZH2 overexpression in patients is associated with a higher risk of relapse.78 PRC2 plays a major role in self-renewal of hematopoietic stem cells;83,84,85,86 its dysregulation is often found in multiple blood cancers87,88,89,90,91,92 in which EZH2 can behave both as a tumor suppressor85,93,94 or an oncogene95,96,97,98 depending on the cell context (reviewed in80) (Fig. 3a).

a Upregulation of PRC2 components results in H3K27 hypermethylation, which, if present in tumor suppressor genes, induces their downregulation. In contrast, downregulation of PRC2 components at oncogenes leads to H3K27 hypomethylation and a switch to acetylation, contributing to the overexpression of specific oncogenes. b GOF mutations (indicated by a star) affecting the SET-domain of EZH2 can lead to overactivation of its H3K27 methyltransferase catalytic activity and to the silencing of tumor suppressor genes. c PTMs of EZH2 participate in tumorigenesis. Left: methylation of K307 of EZH2 by SMYD2 enhances its stability, resulting in a H3K27 hypermethylated state of tumor suppressor genes. Right: on the other hand, methylation of its K735 causes EZH2 degradation. The loss of EZH2 induces the replacement of H3K27me3 by H3K27ac, leading to the transcriptional expression of oncogenes. d Polycomb-independent roles of EZH2 in transcriptional activation. The gene encoding the AR is a direct target of EZH2-mediated transcriptional activation in Androgen-Dependent and Castration-Resistant Prostate Cancers (ADPC and CRPC, respectively). This mechanism is methylation-independent and escapes EZH2 inhibitors. In CRPC, EZH2 acts as a co-factor of AR. This functional transition of EZH2 from a role of repression to a role of activation of transcription depends on its phosphorylation at the level of Ser21. EZH2 and AR directly interact. This interaction inhibits the degradation of the AR and causes the overexpression of the AR target genes. e Under physiological conditions, PRC2 participates in the transcriptional repression of its HOX target genes throughout development. However, oncogenic transformation can redirect PRC2 to new target genes. This PRC2 redistribution, in particular at differentiation-related genes, induces a loss of differentiation and participates in the generation of a pluripotent stem cell-like phenotype. AR, Androgen Receptor; CBP, CREB binding protein; PSA, Prostate-Specific Antigen; SETD2, SET domain-containing 2 (a histone lysine methyltransferase); SMYD2, SET and MYND domain-containing 2.
The onset or cancer progression may be associated with mutations affecting the catalytic SET-domain of EZH2 that is essential for H3K27 methylation (Fig. 3b). An EZH2Y641F/N gain-of-function (GOF) mutation affecting the tyrosine 641 (Y641) located in the SET-domain induces hypermethylation of H3K27.99,100 Particularly, EZH2Y641 has an increased affinity for dimethylated H3K27 form which causes a widespread redistribution of H3K27me3 and a decrease in H3K27me2, leading to transcriptional misregulation of affected genes.100,101,102 Moreover, the higher-order chromatin landscape can also be affected. In recent years, multiple cutting edge approaches have shown that the genome folds into a hierarchy of structures, from nucleosomes, to chromatin loops and nanodomains, Topologically Associating Domain (TADs), chromosome compartments and chromosome territories.103 TADs are particularly interesting since they constitute regulatory landscapes for the genes contained within each TAD.104 Interestingly, co-repression of several tumor suppressors was suggested to participate in tumor growth.105,106 An established tumor state can also participate a posteriori in the redistribution of EZH2 on ectopic targets, triggering changes in cell identity due to misexpression of homeotic genes.107 In addition to GOF effects, loss-of-function (LOF) mutations and deletions affecting EZH2 and SUZ12 in T-cell acute lymphoblastic leukemia (T-ALL) — a hematopoietic cancer — lead to hypomethylation of H3K27 target genes, including Notch, a major player in T-ALL, thereby contributing to oncogenesis108 (Fig. 3a). PRC2 LOF is found in around 25% of T-ALL in association with oncogenic activating mutations of the JAK/STAT signaling pathway and leads to a global epigenetic remodeling towards H3K27Ac. This active histone mark is recognized by Bromodomain and Extraterminal (BET)-domain proteins that act as its specific readers, allowing reactivation of a BET-dependent transcriptional network that triggers stem cell-like programs leading to poor prognosis. PRC2-altered T-ALL being dependent on BET proteins, BET domain protein inhibition is therefore a promising therapeutic avenue in PRC2-associated-T-ALL patients.109
EZH2 post-translational modifications (PTMs) play an additional role in certain type of cancers110,111 (Fig. 3c). In patients with advanced prostate cancer, H3K36me3 and H3K27me3 levels are inversely correlated.78,111 SETD2, the methyltransferase responsible for H3K36me3 deposition, also monomethylates EZH2 on its lysine 735 residue, inducing EZH2 degradation and consequently delaying metastasis. SETD2 is strongly correlated with the presence of EZH2-K735me1 and particularly found in patients with prostate cancer with better clinical outcome.111 On the contrary, EHZ2-K307 methylation by SMYD2 improves its stability and participates in the transcriptional repression of pro-apoptotic, anti-proliferation and anti-invasion target genes112 (Fig. 3c). Multiple EZH2 PTMs play thus a role in EZH2 function and stability that will result in an H3K27 hypermethylation or hypomethylation of the chromatin landscape that favors tumorigenesis (Table 1).
Additionally, SUZ12 is upregulated in a variety of cancers, including ovarian, colorectal and head and neck squamous cell carcinoma.113,114,115 The knockdown of SUZ12 is able to reverse tumor growth by inhibiting proliferation and inducing apoptosis in these contexts.113,115 On the other hand, SUZ12 loss in T-ALL disrupts the PRC2 complex, leading to H3K27me3 decrease which correlates with the opening of chromatin and upregulation of the corresponding genes involved in oncogenic signaling pathways92 (Fig. 3a). Moreover, PRC2 loss induces a genome-wide redistribution of the H3K27Ac mark and the activation of poised enhancers.62 Therefore, similar to EZH2, SUZ12 can act as pro-oncogenic or tumor suppressor depending on the cancer type.
As previously mentioned, PRC2 can be divided into two sub-complexes, PRC2.1 and PRC2.2. While their target genes are overlapping,34,116 their differences rely on their affinity to chromatin.117 Indeed, PRC2.1 tends to have a higher affinity to chromatin, which leads to an increase in H3K27me3 deposition and silencing of PcG target genes in the presence of high ratios of PRC2.1 to 2.2.117 In leukemia, colon and uterine adenocarcinomas, missense mutations of SUZ12, SUZ12(R103P/Q), result in JARID2 depletion, leading to an increase in PRC2.1 formation which enhances PRC2 chromatin occupancy.117 How PRC2.1 could be specifically implicated in cancer remains to be determined.
Although PRC2 dysregulation events have been widely documented in cancer, it is still difficult to decipher whether they are drivers in tumorigenesis. Even if EZH2 is dispensable for the progression of prostate and mammary cancer, it is nonetheless highly expressed.118 In fact, in normally dividing cells, the rate of EZH2 expression correlates with proliferation rates,118 compensating the proliferation-dependent dilution of H3K27me3. In these cancers, even though EZH2 is overexpressed, tumor cells paradoxically fail to maintain a wild-type dose of H3K27me3. The use of EZH2 inhibitors for cancer treatment should therefore carefully take into account the tumor proliferation status.118 With the aim to identify the cancer types in which treatment using PRC2 inhibitors could be beneficial, a genomic and transcriptomic analysis using available databases on clinical tumor samples and a panel of tumor cell lines has been performed, revealing a correlation of EZH2, SUZ12 or EED amplifications with poor prognosis in a subclass of human cancers like renal papillary cell carcinoma, low-grade glioma and hepatocellular carcinoma.119 Interestingly, GOFs of PRC2 subunits are also anti-correlated with poor prognosis in some cancers like gastric cancer and thymoma, suggesting a tumor suppressor function of PRC2 in those cases.
It remains to be understood why certain tumors are addicted to one specific PRC2 subunits but not the others. Clearly, a better understanding of the rate-limiting roles and the cell type-specific functions of each of the PRC2 subunits will require future research.
PRC1 in cancer
Like PRC2, PRC1 components are widely implicated in many types of cancers (Table 2). BMI-1 (PCGF4), a cPRC1.4 subunit, has historically been described as a proto-oncogene that collaborates with the c-Myc oncoprotein to trigger tumorigenesis.120,121,122,123 The INK4a-ARF locus, encoding the tumor suppressors p16Ink4 and p19Arf, is a direct target of PRC1.4.124 BMI-1 deficiency is associated with overexpression of p16 Ink4 and p19Arf and therefore with cell cycle arrest, senescence and apoptosis (Fig. 4a). In contrast, BMI-1 overexpression triggers cell proliferation by repressing ink4a-ARF expression.124 BMI-1 is involved in gastric, pancreatic, breast and ovarian cancer among others.125,126,127,128,129 MEL-18 (PCGF2), a BMI-1 homolog, has a tumor suppressing activity.130,131,132 BMI-1 and MEL-18 expression levels are inversely correlated in various cancers.133,134 BMI-1 expression depends on its counterpart MEL-18 (Fig. 4a). c-Myc is a transcriptional activator of BMI-1. Mel-18 overexpression is linked to c-myc downregulation, leading to BMI-1 decrease, p16 upregulation and ultimately to cell senescence.135 Interestingly, in flies, LOF of cPRC1 members results in upregulation of cancer-related genes, including genes involved in the Notch, JNK and JAK/STAT signaling pathways74,136,137 (Fig. 4b), a difference that might be due in part to the absence of PcG-mediated repression of the INK4a-ARF locus in flies.

a PCGF2 inhibits the transcription of c-myc. Loss of c-Myc results in the decrease of PCGF4 expression, and in the derepression of PCG4 target genes, such as the INK4a-ARF locus. p19 and p16 participate in proliferation control, respectively, by inhibiting MDM2-mediated degradation of p53 and inhibiting CycD/CDK4-mediated phosphorylation of pRb. b PRC1 oncogenic activity may also be PRC2-independent. PRC1 is found on specific targets lacking the H3K27me3 repressive mark. Surprisingly, these genes exhibit active marks such as H3K27Ac and H3K4me1/3. Gene ontology analysis characterized these cancer-related genes as components of cell signaling, like the Notch and JAK/STAT signaling pathways. c PRC1 mutations are rarely found in cancer, although some mutations have been found to impact variant PRC1. Indeed, mutations (indicated by a star) in BCOR, a scaffold protein involved in ncPRC1.1, are found in SHH-driven medulloblastoma. The presence of these mutations promotes a neoplastic state of cancer cells by preventing Polycomb recruitment to its target genes. d PTM of PRC1 subunits can promote tumorigenesis. The deposition of O-GlcNAcylation on PCGF4 (BMI-1) inhibits its degradation. PCGF4 protein levels are increased and participate in the transcriptional silencing of downstream target genes such as the INK4a-ARF locus, thus promoting oncogenic cell proliferation. e In hormone-dependent cancers, PRC1 genes are often amplified. Top: in prostate cancer, the AR promotes the expression of PCGF4. Additionally, it can interact with the PCGF4 protein, resulting in inhibition of AR degradation and transcriptional activation of its downstream target genes. Bottom: cPRC1 can also interact with the ER and its pioneer factor FOXA1 in ER+ breast cancer cells and bind to enhancers that stimulate transcription of cancer-related genes decorated with active histone marks. AR, Androgen Receptor; Cdk4, 6, Cyclin Dependent Kinase 4, 6; ER, Estrogen Receptor; FOXA1, Forkhead Box A1; Igf2, Insulin-like growth factor 2; MDM2, Murine Double Minute 2; PSA, Prostate Specific Antigen; Rb, Retinoblastoma.
Using an in vivo and in vitro approach, ncPRC1.1 was shown to specifically target active genes independently of PRC2.74,138 At a genome-wide level, the correlation between mammalian RING1B and the H3K27me3 mark decreases during lineage decision processes. While PRC1-RING1B targets are clearly enriched for the repressive H3K27me3 mark in ESCs, this is only the case for ~30% of them in differentiated cells.74 While gene ontology categories associated with H3K27me3-dependent targets are linked to developmental pathways, H3K27me3-independent targets are linked to cell cycle regulation, cell polarity, metabolism and signaling pathways74,138 (Fig. 4b). This difference in PRC1 targeting results from major changes in the qualitative and quantitative compositions of the ncPRC1 variant complexes.15,71
Unlike PRC2 mutations, PRC1 mutations are not overrepresented in cancer.139 However, some mutations affecting ncPRC1 have been described.140,141 In SHH-driven medulloblastoma, the PRC1.1 BCOR scaffold protein is mutated at its C-terminal domain that normally interacts with PCGF1,141,142 resulting in loss of PRC1.1 recruitment to genes coding for growth factors that would otherwise be repressed141 (Fig. 4c). Likewise, MGA, a transcription factor that is a member of the Myc network and interacts with ncPRC1.6 subunits, is a tumor suppressor in vivo that acts by recruiting ncPRC1.6 to its target genes.143 Moreover, BAP1, a component of the Polycomb Repressive complex DeUbiquitinase (PR-DUB), is a tumor suppressor.144,145 Recent data suggest that this protein prevents widespread H2AK119ub deposition and chromatin condensation at non-target loci, restricting H2AK119ub to Polycomb target genes. BAP1 may thus prevent inappropriate redistribution of Polycomb complexes away from their targets and play critical roles, particularly by maintaining the appropriate chromatin state of lineage commitment genes.146,147,148,149 It is therefore not surprising that PR-DUB misregulation leads to tumorigenesis. Enhancing deubiquitinase activity leads to a widespread depletion of the H2AK119ub mark.140 Conversely, disruption of its chromatin recruitment or catalytic activity could result in an increase in H2AK119ub and H3K27me3.146,150 Depending on the genes targeted, this might switch the transcriptional state of oncogenes or tumor suppressor genes.
In summary, the implication of PcG components in cancer, either by point mutations or by dysregulation of its components, is widely established. Through tumor suppressor or oncogenic activity in a broad type of cancers, PcG members control tumor growth and survival.151 Targeting PRC2 members or proteins involved in PRC2 stability, either by inhibiting its enzymatic activity or by interfering with PRC2 complex assembly or stability, appears to be a promising strategy to prevent growth of PRC2-dependent tumors79,152,153,154 (Table 3). However, since PRC1 can either repress or activate the transcription of its target genes, it is both the downregulation and/or upregulation of tumor suppressors and oncogenes respectively that might participate in tumorigenicity.69,155,156,157 The exact role of PRC1 complexes in cancer, and in particular the importance of ncPRC1 complexes, remains to be determined. Future work would be important to better characterize the molecular implication of Polycomb complexes and define appropriate therapeutic approaches to rescue their dysregulation in different types of cancer.
Environmental cues and polycomb-dependent oncogenesis
Hormone-dependent cancer
PRC1 genes are significantly amplified in hormone-dependent cancers.139 Since hormone receptors are transcription factors, they might participate in tumorigenesis by triggering ectopic recruitment of Polycomb proteins to a specific set of target genes. In particular, the androgen receptor (AR) and the estrogen receptor (ER) can directly recruit PcG proteins at their response elements in hormone-dependent cancers.139,158,159,160 In prostate cancer, maintenance of AR expression is essential. The overexpression of BMI-1 and its increased protein stability mediated by PTMs, such as O-GlcNAcylation, participate in the self-renewal of cancer cells and the progression of prostate cancer161,162 (Fig. 4d). Furthermore, the binding of BMI-1 to AR inhibits the ubiquitin–proteasome degradation pathway.163 Surprisingly, the AR interacts with BMI-1 in a PRC1-independent manner.163 By coupling ChIP-seq and CRISPR methodologies, it was found that Androgen Response Elements (AREs) are located in the BMI1 locus and enriched for the H3K27Ac active enhancer mark, suggesting that the AR activates transcription of BMI-1.160 Moreover, a positive feedback loop exists in prostate cancer where BMI-1 overexpression stabilizes AR, which in turn transcriptionally activates BMI-1 expression, leading to tumor progression (Fig. 4e). In addition, a PRC2-independent EZH2 oncogenic function relies on its direct interaction with AR, leading to AR transcription and activation of AR downstream targets164,165,166 (Fig. 3d). This PRC2 genome-wide redistribution also results in ectopic targeting, in particular to tumor suppressor genes, particularly those involved in INF-ɣ signaling, that are repressed by the H3K27me3 mark in prostate cancer167,168 (Fig. 3e).
The redistribution of PcG-targets is an important mechanism participating in tumorigenesis and cancer progression. Surprisingly, in breast cancer, ERα, β-catenin and EZH2 interact and target oncogenes, such as c-Myc and Cyclin D1, acting as transcriptional co-activators.169 Furthermore, the redistribution of PRC1 leads to its association with active enhancers enriched for the H3K4me1 mark.139 RING1B was proposed to facilitate ERα recruitment to enhancers and super-enhancers, as well as to promoters of cancer-related genes139,170 (Fig. 4e). However, how RING1B is recruited to open chromatin sites and how it selectively binds to a subset of them is still unclear.
Metabolism
Proliferation and growth of cancer cells are known to be associated with an extensive rewiring of metabolism and energy production networks where Polycomb complexes are clearly involved. As already mentioned, changes in methylation of H3K27 participate in tumor progression.94,108 Tight regulation of the methyl group available for EZH2 activity is essential to maintain a proper chromatin landscape. The catalytic activity of EZH2 depends on the methyl donor S-adenosylmethionine (SAM)171 (Fig. 5e). SAM is formed by the combination of a methionine, which crosses the cell membrane via the LAT1 transporter, and an ATP molecule. Cancer cells with higher levels of LAT1 expression have a more aggressive phenotype.172 Upon LAT1 depletion, the SAM pool is significantly reduced, correlating with a decrease in H3K27me3 deposition even if EZH2 protein concentration is constant.172 In addition, repression of RXRα, a known negative regulator of LAT1, by the PRC2 complex maintains a positive feedback loop between LAT1 and EZH2, enhancing EZH2 methyltransferase activity172 (Fig. 5a). Indeed, EZH2 inhibition via competition with SAM has a potent anti-tumor effect.173

a Left: in a physiological condition, the membrane transporter LAT1 participates in the transport of methionine which reacts with ATP to produce SAM. SAM can in turn be used by PRC2 to induce trimethylation of H3K27, resulting in a PcG-mediated silencing of its targets genes. Lat1 expression depends on RXRα. Right: in cancer cells, Lat1 is overexpressed, enhancing SAM production and inducing H3K27 hypermethylation of the chromatin landscape. The Lat1 negative regulator, RXRα, is thus repressed resulting in a positive feedback loop whereby EAF2 transcriptional silencing dependent on PRC2 results in overexpression of HIF1, which can in turn stimulate Lat1 expression. Therefore, an excess of LAT1 at the cellular membrane increases the transport of BCAAs, thereby enhancing protein synthesis. b Controlling the immune system is of a major importance in cancer. Cancer cells use different mechanisms to do this. First, PRC1 is able to increase the transcriptional expression of CCL2, which will dampen Treg immune response. In addition, PRC2-mediated silencing of the MHC-I antigen processing pathway results in MHC-I absence at the cell membrane, concealing cancer cells from cytotoxic T cells. Finally, PCGF4 overexpression in cancer cells stimulates the expression of GATA2, which will inhibit MICA/B transcription and reduces its presence at the membrane. This prevents the recognition of cancer cells by NK cells. These mechanisms enhance the immunosuppressive response and inhibit the cytotoxic response that would otherwise kill the cancer cells. c Oncohistones are a new line of research, analyzing the effect of mutations on histone genes that could have an impact on tumorigenesis. H3K27M has a dominant negative effect on EZH2 catalytic activity. Left: in a wild-type condition, PRC2 is recruited to nucleation sites that present unmethylated CGIs. Trimethylation of H3K27 occurs and spreads around the nucleation site. The boundaries of Polycomb domains are decorated with H3K36me2. Right: in the presence of the H3K27M oncohistone, that represents 10% of all H3, an epigenetic remodeling occurs. The spreading of H3K27me3 is inhibited and active histone marks, such as H3K27ac, are present on the oncohistone. BCAAs, Branched-chain amino acids; CCL2, C-C motif chemokine ligand 2; CCR2, C-C motif chemokine receptor; EAF2, ELL associated factor 2; GATA2, GATA binding protein 2; HIF1, Hypoxia inducible factor 1; IDH1, Isocitrate dehydrogenase 1; KDM6A/B, Lysine demethylase 6A/B; LAT1, L-type amino acid transporter 1; MHC-I, Major histocompatibility complex I; MHC-I APP, Major histocompatibility complex I antigen processing pathway; MICA/B, MHC I polypeptide-related sequence A/B; RXRα, Retinoid X receptor-alpha; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; TCR, T-cell receptor; SETD2, SET domain containing 2 (histone lysine methyltransferase).
PcG proteins are also involved in the regulation of branched-chain amino acids (BCAAs),174 key regulatory components for protein synthesis and energy production, both of which are also the fuel of cancer progression.175 Enzymes required for BCAA catabolism, known as BCAA aminotransferases (BCATs), are often overexpressed in cancer cells.176 In myeloproliferative neoplasms (MPNs), the combination of partial loss of PRC2 and expression of the constitutively active oncoprotein NRASG12D — a member of the Ras GTPase family — has been shown to lead to BCAT1 expression, which is normally repressed in hematopoietic stem cells.174 The increase in BCAT1 results in a larger pool of BCAAs that activates mTOR, a protein kinase known to participate in tumor growth and proliferation.177 It should be noted that in patients with Acute Myeloid Leukemia (AML), the expression of EZH2 and BCAT1 is inversely correlated, a high expression of BCAT1 being associated with a poor survival outcome.174 In glioblastoma cancer cells, rather than modulating BCAT expression, it is the BCAA pool that increases. In this case, EZH2 represses EAF2 which inhibits Hypoxia-Inducible Factor 1 (HIF1).178 HIF-1 overexpression participates in the Warburg effect by supporting glycolytic metabolism and upregulating expression of LAT1, the main transporter of BCAAs, which results in an increase in BCAA pool178,179 (Fig. 5a, right).
The Warburg effect is the most well-known cancer metabolic alteration, whereby malignant cells use glycolysis rather than oxygen-dependent metabolism. A tight regulation of glucose homeostasis is essential to counter the proliferation of cancer cells. An important node in this pathway is the reaction catalyzed by the enzyme Fructose-1,6-biphosphatase (FBP1). Low FBP1 enzyme activity correlates with higher production of pyruvate, the downstream product of the glycolysis pathway. An over-production of pyruvate corresponds to a greater store of energy available for cancer cell growth. In hepatocellular carcinoma and clear cell renal cell carcinoma, the mRNA levels of Ezh2 and FBP1 are inversely correlated due to the presence of the EZH2-dependent H3K27me3 repressive mark at the promoter of the gluconeogenic enzyme-coding gene.180 Tumor growth was shown to be thwarted either by a short-hairpin RNA (shRNA) directed against Ezh2, or by the reintroduction of FBP1. Interestingly, FBP1 and EZH2 interact directly. In doing so, FBP1 is able to reduce the methyltransferase activity of EZH2 by dissociating the PRC2 complex. This double negative feedback loop provides new insights into the involvement of Polycomb in “oncometabolism”.
Metabolic reprogramming during tumorigenesis is required to better sustain the energy necessary for cancer progression and survival.181,182 PcG proteins have been implicated in the regulation of metabolic genes involved in metabolism of fatty acids and pyruvates among others.139,183 While the link between PRC2 function and metabolism in physiology and cancer is certain, much work remains to be done in order to understand the molecular underpinnings of this link in different cancer types and to harness them to design effective therapeutic strategies. Noteworthy, with most of the current research focusing on the link between PRC2 and metabolism,184 it might also be of interest to examine the involvement of PRC1 in future work.
Immune system
The immune system has a wide array of cells that protect from foreign bodies, also known as non-self. Innate immune cells provide a rapid and nonspecific response while adaptative immune cells have a slower response that relies on a memory process that will be specific to a known foreign object.185 In principle, both innate and adaptative immune cells exert an anti-tumor function.
However, cancer cells can develop multiple mechanisms to evade recognition and destruction by the immune system and become resistant to therapy. In prostate cancer, elevated PRC1 levels and activity coincide with epithelial-to-mesenchymal transition (EMT) and stemness signatures. PRC1 directly promotes metastasis at metastatic initiation sites by controlling self-renewal and both cPRC1 and ncPRC1.1 components directly induce transcriptional expression of CCL2 and other pro-metastatic genes that encode cytokines, which suppress the immune response and promote a pro-angiogenic environment186 (Fig. 5b). CCL2 expression has an oncogenic function by recruiting immune cells such as M2-type Tumor-Associated Macrophages (TAMs) and T-regulatory cells (Tregs), promoting an immunosuppressive microenvironment favorable to tumor progression. Moreover, Natural Killer (NK) cells are also involved in the innate immune response. Upon recognition of MICA/B by NK cells, an immune cytotoxic response is displayed. However, BMI-1 stimulates GATA2 expression which in turn directly inhibits MICA/B expression (Fig. 5b). Reduction of MICA/B expression on the surface of cancer cells prevents NK cell activation and the cytotoxic response.187 The combination of these two escape mechanisms promotes cancer cell progression and metastasis. Pharmacological treatment using a catalytic inhibitor of PRC1 suppresses metastasis by reverting the immunosuppressive microenvironment and promoting the recruitment of NK cells and T effector cells.186,187
Cytotoxic T cells (CD8+ T) identify cancer cells presenting foreign antigens by their Major Histocompatibility Complex I (MHC-I). An IFN-ɣ response is then induced to kill the cancer cells. In order to survive, cancer cells downregulate the MHC-I antigen processing pathway (MHC-I APP), resulting in decreased presentation of foreign antigens to CD8+ T188 (Fig. 5b). PRC2 represses transcription of various MHC-I APP components, participating in cancer cell immunosurveillance escape.189 Furthermore, PRC2 inhibits anti-tumor immunity by altering the transcriptional landscape of Tregs. Indeed, immunocompetent mice bearing tumors treated with an EZH2 inhibitor show a significant decrease in tumor volume compared to mice deficient in T cells, suggesting an interplay between EZH2 and the T cell immune response.190 Tregs promote tumor progression in an EZH2-dependent manner by producing immunosuppressive cytokines and preventing recruitment of T CDC8+.190,191 Pharmacological EZH2 inhibition induces a change in the production of pro-inflammatory cytokines which promotes anti-tumor activity and significantly increases the ratio between CD8+ T and Tregs in the tumor microenvironment.190
Cancer immunotherapy has revolutionized the clinical approach in the field of oncology. However, anti-CTLA4, the first monoclonal antibody used in cancer therapy as an immune checkpoint, induces an upregulation of EZH2 expression191 which may prevent anti-tumor immunity by inducing an immunosuppressive tumor microenvironment.190,191 A synergistic strategy coupling anti-CTLA4 and an EZH2 inhibitor reverses cancer resistance to the immune system.191,192 Moreover, considering the involvement of PcG proteins in pluripotency, it is not surprising that PcG proteins are also involved in cancer stem cell (CSC) development and resistance to treatment. Although anti-PD1 immunotherapy is sufficient to recruit CD8+ T cells into the tumor microenvironment, it is not sufficient to kill BMI-1+ CSCs.193 Inhibition of BMI-1 de-represses H2AK119ub-decorated target genes and increases DNA-damage, stimulating the inflammatory response and CD8+ T cells recruitment.193 In summary, joint targeting of immune checkpoints and PcG proteins appears to be a new promising therapeutic approach to efficiently counter cancer progression by stimulating the immune response.191,192,193
Oncohistones
As already mentioned, the catalytic activities of “writers” and “erasers” enzymes that modify histone PTMs are often dysregulated in cancer where chromatin landscapes are modified, resulting in aberrant transcription of the corresponding genes.99,194 In addition, the lack of recognition of H3K27me3 by the CBX7 “reader” results in a transcriptional de-repression of tumor suppressor genes.195 Likewise, the BAHCC1 mutation in its BAH domain leads to upregulation of tumor suppressor genes that dampen tumor progression.38
These data point to a direct involvement of histone modifications in tumorigenesis. Indeed, somatic mutations in histone genes occur at high frequency in cancer, and they can exhibit oncogenic properties.196 K-to-M/I missense substitutions in histone variants, analyzed from available sequenced genomes of several human cancer types of ~3000 patients, further argue for a driver or contributor effects of the known N-terminal tail mutations affecting H3.196 These mutations are particularly frequent in rare malignancies such as glioma and chondroblastoma. This analysis allowed detection of previously unappreciated situations where histones are mutated at low frequency in common cancers, like H3K27M in melanoma and AML.196
One of the most studied cancer-associated “oncohistones” carries the H3K27M substitution, whereby H3 lysine 27 is mutated to methionine, a missense mutation showing high genetic penetrance in pediatric glioblastomas197,198 (Fig. 5c). It is noteworthy that different H3 mutants are found in distinct locations. Indeed, H3F3A mutations such as H3.3K27M or H3.3G34R/V are found respectively in midline pediatric high-grade gliomas and cortex, whereas HIST1H3B mutations affecting the canonical H3.1 are restricted to the brainstem.199 H3F3A which encodes the histone variant H3.3 is found mutated in 60% of Diffuse Intrinsic Pontine Glioma (DIPG) cases198 and this mutation is suggested to be the first hit in DIPG tumorigenesis.200 This driver mutation is associated with obligate partner mutations throughout tumor progression,200 in particular in the cell cycle regulatory gene TP53 or the chromatin remodeler ATRX. Interestingly, while H3.3K27M represents less than 10% of total H3, this level is sufficient to induce a significant decrease in the trimethylated state of H3K27, leading to a decrease in PcG-dependent transcriptional silencing.201,202
The epigenome is drastically altered in an H3K27M context. Indeed, while H3K27me3 is specifically restricted to unmethylated CGIs and H3K27me3/2 levels are significantly decreased, the monomethylation level of H3K27 remains unchanged.203 Intriguingly, H3K27me1 distribution is completely rewired in an H3K27M context.203 Moreover, just like in a wild-type H3 context, H3K36me2 restricts the spreading of H3K27me2/3.203 Furthermore, H3K27Ac levels are globally increased at the H3K27M location.201,204 This suggests that H3K27M has a dominant negative effect on the catalytic activity of the EZH2 methyltransferase.201,202
There is a strong interest in understanding the molecular mechanisms by which oncohistone mutations change the epigenome and impact gene expression. PRC2 was proposed to have a higher affinity for the mutated histone, which binds the EZH2 enzymatic domain, inhibiting its methyltransferase activity.201,205,206 However, the mechanism by which H3K27M oncohistones inhibits PRC2 activity is still under debate.207 The finding that PRC2 appears to be excluded from the H3K27M-K27Ac domains208 argues against the model of PRC2 sequestration by H3K27M. Moreover, while it is suggested that gliomagenesis is dependent on PRC2 inhibition,201 it has been demonstrated that loss of PRC2 disables growth and colony formation in H3K27M-positive DIPG cells, underlying the importance of PRC2 in tumor maintenance.208
Interestingly, CATACOMB/EZHIP, a PRC2 co-factor, either via its overexpression or a chromosomal translocation inducing its fusion with the NuA4 subunit gene MBTD1, described in low-grade endometrial stromal sarcoma,209 decreases PRC2-dependent methyltransferase activity.9 CATACOMB/EZHIP-dependent hypomethylation is due to a conserved methionine residue M406 which inhibits EZH2, mimicking the H3K27M oncohistone.9 Moreover, H3K27M and CATACOMB/EZHIP are mutually exclusive in gliomas, specifically in Posterior Fossa A (PFA) ependymomas.210 Both are suggested to decrease H3K27 trimethylation by blocking the spreading of the repressive mark from CGIs.211
In Giant Cell Tumor of the bone (GCT), the oncohistone H3.3G34W is encountered in 90% of cases.212 Interestingly, this residue is not post-translationally modified but its impact on the epigenome is undeniable. This mutation leads to loss of H3K36me3 which counteracts H3K27me3 deposition by PRC2.212 As a consequence, a redistribution of the H3K27me3 repressive mark occurs from intergenic to genic regions, resulting in perturbation in Polycomb-mediated silencing and in the maintenance of a progenitor state of the mutated cells.212
As previously mentioned, a crosstalk exists between H3K36me2/3 and H3K27me3 and this interplay remains in the presence of the H3K36M oncohistones, in which lysine 36 of the histone 3 is replaced by a methionine. This mutation is found in 95% of chondroblastomas and 92% of GCT, respectively in the H3F3B and H3F3A genes.213 Following the oncohistone paradigm, the H3K36me2/3 PTMs are reduced due to the inability of specific methyltransferases, namely SETD2, NSD1-NSD3, to deposit their marks.214,215 H3K36M reduces H3K36 methylation and increases nucleosome availability for PRC2 to deposit H3K27me3.214 The genome-wide increase in this repressive mark then induces a PRC1 redeployment which overall dilutes PRC1 at its canonical binding sites, leading to de-repression of self-renewal genes214,216 (Fig. 5c). Similarly, in human papillomavirus (HPV)-negative head and neck squamous cell carcinomas (HNSCCs),217 the H3K36 methylation state is involved in oncogenic promotion.217 NSD1 writer mutations, similarly to H3K36M, remodel the chromatin landscape by decreasing H3K36me2 levels. Considering the interplay between H3K36me2 and H3K27me3, Polycomb components would be expected to be involved in HNSCCs. However, the precise mechanism at play is yet to be characterized.
All these data show that the emerging oncohistone field is an important area of oncology, but much remains to be done and a current strong focus is on the investigation of how histone mutations contribute to epigenome reprogramming and whether these mutations are primarily drivers or contributors of tumorigenesis in a wide range of human cancers.194
Non-genetic drug resistance in cancer
The ability of cancer cells to adapt to or resist anti-cancer therapies may be inherently of a genetic nature or may be acquired during treatment.218 Alongside an undergoing genetic evolution of cancer genomes, cancer cells can also be modified in their epigenetic landscapes and this non-genetic contribution can play a major role in cancer resistance. In fact, relapsing patients often do not present specific mutations that would explain a lower efficiency for the same therapy.219,220
Cancer cells are actually able to evolve and change completely their transcriptional landscape to adapt to treatment-induced stress. Polycomb implication in cancer drug-resistance depends on PRC2 and its catalytic activity as well as on other concomitant mechanisms that can induce transcriptional plasticity.221,222,223
In multiple myeloma (MM), cell adhesion-mediated drug resistance (CAM-DR) develops when malignant plasma cells interact with stromal cells in the bone marrow and become less sensitive to chemotherapy.224 In an in vitro system that recapitulates CAM-DR, anti-MM treatment results in an increase and redistribution of H3K27me3 in a dose-dependent manner in cultured MM cells only when they do not adhere to stromal cells.224 CAM-DR counteracts drug-induced H3K27 hypermethylation via phosphorylation of EZH2 at serine 21, leading to overexpression of anti-apoptotic genes which participate in survival and drug-resistance.224 In addition, miR-15a downregulation triggers PHF19 upregulation in relapsed MM patients.225 The involvement of PHF19 in drug-resistance might depend on its ability to stimulate proliferation by promoting EZH2 serine 21 phosphorylation, which inhibits the H3K27me3 deposition and leads to upregulation of genes linked to cell growth.225
In Testicular Germ Cell Tumors (TGCT), resistance to cisplatin is accompanied by a global decrease in H3K27me3 and H2AK119Ub levels, leading to upregulation of Polycomb target genes.223 Inhibition of the UTX and JMJD3 enzymes, responsible for H3K27 demethylation, is sufficient to increase H3K27me3 and make TGCT cells more sensitive to the initial chemotherapy.223
In AML, therapeutic resistance can arise in the apparent absence of new genetic mutations and is antagonized by inhibiting Lsd1, a demethylase chromatin modulator involved in the regulation of enhancer activity.219 Inhibition of Lsd1 creates enhancer switching, generating new binding sites for pioneer factors that ultimately activate the enhancers of key drug resistance genes. Inhibition of a key chromatin modulator in AML then makes it possible to resensitize cells to the primary treatment.219
Unlike mutations, failed or disrupted epigenetic mechanisms can be quite easily reverted using epidrugs to overcome cancer progression by rewiring malignant epigenomes, either to resensitize tumor cells resistant to conventional therapy or to sensitize them to new therapies. Given the importance of PcG proteins in transcriptional regulation, it will therefore be of great interest to further characterize the mechanisms by which PcG proteins contribute to drug-resistance. In particular, it will be important to expand research aimed at understanding Polycomb functions at enhancers,139,226 since they might be involved in various cancer types and stages.
Conclusive remarks: Polycomb epigenetics in cancer
Although it is commonly assumed that cancer arises from a set of multiple mutations, a pan-cancer analysis established that about 5% of cancer cases did not have driver mutations that could explain tumorigenesis, pointing out that genetics might not be the only player in cancer.227 Non-genetic alterations appear to represent an alternative path toward the development, progression and drug-resistance of cancer cells. In pancreatic ductal adenocarcinoma, metastases do not show driver gene mutations but rather follow drastic epigenomic reprogramming,181,228 suggesting that epigenetic modifiers are mainly involved. Additionally, ependymomas — a childhood brain tumor — are characterized by a very low mutation rate,229 suggesting that cancer is not only a consequence of DNA mutations, but rather emerges and evolves from a crosstalk between genetic and non-genetic processes. In an extreme view, cancer has been defined as an “epigenetic disease”.230 It would therefore not be surprising to find misregulated Polycomb proteins as epi-drivers in tumorigenesis.
PcG proteins have imposed themselves in a wide range of biological processes. Clearly, they are landmark components in the field of cancer research and we have only started to understand the extent of their oncogenic functions. As most research focuses on the EZH2 catalytic subunit of PRC2, it will be interesting to better characterize the involvement of the different PRC2 subunits as well as on the many flavors of PRC1 complexes. Nonetheless, a fascinating part of the oncogenic function of PcG components relies on the fact that some of them can act both in a manner dependent or independent on Polycomb complexes.231 It will be interesting to better characterize these PcG functions at the molecular level in order to have a complete picture of their mode of action. While it is clear that the overexpression or downregulation of PcG proteins is involved in cancer, it will be important to characterize how modifying protein stability by either PTMs and/or interaction with yet unidentified partners might be implicated in tumorigenesis. Finally, context is of paramount importance: misregulation of Polycomb proteins results in different, sometimes even opposing results in different cancer types. Identifying molecular pathways leading to these context-dependent effects will be crucial in order to improve cancer diagnosis, prognosis and therapy.
References
Loubiere, V., Martinez, A. M. & Cavalli, G. Cell fate and developmental regulation dynamics by polycomb proteins and 3D genome architecture. BioEssays 41, 1–15 (2019).
Chan, H. L. & Morey, L. Emerging roles for polycomb-group proteins in stem cells and cancer. Trends Biochem. Sci. 44, 688–700 (2019).
Piunti, A. & Shilatifard, A. The roles of polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 22, 326–345 (2021).
Lewis, E. B. A gene complex controlling segmentation in Drosophila. Nature 276, 565–570 (1978).
Levine, S. S. et al. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol. Cell. Biol. 22, 6070–6078 (2002).
Schuettengruber, B., Bourbon, H. M., Di Croce, L. & Cavalli, G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171, 34–57 (2017).
Kuzmichev, A., Nishioka, K., Erdjument-Bromage, H., Tempst, P. & Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16, 2893–2905 (2002).
Ragazzini, R. et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 10, 3858 (2019).
Piunti, A. et al. Catacomb: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv 5, eaax2887 (2019).
Wang, H. et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878 (2004).
Cohen, I., Bar, C. & Ezhkova, E. Activity of PRC1 and histone H2AK119 monoubiquitination: revising popular misconceptions. BioEssays 42, 1–8 (2020).
Shao, Z. et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98, 37–46 (1999).
Garcia, E., Marcos-Gutiérrez, C., Del Mar Lorente, M., Moreno, J. C. & Vidal, M. RYBP, a new repressor protein that interacts with components of the mammalian Polycomb complex, and with the transcription factor YY1. EMBO J 18, 3404–3418 (1999).
Wang, R. et al. Polycomb group targeting through different binding partners of RING1B C-terminal domain. Structure 18, 966–975 (2010).
Gao, Z. et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 (2012).
Simon, J., Chiang, A., Bender, W., Shimell, M. J. & O’Connor, M. Elements of the Drosophila bithorax complex that mediate repression by Polycomb group products. Dev. Biol. 158, 131–144 (1993).
Van Kruijsbergen, I., Hontelez, S. & Veenstra, G. J. C. Recruiting polycomb to chromatin. Int. J. Biochem. Cell Biol 67, 177–187 (2015).
Kassis, J. A. & Brown, J. L. Polycomb group response elements in Drosophila and vertebrates. Adv. Genet. 81, 83–118 (2013).
Pengelly, A. R., Copur, Ö., Jäckle, H., Herzig, A. & Müller, J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339, 698–699 (2013).
Min, J., Zhang, Y. & Xu, R. M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17, 1823–1828 (2003).
He, J. et al. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 15, 373–384 (2013).
Blackledge, N. P. et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459 (2014).
Cooper, S. et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 7, 13661 (2016).
Tamburri, S. et al. Histone H2AK119 mono-ubiquitination is essential for Polycomb-mediated transcriptional repression. Mol. Cell 77, 840–856.e5 (2020).
Sing, A. et al. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 138, 885–897 (2009).
Woo, C. J., Kharchenko, P. V., Daheron, L., Park, P. J. & Kingston, R. E. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell 140, 99–110 (2010).
Ku, M. et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4, e1000242 (2008).
Mendenhall, E. M. et al. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6, e1001244 (2010).
Riising, E. M. et al. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG Islands genome wide. Mol. Cell 55, 347–360 (2014).
Li, H. et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291 (2017).
Hunkapiller, J. et al. Polycomb-like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PLoS Genet 8, e1002576 (2012).
Perino, M. et al. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 50, 1002–1010 (2018).
Chen, S., Jiao, L., Liu, X., Yang, X. & Liu, X. A dimeric structural scaffold for PRC2-PCL targeting to CpG Island chromatin. Mol. Cell 77, 1265–1268.e7 (2020).
Højfeldt, J. W. et al. Non-core subunits of the PRC2 Complex are collectively required for its target-sitespecificity. Mol. Cell 76, 423–436.e3 (2019).
Bierne, H. et al. Human BAHD1 promotes heterochromatic gene silencing. Proc. Natl. Acad. Sci. USA 106, 13826–13831 (2009).
Zhao, D. et al. The BAH domain of BAHD1 is a histone H3K27me3 reader. Protein Cell 7, 222–226 (2016).
Fan, H. et al. A conserved BAH module within mammalian BAHD1 connects H3K27me3 to Polycomb gene silencing. Nucleic Acids Res 49, 4441–4455 (2021).
Fan, H. et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet. 52, 1384–1396 (2020).
Lebreton, A. et al. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 331, 1319–1321 (2011).
Achour, C. & Aguilo, F. Long non-coding RNA and Polycomb: an intricate partnership in cancer biology. Front. Biosci. 23, 2106–2132 (2018).
Yu, M. et al. Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol. Cell 45, 330–343 (2012).
Cenik, B. K. & Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet. 22, 38–58 (2021).
Douillet, D. et al. Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat. Genet. 52, 615–625 (2020).
Brinkman, A. B. et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22, 1128–1138 (2012).
Streubel, G. et al. The H3K36me2 methyltransferase Nsd1 demarcates PRC2-mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol. Cell 70, 371–379 (2018).
Francis, N. J., Kingston, R. E. & Woodcock, C. L. Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577 (2004).
Trojer, P. et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell 42, 438–450 (2011).
Grau, D. J. et al. Compaction of chromatin by diverse polycomb group proteins requires localized regions of high charge. Genes Dev 25, 2210–2221 (2011).
Cheutin, T. & Cavalli, G. Loss of PRC1 induces higher-order opening of Hox loci independently of transcription during Drosophila embryogenesis. Nat. Commun. 9, 3898 (2018).
Eskeland, R. et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464 (2010).
Blackledge, N. P. et al. PRC1 catalytic activity is central to polycomb system function. Mol. Cell 77, 857–874.e9 (2020).
Zhou, W. et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 29, 69–80 (2008).
Kadoch, C. et al. Dynamics of BAF-polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 49, 213–222 (2017).
Hodges, H. C. et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 25, 61–72 (2018).
Dellino, G. I. et al. Polycomb silencing blocks transcription initiation. Mol. Cell 13, 887–893 (2004).
Stock, J. K. et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 9, 1428–1435 (2007).
Ardehali, M. B. et al. Polycomb repressive complex 2 methylates elongin A to regulate transcription. Mol. Cell 68, 872–884.e6 (2017).
Nakagawa, T. et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev 22, 37–49 (2008).
Tie, F. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 (2009).
Tie, F. et al. Polycomb inhibits histone acetylation by CBP by binding directly to its catalytic domain. Proc. Natl. Acad. Sci. USA 113, E744–E753 (2016).
Tie, F. et al. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize polycomb silencing. Development 141, 1129–1139 (2014).
Ferrari, K. J. et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014).
Lanzuolo, C., Roure, V., Dekker, J., Bantignies, F. & Orlando, V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 9, 1167–1174 (2007).
Ogiyama, Y., Schuettengruber, B., Papadopoulos, G. L., Chang, J. M. & Cavalli, G. Polycomb-dependent chromatin looping contributes to gene silencing during Drosophila development. Mol. Cell 71, 73–88.e5 (2018).
Wani, A. H. et al. Chromatin topology is coupled to polycomb group protein subnuclear organization. Nat. Commun. 7, 10291 (2016).
Kundu, S. et al. Polycomb repressive complex 1 generates siscrete compacted domains that change during differentiation. Mol. Cell 65, 432–446.e5 (2017).
Bonev, B. et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e24 (2017).
Schoenfelder, S. et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 47, 1179–1186 (2015).
Pherson, M. et al. Polycomb repressive complex 1 modifies transcription of active genes. Sci. Adv. 3, e1700944 (2017).
Giner-Laguarda, N. & Vidal, M. Functions of polycomb proteins on active targets. Epigenomes 4, 17 (2020).
Morey, L., Aloia, L., Cozzuto, L., Benitah, S. A. & Di Croce, L. RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep 3, 60–69 (2013).
Frangini, A. et al. The aurora B kinase and the polycomb protein ring1B combine to regulate active promoters in quiescent lymphocytes. Mol. Cell 51, 647–661 (2013).
Schaaf, C. A. et al. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet 9, e1003560 (2013).
Loubiere, V. et al. Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat. Genet. 48, 1436–1442 (2016).
Gao, Z. et al. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354 (2014).
Liu, S et al. NRF1 association with AUTS2-Polycomb mediates specific gene activation in the brain. Mol. Cell 81, 4663–4676.e8 (2021).
Morey, L. et al. Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell 17, 300–315 (2015).
Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002).
Kim, K. H. & Roberts, C. W. M. Targeting EZH2 in cancer. Nat. Med. 22, 128–134 (2016).
Herviou, L., Cavalli, G., Cartron, G., Klein, B. & Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 7, 2284–2296 (2016).
Yamaguchi, H. & Hung, M. Regulation and role of EZH2 in cancer. Cancer Res. Treat. 46, 209–222 (2014).
Safaei, S., Baradaran, B., Hagh, M. F. & Alivand, M. R. Double sword role of EZH2 in leukemia. Biomed. Pharmacother. 98, 626–635 (2018).
Xie, H. et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80 (2014).
Lee, S. C. W. et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 126, 167–175 (2015).
Mochizuki-Kashio, M. et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood 126, 1172–1183 (2015).
Yu, W. et al. Depletion of polycomb repressive complex 2 core component EED impairs fetal hematopoiesis. Cell Death Dis 8, e2744 (2017).
Brecqueville, M. et al. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J 1, e33 (2011).
Béguelin, W. et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692 (2013).
Okosun, J. et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 46, 176–181 (2014).
Ueda, T. et al. Propagation of trimethylated H3K27 regulated by polycomb protein EED is required for embryogenesis, hematopoietic maintenance, and tumor suppression. Proc. Natl. Acad. Sci. USA 113, 10370–10375 (2016).
Béguelin, W. et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 30, 197–213 (2016).
Broux, M. et al. Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia. Blood 134, 1323–1336 (2019).
Muto, T. et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 210, 2627–2639 (2013).
Wang, C. et al. Ezh2 loss propagates hypermethylation at T cell differentiation-regulating genes to promote leukemic transformation. J. Clin. Invest. 128, 3872–3886 (2018).
Yan, J. et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 121, 4512–4520 (2013).
D’Angelo, V. et al. EZH2 is increased in paediatric T-cell acute lymphoblastic leukemia and is a suitable molecular target in combination treatment approaches. J. Exp. Clin. Cancer Res. 34, 83 (2015).
Zhang, H., Gu, H., Li, L., Ren, Y. & Zhang, L. EZH2 mediates ATO-induced apoptosis in acute myeloid leukemia cell lines through the Wnt signaling pathway. Tumor Biol 37, 5919–5923 (2016).
Papakonstantinou, N. et al. The histone methyltransferase EZH2 as a novel prosurvival factor in clinically aggressive chronic lymphocytic leukemia. Oncotarget 7, 35946–35959 (2016).
Sneeringer, C. J. et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 107, 20980–20985 (2010).
Yap, D. B. et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011).
Souroullas, G. P. et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 22, 632–640 (2016).
Oricchio, E. et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci. Transl. Med 9, eaak9969 (2017).
Jerkovic´, I & Cavalli, G Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 511–528 (2021)
Szabo, Q., Bantignies, F. & Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv 5, eaaw1668 (2019).
Cai, Y. et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 12, 719 (2021).
Donaldson-Collier, M. C. et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 51, 517–528 (2019).
Mortimer, T. et al. Redistribution of EZH2 promotes malignant phenotypes by rewiring developmental programmes. EMBO Rep 20, e48155 (2019).
Ntziachristos, P. et al. Genetic inactivation of the PRC2 complex in T-cell acute lymphoblastic leukemia. Nat. Med. 18, 298–301 (2012).
Andrieu, G. P. et al. PRC2 loss of function confers a targetable vulnerability to BET proteins in T-ALL. Blood 138, 1855–1869 (2021).
Li, Z. et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ 27, 3226–3242 (2020).
Yuan, H. et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell 38, 350–365.e7 (2020).
Zeng, Y. et al. Regulation of EZH2 by SMYD2-mediated lysine methylation is implicated in tumorigenesis. Cell Rep 29, 1482–1498.e4 (2019).
Li, H. et al. SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Mol. Cancer Res. 10, 1462–1472 (2012).
Liu, Y. L. et al. Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J. Cancer Res. Clin. Oncol. 141, 661–669 (2015).
Wu, Y. et al. SUZ12 is a novel putative oncogene promoting tumorigenesis in head and neck squamous cell carcinoma. J. Cell. Mol. Med. 22, 3582–3594 (2018).
Healy, E. et al. PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol. Cell 76, 437–452.e6 (2019).
Youmans, D. T., Gooding, A. R., Dowell, R. D. & Cech, T. R. Competition between PRC2.1 and 2.2 subcomplexes regulates PRC2 chromatin occupancy in human stem cells. Mol. Cell 81, 488–501 (2021).
Wassef, M. et al. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev 29, 2547–2562 (2015).
Erokhin, M. et al. Clinical correlations of Polycomb Repressive Complex 2 in different tumor types. Cancers (Basel) 13, 3155 (2021).
Haupt, Y., Alexander, W. S., Barri, G., Peter Klinken, S. & Adams, J. M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in Eμ-myc transgenic mice. Cell 65, 753–763 (1991).
van Lohuizen, M. et al. Identification of cooperating oncogenes in Eμ-myc transgenic mice by provirus tagging. Cell 65, 737–752 (1991).
Haupt, Y., Bath, M. L., Harris, A. W. & Adams, J. M. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene 8, 3161–3164 (1993).
Gunster, M. J. et al. Identification and characterization of interactions between the vertebrate polycomb-group protein BMI1 and human homologs of polyhomeotic. Mol. Cell. Biol. 17, 2326–2335 (1997).
Jacobs, J. L., Kieboom, K., Marino, S., DePinho, R. A. & Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168 (1999).
Li, W., Li, Y., Tan, Y., Ma, K. & Cui, J. Bmi-1 is critical for the proliferation and invasiveness of gastric carcinoma cells. J. Gastroenterol. Hepatol. 25, 568–575 (2010).
Wang, M. C. et al. Polycomb complex protein BMI-1 promotes invasion and metastasis of pancreatic cancer stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro, and in vivo study. Oncotarget 7, 9586–9599 (2016).
Althobiti, M. et al. The prognostic significance of BMI1 expression in invasive breast cancer is dependent on its molecular subtypes. Breast Cancer Res. Treat 182, 581–589 (2020).
Zhao, Q. et al. Role of BMI1 in epithelial ovarian cancer: Investigated via the CRISPR/Cas9 system and RNA sequencing. J. Ovarian Res. 11, 31 (2018).
Wang, M. C. et al. BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 10, 583–588 (2015).
Tetsu, O. et al. mel-18 negatively regulates cell cycle progression upon B cell antigen receptor stimulation through a cascade leading to c-myc/cdc25. Immunity 9, 439–448 (1998).
Won, H. Y. et al. Loss of Mel-18 enhances breast cancer stem cell activity and tumorigenicity through activating Notch signaling mediated by the Wnt/TCF pathway. FASEB J 26, 5002–5013 (2012).
Lee, J. & Kong, G. MEL-18, a tumor suppressor for aggressive breast cancer. Oncotarget 6, 15710–15711 (2015).
Guo, W. J. et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res 67, 5083–5089 (2007).
Zhang, X. W. et al. BMI1 and Mel-18 oppositely regulate carcinogenesis and progression of gastric cancer. Mol. Cancer 9, 40 (2010).
Guo, W.-J., Datta, S., Band, V. & Dimri, G. P. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol. Biol. Cell 18, 536–546 (2007).
Martinez, A.-M. et al. Polyhomeotic has a tumor suppressor activity mediated by repression of Notch signaling. Nat. Genet. 41, 1076–1082 (2009).
Classen, A. K., Bunker, B. D., Harvey, K. F., Vaccari, T. & Bilder, D. A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat. Genet. 41, 1150–1155 (2009).
van den Boom, V. et al. Non-canonical PRC1.1 targets active genes independent of H3K27me3 and is essential for leukemogenesis. Cell Rep 14, 332–346 (2016).
Chan, H. L. et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 9, 3377 (2018).
Balasubramani, A. et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6, 7307 (2015).
Kutscher, L. M. et al. Functional loss of a noncanonical BCOR-PRC1.1 complex accelerates SHH-driven medulloblastoma formation. Genes Dev 34, 1161–1176 (2020).
Waszak, S. M. et al. Germline elongator mutations in Sonic Hedgehog medulloblastoma. Nature 580, 396–401 (2020).
Mathsyaraja, H. et al. Loss of MGA mediated Polycomb repression promotes tumor progression and invasiveness. Elife 10, e64212 (2021).
Harbour, J. W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 330, 1410–1413 (2010).
Wang, S. S. et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc. Natl. Acad. Sci. USA 111, 16538–16543 (2014).
Lafave, L. M. et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 21, 1344–1349 (2015).
Kuznetsov, J. N. et al. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv 5, eaax1738 (2019).
Campagne, A. et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 10, 348 (2019).
Conway, E. et al. BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol. Cell 81, 1–16 (2021).
Xia, Y. K. et al. Tumor-derived neomorphic mutations in ASXL1 impairs the BAP1-ASXL1-FOXK1/K2 transcription network. Protein Cell 12, 557–577 (2021).
Pasini, D. & Di Croce, L. Emerging roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 36, 50–58 (2016).
Zhou, M., Sun, J., Liu, Y. & Ma, J. Suppressor of Zeste 12 homolog RNA interference inhibits retinoblastoma cell invasion. Oncol. Lett. 8, 1933–1936 (2014).
Ciarapica, R. et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 14, 139 (2014).
Ramakrishnan, S. et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ 26, 2100–2114 (2019).
Liu, M. et al. The polycomb group protein PCGF6 mediates germline gene silencing by recruiting histone-modifying proteins to target gene promoters. J. Biol. Chem. 295, 9712–9724 (2020).
Cohen, I. et al. PRC1 fine-tunes gene repression and activation to safeguard skin development and stem cell specification. Cell Stem Cell 22, 726–739.e7 (2018).
Scelfo, A. et al. Functional landscape of PCGF proteins reveals both RING1A/B-dependent-and RING1A/B-independent-specific activities. Mol. Cell 74, 1037–1052.e7 (2019).
Wang, H. et al. Estrogen receptor α-coupled Bmi1 regulation pathway in breast cancer and its clinical implications. BMC Cancer 14, 122 (2014).
Bhan, A. et al. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-a and diethylstilbestrol. J. Mol. Biol. 426, 3426–3441 (2014).
Zhu, S. et al. BMI1 is directly regulated by androgen receptor to promote castration-resistance in prostate cancer. Oncogene 39, 17–29 (2020).
Bansal, N. et al. BMI-1 targeting interferes with patient-derived tumor-initiating cell survival and tumor growth in prostate cancer. Clin. Cancer Res. 22, 6176–6191 (2016).
Li, Y. et al. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene 36, 6293–6305 (2017).
Zhu, S. et al. BMI1 regulates androgen receptor in prostate cancer independently of the polycomb repressive complex 1. Nat. Commun. 9, 500 (2018).
Xu, K. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science (80-.) 338, 1465–1469 (2012).
Kim, J. et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep 25, 2808–2820.e4 (2018).
Liu, Q. et al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int. J. Cancer 145, 415–426 (2019).
Shin, Y. J. & Kim, J. H. The role of EZH2 in the regulation of the activity of matrix metalloproteinases in prostate cancer cells. PLoS One 7, e30393 (2012).
Wee, Z. N. et al. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep 8, 204–216 (2014).
Shi, B. et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 27, 5105–5119 (2007).
Zhang, Y. et al. Estrogen induces dynamic ERα and RING1B recruitment to control gene and enhancer activities in luminal breast cancer. Sci. Adv 6, eaaz7249 (2020).
Pascual, G., Domínguez, D. & Benitah, S. A. The contributions of cancer cell metabolism to metastasis. DMM Dis. Model. Mech 11, dmm032920 (2018).
Dann, S. G. et al. Reciprocal regulation of amino acid import and epigenetic state through Lat1 and EZH2. EMBO J 34, 1773–1785 (2015).
Liu, T. P., Lo, H. L., Wei, L. S., Hsiao, H. H. Y. & Yang, P. M. S-adenosyl-L-methionine-competitive inhibitors of the histone methyltransferase EZH2 induce autophagy and enhance drug sensitivity in cancer cells. Anticancer. Drugs 26, 139–147 (2015).
Gu, Z. et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov 9, 1228–1247 (2019).
Sivanand, S. & Vander Heiden, M. G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell 37, 147–156 (2020).
Ananieva, E. A. & Wilkinson, A. C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 21, 64–70 (2018).
Tian, T., Li, X. & Zhang, J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 20, 755 (2019).
Pang, B. et al. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 7, 45134–45143 (2016).
Zhang, B. et al. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 78, 195–206 (2021).
Liao, K. et al. A feedback circuitry between polycomb signaling and fructose-1, 6-bisphosphatase enables hepatic and renal tumorigenesis. Cancer Res 80, 675–688 (2020).
McDonald, O. G. et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 49, 367–376 (2017).
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
Brookes, E. et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell 10, 157–170 (2012).
Zhang, T., Gong, Y., Meng, H., Li, C. & Xue, L. Symphony of epigenetic and metabolic regulation - Interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenetics 12, 72 (2020).
Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 4, 11–22 (2004).
Su, W. et al. The Polycomb repressor complex 1 drives double-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell 36, 139–155.e10 (2019).
Duan, Q. et al. High glucose promotes pancreatic cancer cells to escape from immune surveillance via AMPK-Bmi1-GATA2-MICA/B pathway. J. Exp. Clin. Cancer Res. 38, 192 (2019).
Burr, M. L. et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36, 385–401.e8 (2019).
Kim, H. J., Cantor, H. & Cosmopoulos, K. Overcoming immune checkpoint blockade resistance via EZH2 inhibition. Trends Immunol 41, 948–963 (2020).
Wang, D. et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep 23, 3262–3274 (2018).
Goswami, S. et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest. 128, 3813–3818 (2018).
Zingg, D. et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep 20, 854–867 (2017).
Jia, L., Zhang, W. & Wang, C.-Y. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell 27, 238–253.e6 (2020).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 21, 381–395 (2011).
Ren, C. et al. Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chem. Biol. 22, 161–168 (2015).
Nacev, B. A. et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478 (2019).
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodeling genes in paediatric glioblastoma. Nature 482, 226–231 (2012).
Wu, G. et al. Somatic histone H3 alterations in paediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012).
Castel, D. et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130, 815–827 (2015).
Nikbakht, H. et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 7, 11185 (2016).
Lewis, P. W. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013).
Chan, K. M. et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27, 985–990 (2013).
Harutyunyan, A. S. et al. H3K27M in gliomas causes a one-step decrease in H3K27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep 33, 108390 (2020).
Herz, H. et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070 (2014).
Justin, N. et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7, 1–11 (2016).
Fang, D. et al. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife 7, e36696 (2018).
Bender, S. et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013).
Piunti, A. et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017).
Dewaele, B. et al. Identification of a novel, recurrent MBTD1-CXorf67 fusion in low-grade endometrial stromal sarcoma. Int. J. cancer 134, 1112–1122 (2014).
Pajtler, K. W. et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136, 211–226 (2018).
Jain, S. U. et al. H3K27M and EZHIP impede H3K27-methylation spreading by inhibiting allosterically stimulated PRC2. Mol. Cell 80, 726–735.e7 (2020).
Khazaei, S. et al. H3.3 G34W promotes growth and impedes differentiation of osteoblast-like mesenchymal progenitors in giant cell tumor of bone. Cancer Discov 10, 1968–1987 (2020).
Behjati, S. et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet 45, 1479–1482 (2013).
Lu, C. et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016).
Abe, S., Nagatomo, H., Sasaki, H. & Ishiuchi, T. A histone H3.3K36M mutation in mice causes an imbalance of histone modifications and defects in chondrocyte differentiation. Epigenetics 16, 1123–1134 (2020).
Fang, D. et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348 (2016).
Papillon-cavanagh, S. et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 49, 180–185 (2017).
Rueff, J. & Rodrigues, A. S. Cancer drug resistance: A brief overview from a genetic viewpoint. Methods Mol. Biol. 1395, 1–18 (2016).
Bell, C. C. et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 10, 2723 (2019).
Bell, C. C. & Gilan, O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer 122, 465–472 (2020).
Hu, S. et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 10, 788–795 (2010).
Hirukawa, A. et al. Reduction of global H3K27me3 enhances HER2/ErbB2 targeted therapy. Cell Rep 29, 249–257.e8 (2019).
Singh, R. et al. Epigenetic remodeling through downregulation of polycomb repressive complex 2 mediates chemotherapy resistance in testicular germ cell tumors. Cancers (Basel) 11, 796 (2019).
Kikuchi, J. et al. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Invest. 125, 4375–4390 (2015).
Yu, T. et al. Polycomb-like protein 3 induces proliferation and drug resistance in multiple myeloma and is regulated by miRNA-15a. Mol. Cancer Res. 18, 1063–1073 (2020).
Loubiere, V., Papadopoulos, G. L., Szabo, Q., Martinez, A. M. & Cavalli, G. Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci. Adv 6, eaax4001 (2020).
Campbell, P. J. et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Makohon-Moore, A. P. et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 49, 358–366 (2017).
MacK, S. C. et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014).
Feinberg, A. P. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med. 378, 1323–1334 (2018).
Cavalli, G. EZH2 Goes Solo. Science 338, 1430–1431 (2012).
Veneti, Z., Gkouskou, K. K. & Eliopoulos, A. G. Polycomb repressor complex 2 in genomic instability and cancer. Int. J. Mol. Sci. 18, 1–16 (2017).
Wan, J. et al. PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res 43, 3591–3604 (2015).
Li, B. et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood 134, 2046–2058 (2019).
Li, Z. et al. Macrophages-stimulated PRMT1-mediated EZH2 methylation promotes breast cancer metastasis. Biochem. Biophys. Res. Commun. 533, 679–684 (2020).
Lo, P. W. et al. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 115, 7302–7307 (2018).
Chu, C. S. et al. O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. USA 111, 1355–1360 (2014).
Cha, T.-L. et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310, 306–310 (2005).
Kim, E. et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852 (2013).
Yan, J. et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 128, 948–958 (2016).
Jin, X. et al. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J. Biol. Chem. 292, 6269–6280 (2017).
Wan, L. et al. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol. Cell 69, 279–291.e5 (2018).
Wu, S. C. & Zhang, Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J. Biol. Chem. 286, 28511–28519 (2011).
Kaneko, S. et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 24, 2615–2620 (2010).
Wei, Y. et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 13, 87–94 (2011).
Ko, H. W. et al. GSK3β inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget 7, 57131–57144 (2016).
Anwar, T et al. P38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 9, (2018).
Nie, L. et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat. Commun. 10, 1–15 (2019).
Yang, C. C. et al. Phosphorylation of EZH2 at T416 by CDK2 contributes to the malignancy of triple negative breast cancers. Am. J. Transl. Res. 7, 1009–1020 (2015).
Sahasrabuddhe, A. A. et al. Oncogenic Y641 mutations in EZH2 prevent Jak2/β-TrCP-mediated degradation. Oncogene 34, 445–454 (2015).
Yu, Y. L. et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol. Med. 5, 531–547 (2013).
Xue, C. et al. The down-regulation of SUZ12 accelerates the migration and invasion of liver cancer cells via activating ERK1/2 pathway. J. Cancer 10, 1375–1384 (2019).
Suh, J. L. et al. Discovery of selective activators of PRC2 mutant EED-I363M. Sci. Rep. 9, 1–10 (2019).
Yu, H. et al. PRC2/EED-EZH2 complex is up-regulated in breast cancer lymph node metastasis compared to primary tumor and correlates with tumor proliferation in situ. PLoS One 7, 1–8 (2012).
Cao, J., Li, H., Liu, G., Han, S. & Xu, P. Knockdown of JARID2 inhibits the proliferation and invasion of ovarian cancer through the PI3K/Akt signaling pathway. Mol. Med. Rep. 16, 3600–3605 (2017).
Lei, X. et al. JARID2 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition through PTEN/AKT signaling. Oncotarget 7, 40266–40284 (2016).
Walters, Z. S. et al. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 33, 1148–1157 (2014).
Celik, H. et al. JARID2 Functions as a tumor suppressor in myeloid neoplasms by repressing self-renewal in hematopoietic progenitor cells. Cancer Cell 34, 741–756.e8 (2018).
Zhang, Q., Wang, W. & Gao, Q. β-TRCP-mediated AEBP2 ubiquitination and destruction controls cisplatin resistance in ovarian cancer. Biochem. Biophys. Res. Commun. 523, 274–279 (2020).
Liefke, R., Karwacki-Neisius, V. & Shi, Y. EPOP interacts with elongin BC and USP7 to modulate the chromatin landscape. Mol. Cell 64, 659–672 (2016).
Hofvander, J. et al. PHF1 fusions cause distinct gene expression and chromatin accessibility profiles in ossifying fibromyxoid tumors and mesenchymal cells. Mod. Pathol. an Off. J. United States Can. Acad. Pathol. Inc 33, 1331–1340 (2020).
Gebre-Medhin, S. et al. Recurrent rearrangement of the PHF1 gene in ossifying fibromyxoid tumors. Am. J. Pathol. 181, 1069–1077 (2012).
Micci, F., Panagopoulos, I., Bjerkehagen, B. & Heim, S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res 66, 107–112 (2006).
Wang, F. et al. Polycomb-like 2 regulates PRC2 components to affect proliferation in glioma cells. J. Neurooncol. 148, 259–271 (2020).
Liang, Y. et al. PCL2 regulates p53 stability and functions as a tumor suppressor in breast cancer. Sci. Bull. 63, 629–639 (2018).
Jain, P., Ballare, C., Blanco, E. & Vizan, P. & Di Croce, L. PHF19 mediated regulation of proliferation and invasiveness in prostate cancer cells. Elife 9, 1–26 (2020).
Ghislin, S., Deshayes, F., Middendorp, S., Boggetto, N. & Alcaide-Loridan, C. PHF19 and Akt control the switch between proliferative and invasive states in melanoma. Cell Cycle 11, 1634–1645 (2012).
Ren, Z. et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 134, 1176–1189 (2019).
Deng, Q. et al. PHF19 promotes the proliferation, migration, and chemosensitivity of glioblastoma to doxorubicin through modulation of the SIAH1/β–catenin axis. Cell Death Dis 9, 1049 (2018).
Xu, H. et al. MicroRNA-195-5p acts as an anti-oncogene by targeting PHF19 in hepatocellular carcinoma. Oncol. Rep. 34, 175–182 (2015).
Hübner, J. M. et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro. Oncol. 21, 878–889 (2019).
Rai, K. et al. Dual roles of RNF2 in melanoma progression. Cancer Discov 5, 1314–1327 (2015).
Bosch, A. et al. The polycomb group protein RING1B is overexpressed in ductal breast carcinoma and is required to sustain FAK steady state levels in breast cancer epithelial cells. Oncotarget 5, 2065–2076 (2014).
Sánchez-Beato, M. et al. Variability in the expression of polycomb proteins in different normal and tumoral tissues. A pilot study using tissue microarrays. Mod. Pathol. 19, 684–694 (2006).
Gong, Y. et al. NSPc1 is a cell growth regulator that acts as a transcriptional repressor of p21Waf1/Cip1 via the RARE element. Nucleic Acids Res 34, 6158–6169 (2006).
Hu, P.-S. et al. NSPc1 promotes cancer stem cell self-renewal by repressing the synthesis of all-trans retinoic acid via targeting RDH16 in malignant glioma. Oncogene 36, 4706–4718 (2017).
Ji, G. et al. PCGF1 promotes epigenetic activation of stemness markers and colorectal cancer stem cell enrichment. Cell Death Dis 12, 633 (2021).
Lee, J. Y. et al. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene 33, 1325–1335 (2014).
Park, J. H. et al. Loss of Mel-18 induces tumor angiogenesis through enhancing the activity and expression of HIF-1α mediated by the PTEN/PI3K/Akt pathway. Oncogene 30, 4578–4589 (2011).
Hu, Y. et al. PCGF3 promotes the proliferation and migration of non-small cell lung cancer cells via the PI3K/AKT signaling pathway. Exp. Cell Res. 400, 112496 (2021).
Srinivasan, M. et al. Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 8, 38731–38742 (2017).
Takamatsu-Ichihara, E. & Kitabayashi, I. The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int. J. Hematol. 103, 634–642 (2016).
Guo, B. H. et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol. Cancer 10, 10 (2011).
Vonlanthen, S. et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br. J. Cancer 84, 1372–1376 (2001).
Rouhigharabaei, L. et al. BMI1, the polycomb-group gene, is recurrently targeted by genomic rearrangements in progressive B-cell leukemia/lymphoma. Genes, Chromosom. Cancer 52, 928–944 (2013).
Lee, J.-H. et al. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov 2, 16025 (2016).
Yang, Y. F., Pan, Y. H., Tian, Q. H., Wu, D. C. & Su, S. G. CBX1 indicates poor outcomes and exerts oncogenic activity in hepatocellular carcinoma. Transl. Oncol. 11, 1110–1118 (2018).
Lee, Y.-H. et al. HP1β is a biomarker for breast cancer prognosis and PARP inhibitor therapy. PLoS One 10, e0121207 (2015).
Zheng, S. et al. Overexpression of CBX2 in breast cancer promotes tumor progression through the PI3K/AKT signaling pathway. Am. J. Transl. Res. 11, 1668–1682 (2019).
Zeng, M., Li, B., Yang, L. & Guan, Q. CBX2 depletion inhibits the proliferation, invasion and migration of gastric cancer cells by inactivating the YAP/β-catenin pathway. Mol. Med. Rep. 23, 137 (2021).
Wheeler, L. J. et al. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis 7, 92 (2018).
Zhang, H., Chen, W., Fu, X., Su, X. & Yang, A. CBX3 promotes tumor proliferation by regulating G1/S phase via p21 downregulation and associates with poor prognosis in tongue squamous cell carcinoma. Gene 654, 49–56 (2018).
Fan, Y., Li, H., Liang, X. & Xiang, Z. CBX3 promotes colon cancer cell proliferation by CDK6 kinase-independent function during cell cycle. Oncotarget 8, 19934–19946 (2017).
Chang, S.-C. et al. CBX3/heterochromatin protein 1 gamma is significantly upregulated in patients with non-small cell lung cancer. Asia. Pac. J. Clin. Oncol. 14, e283–e288 (2018).
Liang, Y. K., Lin, H. Y., Chen, C. F. & Zeng, D. Prognostic values of distinct CBX family members in breast cancer. Oncotarget 8, 92375–92387 (2017).
Chen, L.-Y. et al. CBX3 promotes proliferation and regulates glycolysis via suppressing FBP1 in pancreatic cancer. Biochem. Biophys. Res. Commun. 500, 691–697 (2018).
Jiao, H. K. et al. Prognostic significance of Cbx4 expression and its beneficial effect for transarterial chemoembolization in hepatocellular carcinoma. Cell Death Dis 6, e1689 (2015).
Li, J. et al. Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 25, 118–131 (2014).
Jiang, N. et al. CBX4 transcriptionally suppresses KLF6 via interaction with HDAC1 to exert oncogenic activities in clear cell renal cell carcinoma. EBioMedicine 53, 102692 (2020).
Wang, X. et al. CBX4 suppresses metastasis via recruitment of HDAC3 to the Runx2 promoter in colorectal carcinoma. Cancer Res 76, 7277–7289 (2016).
Wang, J., He, H., Jiang, Q., Wang, Y. & Jia, S. CBX6 promotes HCC metastasis via transcription factors Snail/Zeb1-mediated EMT mechanism. Onco. Targets. Ther. 13, 12489–12500 (2020).
Zheng, H. et al. CBX6 overexpression contributes to tumor progression and is predictive of a poor prognosis in hepatocellular carcinoma. Oncotarget 8, 18872–18884 (2017).
Deng, H. et al. CBX6 is negatively regulated by EZH2 and plays a potential tumor suppressor role in breast cancer. Sci. Rep. 9, 197 (2019).
Ma, R. G., Zhang, Y., Sun, T. T. & Cheng, B. Epigenetic regulation by polycomb group complexes: Focus on roles of CBX proteins. J. Zhejiang Univ. Sci. B 15, 412–428 (2014).
Scott, C. L. et al. Role of the chromobox protein CBX7 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 104, 5389–5394 (2007).
Bernard, D. et al. CBX7 controls the growth of normal and tumor-derived prostate cells by repressing the Ink4a/Arf locus. Oncogene 24, 5543–5551 (2005).
Pallante, P. et al. Loss of the CBX7 gene expression correlates with a highly malignant phenotype in thyroid cancer. Cancer Res 68, 6770–6778 (2008).
Forzati, F. et al. CBX7 is a tumor suppressor in mice and humans. J. Clin. Invest. 122, 612–623 (2012).
Hannafon, B. N., Sebastiani, P., de las Morenas, A., Lu, J. & Rosenberg, C. L. Expression of microRNA and their gene targets are dysregulated in preinvasive breast cancer. Breast Cancer Res 13, R24 (2011).
Lee, S. H., Um, S. J. & Kim, E. J. CBX8 suppresses Sirtinol-induced premature senescence in human breast cancer cells via cooperation with SIRT1. Cancer Lett 335, 397–403 (2013).
Li, G. et al. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One 8, e80970 (2013).
Zhang, C. Z. et al. CBX8 exhibits oncogenic activity via AKT/β-Catenin activation in hepatocellular carcinoma. Cancer Res 78, 51–63 (2018).
Chung, C.-Y. et al. Cbx8 acts non-canonically with Wdr5 to promote mammary tumorigenesis. Cell Rep 16, 472–486 (2016).
Iwata, S. et al. Polycomb group molecule PHC3 regulates polycomb complex composition and prognosis of osteosarcoma. Cancer Sci 101, 1646–1652 (2010).
Deshpande, A. M. et al. PHC3, a component of the hPRC-H complex, associates with E2F6 during G0 and is lost in osteosarcoma tumors. Oncogene2 26, 1714–1722 (2007).
Tan, K. et al. Tumor suppressor RYBP harbors three nuclear localization signals and its cytoplasm-located mutant exerts more potent anti-cancer activities than corresponding wild type. Cell. Signal. 29, 127–137 (2017).
Zhan, S., Wang, T., Ge, W. & Li, J. Multiple roles of Ring 1 and YY1 binding protein in physiology and disease. J. Cell. Mol. Med. 22, 2046–2054 (2018).
Seitz, V. et al. Classical hodgkin’s lymphoma shows epigenetic features of abortive plasma cell differentiation. Haematologica 96, 863–870 (2011).
Zhou, H. et al. RING1 and YY1 binding protein suppresses breast cancer growth and metastasis. Int. J. Oncol. 49, 2442–2452 (2016).
Tong, A.-H. et al. Overexpression of RYBP inhibits proliferation, invasion, and chemoresistance to cisplatin in anaplastic thyroid cancer cells via the EGFR pathway. J. Biochem. Mol. Toxicol. 33, e22241 (2019).
Zhuang, X.-F. et al. miR-34b inhibits the migration/invasion and promotes apoptosis of non-small-cell lung cancer cells by YAF2. Eur. Rev. Med. Pharmacol. Sci. 23, 2038–2046 (2019).
Zhang, S. et al. YAF2 exerts anti-apoptotic effect in human tumor cells in a FANK1- and phosphorylation-dependent manner. Biochem. Biophys. Res. Commun. 554, 99–106 (2021).
Nagel, S. et al. Deregulation of polycomb repressor complex 1 modifier AUTS2 in T-cell leukemia. Oncotarget 7, 45398–45413 (2016).
Coyaud, E. et al. PAX5-AUTS2 fusion resulting from t(7;9)(q11.2;p13.2) can now be classified as recurrent in B cell acute lymphoblastic leukemia. Leukemia research 34, e323–5 (2010).
Astolfi, A. et al. BCOR involvement in cancer. Epigenomics 11, 835–855 (2019).
Panagopoulos, I. et al. Fusion of the ZC3H7B and BCOR genes in endometrial stromal sarcomas carrying an X;22-translocation. Genes. Chromosomes Cancer 52, 610–618 (2013).
Tanaka, T. et al. Internal deletion of BCOR reveals a tumor suppressor function for BCOR in T lymphocyte malignancies. J. Exp. Med. 214, 2901–2913 (2017).
Isshiki, Y. et al. KDM2B in polycomb repressive complex 1.1 functions as a tumor suppressor in the initiation of T-cell leukemogenesis. Blood Adv 3, 2537–2549 (2019).
Kottakis, F. et al. NDY1/KDM2B functions as a master regulator of Polycomb complexes and controls self-renewal of breast cancer stem cells. Cancer Res 74, 3935–3946 (2014).
He, J., Nguyen, A. T. & Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117, 3869–3880 (2011).
Gounder, M. et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet. Oncol. 21, 1423–1432 (2020).
Morschhauser, F. et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet. Oncol. 21, 1433–1442 (2020).
Italiano, A. et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumors: a first-in-human, open-label, phase 1 study. Lancet. Oncol. 19, 649–659 (2018).
Vaswani, R. G. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, Suitab. J. Med. Chem 59, 9928–9941 (2016).
Acknowledgements
Research in the G.C. laboratory was supported by grants from the European Research Council (Advanced Grant 3DEpi, No. 788972), the EU (CHROMDESIGN, No. 813327), the Fondation pour la Recherche Médicale (DEI20151234396), the MSDAVENIR foundation (Project GENE-IGH), the INSERM and the French National Cancer Institute (INCa PLBIO18-362). A-M.M. is supported by a grant of the Fondation ARC pour la Recherche contre le Cancer (No. 216574). V.P. was supported by the Laboratory of Excellence EpiGenMed.
Author information
Authors and Affiliations
Contributions
All authors have written and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parreno, V., Martinez, AM. & Cavalli, G. Mechanisms of Polycomb group protein function in cancer. Cell Res 32, 231–253 (2022). https://doi.org/10.1038/s41422-021-00606-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-021-00606-6
This article is cited by
-
Cohesin mutations in acute myeloid leukemia
Leukemia (2024)
-
The golden key to open mystery boxes of SMARCA4-deficient undifferentiated thoracic tumor: focusing immunotherapy, tumor microenvironment and epigenetic regulation
Cancer Gene Therapy (2024)
-
Understanding the intersection between placental development and cancer: Lessons from the tumor suppressor BAP1
Communications Biology (2024)
-
Polycomb-mediated silencing of miR-8 is required for maintenance of intestinal stemness in Drosophila melanogaster
Nature Communications (2024)
-
Transient loss of Polycomb components induces an epigenetic cancer fate
Nature (2024)
